
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLuminol anchors improve the electrochemical-tyrosine-click labelling of proteins†
Sébastien
Depienne
*a,
Dimitri
Alvarez-Dorta
a,
Mikael
Croyal
bcd,
Ranil C. T.
Temgoua
 a,
Cathy
Charlier
e,
David
Deniaud
a,
Mathieu
Mével
af,
Mohammed
Boujtita
a and
Sébastien G.
Gouin
*a
a,
Cathy
Charlier
e,
David
Deniaud
a,
Mathieu
Mével
af,
Mohammed
Boujtita
a and
Sébastien G.
Gouin
*a
aUniversité de Nantes, CNRS, CEISAM UMR 6230, F-44000 Nantes, France. E-mail: sebastien.gouin@univ-nantes.fr; sebastien.depienne@univ-nantes.fr
bUniversité de Nantes, CNRS, INSERM, L'institut du Thorax, F-44000 Nantes, France
cUniversité de Nantes, CHU Nantes, Inserm, CNRS, SFR Santé, Inserm UMS 016, CNRS UMS 3556, F-44000 Nantes, France
dCRNH-Ouest Mass Spectrometry Core Facility, F-44000 Nantes, France
eIMPACT Platform, Interactions Moléculaires Puces ACTivités, UMR CNRS 6286 UFIP, Université de Nantes, F-44000 Nantes, France
fINSERM UMR 1089, Université de Nantes, CHU de Nantes, 44200 Nantes, France
First published on 10th November 2021
Abstract
New methods for chemo-selective modifications of peptides and native proteins are important in chemical biology and for the development of therapeutic conjugates. Less abundant and uncharged amino-acid residues are interesting targets to form less heterogeneous conjugates and preserve biological functions. Phenylurazole (PhUr), N-methylphenylurazole (NMePhUr) and N-methylluminol (NMeLum) derivatives were described as tyrosine (Y) anchors after chemical or enzymatic oxidations. Recently, we developed the first electrochemical Y-bioconjugation method coined eY-click to activate PhUr in biocompatible media. In this work, we assessed the limitations, benefits and relative efficiencies of eY-click conjugations performed with a set of PhUr, NMePhUr and NMeLum derivatives. Results evidenced a high efficiency of NMeLum that showed a complete Y-chemoselectivity on polypeptides and biologically relevant proteins after soft electrochemical activation. Side reactions on nucleophilic or heteroaromatic amino-acids such as lysine or tryptophan were never observed during mass spectrometry analysis. Myoglobine, bovine serum albumin, a plant mannosidase, glucose oxidase and the therapeutically relevant antibody trastuzumab were efficiently labelled with a fluorescent probe in a two-step approach combining eY-click and strain-promoted azide–alkyne cyclization (SPAAC). The proteins conserved their structural integrity as observed by circular dichroism and the trastuzumab conjugate showed a similar binding affinity for the natural HER2 ligand as shown by bio-layer interferometry. Compared to our previously described protocol with PhUr, eY-click with NMeLum species showed faster reaction kinetics, higher (complete) Y-chemoselectivity and reactivity, and offers the interesting possibility of the double tagging of solvent-exposed Y.
Introduction
Protein bioconjugation is an extensively explored and ever-growing field of research with wide-ranging applications in pharmacology, biotechnology and chemical biology.1–3 These include the development of medically relevant conjugates such as antibody–drug conjugates (ADCs)4 and targeted covalent inhibitors (TCIs),5,6 the improvement of protein-based diagnostics and therapeutics with enhanced solubility and bioavailability,7 and a better understanding of protein–protein interactions (PPIs).5,8Bioorthogonal conjugations can be done with a minutely fine degree of control by site-selective incorporation of an unnatural amino acid in the polypeptidic chain with engineered aminoacyl-tRNA synthetase/tRNA (aaRS/tRNA) pairs.9 Such genetic code expansion techniques are powerful new tools but have to be conducted in specialized laboratories. The chemical modification of native proteins, more easily produced in bulk, remains the most conventional and user-friendly labelling method. Chemical bioconjugations are still mostly performed on nucleophilic lysine (Lys) and cysteine (Cys) amino acids using electrophilic reagents such as NHS-activated esters or maleimides. Modification of the rare Cys precludes a high payload of conjugate and may also be detrimental to protein integrity. Targeting the abundant, surface-exposed and charged Lys residues generally results in the formation of highly heterogeneous mixtures and may significantly impact biological binding. Promising approaches have recently been described to overcome the initial limitations of Lys10,11 and Cys12,13 modifications, and much effort is now dedicated to selectively target less exploited amino acids such as methionine,14 tryptophan,15,16 histidine17,18 or tyrosine (Y).19,20
Y usually occurs at low frequency in proteins (<5%) and is partially buried in the surface, offering unique opportunities for controlled labelling of the most reactive residues.21,22 The phenol side chain of Y is neutral at physiological pH and experiences several biologically relevant post-translational modifications (phosphorylations, sulfations, nitrations, O-glycosylations, oxidations).23 Thus, growing numbers of methods for Y-labelling have been reported over the last decade. Efficient chemical strategies include the use of three-component Mannich-type coupling,24 transition metal complexes,25,26 sulfur(VI) fluoride exchange chemistry (SuFEx),27,28 aryldiazonium salts29,30 and diazodicarboxyamides.31–33 The last of these methods is among the most promising and exemplary. Barbas and co-workers initially showed that silent phenylurazoles (PhUrs) are readily activated to phenyltriazolinediones (PTADs) using chemical oxidizers (Method A, Fig. 1) such as dibromodimethylhydantoin (DBDMH) or N-bromosuccinimide (NBS). Nonetheless, PTADs are known to be unstable in aqueous environment and to decompose into an electrophilic phenylisocyanate specie responsible for undesired side reactions on amino groups.32,34 In a previous study, we developed the first electrochemical tyrosine bioconjugation, named eY-click (Method C, Fig. 1).35 Greater protein labelling and Y-selectivity could be achieved this way than by using the chemical approach, in conventional buffers, without the need for an isocyanate scavenger. Thus, electrochemical methods offer interesting perspectives for site-selective modification of proteins.35–38 Y anchors based on N-methylphenylurazole (NMePhUr) or N-methylluminol (NMeLum) derivatives may also be successfully activated under single-electron transfer (SET) reactions as shown by Nakamura's research group (Method B & C, Fig. 1).37,39–43 NMePhUr and NMeLum were activated using ruthenium photocatalysts,44 hemin and H2O2,40 enzymatic systems (horseradish peroxidase and H2O2, laccase)37,39,45 and electrochemically.43 In the present study, we aimed to evaluate and compare the relative efficiency of new and previously reported urazoles and luminols derivatives in the electrochemical-tyrosine-click labelling strategy (Fig. 1). The scope and efficiency of the eY-click protocol was significantly improved with NMeLum derivatives. Complete Y-selectivity has now been achieved in a fully biocompatible procedure with faster kinetics and a higher conjugate payload.
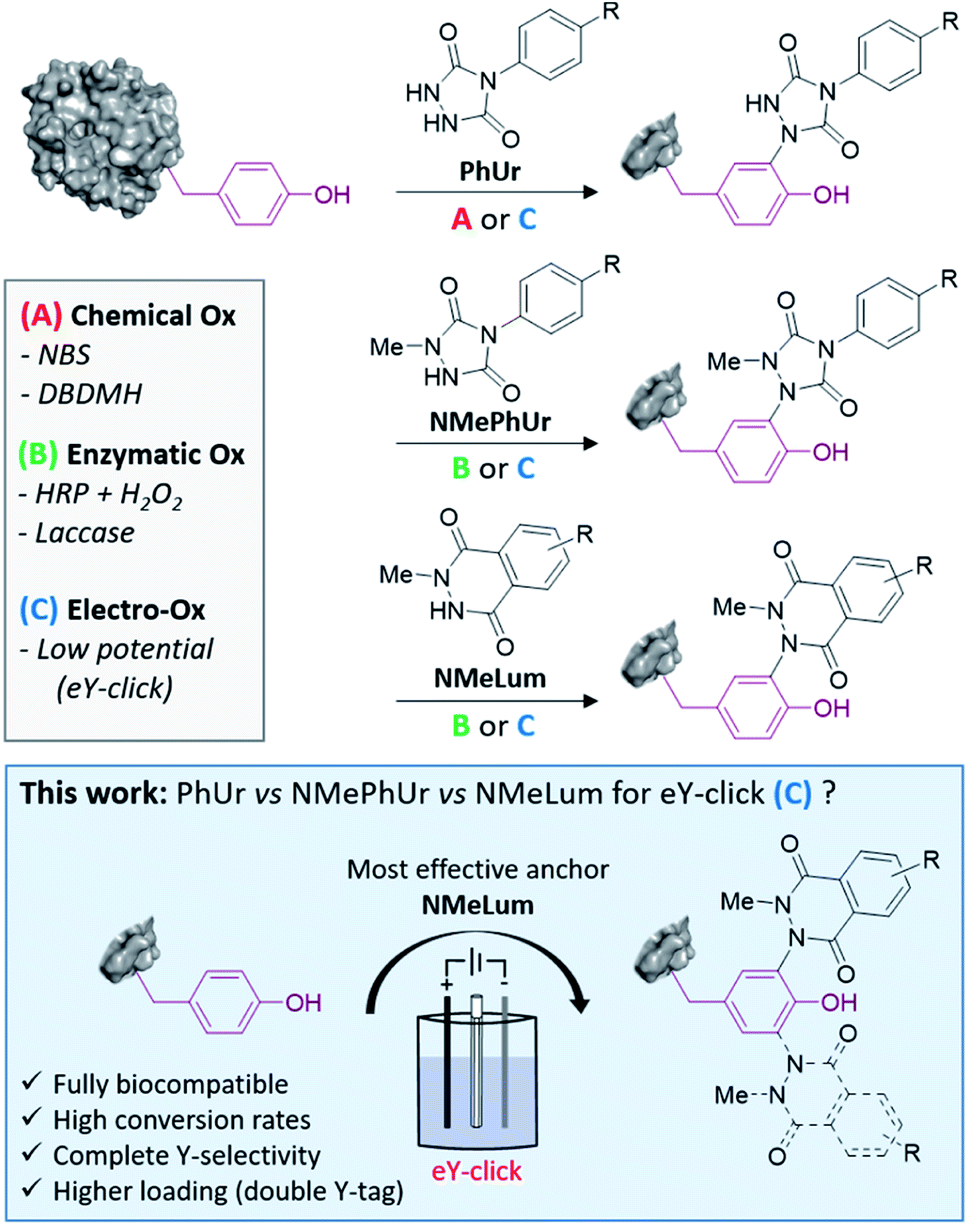 | ||
| Fig. 1 PhUr, NMePhUr and NMeLum may be activated by chemical oxidation (Method A), enzymatically (Method B) or electrochemically (Method C). In this work we found that NMeLum are very efficient anchors for the soft electrochemical Y labelling of proteins. | ||
Results and discussion
We first studied the electrochemical behaviour of new PhUr derivatives 1–6 and previously reported NMePhUr 7-8 and NMeLum 9-10 reagents (Fig. 2a).43 The stability of their oxidized species was compared by cyclic voltammetry. Compounds 1–6 were designed (synthesis described in ESI†) to assess whether electro-donating or electro-withdrawing substituents on the aromatic ring could impact the stability of the electro-generated PTADs, and therefore limit the formation of corresponding isocyanate by-products. The potential impact of a bioorthogonal handle (azidoethyl group) was also investigated. Experiments were performed in Tris/MeCN buffer (pH 7.4) as the electrolyte, with a three-electrode system using a graphite working electrode, a platinum wire as counter electrode, and a saturated calomel electrode (SCE) for reference. The voltammograms (Fig. 2b) showed that substitution on the aromatic ring of PhUr (2–6) did not impact its oxidation potential (O1, 0.33 V vs. SCE) or the intensity of the reduction peak (R1), i.e. the PTADs stability. Thus, aryl substituents do not impact the electro-oxidation process, outlining that a wide range of biorthogonal handles or ligands may be appended to the PhUr anchor. On the contrary, N-methyl substitution of the hydrazide function in 7, 8 (NMePhUr) and 9, 10 (NMeLum) resulted in a shift of the oxidation peak to higher potentials (O2, 0.52 V vs. SCE and O3, 0.62 V vs. SCE) and in substantially higher reduction intensities (R2 and R3) suggesting the formation of much more stable oxidized species. We further compared stabilities of PhUr, NMePhUr and NMeLum oxidized species by examining both oxidation and reduction peaks obtained from 20 repetitive cyclic voltammetry measurements (Fig. 2c). A significant gradual decrease in the peak intensity of 1 highlighted the well-known instability of PTAD, as the oxidation peak of the second cycle (9.5 μA) already lost half the initial intensity (19 μA). This contrasts with the results obtained for 7 and 9 where constant oxidation and reduction peak intensities are observed over 20 cycles, confirming a high stability of the oxidized species.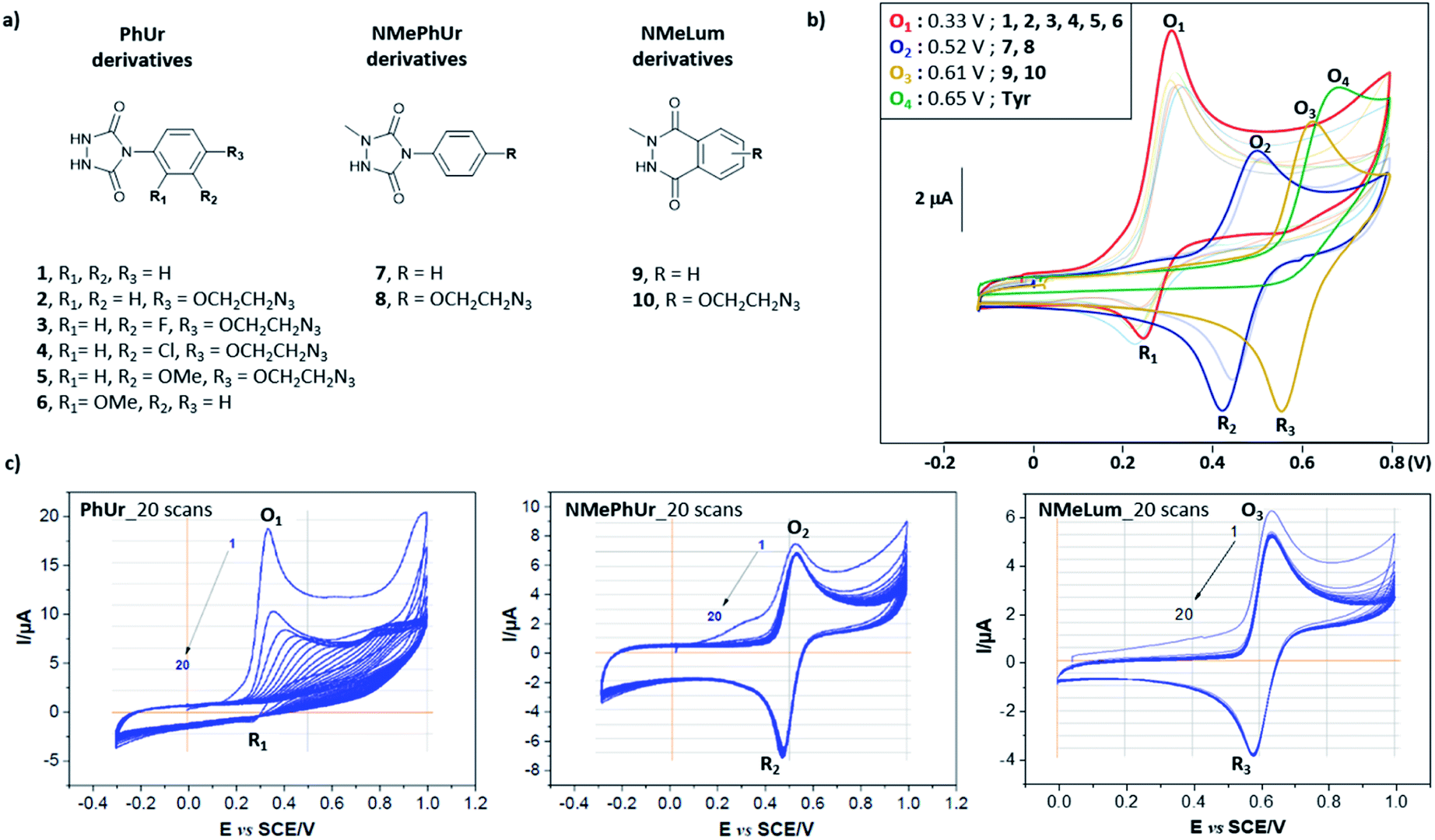 | ||
Fig. 2 Electrochemical behaviour of the studied Y anchors PhUr 1–6, NMePhUr 7, 8 and NMeLum 9, 10. (a) Chemical structures of the compounds 1–10, (b) Cyclic voltammetry of 1–10 and Y (cathode: graphite carbon electrode 2 mm disc, anode: platinum wire, reference: saturated calomel electrode, 100 mV s−1, reagent 1 mM, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 MeCN/Tris 50 mM pH 7.4), (c) Multicyclic voltammetry of 1, 7 and 9 (cathode: graphite carbon electrode 2 mm disc, anode: platinum wire, reference: saturated calomel electrode, 100 mV s−1, reagent 1 mM, 1:1 MeCN/NH4OAc 100 mM pH 7.4). :1 MeCN/Tris 50 mM pH 7.4), (c) Multicyclic voltammetry of 1, 7 and 9 (cathode: graphite carbon electrode 2 mm disc, anode: platinum wire, reference: saturated calomel electrode, 100 mV s−1, reagent 1 mM, 1:1 MeCN/NH4OAc 100 mM pH 7.4). | ||
Of note, PhUr 1 is reported to be chemically or electrochemically two-electron oxidized to generate PTAD for aza–ene reaction with the phenol of Y.31,35 Even though a similar two-electron process cannot be excluded for 7 and 9, relative intensities of the oxidation peak of 1 (O1) vs.7 (O2) support a possible single-electron electro-oxidation process for NMePhUr. The formation of a stable nitrogen-centred radical may also prevail for NMeLum considering the similar N-methylhydrazide function.37,43 Cyclic voltammetry covering higher voltage up to 2 V showed a second non-reversible anodic event at 1.5–1.6 V for 7 and 9 (see ESI†), which could correspond to the second electron oxidation (diazenium formation), as recently proposed.43 Thus, electro-activation of NMePhUr and NMeLum derivatives at low potential could promote radical coupling with Y in an analogous manner as the enzymatic and photo activation protocols.37,44
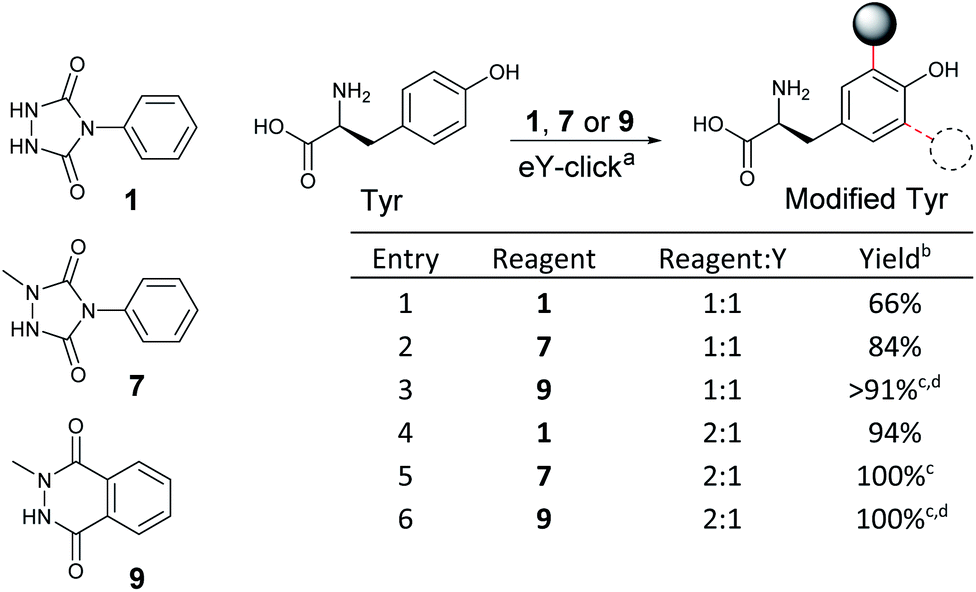
As NMePhUr and NMeLum oxidation potentials shifted to higher values close to that of Y (O4, 0.65 V vs. SCE), we investigated whether these two anchors could be selectively electro-oxidized in the presence of Y. The Y-tagging efficiency was compared with that observed with PhUr in eY-click conditions (Table 1). The electrolysis assays were conducted under stirring conditions, in phosphate buffer (PB) at pH 7.4 (100 mM). Appropriate working potentials were applied until 90% of the theoretical charge given by the Faraday law was generated. For a stoichiometric reagent/Y ratio, overall higher Y conversion was observed with 7 (84%) or 9 (91%) than with 1 (66%). However, the first electrolysis assay with 9 performed in PB at 0.62 V vs. SCE led to partial competitive Y oxidation. This issue was solved by replacing PB with ammonium acetate buffer (pH 7.4). The latter doesn't result in an oxidation potential shift of 9 or Y as confirmed by cyclic voltammetry. We suppose that NMeLum reagent has a better diffusion coefficient in this buffer of different ionic strength, favouring its oxidation in presence of Y. When two equivalents of reagents were used, Y-labelling with 1 reached 94% (entry 4) and a complete conversion was observed with 7 and 9. Interestingly, a double addition adduct was detected by mass spectrometry (MS) for 7(entry 5) and 9 (entry 6). These results show that electro-oxidized species of N-methyl derivatives 7 and 9 can tag Y in higher conversion than PTAD and offer possibility of Y double tagging.
| a Reaction conditions: carbon crucible anode, platinum wire cathode, constant voltage vs. SCE, reagent (0.05–0.10 mmol, 1–2 mM), Tyr (0.05 mmol, 1 mM), phosphate buffer pH 7.4 (50 mL), 2–5 h. b % conv. of Y determined by 1H NMR. c Double modification of Y was detected by MS. d Ammonium acetate pH 7.4 buffer (50 mL) was used. |
|---|
|
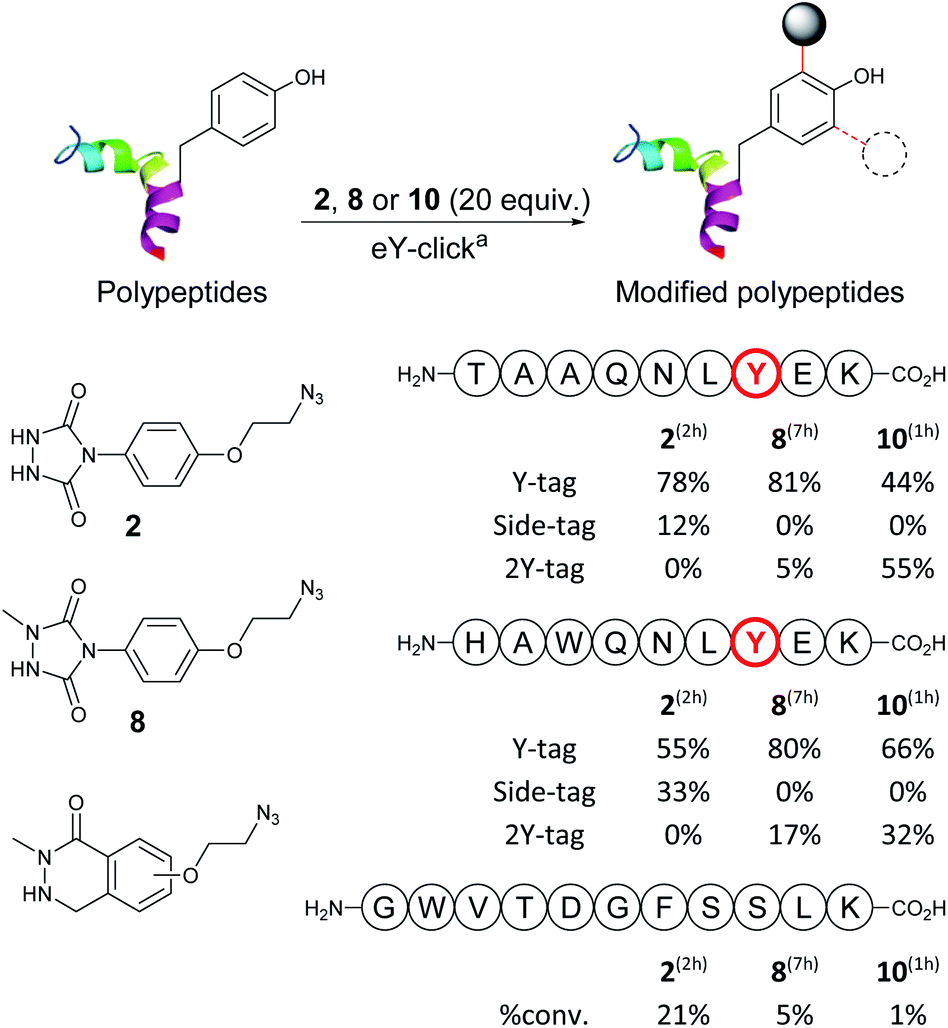
Next, we evaluated and compared the labelling efficiency of azido-armed analogues 2, 8 and 10 (yields, chemo-selectivity and kinetics) on unprotected polypeptides (Table 2). We selected the synthetic nonapeptides TAAQNLYEK, bearing one Y and one lysine (K) and HAWQNLYEK, bearing one Y, one K, a tryptophan (W) and a histidine (H) to evaluate potential side-reactivity on nucleophilic amines and heteroaromatic amino-acids. GWVTDGFSSLK was also selected, as a 11-mer peptide lacking Y, to do a control experiment. The eY-click protocols were performed with 20 equivalents of 2, 8 and 10 (2 mM), as reagent excess are generally needed for protein labelling, and to better assess potential selectivity issues. Samples were periodically collected and analysed by liquid chromatography-tandem mass spectrometry (LC-MS/MS). eY-click performed with 2 on TAAQNLYEK afforded mainly the expected Y-tag conjugate (78%) after 2 h electrolysis as determined by MS/MS fragmentation. N-Terminus urea adducts (12%) from partial decomposition of PTAD into an isocyanate were also observed. Longer electrolysis increased the urea adducts proportion (results not shown). As in previous experiments,35 K adducts were not observed, which could be explained by a lower pKa value of the N-terminus amino groups, due to amide bond proximity.46 NMePhUr derivative 8 required a long reaction time (7 h) but converted the native peptide with high tagging yields (81% Y-tag and 5% 2Y-tag adducts). Side products were not observed, demonstrating complete Y-selectivity. The eY-click with NMeLum derivative 10 was also conducted in PB even though Y oxidation was observed on the free amino acid due to overlapping oxidation potentials (Table 1). Strikingly, by-products from Y oxidation were never observed and the peptide was fully converted into the Y adducts (44% Y-tag and 55% 2Y-tag) after only 1 h.
| a Reaction conditions: graphite plate anode, platinum wire cathode, constant voltage vs. SCE, modification reagent (25 mmol, 2 mM, 20.0 equiv.), polypeptide (1.25 mmol, 0.1 mM, 1.0 equiv.), phosphate buffer (100 mM, 12.5 mL), 700 rpm, room temperature, 2–7 h. Location of conjugation determined by LC-MS/MS analysis. |
|---|
|
Thus, faster kinetics, complete Y selectivity, and higher level of double Y modification were observed with 10. Despite the higher oxidation potential to generate the activated specie (620 mV), the lower diffusion coefficient of the peptide at the anode surface and its lower concentration in solution compared to 10 prevented Y oxidation. A similar trend was observed with HAWQNLYEK. eY-click with 2 led to 55% of the Y-tag peptide after 2 h, with 33% side reactions characterized by MS/MS due to N-terminus modification and W addition. The latter side-reaction has rarely been reported in literature probably because of the low W abundance/accessibility and thermoreversible indole-TAD adduct.47 A recent study showed the TAD-W instability and the kinetically favoured reactivity of TAD for exposed W over Y, allowing a selective W modification on proteins.48 Reagents 8 and 10 efficiently labelled (97–98% overall yields) the peptide with a complete Y-selectivity (K, H, W tagging never observed) and Y-tag/2Y-tag ratios of 80:17 and 66:32, respectively. Again, much faster kinetics were observed with 10. Labelling experiments performed on control peptide GWVTDGFSSLK, which is missing a Y residue, confirmed the good to excellent inertness of electro-activated compounds 2, 8, 10 for other amino-acids with 21%, 5% and 1% of parent peptide conversion, respectively. Thus, peptide experiments revealed a much higher eY-click labelling efficiency with NMeLum 10 and, to a lesser extent, NMePhUr 8 compared with the original method using PhUr 2.
Next, 8 and 10 were investigated for protein labelling. α-Chymotrypsinogen A (α-Chymo), a 25.6 kDa pre-digestive enzyme, was selected as a first model. As α-Chymo has a limited number of four accessible Y, it enabled us to easily track and analyse by mass spectrometry the Y adducts. The eY-click conjugations were performed on a low concentration of α-Chymo (1 μM) during fixed times of 4 and 1 h for 8 and 10, respectively (Fig. 3a), according to the relative compounds reactivity on peptides (Table 2). Mass profiles showed a clean labelling distribution of protein adducts after deconvolution. As observed with peptides, 10 exhibited a high tagging potency as +1 to +5 adducts of 10-Chymo could be revealed by MS after 1 h with near complete disappearance of the unmodified protein peak (Fig. 3c). In stark contrast, 8 only partially labelled the protein batch up to +2-tags in a 4 h electrolysis (Fig. 3b). Of note, a poor labelling efficiency of α-Chymo was reported with PhUr species after chemical activation.32 Interestingly, formation of the +5 tag adduct with 10 supports a partial double tagging of a single Y, as observed on polypeptides (Table 2). We further studied the 10-Chymo sample and performed enzymatic proteolysis with pepsin (pH 3) to detect peptide fragments and identify location and nature of the modifications. Satisfyingly, both single and double Y-modification of the proteolytic peptide TRYTNA containing 146Y could be evidenced by LC-MS analysis and MS/MS fragmentation, which confirmed the ability of 10 to double-tag solvent-exposed Y at protein scale.
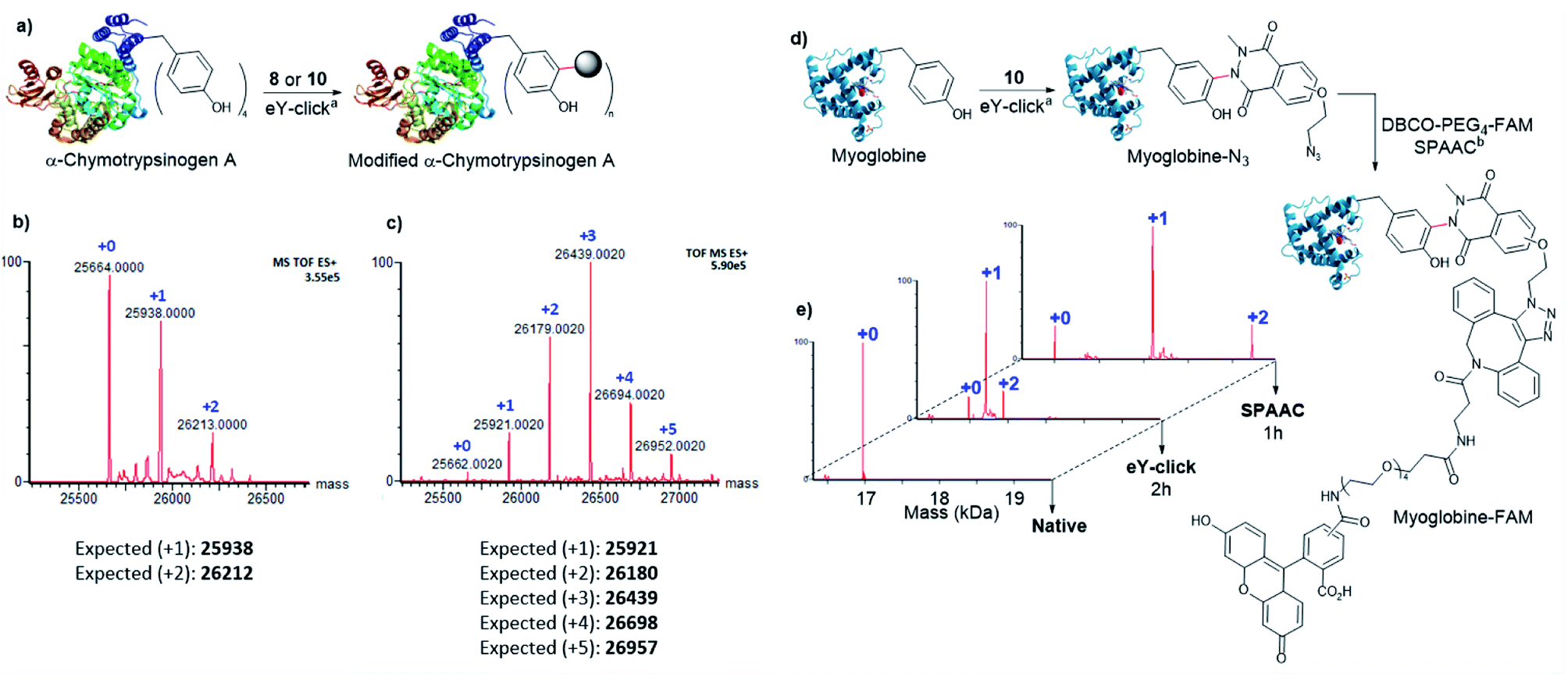 | ||
| Fig. 3 eY-click protocol on proteins. (a) Modification of α-chymotrypsinogen A, areaction conditions: graphite plate anode, platinum plate cathode, constant voltage vs. Ag/AgCl, modification reagent (5.0 μmol, 1.0 mM), α-Chymo (5.0 nmol, 1.0 μM), phosphate buffer (100 mM, 5.0 mL), 500 rpm, room temperature, 4 h (8) or 1 h (10), (b) Deconvoluted profile (MS) for α-Chymo modification by 8 after 4 h eY-click, (c) deconvoluted profile (MS) for α-Chymo modification by 10 after 1 h eY-click, (d) two-steps modification of myoglobin, areaction conditions: graphite plate anode, platinum plate cathode, constant voltage vs. Ag/AgCl, 10 (5.0 μmol, 1.0 mM), Myo (5.0 nmol, 1.0 μM), phosphate buffer (100 mM, 5.0 mL), 500 rpm, room temperature, 1 h, breaction conditions: Myo-N3 (2.0 nmol, 1.0 equiv.), DBCO-PEG4-5/6-FAM (100.0 nmol, 50.0 equiv.), distillated water (0.5 mL), DMF (3 μL), 37 °C, 1 h, (e) deconvoluted profiles (MS) evolution of Myo two-steps modification. | ||
Next, the capacity of 10 to label Y with a low accessibility (partially buried in the protein surface) was assessed on myoglobin (Myo), a 17 kDa oxygen-binding protein with two Y, one being inaccessible (deeply buried) and the other only poorly exposed to the solvent (Fig. 3d). A high conversion was observed after 2 h with the formation of the +1 and +2 tags protein adducts (Fig. 3e). To evaluate if both +1 and +2 10-Myo adducts were indeed functional for bioconjugation, the samples were engaged in a strain promoted azide–alkyne cycloaddition (SPAAC) with the fluorescent probe DBCO-PEG4-5/6-FAM during 1 h at 37 °C. To our delight, the deconvoluted MS profile showcased the two functionalized adducts with the expected +1 (+880 Da) and +2 (+1760 Da) mass shift (Fig. 3e).
The softness of the eY-click protocol with 10 was next evaluated on a set of proteins including Bovine Serum Albumin (BSA), Jack bean α-mannosidase (α-ManJB), and Glucose Oxidase enzyme (GOx) which is used for blood glucose monitoring and in cancer diagnosis and treatment (Fig. 4a). Structural integrity of the proteins after eY-click conjugations of 10 was confirmed by circular dichroism analysis, where conjugated proteins showed no alteration in secondary structural contents, highlighting the softness of the method (Fig. 4b). The obtained azido-armed proteins were further functionalized by SPAAC with DBCO-PEG4-5/6-FAM for 1 h at 37 °C and analyzed by SDS-PAGE. Brilliant Blue Coomassie detection of native proteins and FAM-protein adducts showed a unique band at the expected molecular weight for BSA and BSA-FAM (∼65–70 kDa), GOx and GOx-FAM (∼80–85 kDa) and two bands for the heterodimeric α-ManJB and α-ManJB-FAM (∼45–50 kDa and ∼60–65 kDa). Fluorescent detection at 492 nm unambiguously proved the formation of protein-FAM adducts without protein degradation (Fig. 4c).
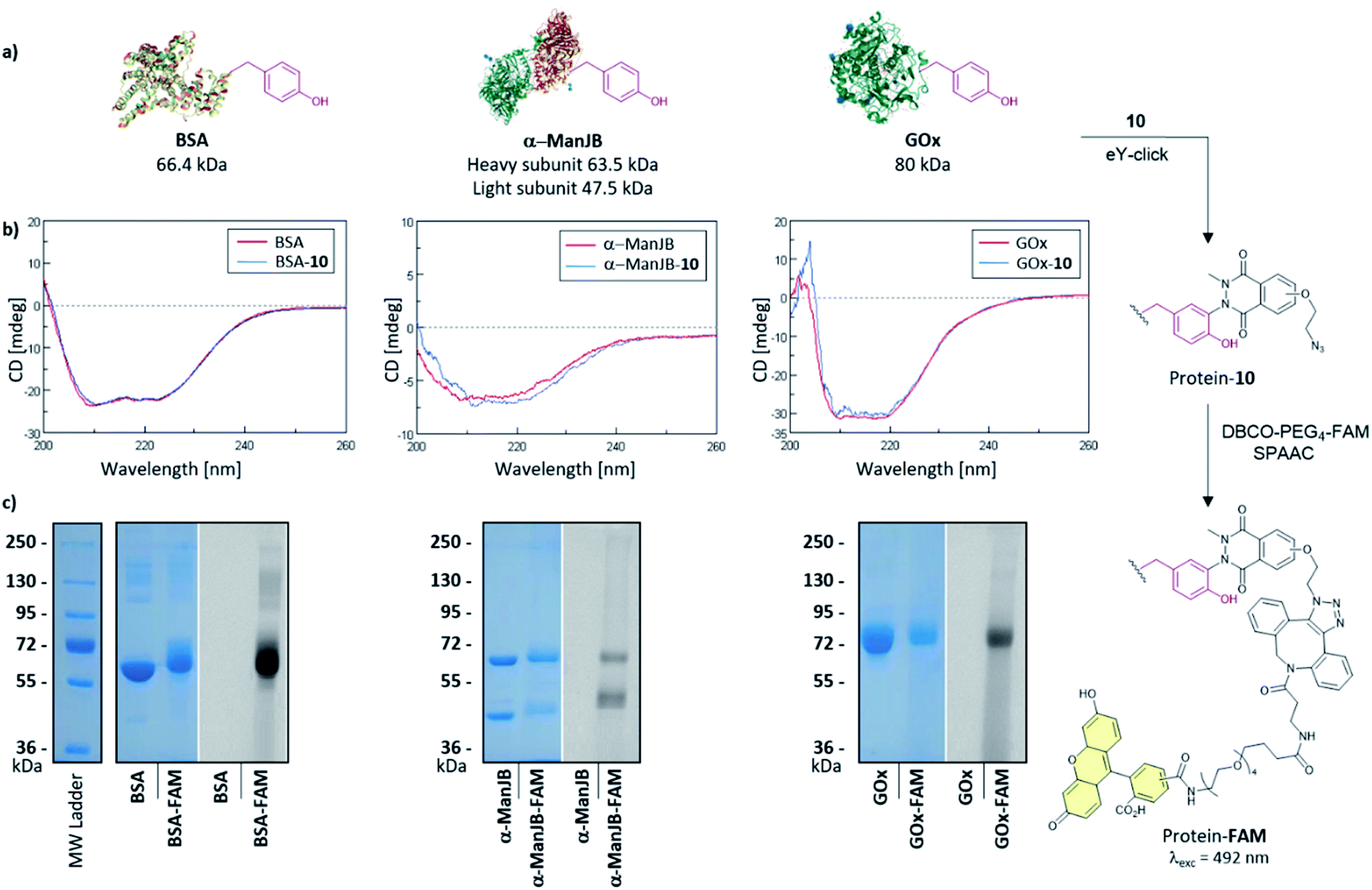 | ||
| Fig. 4 (a) Two-steps modification of BSA, α-ManJB and GOx. (b) CD analysis of the native (red) and eY-click modified (blue) proteins with 10. (c) SDS-PAGE analysis of native proteins (from left to right: BSA, α-ManJB and GOx) and their FAM conjugates after eY-click with 10 followed by SPAAC with DBCO-PEG4-5/6-FAM. Native and conjugated proteins were revealed by coomassie brilliant blue (left side) and fluorescence was detected at 492 nm (right side). | ||
Suitable bioconjugation techniques for antibody-conjugates development are still being extensively explored and are required for vectorized immunotherapies or cancer diagnostics.49–51 We finally evaluated eY-click with 10 for antibody labelling. Trastuzumab (Tras, ∼150 kDa) was selected as a model monoclonal antibody referenced for breast cancer therapies due to its high affinity for the overexpressed Human Epidermal Growth Factor Receptor-2 (HER2). Structural integrity of the Tras-10 conjugate was confirmed by circular dichroism (Fig. 5a), and SDS-PAGE revealed successful subsequent FAM labelling with DBCO-PEG4-5/6-FAM (Fig. 5c). The functional activity of the Tras-10 conjugate for HER2 receptor was also investigated by bio-layer interferometry (BLI). HER2 was immobilized on the biosensor tip surface and the binding affinity was measured with solutions of Tras and Tras-10. Tras showed a nanomolar affinity for HER2 (dissociation constant KD = 1.2 nM). A high affinity for HER2 was also measured for Tras-10 which showed a KD in the same order as Tras (KD = 3.8 nM). Thus, eY-click with 10 did not compromise HER2 binding and represents a soft and effective methodology for antibody labelling.
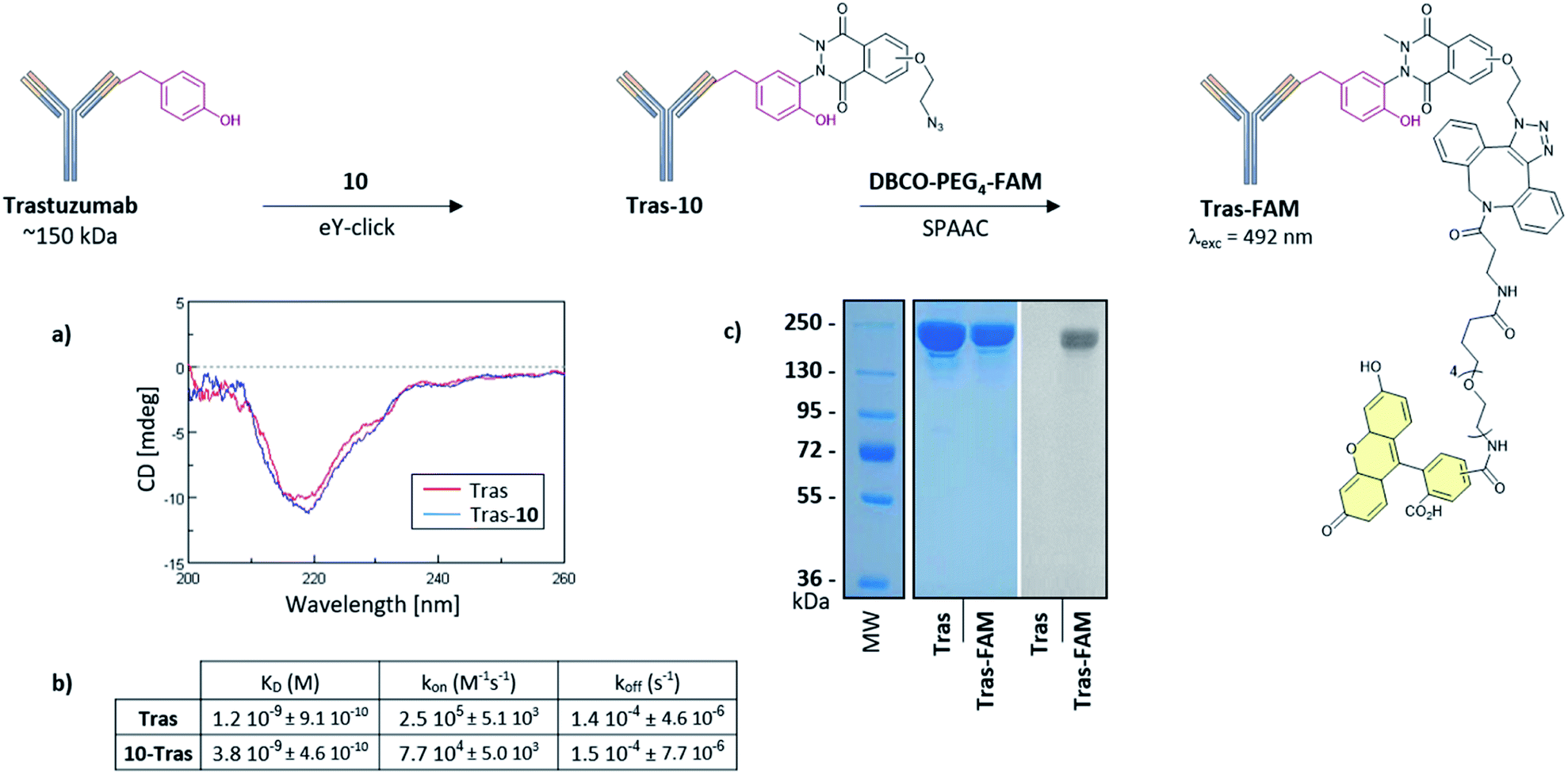 | ||
| Fig. 5 Two-steps modification of Tras. (a) CD analysis of the native (red) and eY-click modified (blue) Tras with 10. (b) Affinities of Tras and Tras-10 for HER2. Dissociation constants (KD) and binding kinetic parameters (kon, koff) were measured and plotted by bio-layer interferometry. (c) SDS-PAGE analysis of native Tras and its FAM conjugate after eY-click with 10 followed by SPAAC with DBCO-PEG4-5/6-FAM. Native and conjugated Tras were revealed by coomassie brilliant blue (left side) and fluorescence was detected at 492 nm (right side). | ||
Conclusions
Y-labelling methods have been extensively explored to study Y post-translational modifications and protein conformations,52 and to design more defined protein conjugates. Chemical, enzymatic and electrochemical activations of Y anchors are complementary tools with specific advantages and limitations. Our initial electrochemical protocol for the activation of PhUr anchors is an efficient strategy for soft, time-controlled Y-labelling of native protein, not requiring chemical oxidant or enzymatic catalysis. Nevertheless, reaction kinetics were slower than with chemical approaches and the present work revealed that complete Y-selectivity may not be fully achieved depending on peptide substrates. Even though the well-known PhUr degradation into electrophilic phenylisocyanate is considerably limited during electrochemical activation, preventing lysine modification, a partial side-tagging of the N-terminus amine of peptide was observed here with PhUr 2. The heteroaromatic amino acid side-chain of tryptophan was also not fully inert towards electrogenerated PTAD, as previously shown after chemical activation.48In this work, we showed that complete Y-chemoselectivity can now be achieved by electrochemical activation of NMeLum derivatives such as 10. Although a higher oxidation potential is required for electro-activation of NMeLum compared to PhUr species, this has virtually no impact on peptides and proteins due to their lower diffusion coefficient at the electrode surface. NMeLum derivatives provide complete Y-selectivity, faster reaction kinetics, and display a higher reactivity for less surface-exposed Y, with possible double Y modification for a higher payload. Altogether, these results expand the scope of eY-click as a chemoselective, soft and user-friendly method for peptides and proteins bioconjugations.
Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
Conceptualization, supervision and writing: S. D., M. B. and S. G. Revising: D. D, Acquisition and formal analysis: S. D.; D. A-D.; M. M. and R. C. T. T. (chemistry & electrochemistry), M. C. (MS analysis), C. C. (DC & BLItz).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was carried out with financial support from the Centre National de la Recherche Scientifique (CNRS), the Ministère de l’Enseignement Supérieur et de la Recherche in France and the National Agency for Research (ANR project ECLICK).Notes and references
- O. Boutureira and G. J. L. Bernardes, Chem. Rev., 2015, 115, 2174–2195 CrossRef CAS.
- C. D. Spicer and B. G. Davis, Nat. Commun., 2014, 5, 4740 CrossRef CAS.
- Q.-Y. Hu, F. Berti and R. Adamo, Chem. Soc. Rev., 2016, 45, 1691–1719 RSC.
- V. Chudasama, A. Maruani and S. Caddick, Nat. Chem., 2016, 8, 114–119 CrossRef CAS PubMed.
- T. Ueda, T. Tamura, M. Kawano, K. Shiono, F. Hobor, A. J. Wilson and I. Hamachi, J. Am. Chem. Soc., 2021, 143, 4766–4774 CrossRef CAS PubMed.
- T. Tamura, T. Ueda, T. Goto, T. Tsukidate, Y. Shapira, Y. Nishikawa, A. Fujisawa and I. Hamachi, Nat. Commun., 2018, 9, 1870 CrossRef PubMed.
- F. M. Veronese and A. Mero, BioDrugs, 2008, 22, 315–329 CrossRef CAS.
- M. A. Trakselis, S. C. Alley and F. T. Ishmael, Bioconjugate Chem., 2005, 16, 741–750 CrossRef CAS PubMed.
- J. C. M. van Hest, K. L. Kiick and D. A. Tirrell, J. Am. Chem. Soc., 2000, 122, 1282–1288 CrossRef CAS.
- A.-D. Guo, D. Wei, H.-J. Nie, H. Hu, C. Peng, S.-T. Li, K.-N. Yan, B.-S. Zhou, L. Feng, C. Fang, M. Tan, R. Huang and X.-H. Chen, Nat. Commun., 2020, 11, 5472 CrossRef CAS PubMed.
- M. J. Matos, B. L. Oliveira, N. Martínez-Sáez, A. Guerreiro, P. M. S. D. Cal, J. Bertoldo, M. Maneiro, E. Perkins, J. Howard, M. J. Deery, J. M. Chalker, F. Corzana, G. Jiménez-Osés and G. J. L. Bernardes, J. Am. Chem. Soc., 2018, 140, 4004–4017 CrossRef CAS PubMed.
- V. F. C. Ferreira, B. L. Oliveira, A. D'Onofrio, C. M. Farinha, L. Gano, A. Paulo, G. J. L. Bernardes and F. Mendes, Bioconjugate Chem., 2021, 32, 121–132 CrossRef CAS PubMed.
- B. Bernardim, M. J. Matos, X. Ferhati, I. Compañón, A. Guerreiro, P. Akkapeddi, A. C. B. Burtoloso, G. Jiménez-Osés, F. Corzana and G. J. L. Bernardes, Nat. Protoc., 2019, 14, 86–99 CrossRef CAS PubMed.
- A. H. Christian, S. Jia, W. Cao, P. Zhang, A. T. Meza, M. S. Sigman, C. J. Chang and F. D. Toste, J. Am. Chem. Soc., 2019, 141, 12657–12662 CrossRef CAS PubMed.
- Y. Seki, T. Ishiyama, D. Sasaki, J. Abe, Y. Sohma, K. Oisaki and M. Kanai, J. Am. Chem. Soc., 2016, 138, 10798–10801 CrossRef CAS PubMed.
- J. M. Antos and M. B. Francis, J. Am. Chem. Soc., 2004, 126, 10256–10257 CrossRef CAS PubMed.
- X. Chen, F. Ye, X. Luo, X. Liu, J. Zhao, S. Wang, Q. Zhou, G. Chen and P. Wang, J. Am. Chem. Soc., 2019, 141, 18230–18237 CrossRef CAS PubMed.
- K. Nakane, S. Sato, T. Niwa, M. Tsushima, S. Tomoshige, H. Taguchi, M. Ishikawa and H. Nakamura, J. Am. Chem. Soc., 2021, 143, 7726–7731 CrossRef CAS PubMed.
- P. A. Szijj, K. A. Kostadinova, R. J. Spears and V. Chudasama, Org. Biomol. Chem., 2020, 18, 9018–9028 RSC.
- D. Alvarez Dorta, D. Deniaud, M. Mével and S. G. Gouin, Chem.–Eur. J., 2020, 26, 14257–14269 CrossRef CAS PubMed.
- H. Ischiropoulos, Arch. Biochem. Biophys., 1998, 356, 1–11 CrossRef CAS.
- S. Miller, J. Janin, A. M. Lesk and C. Chothia, J. Mol. Biol., 1987, 196, 641–656 CrossRef CAS.
- L. H. Jones, A. Narayanan and E. C. Hett, Mol. BioSyst., 2014, 10, 952–969 RSC.
- N. S. Joshi, L. R. Whitaker and M. B. Francis, J. Am. Chem. Soc., 2004, 126, 15942–15943 CrossRef CAS PubMed.
- S. D. Tilley and M. B. Francis, J. Am. Chem. Soc., 2006, 128, 1080–1081 CrossRef CAS PubMed.
- J. Ohata, M. K. Miller, C. M. Mountain, F. Vohidov and Z. T. Ball, Angew. Chem., Int. Ed., 2018, 57, 2827–2830 CrossRef CAS PubMed.
- N. P. Grimster, S. Connelly, A. Baranczak, J. Dong, L. B. Krasnova, K. B. Sharpless, E. T. Powers, I. A. Wilson and J. W. Kelly, J. Am. Chem. Soc., 2013, 135, 5656–5668 CrossRef CAS.
- E. C. Hett, H. Xu, K. F. Geoghegan, A. Gopalsamy, R. E. Kyne, C. A. Menard, A. Narayanan, M. D. Parikh, S. Liu, L. Roberts, R. P. Robinson, M. A. Tones and L. H. Jones, ACS Chem. Biol., 2015, 10, 1094–1098 CrossRef CAS.
- J. M. Hooker, E. W. Kovacs and M. B. Francis, J. Am. Chem. Soc., 2004, 126, 3718–3719 CrossRef CAS PubMed.
- F. W. Kimani and J. C. Jewett, Angew. Chem., Int. Ed., 2015, 54, 4051–4054 CrossRef CAS PubMed.
- H. Ban, J. Gavrilyuk and C. F. Barbas, J. Am. Chem. Soc., 2010, 132, 1523–1525 CrossRef CAS PubMed.
- H. Ban, M. Nagano, J. Gavrilyuk, W. Hakamata, T. Inokuma and C. F. Barbas, Bioconjugate Chem., 2013, 24, 520–532 CrossRef CAS.
- D. M. Bauer, I. Ahmed, A. Vigovskaya and L. Fruk, Bioconjugate Chem., 2013, 24, 1094–1101 CrossRef CAS PubMed.
- Q.-Y. Hu, M. Allan, R. Adamo, D. Quinn, H. Zhai, G. Wu, K. Clark, J. Zhou, S. Ortiz, B. Wang, E. Danieli, S. Crotti, M. Tontini, G. Brogioni and F. Berti, Chem. Sci., 2013, 4, 3827 RSC.
- D. Alvarez-Dorta, C. Thobie-Gautier, M. Croyal, M. Bouzelha, M. Mével, D. Deniaud, M. Boujtita and S. G. Gouin, J. Am. Chem. Soc., 2018, 140, 17120–17126 CrossRef CAS PubMed.
- C. Song, K. Liu, Z. Wang, B. Ding, S. Wang, Y. Weng, C.-W. Chiang and A. Lei, Chem. Sci., 2019, 10, 7982–7987 RSC.
- S. Sato, K. Nakane and H. Nakamura, Org. Biomol. Chem., 2020, 18, 3664–3668 RSC.
- L. Cui, Y. Ma, M. Li, Z. Wei, Y. Huan, H. Li, Q. Fei and L. Zheng, Anal. Chem., 2021, 93, 4434–4440 CrossRef CAS PubMed.
- S. Sato, K. Nakamura and H. Nakamura, ChemBioChem, 2017, 18, 475–478 CrossRef CAS PubMed.
- S. Sato, K. Nakamura and H. Nakamura, ACS Chem. Biol., 2015, 10, 2633–2640 CrossRef CAS PubMed.
- J. Ertl, M. E. Ortiz-Soto, T. A. Le, J. Bechold, J. Shan, J. Teßmar, B. Engels and J. Seibel, Chem.–Eur. J., 2019, 25, 6533–6541 CrossRef CAS.
- M. E. Ortiz-Soto, J. Ertl, J. Mut, J. Adelmann, T. A. Le, J. Shan, J. Teßmar, A. Schlosser, B. Engels and J. Seibel, Chem. Sci., 2018, 9, 5312–5321 RSC.
- S. Sato, M. Matsumura, T. Kadonosono, S. Abe, T. Ueno, H. Ueda and H. Nakamura, Bioconjugate Chem., 2020, 31, 1417–1424 CrossRef CAS PubMed.
- S. Sato, K. Hatano, M. Tsushima and H. Nakamura, Chem. Commun., 2018, 54, 5871–5874 RSC.
- T. A. Wang, R. Wu, Y. Hong, Z. Wang, T. Li, J. Shie and C. Hsu, ChemBioChem, 2021, 22, 2415–2419 CrossRef CAS.
- C. B. Rosen and M. B. Francis, Nat. Chem. Biol., 2017, 13, 697–705 CrossRef CAS PubMed.
- S. Billiet, K. De Bruycker, F. Driessen, H. Goossens, V. Van Speybroeck, J. M. Winne and F. E. Du Prez, Nat. Chem., 2014, 6, 815–821 CrossRef CAS PubMed.
- K. Decoene, K. Unal, A. Staes, K. Gevaert, J. M. Winne and A. Madder, DOI:10.26434/chemrxiv.13739320.v1.
- C. Sornay, S. Hessmann, S. Erb, I. Dovgan, A. Ehkirch, T. Botzanowski, S. Cianférani, A. Wagner and G. Chaubet, Chem.–Eur. J., 2020, 26, 13797–13805 CrossRef CAS PubMed.
- S. J. Walsh, S. Omarjee, W. R. J. D. Galloway, T. T.-L. Kwan, H. F. Sore, J. S. Parker, M. Hyvönen, J. S. Carroll and D. R. Spring, Chem. Sci., 2019, 10, 694–700 RSC.
- J. J. Bruins, A. H. Westphal, B. Albada, K. Wagner, L. Bartels, H. Spits, W. J. H. van Berkel and F. L. van Delft, Bioconjugate Chem., 2017, 28, 1189–1193 CrossRef CAS PubMed.
- M. Moinpour, N. K. Barker, L. E. Guzman, J. C. Jewett, P. R. Langlais and J. C. Schwartz, Protein Sci., 2020, 29, 1784–1793 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc04809k |
| This journal is © The Royal Society of Chemistry 2021 |