
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnexpected high selectivity for acetate formation from CO2 reduction with copper based 2D hybrid catalysts at ultralow potentials†
Rongming
Cai
ab,
Mingzi
Sun
 c,
Jiazheng
Ren
a,
Min
Ju
a,
Xia
Long
*a,
Bolong
Huang
*c and
Shihe
Yang
*ab
c,
Jiazheng
Ren
a,
Min
Ju
a,
Xia
Long
*a,
Bolong
Huang
*c and
Shihe
Yang
*ab
aGuangdong Provincial Key Lab of Nano-Micro Material Research, School of Chemical Biology and Biotechnology, Shenzhen Graduate School, Peking University, Shenzhen 518055, China. E-mail: xialong@pku.edu.cn; chsyang@pku.edu.cn
bInstitute of Biomedical Engineering, Shenzhen Bay Laboratory, Shenzhen 518107, China
cDepartment of Applied Biology and Chemical Technology, The Hong Kong Polytechnic University, Hung Hom, Kowloon, Hong Kong SAR, China. E-mail: bhuang@polyu.edu.hk
First published on 9th November 2021
Abstract
Copper-based catalysts are efficient for CO2 reduction affording commodity chemicals. However, Cu(I) active species are easily reduced to Cu(0) during the CO2RR, leading to a rapid decay of catalytic performance. Herein, we report a hybrid-catalyst that firmly anchors 2D-Cu metallic dots on F-doped CuxO nanoplates (CuxOF), synthesized by electrochemical-transformation under the same conditions as the targeted CO2RR. The as-prepared Cu/CuxOF hybrid showed unusual catalytic activity towards the CO2RR for CH3COO− generation, with a high FE of 27% at extremely low potentials. The combined experimental and theoretical results show that nanoscale hybridization engenders an effective s,p-d coupling in Cu/CuxOF, raising the d-band center of Cu and thus enhancing electroactivity and selectivity for the acetate formation. This work highlights the use of electronic interactions to bias a hybrid catalyst towards a particular pathway, which is critical for tuning the activity and selectivity of copper-based catalysts for the CO2RR.
Introduction
The electrocatalytic reduction of CO2 (CO2RR) with low-cost catalysts holds great promise as a viable CO2 fixation process.1,2 Among the catalysts that have been extensively investigated so far, copper is unique in that it is the only metal with a negative adsorption energy for *CO but a positive adsorption energy for *H, favoring the formation of CO2RR products beyond CO*.3,4 In recent years, various Cu-based electrocatalysts have been developed including metallic Cu and Cu alloys, as well as Cu compounds,5,6 and their catalytic performance has been continuously improved by surface faceting, nanostructuring, doping, etc.7–15 It is now widely accepted that the oxidation states of Cu have significant effects on the products of the CO2RR. For instance, metallic Cu is found to produce CO and HCOOH as the main products at low overpotentials but CH4 or C2H4 at higher overpotentials. For Cu2O, on the other hand, CH3OH is the dominant product,16 and the catalytic activity of Cu2O decreases quickly due to the decomposition of Cu2O to Cu.Several approaches have been reported to stabilize Cu(I) species, such as introduction of the copper nitride (Cu3N) support,17,18 electro-redeposition of catalysts,19,20 and doping.7 Moreover, the electronic and the crystalline structures of Cu compounds with multiple-anions could be greatly modulated by different charges, ionic radii and electronegativities of anions,21 leading to different catalytic performances. In fact, it has been shown theoretically and experimentally that the modulation of surface Cu(I) active sites on copper based catalysts with non-metal elements could improve the selectivity of C2 products by changing the reaction pathways.8,9,11,22 More recently, hybrid catalysts of Cu(0) and Cu(I) synthesized by electrochemical treatment of physically mixed Cu and CuI powder were found to enhance CO2 reduction and C–C coupling to generate alcohols.10,23 However, important information on active sites and effects of anions on catalytic activity and selectivity of hybrid catalysts is still largely missing, hindering the design of efficient CO2RR catalysts by tailoring their surface/interface properties.
Herein, we report the synthesis and the unusual CO2RR activity/selectivity of a novel two-dimensional (2D) copper-based hybrid catalyst featuring distinctive F− anion coordination. First, we systematically investigated the chemical and structural transformation of the 2D Cu(OH)F precursor into the hybrid catalyst under the same conditions as those for the alkaline CO2RR (Fig. 1A). The introduction of F not only afforded the exposed high energy facets of 2D Cu(0) that were well dispersed on 2D CuxOF (named Cu/CuxOF), but also played a critical role in protecting Cu(I) from being fully reduced to Cu(0) under the reductive CO2RR conditions. Secondly, the highly active sites allowed the as-formed Cu/CuxOF to catalyze the CH3COO− formation via the CO2RR at an extremely low potential of −0.3 V (vs. RHE), outperforming most of the electrocatalysts reported to date for this reaction.24–27 In combination with theoretical calculations, we revealed the importance of the electronic interaction in terms of the s,p-d coupling between the Cu species and the hetero-anions in promoting the activity/selectivity towards the acetate formation via the CO2RR.
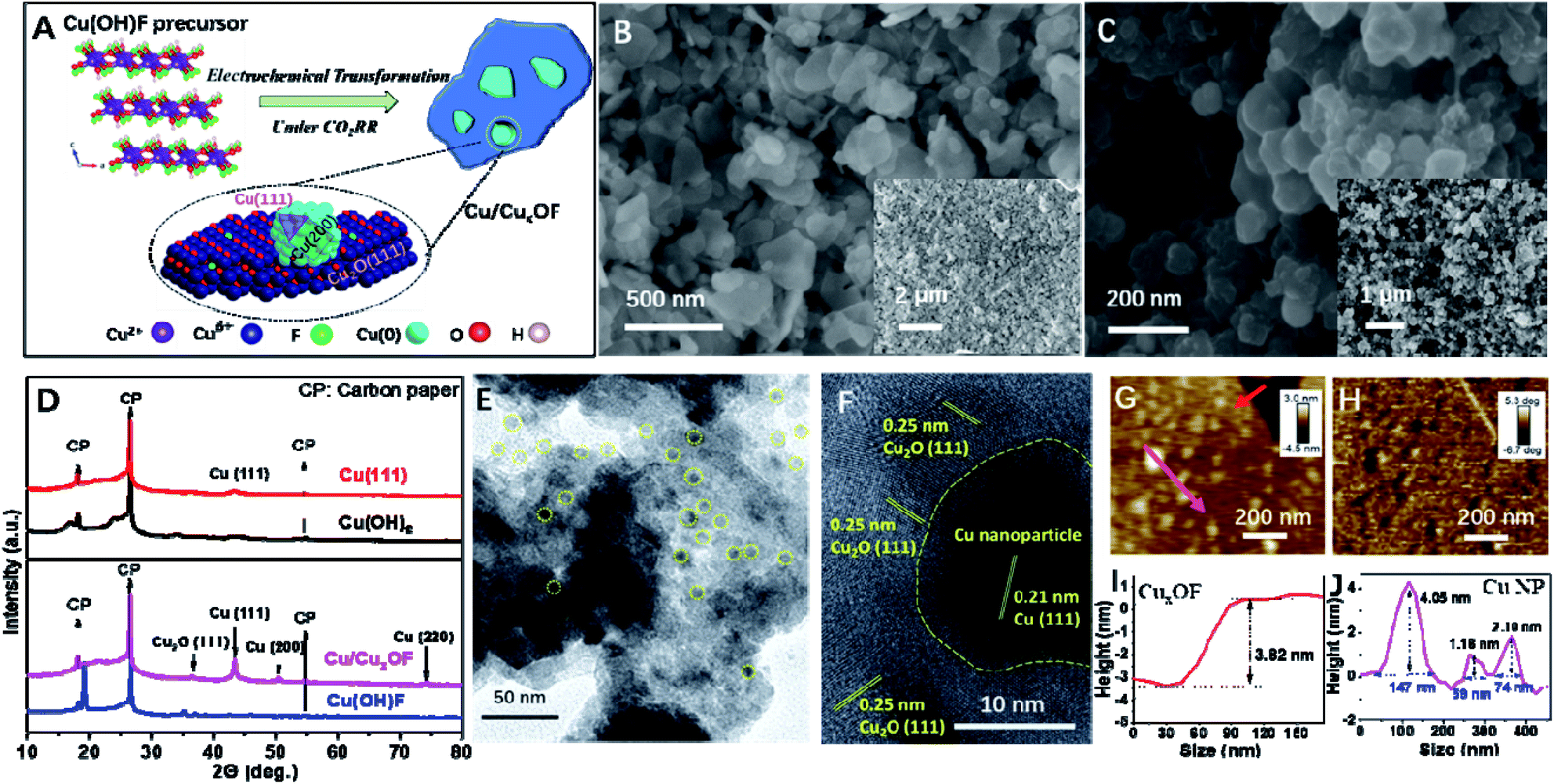 | ||
| Fig. 1 Morphology and structural characterization of the as-prepared copper hybrid catalyst. (A) Schematic atomic structures of Cu(OH)F and the transformed Cu/CuxOF hybrid. (B and C) SEM images of (B) Cu(OH)F precursor and (C) as-formed Cu/CuxOF. (D) XRD patterns of Cu(OH)F and Cu(OH)2, and the corresponding transformed copper catalysts of Cu/CuxOF and Cu-(111). (E) TEM image and (F) high resolution TEM image of Cu/CuxOF. (G–J) AFM height image (G) and the corresponding height profiles of CuxOF nanoplates (I) and metallic Cu nanoparticles (J), as well as the AFM phase image (H). | ||
Results and discussion
The Cu(OH)F precursor was synthesized via a modified hydrothermal method reported previously,28 and turned out to be a well crystallized 2D compound according to the XRD patterns (Fig. S1†) and the SEM (Fig. S2† and 1B), TEM and HRTEM (Fig. S3†) images. More interestingly, the 2D microstructure was well-retained after the formation of Cu/CuxOF via electrochemical transformation, although the surface showed signs of roughening (Fig. 1C). Fig. S4† shows the elemental mapping image from energy dispersive X-ray analysis (EDX), and a uniform distribution of Cu, O and F can be observed. From the X-ray diffraction (XRD) pattern of Cu/CuxOF (Fig. 1D), typical diffraction peaks of both Cu2O and metallic Cu are observed. Apart from the strong diffraction peaks of Cu (111), other smaller peaks corresponding to (200) and (220) were also observed. Importantly, on the larger 2D nanoplates with lateral sizes of several hundreds of nanometers, there lay much smaller ones with sizes of 20–50 nm as found in the transmission electron microscopy (TEM, Fig. 1E, S5 and Table S1†), high resolution TEM (HRTEM, Fig. 1F) and atomic force microscopy (AFM, Fig. 1G–J and S6†) images. Such an overall morphology of Cu/CuxOF is illustrated in Fig. 1A. The lattice fringes of ∼0.25 nm, ∼0.18 nm and 0.21 nm in the HRTEM images (Fig. 1F and S7†) further suggest that the larger nanoplates are made of Cu2O with the lattice fringe of (111), and the smaller 2D nanoparticles are actually metallic Cu with the lattice fringes of (200) and (111), respectively, in accordance with the XRD results. Note that the as-formed CuxOF nanoplates comprise both well crystallized Cu2O grains and amorphous regions, which could be well distinguished from each other in the HRTEM images (Fig. 1F and S8†). This is also consistent with the low peak intensity of Cu2O in the XRD patterns (Fig. 1D).In a control experiment, we also investigated the structural change of Cu(OH)F in 1 M KOH electrolyte, as opposed to the abovementioned electrochemical transformation. From the XRD patterns shown in Fig. S9,† one can see that after the KOH treatment, Cu(OH)F was transformed into Cu(OH)2 with a typical nanowire microstructure (Fig. S10 and S11†). This is supported by the greatly reduced content of F (∼0.24%, see Fig. S12†). Of note, the much higher F content in Cu/CuxOF is probably due to the fast transformation process as confirmed by in situ electrochemical quartz crystal microbalance with dissipation (EQCM-D) (Fig. S13†). These results indicate that the direct electrochemical treatment was the main driving force for the successful formation of Cu/CuxOF (with the F content as high as ∼5.92%) from the Cu(OH)F precursor (Fig. S14†). Further, Cu(OH)2 without any F (Fig. S15†) and KOH-treated Cu(OH)F (with ∼0.24% F) were subsequently treated under the CO2RR conditions. As can be seen from the XRD patterns (Fig. 1D and S16†), HRTEM (Fig. S17 and S18†) and AFM (Fig. S19†) images, only 2D metallic Cu(0) with the low-energy facet of (111) could be obtained (named Cu-(111)) from the Cu(OH)2 precursor, while Cu/CuxO could be formed by using the KOH-treated Cu(OH)F as the precursor, further confirming the critical role of F in stabilizing the oxidized Cu species.
Then X-ray photoelectron spectroscopy (XPS), Auger electron spectroscopy (AES), and electron paramagnetic resonance (EPR) spectroscopy were combined to get full information on the electronic structure of the metal and anions in the as-prepared copper catalysts. First, the XPS peak of F in Cu/CuxOF was much sharper than that of the Cu(OH)F precursor (Fig. 2A), suggesting a different electronic environment of F in Cu/CuxOF arising from the decreased content of F and the absence of H+ ions. The binding energy of O 1s in Cu/CuxOF was slightly larger than that in Cu(OH)2 while smaller than that in Cu(OH)F (Fig. 2B), signifying the electron withdrawing and polarization effects of F on O 1s, and expectedly, on the directly bonded metal ions, i.e., Cuδ+.29 Moreover, negligible peaks could be found for the O spin-orbital of Cu-(111) (Fig. S20†), indicating the existence of only metallic Cu without any oxidized copper species, in accordance with the results of XRD (Fig. 1D), HRTEM (Fig. S17†) and XPS of Cu-(111) in the Cu 2p spin-orbital (Fig. 2C). As expected, for the Cu(OH)2 precursor, Cu2+ located at 935.5 eV was clearly observed. Notably, a large positive shift of Cu 2p was observed for Cu(OH)F (Fig. S21†) due to the strong polarization and electron withdrawing effects of F as mentioned above. For the as-prepared Cu/CuxOF, however, we found that besides Cu(0), there also existed moieties between Cu(0) and Cu(I), hereafter denoted as Cuδ+ (to distinguish them from Cu and Cu(I) in Cu/Cu2O), as shown in Fig. 2C, D and S22.† The LMM Auger spectra of copper shown in Fig. 2E further confirm the existence of metallic Cu(0) and Cuδ+ in the as-prepared Cu/CuxOF. These results suggest that in addition to the electronic interactions between O and F, the two anions together also impact the Cu species, and thus would influence the catalytic performance of the hybrid catalyst.
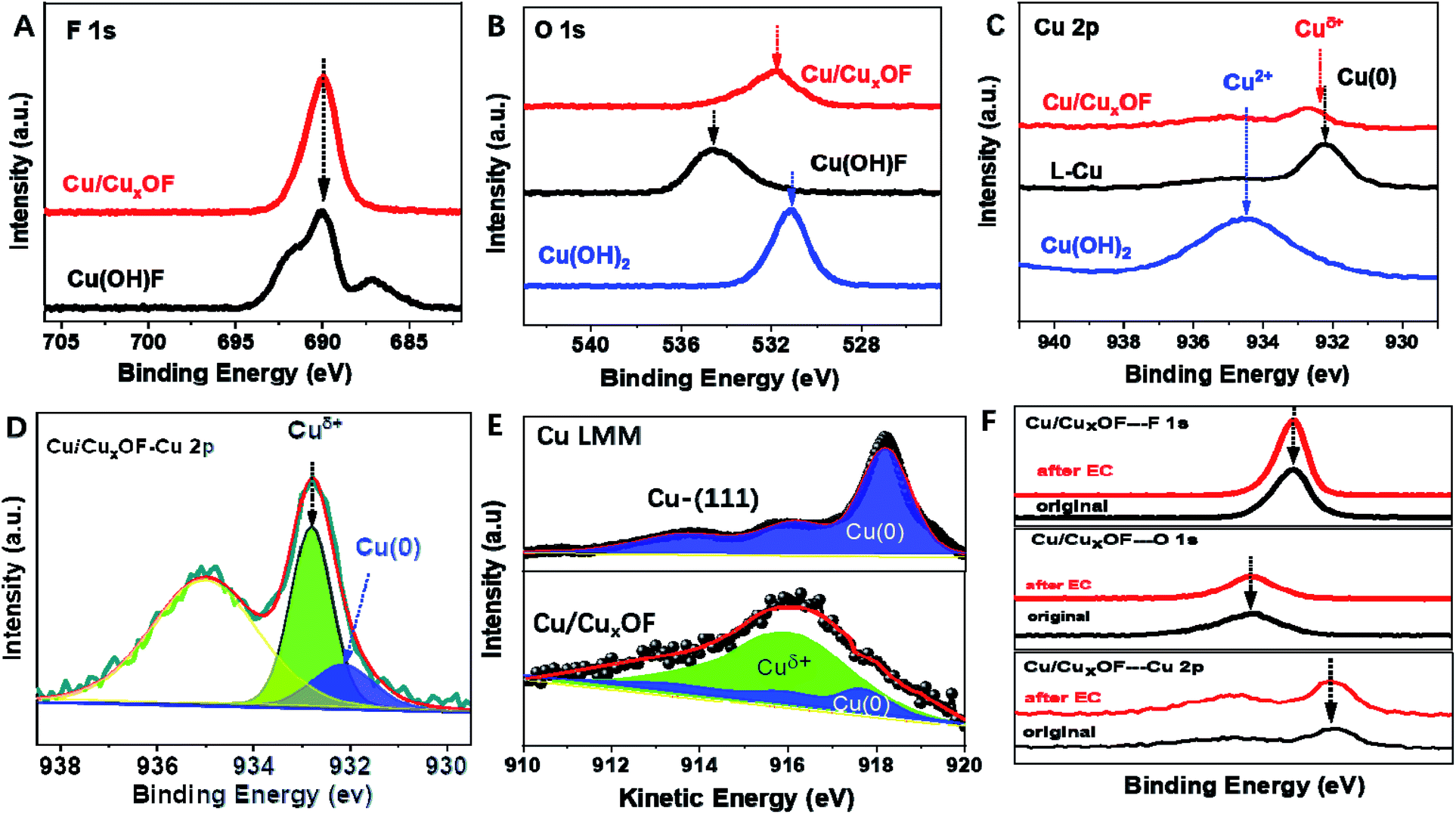 | ||
| Fig. 2 XPS analysis of the copper catalysts and precursors. (A) F 1s of Cu/CuxOF and the Cu(OH)F precursor, (B) O 1s of Cu/CuxOF, the Cu(OH)F precursor and Cu(OH)2 precursor, (C) Cu 2p of Cu/CuxOF, Cu-(111) and the Cu(OH)2 precursor, (D) de-convoluted XPS spectrum of Cu 2p for Cu/CuxOF, (E) Cu LMM Auger spectra of Cu/CuxOF and Cu-(111), and (F) comparison of the XPS spectra of Cu/CuxOF at F 1s, O 1s, and Cu 2p spin-orbitals before and after the electrochemical tests at the stepped potentials from −0.3 V to −0.7 V vs. RHE for more than 28 h. | ||
Further, EPR spectra were collected and analyzed in order to get detailed information on the unpaired electrons in the material. The g-factor is a dimensionless measure of the intrinsic magnetic moments of the electron, which is 2.0023 for a free electron but can vary for unpaired electrons in different molecules and materials. Here Cu(I) has a d10 electronic configuration with no unpaired electrons and thus is EPR silent. However, EPR is a useful tool for both structural and mechanistic studies of Cu(II) and Cu(0) on account of their d9 and d10s1 configurations, respectively. As can be seen from Fig. S23,† Cu-(111) and Cu/CuxOF show a g value close to ∼2.63, indicating the presence of unpaired electrons in both the materials of similar origin, namely the metallic Cu species.
We examined the stability of the catalysts by checking any possible structural changes. As shown in Fig. 2F, all the XPS peaks for F, O, and Cu spin-orbitals of Cu/CuxOF showed a negligible change after the electrochemical CO2RR evaluation at the stepped potentials from −0.3 V to −0.7 V vs. RHE for more than 28 h. Moreover, based on the XRD (Fig. S24†), TEM (Fig. S25†), HRTEM (Fig. S26†) and elemental analysis (Fig. S27 & S28†) results, we found that both the metallic Cu and 2D CuxOF with ∼5.83% of F (close to the initial value of ∼5.92%) were retained after the ∼28 h stability tests, indicating the good working stability of the as-prepared Cu/CuxOF for the CO2RR. In addition, the in situ EQCM-D further suggests that the surface of Cu/CuxOF is actually in a dynamic equilibrium as characterized by periodic adsorption–desorption processes during the CV cycles (Fig. S29†).30,31
Then we proceeded to the electrochemical performance of the as-prepared copper catalysts towards the CO2RR, which was evaluated in a flow cell with 1 M KOH as the electrolyte (see the ESI† for details). From the J–V curves (Fig. 3A), it is clear that Cu/CuxOF showed larger current densities than Cu-(111) and Cu/CuxO at the same applied potentials. Moreover, the partial current density (Fig. S30†) and faradaic efficiency (FE, Fig. 3B) of CH3COO− on Cu/CuxOF were found to be ∼4.0 mA cm−2 and 27% at a low potential of −0.3 V (vs. RHE), which differs from those of the main C2 products of C2H5OH and C2H4 on Cu-(111) (Fig. S31†) and Cu/CuxO (Fig. S32†), and also competes with the best noble-metal free electrocatalysts for the CO2RR with the selective product of CH3COO− that usually required more negative potentials (Table S2†).16,24–27,32 We noted that Cu/CuxOF and Cu/CuxO have a similar microstructure including both nanoparticle size and hierarchical structure (Fig. S33–S35†), thus the distinctive catalytic selectivity of Cu/CuxOF for the CO2RR (Fig. 3E–I) probably results from the F-doping induced modification of surface physicochemical properties of Cu/CuxOF, which would favor the acetate formation. Moreover, the current density of the CO2RR reached ∼56 mA cm−2 at E = −0.5 V (vs. RHE) (Fig. 3A), larger than the reported values collected on copper based catalysts under similar reaction conditions.13,33,34 In addition, compared with Cu-(111) and Cu/CuxO, Cu/CuxOF showed much higher FE for the CO2RR (Fig. 3C) and lower FE for the HER (Fig. 3D) in the whole potential range from −0.3 to −0.7 V vs. RHE, demonstrating much better CO2RR activity of Cu/CuxOF than Cu-(111) and Cu/CuxO catalysts. The long-term stability at stepped potentials was also estimated. From Fig. S36,† it is clear that the change of the CO2RR current density at each stage is less than 5% even at a deep potential down to −0.7 V vs. RHE. What's more, the FE of acetate during the stability tests was essentially unchanged at each potential, evidencing the good catalytic stability of the as-prepared Cu/CuxOF for the CO2RR. In conjunction with the structural stability discussed earlier in the previous paragraph, we can conclude that the as-prepared Cu/CuxOF is indeed an advanced electrocatalyst for the CO2RR with excellent catalytic selectivity and stability toward acetate generation.
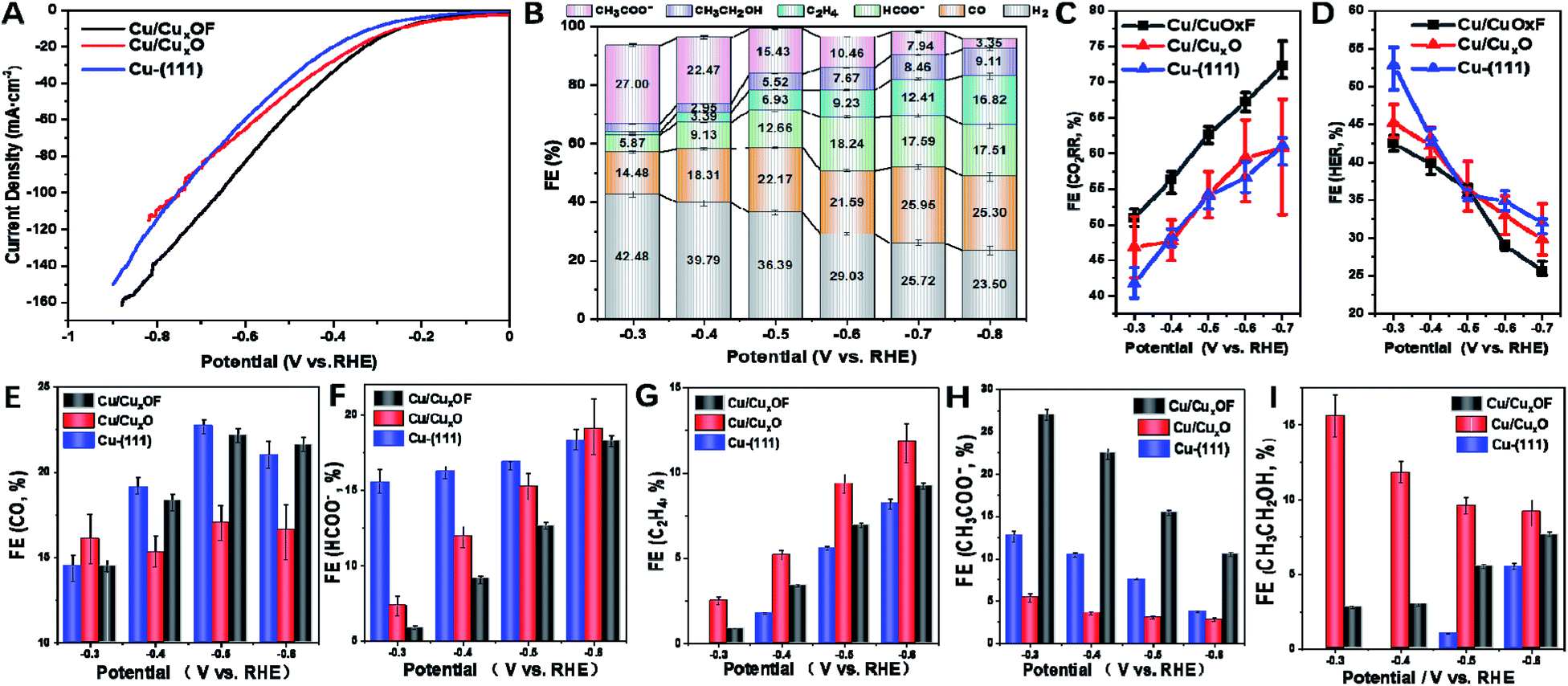 | ||
| Fig. 3 Electrocatalytic performance towards the CO2RR in 1 M KOH. (A) J–V curves collected on Cu-(111), Cu/CuxO and Cu/CuxOF. (B) FE of all the products on Cu/CuxOF at various applied potentials. (C and D) FE for (C) the CO2RR and (D) HER on Cu-(111), Cu/CuxO and Cu/CuxOF. (E–I) comparison of FE on Cu-(111), Cu/CuxO and Cu/CuxOF for all the CO2RR products of (E) CO, (F) HCOO−, (G) C2H4, (H) CH3COO− and (I) CH3CH2OH. | ||
As is widely known, the CO2RR involves multiple electron-transfer and chemical reaction steps, and this complex process strongly depends on the physiochemical and surface properties of the catalysts. Previous theoretical and experimental studies have demonstrated that the products of the CO2RR at low overpotentials were generally limited to C1 species on metallic Cu nanocatalysts, especially on their low-energy facets, such as CO and HCOOH.35,36 This was in accordance with the results observed for Cu-(111) prepared in this work. At high potentials, the FE of C2 increased along with the suppression of the HER, which again agrees with what we observed for Cu-(111) (Fig. 3G, I and S37†).37,38 However, markedly different catalytic selectivities were observed for Cu/CuxOF. Specifically, the FE of C2 products was maintained at ∼30% in almost the whole potential range we studied, while the FE of C1 products increased (Fig. 3G and F) in parallel with the suppression of the HER when the applied potential increased (Fig. 3E–I), amounting to ∼72% FE at −0.7 V vs. RHE (Fig. 3B and C). So what is the explanation for this striking catalytic performance? Plausibly, in Cu/CuxOF with F-coordination, multi-copper oxidation states and the hierarchical 2D–2D microstructure, could provide multiple sites with different adsorption–desorption characteristics for the reactants and intermediates of the CO2RR (Scheme S1†), promoting C–C coupling9,39–41 to form the CH3COO− product even at very low overpotentials. More detailed discussions on the reaction mechanism along with theoretical calculations will be provided below.
To better understand the CO2RR process on the copper-based hybrid catalysts, we carried out the DFT calculations of their electronic structures and energy trends in the reaction process of interest here, and then explored the effects of the electronic structure and energy on electroactivity of the catalysts (Fig. S38†). Clearly, the Cu-(111) surface shows a highly ordered electronic distribution guided by the Cu sites (Fig. 4A). Such a surface electronic structure usually leads to low product selectivity due to the strong competition between different reaction pathways. By contrast, the Cu/CuxOF surface shows a strong perturbation of the electronic distribution near the Fermi level (EF) (Fig. 4B) due to hybridization with the CuxOF nanoplate. We believe that the resulting strong bonding orbitals of the metallic Cu nanoparticle surfaces played an important role in forming the highly electroactive region for the CO2RR. To illustrate this point more clearly on Cu/CuxOF, Fig. 4C displays the projected partial density of states (PDOS). Notably, the Cu-3d orbitals in Cu/CuxOF are now located at a position much closer to the EF than those in Cu-(111), meaning a higher d-band center and thus an improved electroactivity. Meanwhile, both the O-s,p, and the F-s,p orbitals are located deeper in energy, which acted as the electron reservoir. Compared to Cu/CuxOF, Cu/CuxO shows a slightly lower d-band center, which leads to decreased electroactivity and FE of the C2 products (Fig. S39†).
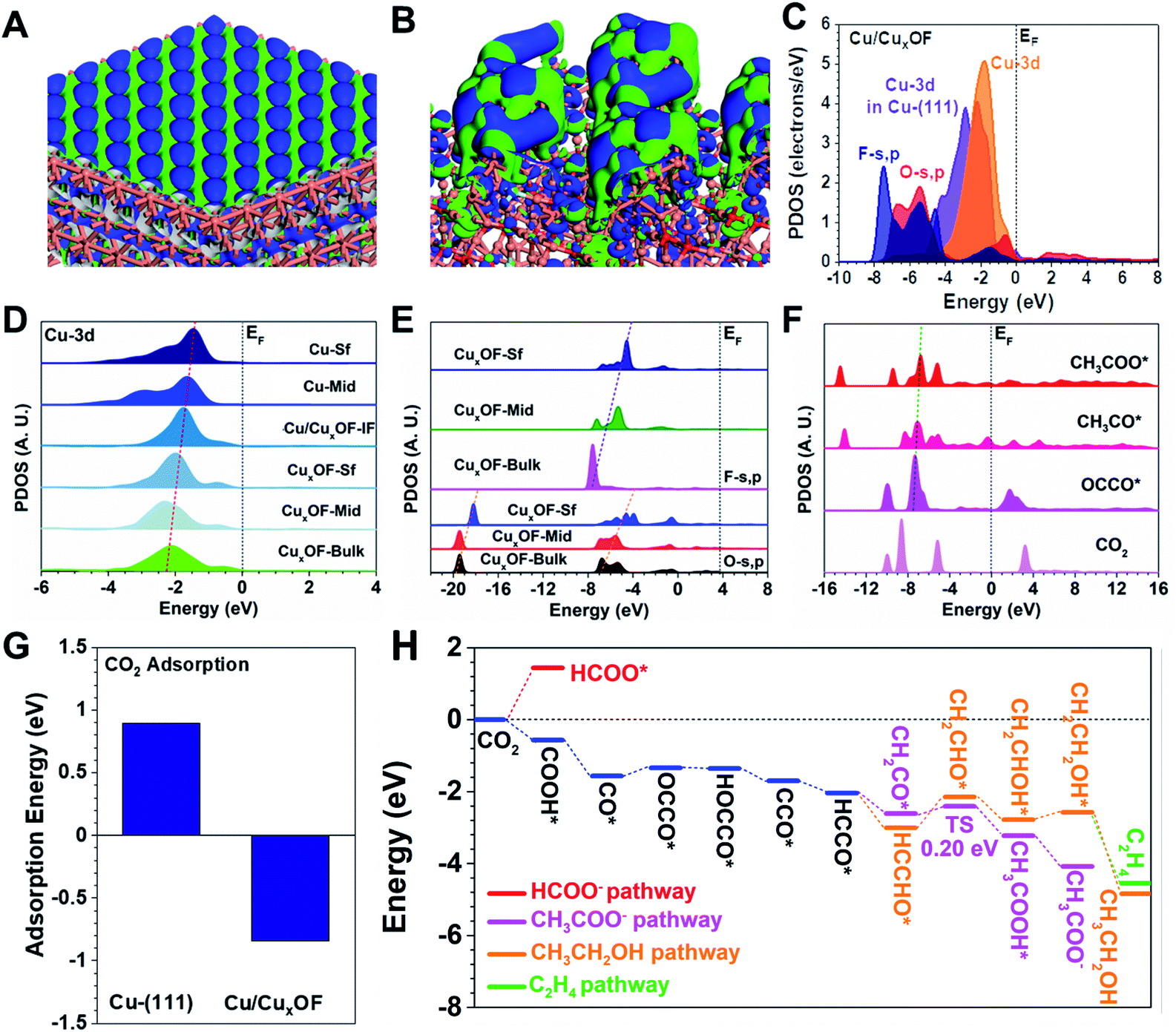 | ||
| Fig. 4 The 3D contour plot for electronic distributions of (A) Cu-(111) and (B) Cu/CuxOF. (C) PDOS of Cu/CuxOF. Site-dependent PDOS of (D) Cu-3d, and (E) O-s,p and F-s,p in Cu/CuxOF. (F) The PDOS of key adsorbates during the CO2RR process. (G) The comparison of CO2 adsorption on Cu-(111) and Cu/CuxOF. (H) The CO2RR reaction pathways on Cu/CuxOF. The orange, red, and cyan balls in (A) and (B) represent Cu, O and F atoms, respectively, and the blue and green isosurfaces indicate the bonding and anti-bonding orbitals, respectively. The dashed lines in (D and E) represent the higher position of the band centers for Cu and O-s,p, and F-s,p, respectively. | ||
We next unravel the site-dependent PDOS of the different elements in the catalysts. Focusing now on the Cu sites, we notice that from the bulk CuxOF nanoplate to the surface of metallic Cu with high-energy facets, Cu-3d orbitals exhibited a nearly linear upshifting trend toward the EF (Fig. 4D). Consequently, the surface Cu nanoparticles with high electroactivity were found to be the active sites for the CO2RR process. Then, the electronic structures of both O and F sites in the CuxOF nanoplates are investigated (Fig. 4E). Notably, from the bulk to the nanoplate surface, the s,p orbitals exhibit a gradual upshifting trend, which supports the improved electron transfer capability from the CuxOF surface to the Cu nanoparticle surface, and ultimately to the intermediates during the CO2RR. To follow up the intermediate conversion process, the PDOS of key adsorbates during the CO2RR process are displayed in Fig. 4F. The structural configurations of these key adsorbates are displayed in Fig. S40.† Compared to the free CO2, the s,p orbitals of the intermediates have shown a slight upshift, confirming the successful reduction of CO2. In the reduction of OCCO* to CH3COO*, we notice a nearly linear correlation of the s,p orbital energies, a sign that would guarantee efficient electron transfer and the intermediate conversions. The linear correlation of the σ orbitals with an upshifting trend is well preserved, which not only indicates the most efficient electron transfer but also the optimal adsorption strength of intermediates for the CO2RR.
Finally, the adsorption of CO2 on the catalysts was also investigated. Compared to the Cu-(111) surface, the adsorption on Cu/CuxOF became much more energetically favorable (Fig. 4G). While the CO2 adsorption on the Cu-(111) surface showed an energy cost as high as 0.89 eV, Cu/CuxOF demonstrates a highly exothermic adsorption energy of −0.84 eV, accounting for the high electroactivity towards the CO2RR. The energy evolution of the CO2RR process is shown in Fig. 4H. For the initial hydrogenation, the strong preference of O–H over C–H shows a low selectivity towards the formation of HCOO− on Cu/CuxOF. The key reaction step in formation of C2 products usually relies on the coupling of the CO* intermediates to form OCCO*, which shows only a minor energy barrier of 0.22 eV. The following reduction steps are all energetically favorable until the formation of HCCO*. In further hydrogenation, the formation of both CH2CO* and HCCHO* becomes exothermic. The key step in acetic acid formation is the incorporation of water in CH2CO*, which is energetically favorable with a small activation barrier of 0.20 eV. This low barrier guarantees the efficient conversion towards CH3COOH*. Remarkably, the strong reaction trend with an energy release of −4.08 eV makes the formation of CH3COO− the most preferred of all the reaction pathways at a very low potential, in good agreement with the foregoing experimental findings. Proceeding with the CH3CH2OH and C2H4 reaction pathways, the hydrogenation steps from HCCHO* to CH2CHO* and from CH2CHOH* to CH2CH2OH* have an energy barrier of 0.87 and 0.22 eV, respectively. These substantial energy barriers, especially the first one, largely limit the formation of both CH3CH2OH and C2H4 at low potentials. However, by increasing the applied potential, the contents of CH3CH2OH and C2H4 will gradually increase, as we observed experimentally. Therefore, both electronic structures and reaction trends have confirmed the high electroactivity and selectivity of Cu/CuxOF towards the generation of C2 products during the CO2RR.
Conclusions
In summary, by exploiting 2D Cu(OH)F nanoplates as a precursor, we successfully synthesized a copper based hybrid catalyst, by coupling 2D metallic Cu nanoparticles with high-energy facets and F-doped CuxO (CuxOF) nanoplates. For the catalyst synthesis with in situ electrochemical transformation, we purposely used the same conditions as those for the alkaline CO2RR in order to ensure a durable catalytic operation. The critical roles of F-modification have been revealed to effectively tailor the exposed facets of metallic Cu nanoparticles, stabilize the oxidized copper active species under the CO2RR conditions, and more importantly, to purposely induce the s,p-d coupling between the metal and hetero-anions tending heavily towards the acetate pathway. Consequently, the as-prepared 2D Cu/CuxOF hybrid catalyst creates an electronic environment leading to high electroactivity, particularly the unexpected CH3COO− selectivity at an extremely low overpotential. This work unravels interesting electronic interactions between Cu species and different anions in copper-based hybrid materials, and provides an efficient strategy to construct more efficient catalysts for the CO2RR.Data availability
All experimental and computational data are available within the article or in the ESI file.†Author contributions
X. L. and S. Y. conceived the idea. R. C. synthesized the catalysts. R. C. and J. R. carried out structural and electrochemical characterization. R. C. and X. L. performed EQCM-D experiments. R. C., M. J. and X. L. performed AFM characterization experiments. R. C., M. J., and J. R. performed morphology characterization. X. L. and R. C. prepared the figures and analyzed the data. M. S. and B. H. performed the DFT calculations. X. L., R. C. B. H. and S. Y. wrote the manuscript. All the authors discussed the results and commented on the manuscript at all stages.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors appreciate the financial support from the National Natural Science Foundation of China (21703003, and 21972006), Shenzhen Peacock Plan (KQTD2016053015544057), Shenzhen Science and Technology Innovation Commission (JCYJ20180302153417057, and JCYJ20190808155413194), and Nanshan Pilot Plan (LHTD20170001).Notes and references
- R. Cai, M. Ju, J. Chen, J. Ren, J. Yu, X. Long and S. Yang, Mater. Chem. Front., 2021, 5, 3576–3592 RSC
.
- Y. Liu, S. Chen, X. Quan and H. Yu, J. Am. Chem. Soc., 2015, 137, 11631–11636 CrossRef CAS PubMed
- A. Bagger, W. Ju, A. S. Varela, P. Strasser and J. Rossmeisl, ChemPhysChem, 2017, 18, 3266–3273 CrossRef CAS PubMed
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Norskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed
- M. B. Gawande, A. Goswami, F. X. Felpin, T. Asefa, X. Huang, R. Silva, X. Zou, R. Zboril and R. S. Varma, Chem. Rev., 2016, 116, 3722–3811 CrossRef CAS PubMed
- D. Raciti and C. Wang, ACS Energy Lett., 2018, 3, 1545–1556 CrossRef CAS
- Y. Zhou, F. Che, M. Liu, C. Zou, Z. Liang, P. De Luna, H. Yuan, J. Li, Z. Wang, H. Xie, H. Li, P. Chen, E. Bladt, R. Quintero-Bermudez, T. K. Sham, S. Bals, J. Hofkens, D. Sinton, G. Chen and E. H. Sargent, Nat. Chem., 2018, 10, 974–980 CrossRef CAS PubMed
- M. B. Ross, P. De Luna, Y. F. Li, C. T. Dinh, D. Kim, P. Yang and E. H. Sargent, Nat. Catal., 2019, 2, 648–658 CrossRef CAS
- H. Mistry, A. S. Varela, C. S. Bonifacio, I. Zegkinoglou, I. Sinev, Y. W. Choi, K. Kisslinger, E. A. Stach, J. C. Yang, P. Strasser and B. R. Cuenya, Nat. Commun., 2016, 7, 12123 CrossRef PubMed
- H. Xiao, W. A. Goddard III, T. Cheng and Y. Liu, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 6685–6688 CrossRef CAS PubMed
- F. L. Ni, H. Yang, Y. Z. Wen, H. P. Bai, L. S. Zhang, C. Y. Cui, S. Y. Li, S. S. He, T. Cheng, B. Zhang and H. S. Peng, Sci. China Mater., 2020, 63, 2606–2612 CrossRef
- K. Sun, T. Cheng, L. Wu, Y. Hu, J. Zhou, A. Maclennan, Z. Jiang, Y. Gao, W. A. Goddard III and Z. Wang, J. Am. Chem. Soc., 2017, 139, 15608–15611 CrossRef CAS PubMed
- J. J. Lv, M. Jouny, W. Luc, W. Zhu, J. J. Zhu and F. Jiao, Adv. Mater., 2018, 30, e1803111 CrossRef PubMed
- E. Landaeta, R. A. Masitas, T. B. Clarke, S. Rafacz, D. A. Nelson, M. Isaacs and Z. D. Schultz, ACS Appl. Nano Mater., 2020, 3, 3478–3486 CrossRef CAS
- X. Yang, E. A. Fugate, Y. Mueanngern and L. R. Baker, ACS Catal., 2017, 7, 177–180 CrossRef CAS
- M. Le, M. Ren, Z. Zhang, P. T. Sprunger, R. L. Kurtz and J. C. Flake, J. Electrochem. Soc., 2011, 158, E45–E49 CrossRef CAS
- Z. Y. Yin, C. Yu, Z. L. Zhao, X. F. Guo, M. Q. Shen, N. Li, M. Muzzio, J. R. Li, H. Liu, H. H. Lin, J. Yin, G. Lu, D. Su and S. H. Sun, Nano Lett., 2019, 19, 8658–8663 CrossRef CAS PubMed
- C. Giordano and M. Antonietti, Nano Today, 2011, 6, 366–380 CrossRef CAS
- D. Wakerley, S. Lamaison, F. Ozanam, N. Menguy, D. Mercier, P. Marcus, M. Fontecave and V. Mougel, Nat. Mater., 2019, 18, 1222–1227 CrossRef CAS PubMed
- W. C. Ma, S. J. Xie, T. T. Liu, Q. Y. Fan, J. Y. Ye, F. F. Sun, Z. Jiang, Q. H. Zhang, J. Cheng and Y. Wang, Nat. Catal., 2020, 3, 478–487 CrossRef CAS
- H. Kageyama, K. Hayashi, K. Maeda, J. P. Attfield, Z. Hiroi, J. M. Rondinelli and K. R. Poeppelmeier, Nat. Commun., 2018, 9, 772 CrossRef PubMed
- S. Popovic, M. Smiljanic, P. Jovanovic, J. Vavra, R. Buonsanti and N. Hodnik, Angew. Chem., Int. Ed., 2020, 59, 14736–14746 CrossRef CAS PubMed
- H. Li, T. Liu, P. Wei, L. Lin, D. Gao, G. Wang and X. Bao, Angew. Chem., Int. Ed., 2021, 60, 14329–14333 CrossRef CAS PubMed
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Energy Environ. Sci., 2012, 5, 7050–7059 RSC
- C. Genovese, M. E. Schuster, E. K. Gibson, D. Gianolio, V. Posligua, R. Grau-Crespo, G. Cibin, P. P. Wells, D. Garai, V. Solokha, S. K. Calderon, J. J. Velasco-Velez, C. Ampelli, S. Perathoner, G. Held, G. Centi and R. Arrigo, Nat. Commun., 2018, 9, 935 CrossRef PubMed
- Y. M. Liu, S. Chen, X. Quan and H. T. Yu, J. Am. Chem. Soc., 2015, 137, 11631–11636 CrossRef CAS PubMed
- Y. Wang, D. G. Wang, C. J. Dares, S. L. Marquard, M. V. Sheridan and T. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 278–283 CrossRef CAS PubMed
- H. Tian, Y. Wang, J. Zhang, Y. Ma, H. Cui, Q. Cui and Y. Ma, J. Phys. Chem. C, 2019, 123, 25492–25500 CrossRef CAS
- Y. Kawamoto, K. Ogura, M. Shojiya, M. Takahashi and K. Kadono, J. Fluorine Chem., 1999, 96, 135–139 CrossRef CAS
- N. Shpigel, M. D. Levi, S. Sigalov, O. Girshevitz, D. Aurbach, L. Daikhin, P. Pikma, M. Marandi, A. Jänes, E. Lust, N. Jäckel and V. Presser, Nat. Mater., 2016, 15, 570–575 CrossRef CAS PubMed
- M. Ju, R. Cai, J. Ren, J. Chen, L. Qi, X. Long and S. Yang, ACS Appl. Mater. Interfaces, 2021, 13, 37063–37070 CrossRef CAS PubMed
- R. N. De, S. Gonglach, S. Paul, M. Haas, S. S. Sreejith, P. Gerschel, U. P. Apfel, T. H. Vuong, J. Rabeah, S. Roy and W. Schofberger, Angew. Chem., Int. Ed., 2020, 59, 10527–10534 CrossRef CAS PubMed
- C. T. Dinh, T. Burdyny, M. G. Kibria, A. Seifitokaldani, C. M. Gabardo, F. P. Garcia de Arquer, A. Kiani, J. P. Edwards, P. De Luna, O. S. Bushuyev, C. Zou, R. Quintero-Bermudez, Y. Pang, D. Sinton and E. H. Sargent, Science, 2018, 360, 783–787 CrossRef CAS PubMed
- T.-T. Zhuang, Y. Pang, Z.-Q. Liang, Z. Wang, Y. Li, C.-S. Tan, J. Li, C. T. Dinh, P. De Luna, P.-L. Hsieh, T. Burdyny, H.-H. Li, M. Liu, Y. Wang, F. Li, A. Proppe, A. Johnston, D.-H. Nam, Z.-Y. Wu, Y.-R. Zheng, A. H. Ip, H. Tan, L.-J. Chen, S.-H. Yu, S. O. Kelley, D. Sinton and E. H. Sargent, Nat. Catal., 2018, 1, 946–951 CrossRef CAS
- F. Calle-Vallejo and M. T. Koper, Angew. Chem., Int. Ed., 2013, 52, 7282–7285 CrossRef CAS PubMed
- C. W. Li, J. Ciston and M. W. Kanan, Nature, 2014, 508, 504–507 CrossRef CAS PubMed
- Y. Yoon, A. S. Hall and Y. Surendranath, Angew. Chem., Int. Ed., 2016, 55, 15282–15286 CrossRef CAS PubMed
- A. S. Hall, Y. Yoon, A. Wuttig and Y. Surendranath, J. Am. Chem. Soc., 2015, 137, 14834–14837 CrossRef CAS PubMed
- S. Ma, M. Sadakiyo, R. Luo, M. Heima, M. Yamauchi and P. J. A. Kenis, J. Power Sources, 2015, 301, 219–228 CrossRef
- A. A. Peterson and J. K. Nørskov, J. Phys. Chem. Lett., 2012, 3, 251–258 CrossRef CAS
- K. J. P. Schouten, Y. Kwon, C. J. M. v. d. Ham, Z. Qin and M. T. M. Koper, Chem. Sci., 2011, 2, 1902–1909 RSC
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, characterization and performance data of this work. See DOI: 10.1039/d1sc05441d |
| This journal is © The Royal Society of Chemistry 2021 |