
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSkeletal remodeling of chalcone-based pyridinium salts to access isoindoline polycycles and their bridged derivatives†
Lele
Wang‡
a,
Huabin
Han‡
a,
Lijie
Gu
a,
Wenjing
Zhang
 *b,
Junwei
Zhao
*a and
Qilin
Wang
*a
*b,
Junwei
Zhao
*a and
Qilin
Wang
*a
aCollege of Chemistry and Chemical Engineering, Henan University, Kaifeng 475004, China. E-mail: zhaojunwei@henu.edu.cn; wangqilin@henu.edu.cn
bGreen Catalysis Center, College of Chemistry, Zhengzhou University, Zhengzhou 450001, China. E-mail: zhangwj@zzu.edu.cn
First published on 9th November 2021
Abstract
Simultaneous deconstructive ring-opening and skeletal reconstruction of an inert, aromatic pyridinium ring is of great importance in synthetic communities. However, research in this area is still in its infancy. Here, a skeletal re-modeling strategy was developed to transform chalcone-based pyridinium salts into structurally intriguing polycyclic isoindolines through a dearomative ring-opening/ring-closing sequence. Two distinct driving forces for the deconstruction of the pyridinium core were involved in these transformations. One was the unprecedented harnessing of the instability of in situ generated cyclic β-aminoketones, and the other was the instability of the resultant N,N-ketals. The desired isoindoline polycycles could undergo the Wittig reaction with various phosphorus ylides to achieve structural diversity and complexity. Notably, by tuning the Wittig conditions by addition of one equivalent of base, an additional bridged ring was introduced. A plausible mechanism was proposed on the basis of control experiments and theoretical calculations.
Introduction
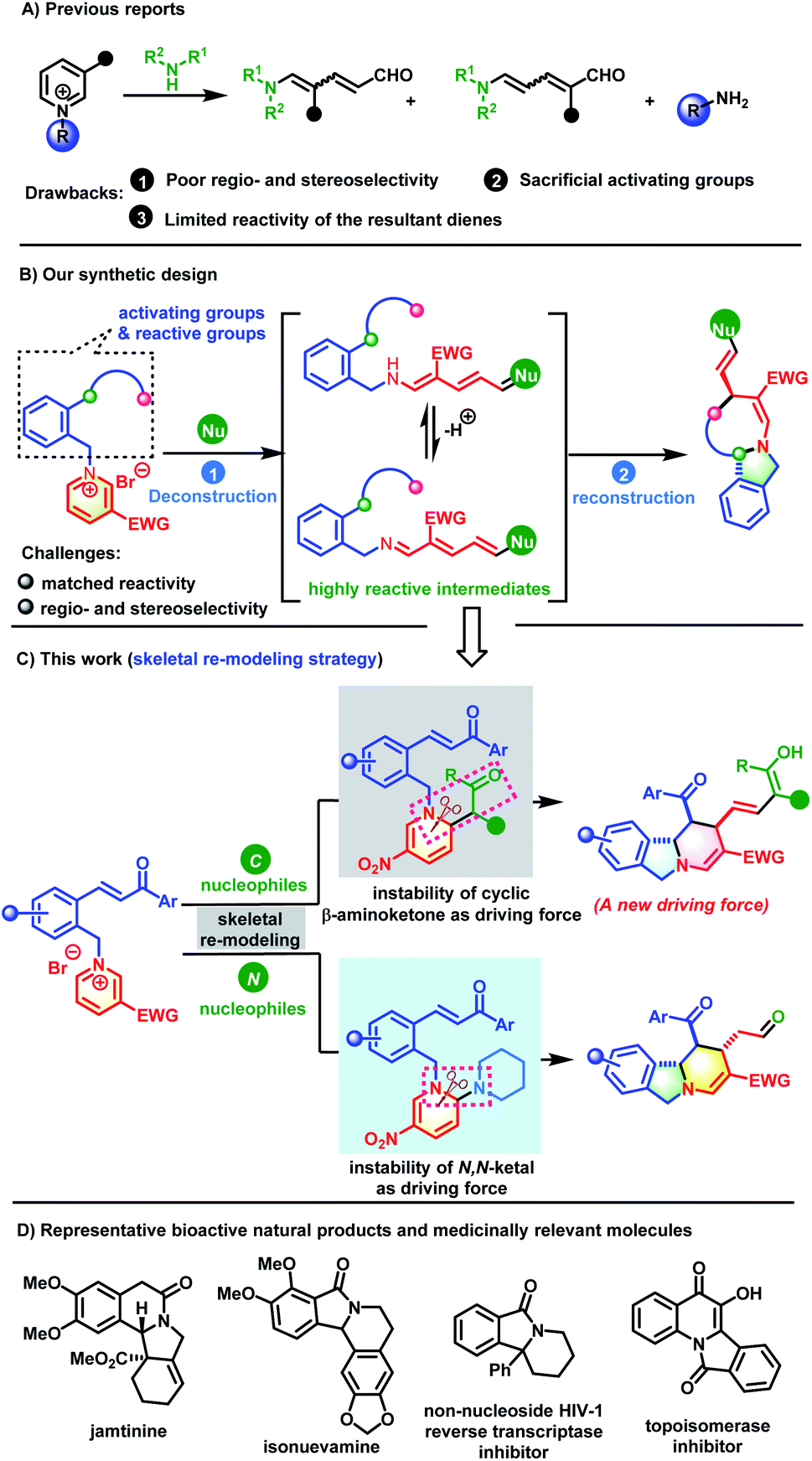
Aza-heterocycles are frequently encountered in natural products, bioactive molecules and functional materials.1 Among the aza-heterocycles, pyridine rings, which are readily accessible feedstock chemicals, are the most ubiquitous core structures. Therefore, the exploration of versatile pyridyl ring based chemical transformations is of great importance, especially for unlocking new reactivities to achieve the synthesis of high-value-added products that are otherwise difficult to access. Recently, the dearomatization of activated pyridines has emerged as a booming research area that is capable of converting flat pyridine cores into complex three-dimensional fully or partially saturated azaheterocycles.2 Despite substantial advances, this strategy only modifies the periphery of the dearomatized pyridine molecules by tuning the substituents while retaining the six-membered cyclic structure.3 Skeletal re-editing is an important complementary method that comprises two elementary steps: deconstruction of the underlying molecular skeleton to generate reactive species and subsequent reconstruction to deliver structurally distinct molecules. This method rapidly and reliably achieves skeletal diversity by modifying the core framework.4 However, the skeletal remodeling of aromatic pyridine cores remains underdeveloped, because their high resonance stabilization energy needs to be overcome first to dearomatize the aromatic ring. In addition, dearomatized rings contain unstrained six-membered rings that are very difficult to dissociate.Stemming from the pioneering work of Zincke and König,5 dearomative ring-opening of pyidinium salts has received increasing attention (Scheme 1A). This strategy is very reliable for the construction of structurally interesting 5-amino-2,4-pentadienals, a new class of D–π–A dienes, using secondary amines as nucleophiles. The pentadienals can undergo further cyclization to generate valuable molecules with increasing complexity and diversity.6 Although valuable progress has been made, there are still some challenges to be overcome, including the following: (1) the driving force for the deconstruction needs to be further explored. Currently, methods for the ring-opening of pyridinium salts are dominated by N,O- or N,N-ketal formation via nucleophilic attack by an amine or hydroxyl group followed by heterolytic C–N bond cleavage by taking advantage of the instability of the in situ generated aminals. (2) The reactions are often associated with low regio- and stereo-selectivity issues.7 When unsymmetrical pyridiniums are employed as substrates, their C2-, C4- and C6-positions are all potential electrophilic sites and it is quite difficult to identify subtle differences in reactivity. In addition, both Z- and E-isomers of the resulting diene intermediates are generated concomitantly. (3) The reactivity of the activating groups is underutilized. After the reaction reaches completion, the pre-installed activating groups are released without participating in the subsequent reaction. Rational reaction design to allow for the utilization of the activation group would be of great value to increase structural complexity. (4) The reactivity of the resultant 5-amino-2,4-pentadienals has not been fully exploited. Structurally, they possess a D–π–A conjugated system. In addition to their identity as electron-deficient all-carbon dienes that can participate in inverse-electron-demand [4+2] cycloadditions,6c–f they could also serve as azadienes to undergo normal [4+2] cycloaddition with electron-deficient dienophiles because of their structural characteristics with “push–pull” electrons. However, research in this area is lacking.
 | ||
| Scheme 1 Skeletal remodeling of pyridiniums through dearomative ring-opening/ring-closing. | ||
On the basis of fully understanding the above challenges, we envisioned that if a reactive group with appropriate reactivity was attached on the activating group, we would solve the activating-group utilization and product-diversity problems. Once the D–π–A diene intermediate was obtained after the dearomative ring-opening of the pyridinium core, it could be intercepted by the reactive sites on the activating group. This would achieve skeletal re-modeling of the pyridinium salts to assembly novel and complex molecules (Scheme 1B). The key to the success of this strategy is the identification of a suitable reactive group. The electronic characteristics of the dienes provide an important clue that incorporating a Michael acceptor on the activating group could meet the need for subsequent reconstruction. Based on this analysis, our previously designed chalcone-based pyridinium salts came into our sight.8 If successful, this skeletal re-modeling strategy would enable scaffold hopping from one planar and aromatic pyridinium ring to another three-dimensional azaheteropolycycle without the need for costly reagents and harsh conditions.
As a continuation of our research into the dearomatization of six-membered oxonium and pyridinium salts to construct various novel and complex molecules,9 herein, we wish to disclose a skeletal re-modeling strategy for pyridinium salts to assemble polycyclic isoindolines and their bridged derivatives. Our skeletal re-modeling strategy has two distinct driving forces (Scheme 1C). When active methylene compounds such as 1,3-diketone are used as nucleophiles, the cyclic architecture of the pyridine ring was broken by harnessing the instability of the β-aminoketone. When secondary amines were used as nucleophiles, the driving force for deconstruction was the instability of the in situ generated N,N-ketals. This strategy would enable the synthesis of isoindoline-fused polycyclic systems, which are the core structures of many biologically active natural products and medicinally relevant molecules (Scheme 1D).10 Although great advancement has been made toward their synthesis, current methods often need tedious procedures and custom reagents.11
Results and discussion
Proof-of-principle investigation
To commence this study, chalcone-based pyridinium salt 1 and acetylacetone 2 were selected as model substrates to verify the feasibility of the synthetic strategy. Delightfully, this reaction proceeded smoothly in the presence of 2.0 equivalents of 1,1,3,3-tetramethyl guanidine (TMG) to afford polycyclic isoindoline 3 in 84% yield (determined by 1H NMR) in only 5 min. To further improve the synthetic efficiency, various organic and inorganic bases were evaluated (Table 1, entries 2–4). However, all were less productive than TMG. Subsequently, the effect of solvents was investigated. Among the solvents, acetone exhibited the best efficiency, in which the reaction reached completion within 5 min and produced 3 in 90% isolated yield. The temperature also affected the yields, with a lower temperature not beneficial to the reaction outcome (Table 1, entry 7 vs. entries 8 and 9). The optimum reaction conditions for the formation of 3 were as follows: 0.15 mmol of 2 and 1.5 equivalents of 1 in the presence of 2.0 equivalents of TMG in 1.0 mL of acetone at 60 °C.|
|
||||
|---|---|---|---|---|
| Entry | Base | Solvent | Time | Yieldb (%) |
| a Reactions performed at 60 °C on a 0.15 mmol scale using 1.5 equivalents of 1 in the presence of 2.0 equivalents of base in 1.0 mL of solvent. b Yields determined by 1H NMR analysis of the crude mixture using 1,3,5-trimethoxybenzene as the internal standard. c Isolated yield obtained by silica gel column chromatography. d At 50 °C. e At 30 °C. TMG = 1,1,3,3-tetramethyl guanidine. DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene. n.r. = no reaction. | ||||
| 1 | TMG | CH3CN | 5 min | 84 |
| 2 | DBU | CH3CN | 1 h | 63 |
| 3 | NEt3 | CH3CN | 24 h | n.r. |
| 4 | Cs2CO3 | CH3CN | 1 h | 78 |
| 5 | TMG | CHCl3 | 5 min | 57 |
| 6 | TMG | DMF | 5 min | 93 |
| 7 | TMG | Acetone | 5 min | 95 (90)c |
| 8d | TMG | Acetone | 5 min | 87 |
| 9e | TMG | Acetone | 5 min | 77 |
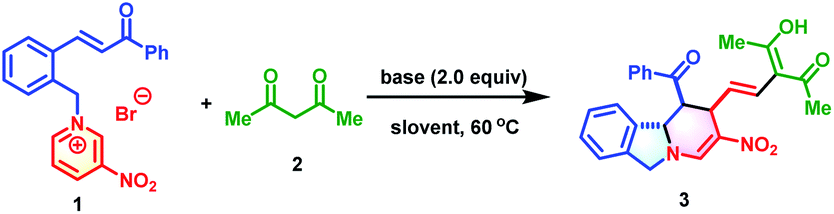
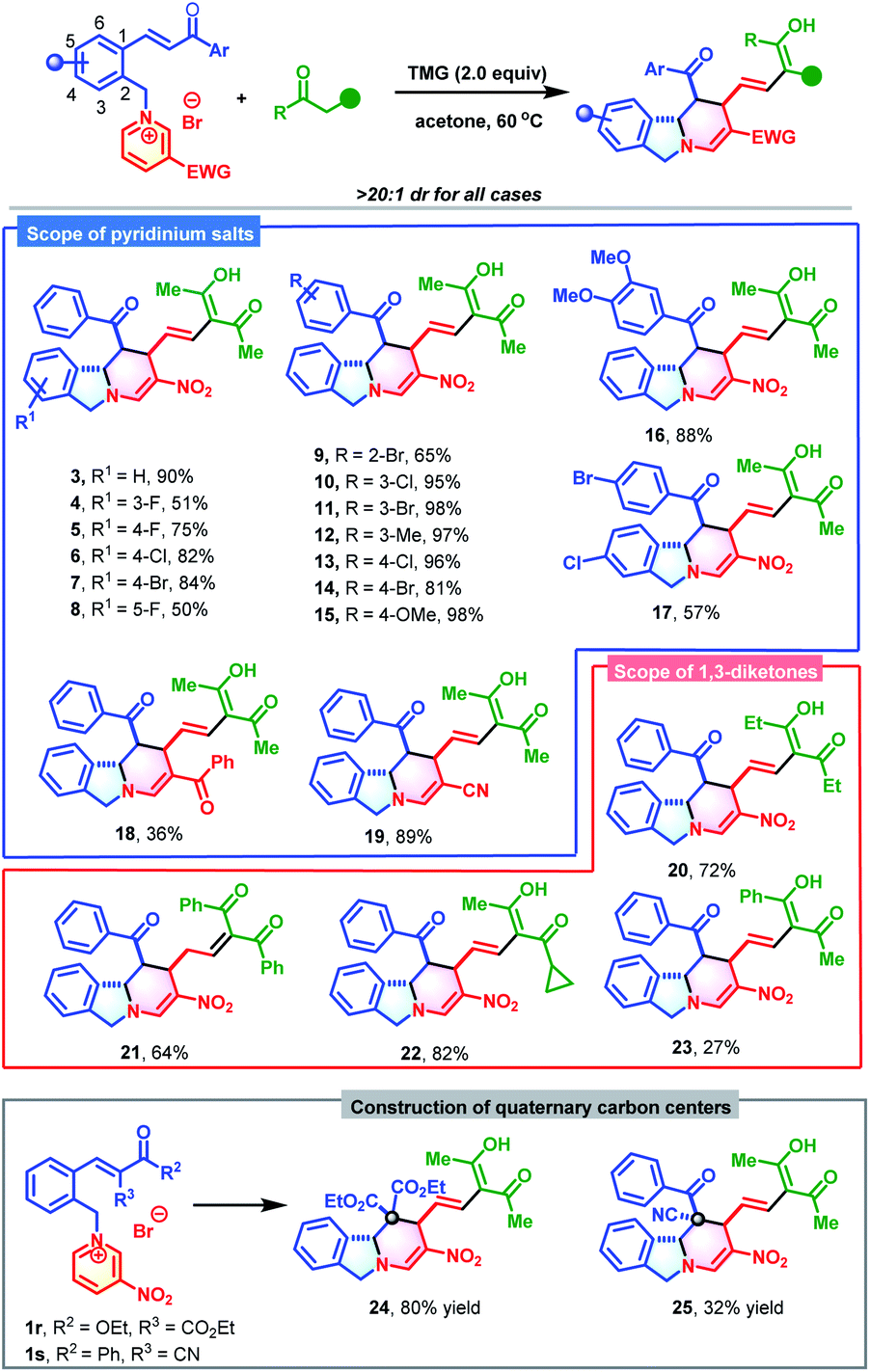
With the optimized conditions established, the substrate scope of this dearomative ring-opening/reconstruction sequential synthetic strategy was explored. A wide range of pyridinium salts bearing substituents with different electronic characters and varied positions were all compatible (Table 2), which allowed for the synthesis of polycyclic isoindolines 3–17 in 50–98% yields. Notably, the nitro group on the pyridine ring was not indispensable, and replacement of it with benzoyl or cyano groups produced 18 in 36% yield and 19 in 89% yields, respectively. Next, the generality of 1,3-dicarbonyl compounds was investigated. More steric 1,3-diketones, such as heptane-3,5-dione, 1,3-diphenylpropane-1,3-dione, and even unsymmetrical 1-cyclopropylbutane-1,3-dione and 1-phenylbutane-1,3-dione were all suitable reaction partners. These compounds generated the desired products 20–23 in 27–82% yields. Remarkably, when 1,3-diphenylpropane-1,3-dione was used, the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond migrated. In view of the significance and synthetic challenges of quaternary carbon centers,12 the applicability of our strategy in the construction of isoindoline polycycles with a tetrasubstituted quaternary carbon center was examined. The more hindered pyridinium salts 1r and 1s could also participate in this dearomative ring-opening/reconstruction cascade reaction successfully to deliver 24 in 80% yield and 25 in 32% yield as sole diastereomers with tetrasubstituted quaternary carbon centers.
C double bond migrated. In view of the significance and synthetic challenges of quaternary carbon centers,12 the applicability of our strategy in the construction of isoindoline polycycles with a tetrasubstituted quaternary carbon center was examined. The more hindered pyridinium salts 1r and 1s could also participate in this dearomative ring-opening/reconstruction cascade reaction successfully to deliver 24 in 80% yield and 25 in 32% yield as sole diastereomers with tetrasubstituted quaternary carbon centers.
| a Reactions performed on a 0.15 mmol scale using 1.5 equivalents of 1 with 2.0 equivalents of TMG in 1.0 mL of acetone at 60 °C. Yields refer to the isolated products after column chromatography. |
|---|
|
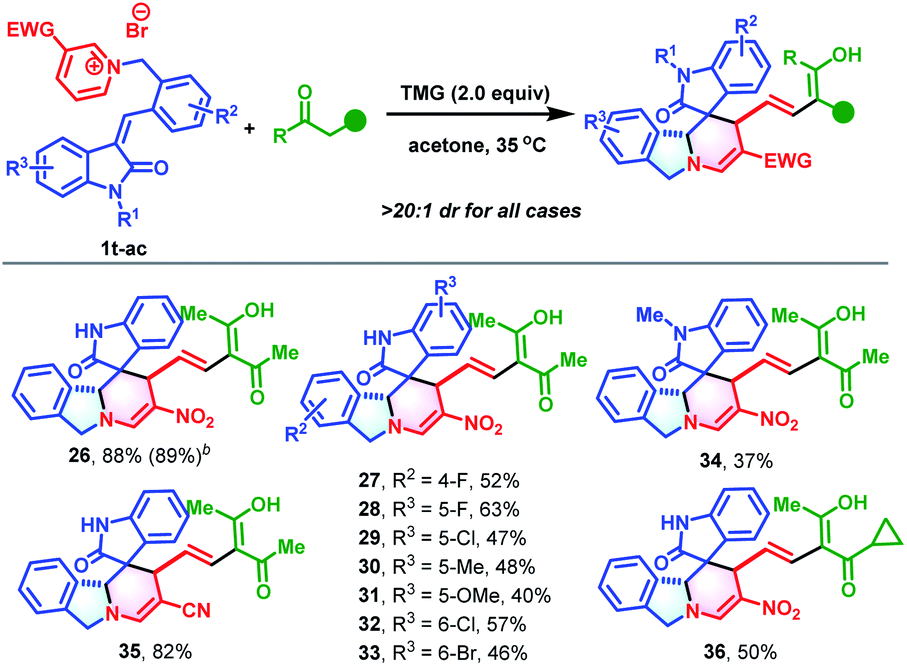
Encouraged by the success of this synthetic strategy, a series of 3-alkenyl oxindile-based pyridinium salts (1t-ac, for details, see Section S2†) were prepared and evaluated in the cascade reaction using a similar process to construct polycyclic isoindoline spirooxindoles. As anticipated, the dearomative ring-opening/reconstruction cascade reaction between 1t and 2 took place successfully under slightly modified conditions (0.10 mmol of 1t and 1.2 equivalents of 2 at 35 °C), and produced polycyclic isoindoline spirooxindole 26 in 88% yield (Table 3). It should be noted that this reaction could also be scaled up without compromising the efficiency (2.5 mmol and 89% yield). Next, the substrate scope was evaluated. Generally, a wide range of 3-alkenyl oxindole-based pyridinium salts were tolerable in these transformations. Regardless of their substitution patterns and electronic nature, all reactions proceeded smoothly to produce 27–35 in 37–82% yields. In addition to acetylacetone, 1-cyclopropylbutane-1,3-dione was also an appropriate reaction partner to give 36 in 50% yield.
Secondary amine-promoted skeletal remodeling of chalcone-based pyridiniums
Motivated by the above exciting results, the secondary amine-promoted skeletal re-modeling strategy was investigated (Scheme 2). On treatment with 2.5 equivalents of piperidine in 1.0 mL of water at 80 °C for 2 h, pyridinium salt 1 was successfully converted into the desired polycyclic isoindoline 37 in 89% isolated yield (Table S1,† entry 1). After extensive experimentation, the optimum conditions were established (Table S1,† entry 1). We also attempted the asymmetric synthesis of polycyclic isoindoline 37 by using 2.5 equivalents of secondary amines. Proline could not promote this reaction efficiently (Table S2,† entry 1). Among the amines tested, proline-derived trifluoromethanesulfonamide gave the best yield and stereoselectivity (Table S2,† entry 2, 89% yield, and 68% ee). Remarkably, the product precipitated out from the sustainable water system and required only simple filtration for purification. To enrich the diversity of the obtained product 37 and improve its solubility, a sequential Wittig reaction was conducted in toluene at 110 °C. The desired product 38 was obtained in 74% yield with a 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 d.r.
:1 d.r.
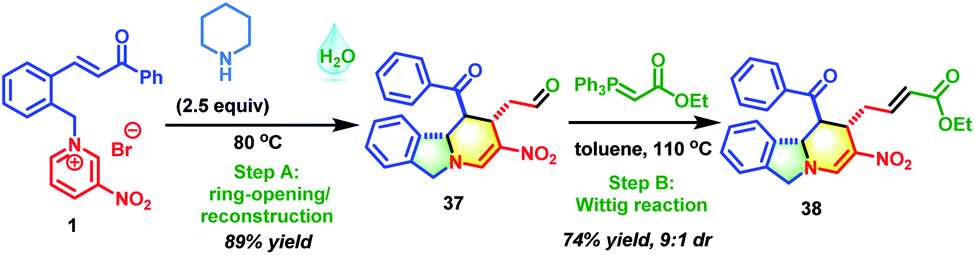 | ||
| Scheme 2 Piperidine-promoted skeletal re-modeling of 1. | ||
With the proof of concept established, the substrate scope of this skeletal re-editing strategy was explored (Table 4). Initially, the effects of Ar groups neighboring the carbonyl group with different substitution patterns were evaluated. Broadly, a variety of pyridinium salts were applicable in these transformations, successfully producing 39–46 in satisfactory yields. The reactions were apparently sensitive to the substituent positions. Compared with meta- or para-Br substituted substrates, the ortho-Br substituted substrate gave a much lower yield (see 39vs.41 and 44). The electronic characteristics also affected the reaction outcomes. The Ar groups with electron-withdrawing groups performed much better than those with electron-donating groups (39–41 and 43–44vs.42 and 45–46). The Ar groups were not crucial, and they could be replaced by methyl, for which an additional aldol reaction took place with the formation of 47 in 10% yield. The introduction of a halogen on the phenyl group adjacent to the pyridine ring was also compatible, delivering 48–51 in acceptable yields. The substituents on the pyridine ring were not limited to nitro groups, and cyano, benzoyl and ester groups were also tolerable (CN, 52; Bz, 53; and CO2Et, 54). Next, the scope of phosphorus ylides was evaluated. Except for ester-derived ylides, acetophenone-derived ylide could also participate in this cascade process successfully, enabling the formation of 54 in excellent yield with complete diastereocontrol. Encouragingly, yildes containing natural products and drug molecules were also applicable in this reaction and produced 56–60 in 60–82% yields.
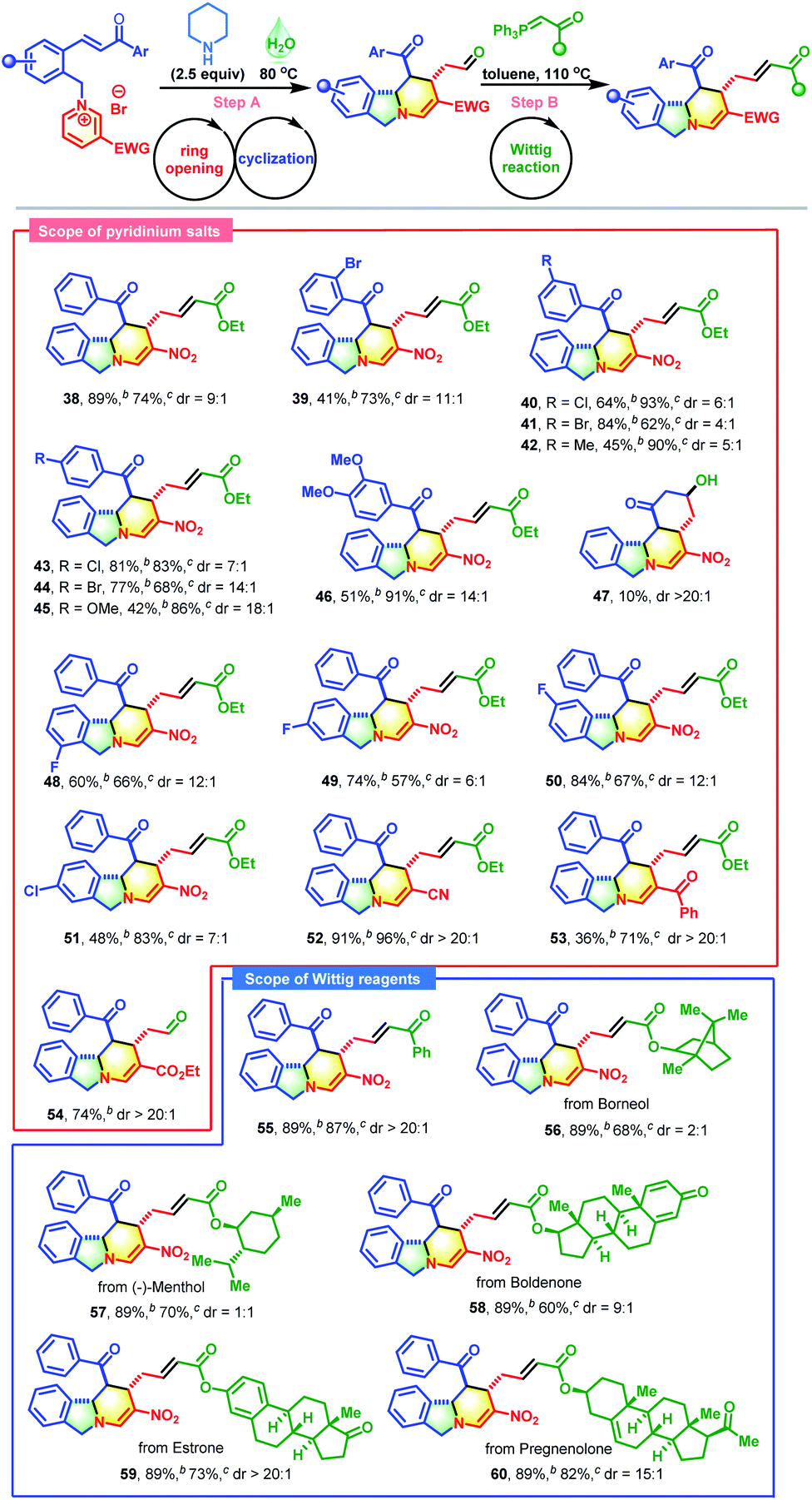
Inspired by the success of the above two-pot reactions, the reduction of this cascade process to a one-pot reaction was investigated. When the reaction of pyridinium salt 1, piperidine and Wittig reagent was performed in water at 100 °C, an unexpected bridged isoindoline polycycle 61 was obtained in 25% yield with complete diastereocontrol and no traces amount of 38 (Scheme 3). This transformation might proceed through an additional piperidine-promoted Michael addition, as evidenced by the fact that 38 could be smoothly transformed into 61 in the presence of one equivalent of TMG in 61% yield with >20:1 d.r. (Table S4,† entry 1). As shown in Scheme 3, this reaction was presumably initiated by the deprotonation of the benzylic proton of 38 with the help of a base to generate nucleophilic reactive sites, followed by an intramolecular Michael addition with α,β-unsaturated ester. Further investigations revealed that water was beneficial to the first ring-opening/reconstruction but detrimental to the subsequent Wittig/cyclization reaction (Table S3†). Solvent mixtures also failed to improve the synthetic efficiency (Table S2,† entry 3). Therefore, this reaction was conducted in a two-step fashion. After some experimentation, the optimum conditions were established (Table S4,† entry 1).
 | ||
| Scheme 3 One-pot approach for the synthesis of bridged isoindole polycycle 61. | ||
Next, the generality and robustness of this approach were investigated (Table 5). Broadly, this transformation could tolerate a plethora of pyridinium salts with different electronic and steric effects, affording 62–69 in 35–59% yields (for step B). Notably, although the products contained four stereocenters and a challenging rigid bridged ring, only one diastereomer was obtained in all cases. In addition, isoindoline polycycle-based aldehyde 37 could undergo an intramolecular Aldol reaction with the help of TMG, which successfully produced 70 and its diastereomer 70′ in 83% yield with 2:1 d.r.
| a Reaction conditions: Step A: pyridinium salts (0.2 mmol), piperidine (2.5 equivalents), H2O (1.0 mL), and 80 °C. Step B: ylides (1.5 equivalents based on the products of step A), TMG (1.0 equivalent), toluene (1.0 mL), and 110 °C. The d.r. value was determined by 1H NMR. b The yields of step A. c The yields of step B. |
|---|

|
Synthetic applicability
To highlight the synthetic utility and practicability of this strategy, a gram-scale preparation of 3 was performed on a 3.0 mmol scale (Scheme 4). To our delight, 3 was obtained in acceptable yield (65%). Next, some chemical conversions of 3 were carried out to further expand the synthetic value. For instance, a reaction with phenylhydrazine or hydroxylamine hydrochloride could be used to easily install an imidazole or oxazole ring onto the molecular scaffold of 3 to generate 71 and 72 in 82% and 53% yields. The brominaion of 3 with N-bromosuccinimide afforded 73 in 43% yield. In addition, I2- or DDQ-mediated cyclization generated 74 with a newly formed furan ring. One acetyl group of 3 could be removed by subjecting it to InCl3 catalysis, which produced 75 in 76% yield. By harnessing the condensation between aldehyde 37 and various primary amines, some imines bearing either the benzyl group (76) or other biologically active pharmaceutical ingredients (77–80) were smoothly synthesized. The treatment of 38 with NaBH4 resulted in a reduction/intramolecular Michael addition to produce 81 with a newly formed tetrahydropyran ring in 55% yield with 3:1 d.r.
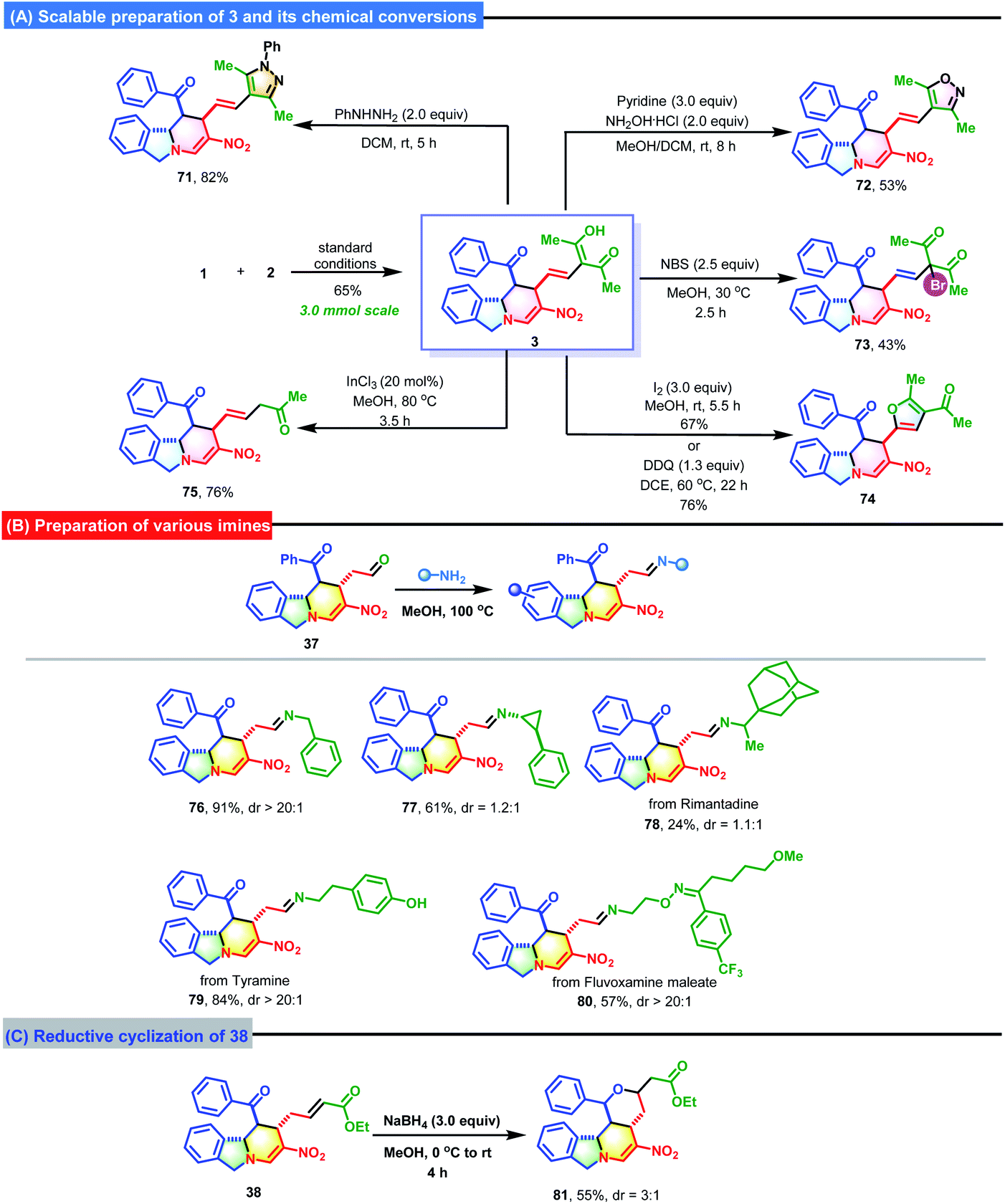 | ||
| Scheme 4 Synthetic applicability. | ||
The structures and relative configurations of 3, 21, 25, 26, 47, 48, 52, 61, 70, 72 and 74 were unequivocally determined by X-ray single crystal diffraction.13 The relative configurations of other products were determined by analogy.
Mechanistic studies
To understand the mechanism in depth, some control experiments were conducted (Scheme 5). First, the role of piperidine was studied. Using the 3-alkenyl oxindole-based pyridinium salt 1t as the substrate, the deconstruction/reconstruction product 82 with an enamine skeleton was obtained. The enamine could be converted into aldehyde 83 in nearly quantitative yield by subjecting it to silica gel chromatography (Scheme 5A). This case and the data shown in Table S1† jointly confirmed the indispensable role of secondary amines in attacking the C-6 position of the pyridine core to form an unstable N,N-ketal. This was the driving force behind the opening of the pyridine core and subsequent reconstruction. In addition, two deuterium scrambling experiments were performed with D2O and H218O (Scheme 5B). The results clearly indicated that the aldehyde stemmed from the hydrolysis of enamine. The formation of bridged isoindoline polycycle 61 might proceed through a radical process or two-electron Michael addition process. To differentiate between these mechanisms, a one-pot reaction of pyridinium salt 1, piperidine and Wittig reagent was performed in D2O (Scheme 5C). This gave the product (61-D) with deuterium incorporated at the carbon adjacent to the ester group. In addition, the bridged product 61 was obtained from 38 in the presence of one equivalent of 2,2,6,6-tetramethyl-1-piperidinyloxy with only a slight reduction in the yield (50% vs. 61%) (Scheme 5D). Furthermore, the addition of 20 equivalents of D2O to the reaction system of 38 and TMG in toluene gave the same yield (61%) with 76% D-incorporation at the carbon linked to the ester group (Scheme 5E). These results strongly suggest that this transformation from 38 into 61 proceeds through a two-electron intramolecular Michael addition rather than a radical process.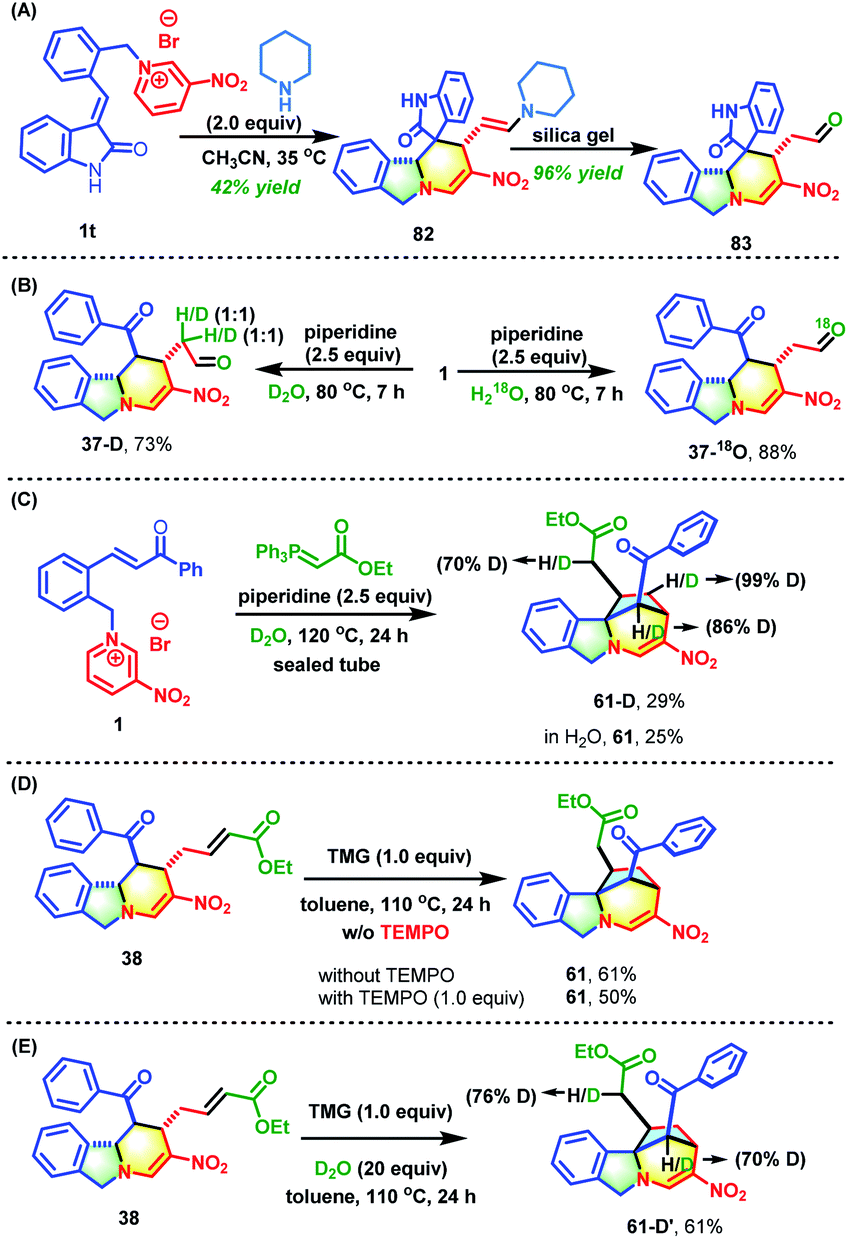 | ||
| Scheme 5 Control experiments. | ||
On the basis of the experimental results, a tentative reaction pathway was proposed as shown in Scheme 6. Generally, two successive processes of deconstructive ring-opening and reconstruction were involved in these transformations. The reactions began with dearomative addition by nucleophiles such as acetylacetone and piperidine to generate intermediate Int-A, which was not stable and was prone to undergo ring-opening to form the reactive D–π–A intermediate. In this process, two distinct driving forces for the deconstruction of the pyridine core were utilized. When acetylacetone was used as the nucleophile, the driving force was the instability of the cyclic β-aminoketones. When piperidine was used as the nucleophile, the driving force was the instability of the in situ generated N,N-ketals. Followed by intramolecular [4+2] cycloaddition, the desired isoindoline polycycles were afforded. When piperidine was used as the nucleophile, an additional hydrolysis from Int-B occurred to produce 37. By reacting with phosphorus ylide, 37 was smoothly converted into 38 bearing an α,β-unsaturated ester group. With the help of TMG, an intramolecular Michael addition of 38 occurred to deliver the desired bridged isoindoline polycycle 61. This product could also be obtained from 37 in a one-pot fashion.
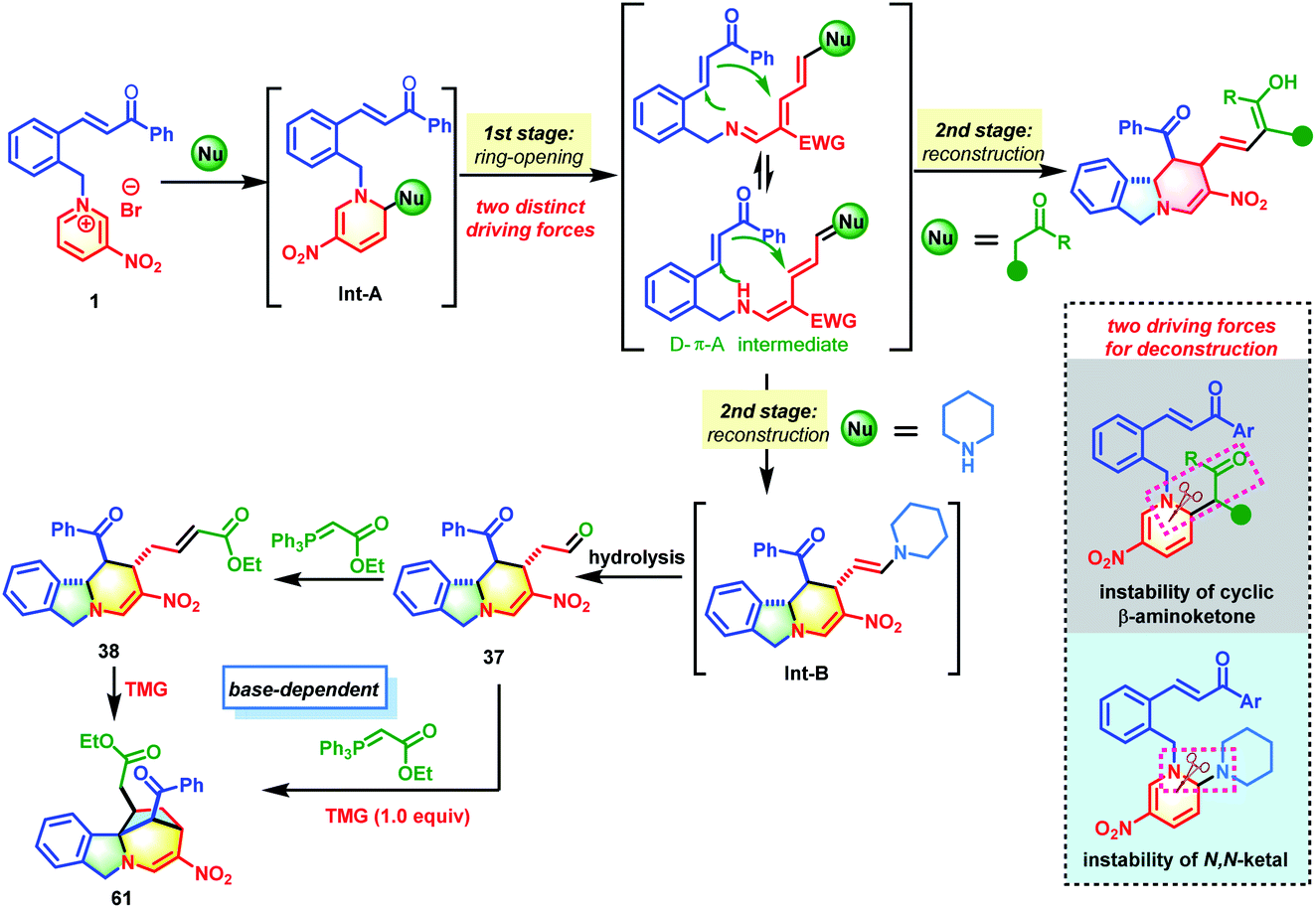 | ||
| Scheme 6 Proposed mechanism. | ||
A mechanistic study using density functional theory (DFT)14 provided further verification. When acetylacetone was used as the nucleophile, it could be activated through deprotonation by a reaction with TMG, which produced the carbon anion R2 (Fig. 1). The combination with pyridinium R1 at the C(6)-position was slightly endothermic by 1.1 kcal mol−1. After neutral compound Int1 was activated by proton transfer to the TMG base, it underwent successive ring opening, [4+2] cycloaddition, and protonation. The energy barrier for the ring opening via transition state TS2 was 17.3 kcal mol−1, and the generated D–π–A intermediate Int3 was 11.0 kcal mol−1 higher in energy than cyclic compound Int2. The direct [4+2] cycloaddition from Int3via transition state TS3 would result in an energy barrier of 24.7 kcal mol−1. This could be attributed mainly to the instability of the cis configuration of Int3 and TS3. The conformation transformation from cis-Int3 to trans-Int4 led to an exothermic release of 7.8 kcal mol−1. The following [4+2] cycloaddition occurred through a stepwise mechanism because the energy barrier of the concerted reaction viaTS4 was 2.4 kcal mol−1 higher than that of the nucleophilic addition viaTS5. The protonated TMG played an important role in stabilizing the enolate anion in intermediate Int5. The energy barriers for the two steps of cycloaddition were 15.2 kcal mol−1 and 6.3 kcal mol−1, respectively, and the resultant isoindoline polycycle compound Int6 was 30.9 kcal mol−1 lower in energy than TS5, indicating an irreversible process. Finally, the protonation of the terminal enolate anion by proton transfer from protonated TMG gave the stable neutral product 3 with an energy barrier of 1.9 kcal mol−1 and exothermic release of 10.1 kcal mol−1.
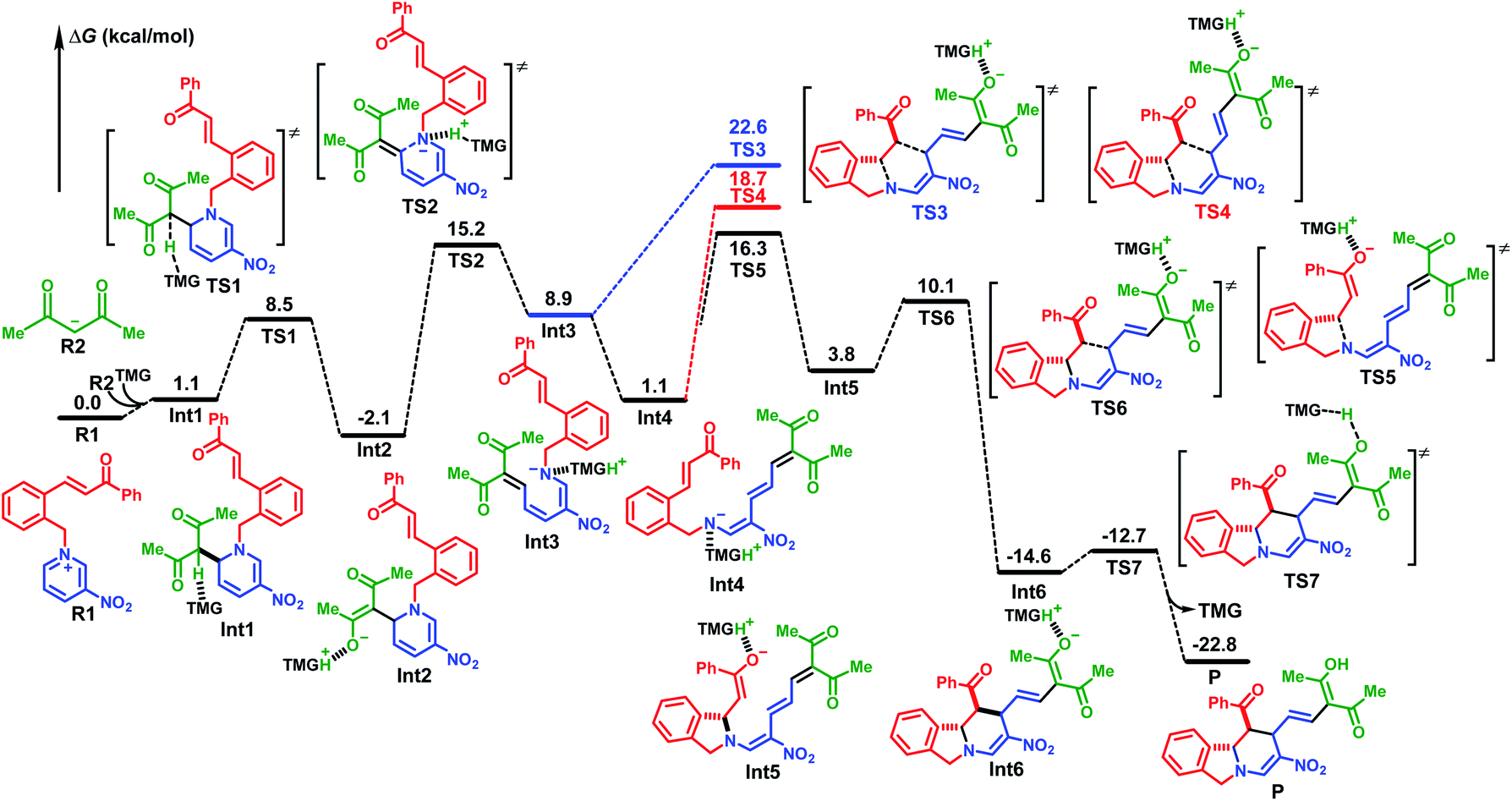 | ||
| Fig. 1 Free-energy profiles for the reaction with acetylacetone added to the C(6)-position of pyridinium R1. Free energies are given in kcal mol−1 and represent relative free energies calculated by using the M06-2X functional in acetone. | ||
The reaction with acetylacetone anion R2 added to the C(2)-position of pyridinium R1 was also investigated in detail. The computational results indicated that the energy barrier of the initial deprotonation activation was 9.6 kcal mol−1 higher than that of the corresponding reaction at the C(6)-position. In the following step, the barrier for either the ring opening or the Michael addition was more than 4.5 kcal mol−1 higher than that of the stepwise [4+2] cycloaddition as illustrated above. Therefore, the regioselectivity was at the C(6)-position rather than the C(2)-position (for more details, see Fig. S5†).
Conclusion
In conclusion, an efficient synthetic strategy merging deconstructive ring-opening and reconstruction was developed for skeletal re-modeling of chalcone-based pyridinium salts. This reaction proceeds smoothly to produce isoindoline polycycles, which are otherwise difficult to access in good yields with precise regio- and diastereocontrol. In this transformation, activating groups are efficiently used to construct structurally novel and complex molecules. In addition to the instability of N,N-ketals as a driving force for deconstruction, the instability of cyclic β-aminoketones could be utilized as a new and efficient driving force. Remarkably, structurally challenging and rigid bridged isoindoline polycycles could be prepared. A combination of control experiments and theoretical calculations deduced the plausible mechanism of this skeletal remodeling strategy. Further investigations on the skeletal re-modeling of pyridinium salts and related bioassays are currently underway.Data availability
Experimental procedures, condition optimization, characterisation data, mechanistic studies, computational details, and copies of 1H, 13C and 19F NMR spectra for all compounds are provided in the ESI.†Author contributions
Reaction design, supervision and writing – Q. W. and J. Z.; investigation and methodology – L. W., H. H., and L. G.; theoretical calculation – W. Z.; all authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (22101073 and U1504206), China Postdoctoral Science Foundation (2020M672200), the Program for Innovation Teams in Science and Technology in Universities of Henan Province (20IRTSTHN004), and Henan University (SYL19030204, SYL19060137, and SYL20060149). We also thank Dr Zhenhua Wang and Dr Peihao Dou from the Chengdu Institute of Organic Chemistry, Chinese Academy of Sciences for chiral HPLC analysis.Notes and references
- For selected reviews, see: (a) P. Bhutani, G. Joshi, N. Raja, N. Bachhav, P. K. Rajanna, H. Bhutani, A. T. Paul and R. Kumar, J. Med. Chem., 2021, 64, 2339–2381 CrossRef CAS PubMed; (b) E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed; (c) X.-X. Jiang and R. Wang, Chem. Rev., 2013, 113, 5515–5546 CrossRef CAS PubMed; (d) J. P. Michael, Nat. Prod. Rep., 2008, 25, 166–187 RSC; (e) Y.-G. Zhou, Acc. Chem. Res., 2007, 40, 1357–1366 CrossRef CAS PubMed.
- For selected reviews, see: (a) G. Bertuzzi, L. Bernardi and M. Fochi, Catalysts, 2018, 8, 632–665 CrossRef; (b) S. Sowmiah, J. M. S. S. Esperança, L. P. N. Rebelo and C. A. M. Afonso, Org. Chem. Front., 2018, 5, 453–493 RSC; (c) Q.-P. Ding, X.-L. Zhou and R.-H. Fan, Org. Biomol. Chem., 2014, 12, 4807–4815 RSC; (d) J. A. Bull, J. J. Mousseau, G. Pelletier and A. B. Charette, Chem. Rev., 2012, 112, 2642–2713 CrossRef CAS PubMed; (e) Z.-Y. Zhang, H.-B. Han, L.-L. Wang, Z.-W. Bu, Y. Xie and Q.-L. Wang, Org. Biomol. Chem., 2021, 19, 3960–3982 RSC.
- For selected examples, see: (a) B. Cheng, X.-P. Zhang, Y.-T. Li, H. Li, Y.-X. He, Y. Li, T.-M. Wang and H.-B. Zhai, Chem. Commun., 2020, 56, 8396–8399 RSC; (b) T. Wagener, L. Lückemeier, C. G. Daniliuc and F. Glorius, Angew. Chem., Int. Ed., 2021, 60, 6425–6429 CrossRef CAS PubMed; (c) X. Song, R.-J. Yan, W. Du and Y.-C. Chen, Org. Lett., 2020, 22, 7617–7621 CrossRef CAS PubMed; (d) A. Grozavu, H. B. Hepburn, P. J. Smith, H. K. Potukuchi, P. J. Lindsay-Scott and T. J. Donohoe, Nat. Chem., 2019, 11, 242–247 CrossRef CAS PubMed; (e) D. Zhang, L.-L. Lin, J. Yang, X.-H. Liu and X.-M. Feng, Angew. Chem., Int. Ed., 2018, 57, 12323–12327 CrossRef CAS PubMed; (f) S.-Y. Baek, J.-Y. Lee, D. Ko, M.-H. Baik and E.-J. Yoo, ACS Catal., 2018, 8, 6353–6361 CrossRef CAS; (g) Z.-P. Yang, Q.-F. Wu, W. Shao and S.-L. You, J. Am. Chem. Soc., 2015, 137, 15899–15906 CrossRef CAS PubMed; (h) Z.-P. Yang, Q.-F. Wu and S.-L. You, Angew. Chem., Int. Ed., 2014, 53, 6986–6989 CrossRef CAS PubMed; (i) B.-S. Mu, X.-Y. Cui, X.-P. Zeng, J.-S. Yu and J. Zhou, Nat. Commun., 2021, 12, 2219–2226 CrossRef CAS PubMed.
- (a) J. B. Roque, Y. Kuroda, L. T. Göttemann and R. Sarpong, Nature, 2018, 564, 244–248 CrossRef CAS PubMed; (b) Y. Xu, X.-T. Qi, P.-F. Zheng, C. C. Berti, P. Liu and G.-B. Dong, Nature, 2019, 567, 373–378 CrossRef CAS PubMed; (c) A. J. Smaligo, M. Swain, J. C. Quintana, M. F. Tan, D. A. Kim and O. Kwon, Science, 2019, 364, 681–685 CrossRef CAS PubMed; (d) S. H. Kennedy, B. D. Dherange, K. J. Berger and M. D. Levin, Nature, 2021, 593, 223–227 CrossRef CAS PubMed.
- (a) T. Zincke and F. Schreyer, Liebigs Ann., 1907, 353, 380–385 CrossRef CAS; (b) T. Zincke and W. Würker, Liebigs Ann., 1904, 338, 107–141 CrossRef; (c) T. Zincke, G. Heuser and W. Möller, Liebigs Ann., 1904, 333, 296–345 CrossRef; (d) W. König, J. Prakt. Chem., 1904, 69, 105–137 CrossRef.
- (a) A. M. Kearney and C. D. Vanderwal, Angew. Chem., Int. Ed., 2006, 45, 7803–7806 CrossRef CAS PubMed; (b) T. D. Michels, M. J. Kier, A. M. Kearney and C. D. Vanderwal, Org. Lett., 2010, 12, 3093–3095 CrossRef CAS PubMed; (c) D. B. C. Martin and C. D. Vanderwal, Chem. Sci., 2011, 2, 649–651 RSC; (d) D. B. C. Martin, L. Q. Nguyen and C. D. Vanderwal, J. Org. Chem., 2012, 77, 17–46 CrossRef CAS PubMed; (e) D. B. C. Martin and C. D. Vanderwal, J. Am. Chem. Soc., 2009, 131, 3472–3473 CrossRef CAS PubMed; (f) A. Y. Hong and C. D. Vanderwal, J. Am. Chem. Soc., 2015, 137, 7306–7309 CrossRef CAS PubMed; (g) K. Xu, W.-J. Li, R. Sun, L.-H. Luo, X. Chen, C.-C. Zhang, X.-L. Zheng, M.-L. Yuan, H.-Y. Fu, R.-X. Li and H. Chen, Org. Lett., 2020, 22, 6107–6111 CrossRef CAS PubMed; (h) Z.-D. Tan, C.-G. Ci, J. Yang, Y. Wu, L. Cao, H.-F. Jiang and M. Zhang, ACS Catal., 2020, 10, 5243–5249 CrossRef CAS.
- (a) L. V. Andriyankova, L. P. Nikitina, K. V. Belyaeva, A. G. Mal'kina, A. V. Afonin, V. M. Muzalevskii, V. G. Nenaidenko and B. A. Trofimov, Russ. J. Org. Chem., 2016, 52, 1857–1860 CrossRef CAS; (b) B. A. Trofimov, L. V. Andriyankova, K. V. Belyaeva, L. P. Nikitina, A. V. Afonin and A. G. Mal'kina, Eur. J. Org. Chem., 2015, 7876–7879 CrossRef CAS; (c) T. D. Michels, J. U. Rhee and C. D. Vanderwal, Org. Lett., 2008, 10, 4787–4790 CrossRef CAS PubMed.
- L.-L. Wang, H.-B. Han, Z.-H. Cui, J.-W. Zhao, Z.-W. Bu and Q.-L. Wang, Org. Lett., 2020, 22, 873–878 CrossRef CAS PubMed.
- (a) H.-J. Miao, L.-L. Wang, H.-B. Han, Y.-D. Zhao, Q.-L. Wang and Z.-W. Bu, Chem. Sci., 2020, 11, 1418–1424 RSC; (b) S.-J. Jin, L.-L. Wang, H.-B. Han, X.-L. Liu, Z.-W. Bu and Q.-L. Wang, Chem. Commun., 2021, 57, 359–362 RSC; (c) H.-J. Miao, X.-G. Bai, L.-L. Wang, J.-H. Yu, Z.-W. Bu and Q.-L. Wang, Org. Chem. Front., 2021, 8, 204–211 RSC; (d) X.-G. Bai, H.-J. Miao, Y. Zhao, Q.-L. Wang and Z.-W. Bu, Org. Lett., 2020, 22, 5068–5073 CrossRef CAS PubMed; (e) J.-M. Guo, H.-J. Miao, Y. Zhao, X.-G. Bai, Y.-S. Zhu, Q.-L. Wang and Z.-W. Bu, Chem. Commun., 2019, 55, 5207–5210 RSC; (f) W.-B. Wang, X.-G. Bai, S.-J. Jin, J.-M. Guo, Y. Zhao, H.-J. Miao, Y.-S. Zhu, Q.-L. Wang and Z.-W. Bu, Org. Lett., 2018, 20, 3451–3454 CrossRef CAS PubMed; (g) Y.-S. Zhu, J. Zhou, S.-J. Jin, H.-H. Dong, J.-M. Guo, X.-G. Bai, Q.-L. Wang and Z.-W. Bu, Chem. Commun., 2017, 53, 11201–11204 RSC.
- For selected examples, see: (a) T. Rasheed, M. N. I. Khan, S. S. A. Zhadi and S. Durrani, J. Nat. Prod., 1991, 54, 582–584 CrossRef CAS; (b) A. Mertens, H. Zilch, B. Koenig, W. Schaefer, T. Poll, W. Kampe, H. Seidel, U. Leser and H. Leinert, J. Med. Chem., 1993, 36, 2526–2535 CrossRef CAS PubMed; (c) Z.-H. Sun, J. Altom, V. N. Nguyen, J. Fernandez, J. I. Bernstein, J. J. Hiliard, J. F. Barrett, B. L. Podlogar and K. A. Ohemeng, Bioorg. Med. Chem., 1998, 6, 735–742 CrossRef.
- For selected examples, see: (a) Y. Quevedo-Acosta, I. D. Jurberg and D. Gamba-Sánchez, Org. Lett., 2020, 22, 239–243 CrossRef CAS PubMed; (b) J. Li, S.-H. Bai, Y. Li, Z.-B. Wang, X.-Y. Huo and L. Liu, J. Org. Chem., 2018, 83, 8780–8785 CrossRef CAS PubMed; (c) D. Grosheva and N. Cramer, ACS Catal., 2017, 7, 7417–7420 CrossRef CAS; (d) S.-C. Shi, R. Lalancette, R. Szostak and M. Szostak, Chem.–Eur. J., 2016, 22, 11949–11953 CrossRef CAS PubMed; (e) M. Janjić, R. Prebil, U. Grošelj, D. Kralj, Č. Malavašič, A. Golobič, K. Stare, G. Dahmann, B. Stanovnik and J. Svete, Helv. Chim. Acta, 2011, 94, 1703–1717 CrossRef; (f) G. Satyanarayana, C. Maichle-Mössmer and M. E. Maier, Chem. Commun., 2009, 1571–1573 RSC; (g) J. J. Sahn, J. Y. Su and S. F. Martin, Org. Lett., 2011, 13, 2590–2593 CrossRef CAS PubMed.
- For selected reviews, see: (a) F. Zhou, L. Zhu, B.-W. Pan, Y. Shi, Y.-L. Liu and J. Zhou, Chem. Sci., 2020, 11, 9341–9365 RSC; (b) C.-X. Li, S. S. Ragab, G.-D. Liu and W.-J. Tang, Nat. Prod. Rep., 2020, 37, 276–292 RSC; (c) L. Süsse and B. M. Stoltz, Chem. Rev., 2021, 121, 4084–4099 CrossRef PubMed; (d) W.-C. Xue, X. Jia, X. Wang, X.-H. Tao, Z.-G. Yin and H.-G. Gong, Chem. Soc. Rev., 2021, 50, 4162–4184 RSC; (e) T. T. Talele, J. Med. Chem., 2020, 63, 13291–13315 CrossRef CAS PubMed; (f) F. Ye, Z. Xu and L.-W. Xu, Acc. Chem. Res., 2021, 54, 452–470 CrossRef CAS PubMed; (g) X.-P. Zeng, Z.-Y. Cao, Y.-H. Wang, F. Zhou and J. Zhou, Chem. Rev., 2016, 116, 7330–7396 CrossRef CAS PubMed.
- CCDC 2071226 (3), 2071227 (21), 2071228 (25), 2071230 (26), 2116271 (47), 2086413 (48), 2086415 (52), 2086416 (61), 2086417 (70), 2071232 (72), 2071233 (74) and 2086418 (82).†.
- (a) W. Kohn and L. Sham, J. Phys. Rev., 1965, 140, A1133–A1138 CrossRef; (b) W. Kohn, Rev. Mod. Phys., 1999, 71, 1253–1266 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures; characterization data of all the new products; detailed mechanistic studies; copies of 1H NMR and 13C NMR. CCDC 2071226 (3), 2071227 (21), 2071228 (25), 2071230 (26), 2116271 (47), 2086413 (48), 2086415 (52), 2086416 (61), 2086417 (70), 2071232 (72), 2071233 (74) and 2086418 (82). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc05741c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |