
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCO/light dual-activatable Ru(II)-conjugated oligomer agent for lysosome-targeted multimodal cancer therapeutics†
Min
Yang‡
a,
Hao
Zhao‡
 b,
Ziqi
Zhang
a,
Qiong
Yuan
a,
Qian
Feng
a,
Xinrui
Duan
a,
Shu
Wang
b and
Yanli
Tang
*a
b,
Ziqi
Zhang
a,
Qiong
Yuan
a,
Qian
Feng
a,
Xinrui
Duan
a,
Shu
Wang
b and
Yanli
Tang
*a
aKey Laboratory of Analytical Chemistry for Life Science of Shaanxi Province, Key Laboratory of Applied Surface and Colloid Chemistry, Ministry of Education, School of Chemistry and Chemical Engineering, Shaanxi Normal University, Xi'an, Shaanxi Province 710119, P. R. China. E-mail: yltang@snnu.edu.cn
bBeijing National Laboratory for Molecular Sciences, Key Laboratory of Organic Solids, Institute of Chemistry, Chinese Academy of Sciences, P. R. China
First published on 21st July 2021
Abstract
Stimuli-activatable and subcellular organelle-targeted agents with multimodal therapeutics are urgently desired for highly precise and effective cancer treatment. Herein, a CO/light dual-activatable Ru(II)-oligo-(thiophene ethynylene) (Ru-OTE) for lysosome-targeted cancer therapy is reported. Ru-OTE is prepared via the coordination-driven self-assembly of a cationic conjugated oligomer (OTE-BN) ligand and a Ru(II) center. Upon the dual-triggering of internal gaseous signaling molecular CO and external light, Ru-OTE undergoes ligand substitution and releases OTE-BN followed by dramatic fluorescence recovery, which could be used for monitoring drug delivery and imaging guided anticancer treatments. The released OTE-BN selectively accumulates in lysosomes, physically breaking their integrity. Then, the generated cytotoxic singlet oxygen (1O2) causes severe lysosome damage, thus leading to cancer cell death via photodynamic therapy (PDT). Meanwhile, the release of the Ru(II) core also suppresses cancer cell growth as an anticancer metal drug. Its significant anticancer effect is realized via the multimodal therapeutics of physical disruption/PDT/chemotherapy. Importantly, Ru-OTE can be directly photo-activated using a two-photon laser (800 nm) for efficient drug release and near-infrared PDT. Furthermore, Ru-OTE with light irradiation inhibits tumor growth in an MDA-MB-231 breast tumor model with negligible side effects. This study demonstrates that the development of an activatable Ru(II)-conjugated oligomer potential drug provides a new strategy for effective subcellular organelle-targeted multimodal cancer therapeutics.
Introduction
Cancer, with its high mortality and low therapeutic treatment efficiency, is one of the most serious healthcare problems that threatens human health worldwide.1 The development of highly precise and effective agents, as well as therapeutic methods, is urgently desired for cancer treatments.2,3 Photodynamic therapy (PDT) has been clinically approved as a non-invasive and effective alternative approach to traditional treatments for various types of cancer due to its negligible drug resistance and minimal side effects.4 In PDT, photosensitizers (PSs) can be excited by light and sensitize the surrounding oxygen to produce cytotoxic reactive oxygen species (ROS), which lead to cell death through vasculature damage, apoptosis and/or necrosis pathways, and acute inflammatory reactions.5–8 However, conventional PSs may suffer low effectiveness because of their strict dependence on an abundant oxygen supply, undesirable nonspecific photodamage and simple therapeutic model.9 Stimuli-activatable PSs for multimodal therapeutics provide a new opportunity to cancer therapy, aimed at improving their therapeutic precision and efficiency.Stimuli-activatable agents combined with PSs and drugs, have attracted increasing attention in cancer therapy in virtue of their selective activation, targeted delivery, high systemic safety, and enhanced therapeutic efficiency.10–13 Upon selective activation by external and/or internal stimulations, stimuli-activatable agents release active therapeutic moieties (PSs and drugs) that exercise multi-modal PDT and chemotherapy functions, thus improving selectivity and reducing toxicity towards non-triggered normal tissues. Importantly, subcellular organelle-targeted PDT can enhance the therapeutic efficiency as ROS work robustly in a small radius in biological systems.9,14–16 In particular, lysosomes have emerged as a target for PDT owing to their close relationship with apoptosis. It has been demonstrated that lysosome damage caused by PDT is more efficacious in leading to cell death rather than other organelles.17–20 Lysosome-targeted PSs are promising agents for highly efficient and precise PDT. To date, the reported stimuli-activatable PSs were prepared via non-covalent interactions (electrostatic interactions, hydrophobic interactions and van der Waals force) and were initiated by only one type of stimulation (e.g., light or pH). PSs that are dual-activated via internal gaseous signaling molecules and external light with robust spatiotemporal precision further increases the biosafety and reduces the risk of systemic toxicity. The construction of covalently stable and dual-activatable systems for lysosome-targeted multimodal cancer treatment remains a big challenge to augment therapeutics controllability, precision and efficiency.
Ru(II) complexes have proven to be promising anticancer metal drugs by taking advantages of their improved selectivity, preferable biocompatibility, and better therapeutic effects.21,22 Generally, some Ru(II) complexes can produce 1O2 and undergo ligand substitution under light irradiation to release Ru(II) species and ligands for chemotherapy and PDT.23–29 In terms of Ru(II) complexes, an activated fluorescence system as a therapeutic platform for imaging cancer cells, monitoring drug delivery and multimodal cancer treatment has been rarely reported. Recently, cationic conjugated oligomers (including cationic oligo(p-phenylene vinylene), oligo(p-phenylene ethynylene)s, and oligo-(thiophene ethynylene) (OTE)), have been shown to exhibit high antibacterial and antitumor activity on account of their noticeable PDT effect.30–32 In addition, cationic conjugated oligomers usually exhibit strong light-harvesting ability and high fluorescence quantum yield,33,34 which form a solid base for investigating the dynamic distribution of drugs inside cells.35 In particular, OTE can physically break the membrane structure of microorganisms efficiently.36 Thus, it would be appealing to design and construct a cationic conjugated oligomer-based dual-activatable potential drug system for lysosome-targeted multimodal cancer treatment together with fluorescence monitored drug delivery.
In this work, we designed and synthesized a new dual-activatable Ru(II)-conjugated oligomer agent (Ru-OTE) through the coordination-driven self-assembly of a cationic conjugated oligomer ligand (OTE-BN) and a Ru(II) center. Carbon monoxide (CO) is an intracellular gaseous signaling molecule and an intrinsic metal ligand in enzymes,37 the production of which will increase in the body under certain pathophysiological conditions (e.g. inflammation).38 Herein, CO and light as internal and external stimulants, respectively, are applied to trigger the release of OTE-BN (PSs) and Ru(II) agents. As shown in Scheme 1, Ru-OTE has weak fluorescence due to the heavy atom effect, and can be uptaken by cancer cells. Subsequently, OTE-BN is released from Ru-OTE via the dual-activation of CO and light. The released OTE-BN then emits bright fluorescence and selectively accumulates in lysosomes, physically breaking their integrity. Besides this, the generated cytotoxic 1O2 causes severe lysosome damage, thus leading to cancer cell death via PDT. Meanwhile, the release of Ru(II) anticancer agents also suppresses cancer cell growth as anticancer metal drugs.39–41 Significant anti-cancer effect was realized by the multimodal therapeutics of physical disruption/PDT/chemotherapy. This study reports a cationic conjugated oligomer-based dual-activatable system for lysosome-targeted multimodal cancer treatment together with fluorescence monitored drug delivery, which paves the way to the development of a more secure, effective and intelligent therapeutic potential drug.
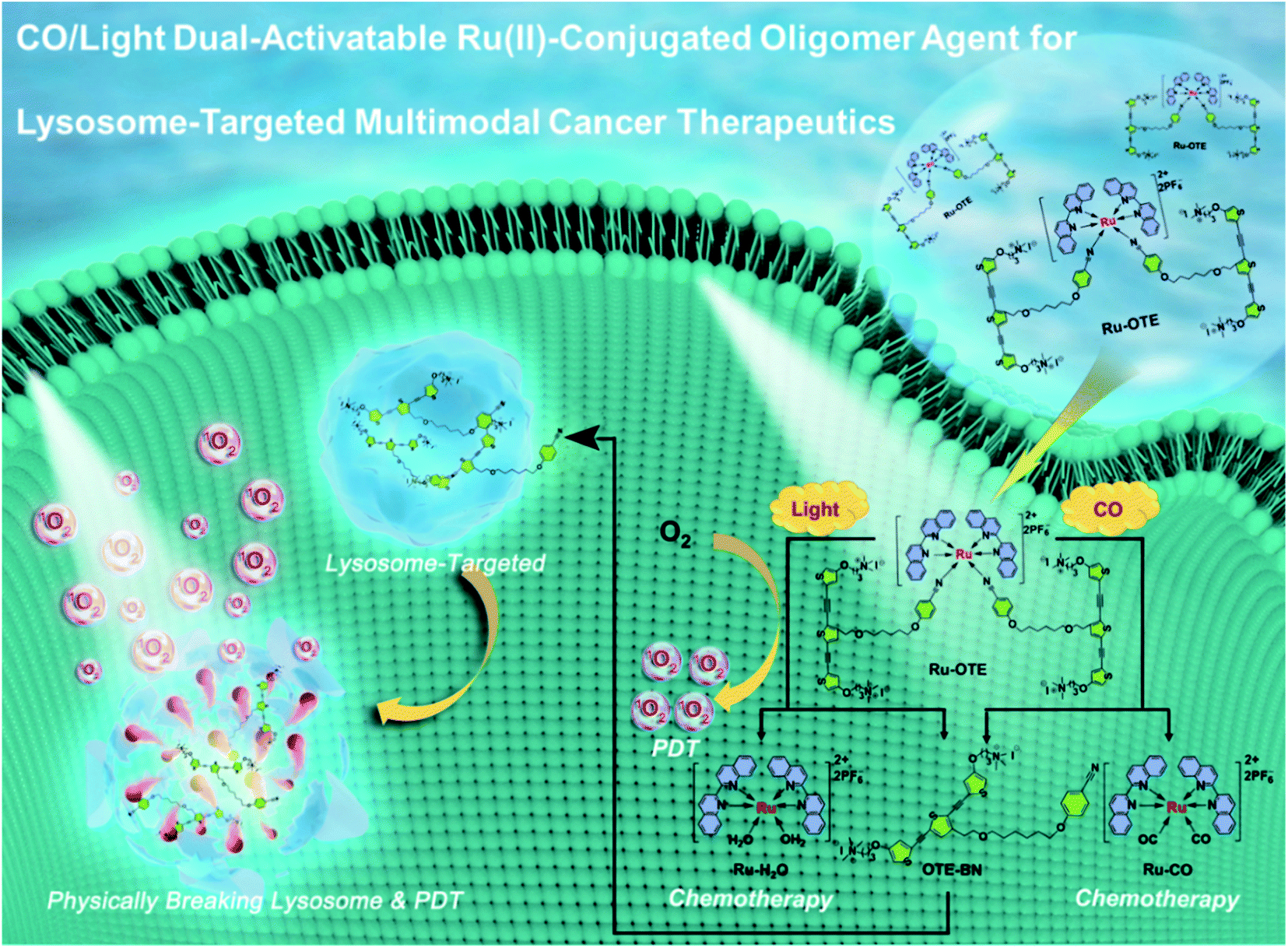 | ||
| Scheme 1 Schematic illustration of CO/light dual-activatable Ru-OTE agent for lysosome-targeted multimodal cancer therapeutics. | ||
Results and discussion
Design, synthesis and characterization of Ru-OTE
To obtain a multifunctional agent for synergistic cancer therapy, a novel cationic conjugated oligomer-Ru(II) complex Ru-OTE was designed. The synthesis route of Ru-OTE is displayed in Fig. 1 and the detailed synthesis procedures are shown in the ESI.† Conjugated oligomer 9 with a cyano group was obtained via a Sonogashira coupling reaction of compound 3 and 8 in a yield of 52%. Ru(biq)2Cl2 and Ru-H2O were synthesized according to the literature.24 The reaction of Ru-H2O with 9 afforded Ru-OTE-pre in a yield of 11% through coordination between Ru and the cyano group on compound 9. Then, cationic Ru-OTE was prepared via the quaternization reaction of Ru-OTE-Pre with CH3I in a yield of 98%, which imparted Ru-OTE with water-solubility and the benefit of being able to be applied in biomedical applications without the involvement of harmful organic solvents. Additionally, OTE-BN was obtained via a quaternization reaction as a control molecule in following investigations. All intermediates and the final product Ru-OTE were fully characterized by nuclear magnetic resonance (NMR) spectroscopy and mass spectrometry (Fig. S12–S23, ESI†). The photophysical properties, ROS generation ability and optical response of Ru-OTE towards CO and light were studied prior to exploring the biomedical applications. As shown in Fig. 2a and S1,† Ru-OTE exhibits a broad metal-to-ligand charge transfer (MLCT) band, with three absorption peaks at 348, 556 and 674 nm, respectively, indicating that the photocleavage of the Ru-cyano coordination bond in Ru-OTE could be induced in the whole white light region. Besides this, Ru-OTE displays two emission peaks at 412 and 440 nm. The maximal absorption of OTE-BN is located at 371 nm, while two absorption peaks at 344 and 593 nm are observed for Ru-H2O. The emission peaks of OTE-BN and Ru-H2O appear at 438 and 445 nm, respectively.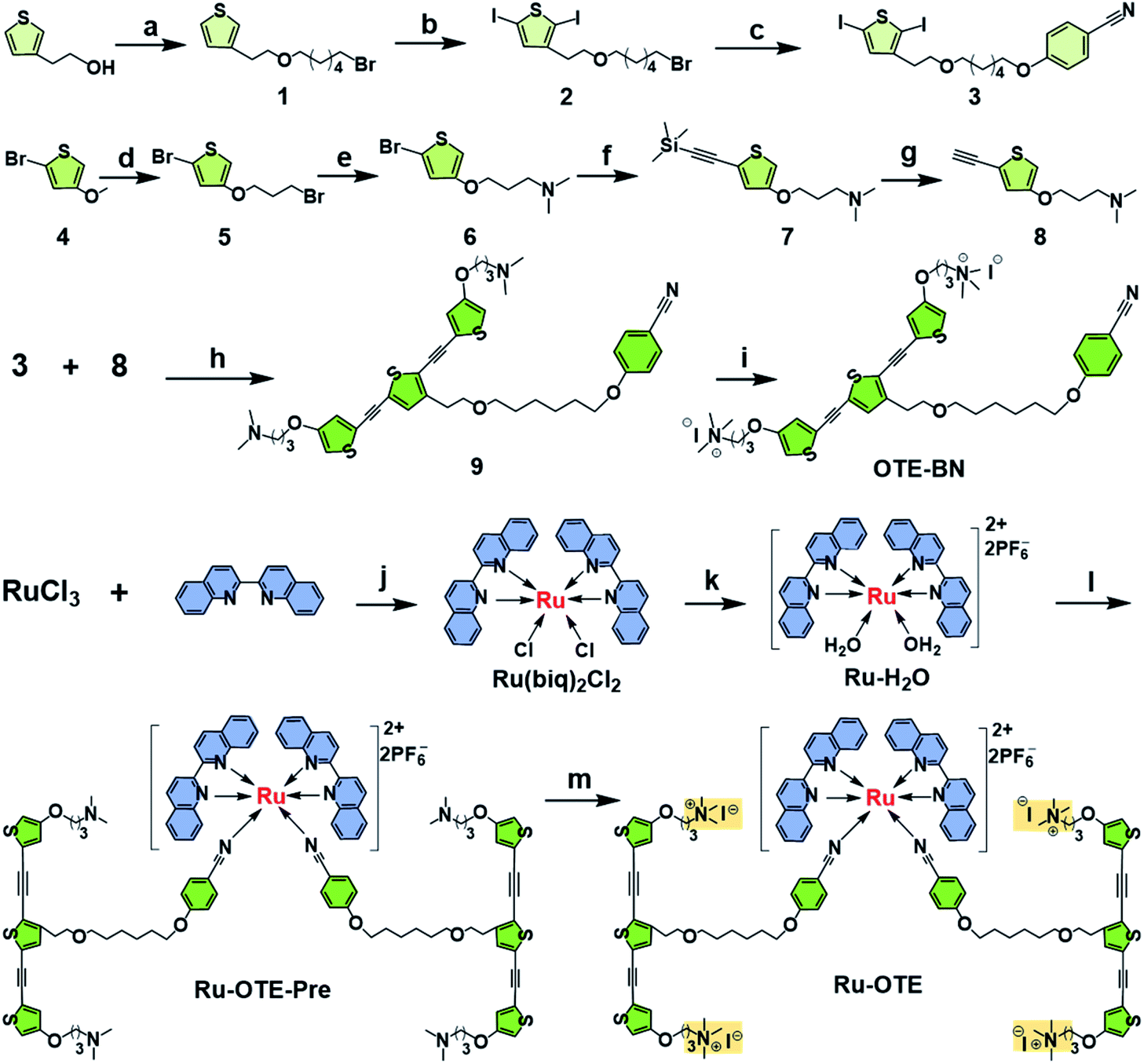 | ||
| Fig. 1 The synthesis route of Ru-OTE. (a) Br(CH2)6Br, THF, KOH, 85 °C, 12 h; (b) NIS, CH2Cl2/CH3COOH, r.t., 8 h; (c) 4-hydroxybenzonitrile, K2CO3, 18-crown-6, acetone, 80 °C, 8 h; (d) Br(CH2)3OH, toluene, NaHSO4, 100 °C, 12 h; (e) NH(CH3)2, THF, r.t., 24 h; (f) TMSA, Pd(PPh3)2Cl2/CuI, triethylamine, 100 °C, 12 h; (g) THF, CH3OH, K2CO3, r.t. 3 h; (h) TMSA, diethylamine/CHCl3, Pd(PPh3)2Cl2/CuI, 35 °C, 2 h; (i) CH3I, CHCl3, 48 h; (j) LiCl, N,N-dimethylformamide, 135 °C, overnight; (k) AgPF6/KPF6, ethanol/H2O, 90 °C, 5 h; (l) OTE-BN, acetone, 56 °C, 12 h; (m) CH3I, CHCl3/CH3OH, 48 h. | ||
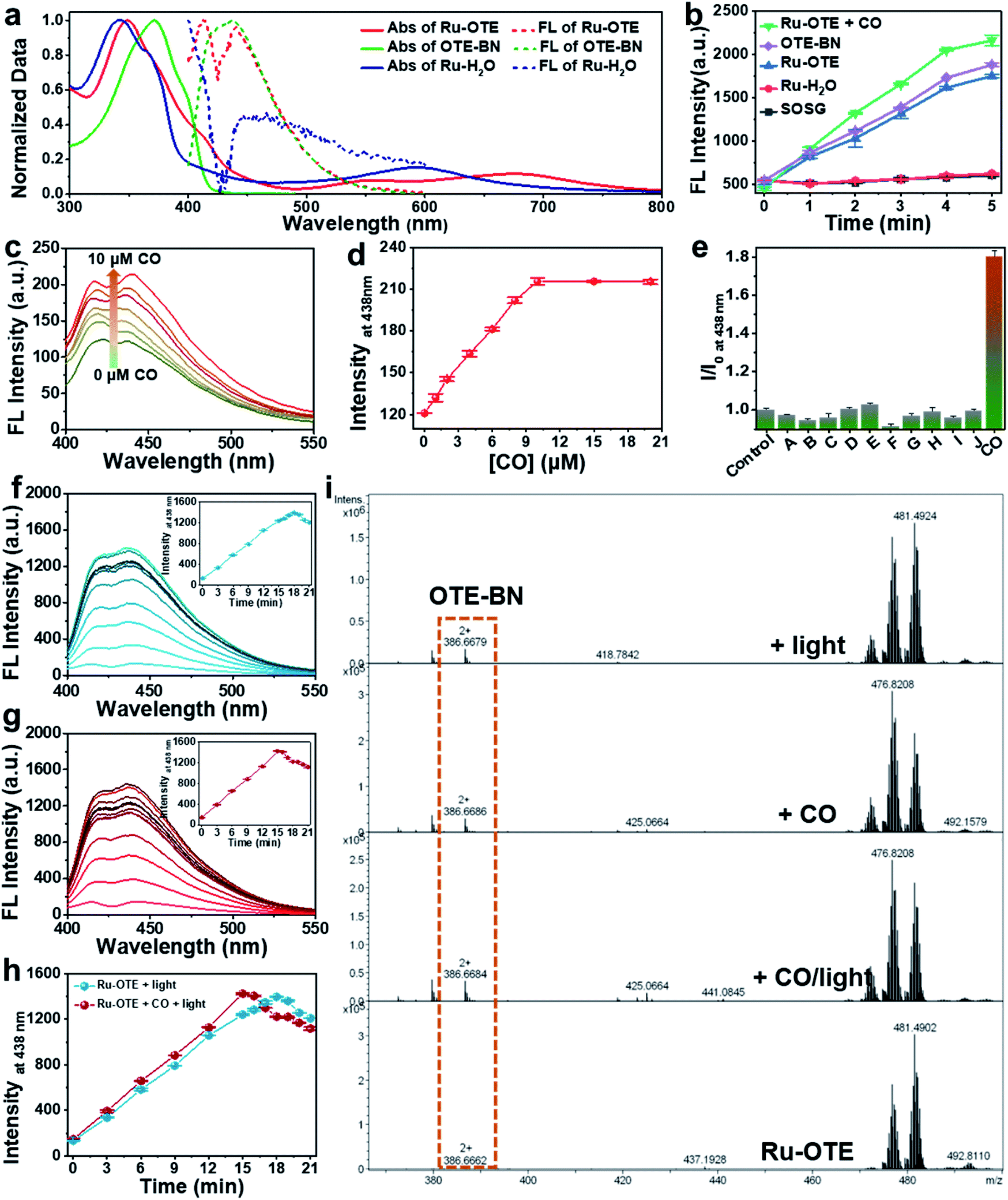 | ||
| Fig. 2 The photophysical properties, ROS generation ability and optical response of Ru-OTE towards CO and light. (a) Normalized absorption and emission spectra of Ru-OTE, OTE-BN and Ru-H2O in aqueous solution. (b) Fluorescence intensity of SOSG at 525 nm in tris (100 mM, pH 7.5) in the presence of different agents under white light irradiation (5 mW cm−2, 5 min) with an excitation wavelength of 504 nm [SOSG] = [other agents] = 1.0 μM. (c) Fluorescence emission spectra and (d) intensity at 438 nm of Ru-OTE (1.0 μM) in PBS (10 mM, pH 7.4) in the presence of different concentrations of CO. (e) Selectivity of Ru-OTE towards CO. The concentration of all of the analytes is 10 μM. The excitation wavelength is 371 nm. The error bars represent the standard deviations of three parallel assays. (A–J) H2O2, H2S, NO, Cys, GSH, ONOO−, HCO3−, NaCit, AA and imidazole. (f) Fluorescence emission spectra of Ru-OTE (1.0 μM) in PBS after white light irradiation (25 mW cm−2). (g) Fluorescence emission spectra of Ru-OTE (1.0 μM) in PBS in the presence of the CO after white light irradiation (25 mW cm−2). (h) Fluorescence intensity at 438 nm with respect to light irradiation time. λex: 371 nm. (i) High-resolution mass spectra of Ru-OTE before and after activation by CO and/or light. | ||
CO/light dual-activatable response
Furthermore, the optical response of Ru-OTE towards CO and light was explored to prove its dual-activation properties. CORM-3 was chosen as a CO release agent in the following studies. CO coordinates with Ru(II) and undergoes ligand exchange so that OTE-BN is released from Ru-OTE. The fluorescence of OTE-BN is subsequently recovered due to the inhibited heavy atomic effect. As shown in Fig. 2c, the fluorescence intensity indeed increased after the addition of CO. A 1.76-fold intensity enhancement was observed and the fluorescence reached a plateau upon the addition of 10 μM CORM-3 (Fig. 2d). The low detection limit was calculated to be 98 nM (S/N = 3). Fig. S2† displays the rapid response of Ru-OTE to CO, where the ligand substitution of water by CO at the Ru(II) center was achieved within 15 min. Then, the selectivity of Ru-OTE towards CO was investigated by assaying the fluorescence of Ru-OTE in the presence of various species, including hydrogen peroxide (H2O2), hydrogen sulfide (H2S), nitric oxide (NO), cysteine, glutathione (GSH), peroxynitrite anion (ONOO−), carbonate anionnitrite (HCO3−), sodium citrate (NaCit), ascorbic acid (AA) and imidazole. As shown in Fig. 2e, negligible fluorescence changes were observed for the interferents, except for CO. This result distinctly demonstrates that Ru-OTE can be specifically activated by CO.The optical response of Ru-OTE to light was also studied by measuring its fluorescence intensity after irradiation with white light. Light illumination breaks the Ru(II)–cyano bonds in Ru-OTE, then OTE-BN is quickly and efficiently released, leading to an enhancement in fluorescence intensity (Fig. 2f). Around 10.8-fold increase in intensity was achieved when Ru-OTE was irradiated by white light (25 mW cm−2) for 18 min, which was higher than that of CO. To investigate the synergistic response of Ru-OTE co-activated by CO and light, Ru-OTE was incubated with CO for 15 min followed by irradiation with light. As shown in Fig. 2g, the fluorescence intensity increased rapidly and reached maximum at 15 min, which was faster than that only by light irradiation (Fig. 2h). This result indicates that the synergistic effect of CO and light accelerates the release of OTE-BN. To directly confirm the release of OTE-BN from Ru-OTE, high-resolution mass spectra of Ru-OTE before and after activation by CO and/or light were measured. As shown in Fig. 2i, there was no peak of OTE-BN at 386.6662 (m/z) in the spectrum of the Ru-OTE group. While a peak for OTE-BN obviously appeared after Ru-OTE was activated by CO, light and CO + light, which solidly confirmed the valid activation of Ru-OTE by CO and light.
1O2 generation capacity of Ru-OTE
Additionally, the 1O2 generation capacity of the Ru-OTE system in the presence of CO and light was explored using a singlet oxygen sensor green reagent (SOSG). As shown in Fig. 2b, the fluorescence of SOSG dramatically increased upon the addition of Ru-OTE, which was similar to that of OTE-BN. Interestingly, higher fluorescence intensity was detected after Ru-OTE was incubated with CO. When SOSG was mixed with Ru-H2O, the fluorescence remained the same as the control. In addition, as displayed in Fig. S3,† the 1O2 quantum yield of OTE-BN was measured to be 1.05 using 9,10-anthracenediyl-bis(methylene)dimalonic acid (ABDA) as an indicator and rose bengal (RB) as a standard reference (1O2 quantum yield = 0.75).42,43 These results reasonably confirm that Ru-OTE can be regarded as an effective PDT agent for antitumor treatment, which is probably due to the release of OTE-BN from Ru-OTE. Importantly, the synergistic release of OTE-BN via the dual activation of CO and light lead to the higher production of 1O2.Intracellular response of Ru-OTE towards light/CO
The excellent CO/light dual-activatable system, desirable turn-on fluorescence properties and high 1O2 generation capacity of the activatable Ru-OTE system paved the way for exploring the bioapplications of activable drug delivery monitoring and multimodal cancer therapeutics. Firstly, the intracellular response of Ru-OTE towards light (as an external stimulus) and CO (as an internal stimulus) in MDA-MB-231 human breast cancer cells was studied. MDA-MB-231 cells were incubated with Ru-OTE in the dark for 2, 4, 6, and 8 h, followed by irradiation with white light (25 mW cm−2) for 30 min. As shown in Fig. S4a and b,† the signal of Ru-OTE in cells gradually increased with incubation time, reaching a maximum at 6 h, indicating the complete cellular uptake of Ru-OTE. Then, the response of Ru-OTE to endogenous CO was next investigated. As heme induces endogenous CO production, MDA-MB-231 cells were treated with heme for 2, 4, 6, and 8 h, then incubated with Ru-OTE for 6 h. As shown in Fig. S4c and d,† the fluorescence intensity of Ru-OTE inside cells indeed increased along with the heme treatment time, which was due to endogenous CO coordinating with Ru-OTE and releasing fluorescent OTE-BN. Furthermore, the dual-activation of Ru-OTE by CO/light was studied after cells being treated with heme and irradiated by light. As shown in Fig. S5,† bright blue fluorescence was observed when cells were treated by both hemin and light, indicating that dual-stimulation generates stronger fluorescence than only one treatment. These results verify that the release of OTE-BN and Ru(II) species from Ru-OTE can be dual-activated via external and internal stimulation, thus improving the delivery efficacy and accuracy, and reducing the possible deficiency of single stimulus. Such release was also monitored by fluorescence imaging. Additionally, the morphological structure of the cells was gradually destroyed, which may result from the 1O2 produced by OTE-BN under light irradiation (Fig. S5†).CO/light dual-activation drug delivery and cell cytotoxicity
A schematic diagram of a CO/light dual-activatable Ru-OTE agent for lysosome-targeted cancer therapeutics is illustrated in Fig. 3a. Upon the CO/light dual-activation of Ru-OTE, OTE-BN and Ru(II) species were released, which was accompanied by fluorescence turn-on. OTE-BN undergoes targeted accumulation in lysosomes and physically breaks lysosomes,36,44,45 meanwhile 1O2 is robustly generated for PDT under light irradiation. Besides this, the Ru(II) species also suppressed cancer cell growth as an anticancer metal drug. The half-maximal inhibitory concentration (IC50) values of Ru-OTE, Ru-H2O and OTE-BN under dark and light irradiation conditions were measured. As shown in Fig. 3b and S6,† the IC50dark values of Ru-OTE, Ru-H2O and OTE-BN were determined to be 12.6, 13.5 and 8.0 μM, respectively. The IC50light values of Ru-OTE, Ru-H2O and OTE-BN were as low as 1.2, 7.2 and 3.8 μM, respectively. In addition, the phototoxicity indexes (PI) of Ru-OTE, Ru-H2O and OTE-BN were calculated be 10.5, 1.9 and 2.1. It should be noted that the PI of Ru-OTE is 10.5, which is significantly higher than that of the photoreleased products Ru-H2O and OTE-BN, indicating the good performance of Ru-OTE in CO/light dual-activatable multimodal cancer therapeutics. Subsequently, the anticancer efficiency of Ru-OTE after activation by CO and light was explored. As shown in Fig. 3c, the Ru-OTE + CO group only demonstrated slightly stronger cytotoxicity than the Ru-OTE group, which resulted from the partial delivery and good biocompatibility of OTE-BN at lower concentration. When cells were incubated with Ru-OTE and then illuminated with light for 30 min, the cell viability dramatically decreased. Under the conditions of Ru-OTE (8.0 μM), the cell viabilities of Ru-OTE + light group and Ru-OTE + light + CO were 15 ± 2.0% and 6.9 ± 0.4%, respectively, around an 8% improvement in the cell killing rate. Besides, the data differences of the mentioned two groups were statistically significant. It should be noted that the improved cell killing rates were higher than the data difference between the Ru-OTE group and Ru-OTE + CO. So, the improved cell killing rate was found to be due to the synergistic effect of light/CO, rather than an additive effect, indicating the multimodal antitumor effect of Ru-OTE. Finally, Ru-OTE showed good biocompatibility towards human normal liver HL7702 cell over some concentration ranges (Fig. S7†), representing potential for use in biomedical applications. | ||
| Fig. 3 Schematic illustration, anticancer efficiency and therapeutic mechanism of the CO/light dual-activatable Ru-OTE system. (a) Illustration of the chemical changes after CO/light dual-activation and explanation of the multimodal anticancer therapeutics. (b) Half-maximal inhibitory concentration (IC50) values of Ru-OTE, Ru-H2O and OTE-BN against MDA-MB-231 cells in dark or white light irradiation conditions. (c) Cytotoxicity of Ru-OTE against MDA-MB-231 in the absence and presence of CO, light, or CO + light. n = 6, mean ± SD, *P < 0.05; **P < 0.01; ***P < 0.001. (d) CLSM images of MDA-MB-231 cells incubated with Ru-OTE (4.0 μM) and stained by LysoTracker® Red DND-99 in the absence and presence of CO or light. The fluorescence imaging of Ru-OTE and LysoTracker® Red DND-99 were collected at 420–460 nm (λex: 405 nm), 570–670 nm (λex: 559 nm), respectively. Scale bar: 10 μm. (e) CLSM images of MDA-MB-231 cells after incubation with Ru-OTE and stained by AO in the absence and presence of CO or light. Fluorescence imaging of AO was collected at 515–545 nm in the green channel and 610–640 nm in the red channel (λex: 488 nm). Scale bar: 10 μm. | ||
Localization and anticancer mechanism of Ru-OTE
To determine the anticancer mechanism of Ru-OTE, cell colocation experiments were conducted accordingly. MDA-MB-231 cells were cultured with Ru-OTE at 37 °C for 6 h, then they were dyed with LysoTracker® Red DND-99. As shown in Fig. 3d and S8a,† after CO activation, blue fluorescence started to be emitted and the red fluorescence of the lysosome tracker weakened compared to the cell and Ru-OTE groups, indicating that OTE-BN was released and interacted with lysosomes, leading to the physical disruption of lysosomal structures. Notably, when the cells (incubated with Ru-OTE) were illuminated with white light, bright luminescence in the blue channel and a negligible signal in the red channel were observed, implying that more OTE-BN was photoreleased and cytotoxic 1O2 almost completely destroyed the lysosomes in the cells. To confirm these two phenomena, the synthesized OTE-BN was incubated with cells for cell imaging. Fig. S9† showed that the fluorescence signal in the red channel became weaker after incubation with OTE-BN. Importantly, the red fluorescence even disappeared completely upon white light irradiation. However, when the cells were incubated with Ru-H2O, the bright red fluorescence of the lysosome tracker was observed in the absence of irradiation. The red color still remained, even after illumination with light (Fig. S8a†), which demonstrated that Ru-H2O has no influence on the structure of the lysosomes. These results confirm that the CO/light dual-activation of Ru-OTE was lysosome-targeted and the disruption of lysosomal structure mainly resulted from the released OTE-BN and its generated 1O2. The ROS generation ability of Ru-OTE within tumor cancers was verified by incubating it with 2′,7′-dichlorofluorescin diacetate (DCFH-DA) probe. After white light irradiation for 30 min, intracellular green emission originating from the oxidation of DCFH-DA by 1O2 was observed, while no red fluorescence of LysoTracker was observed (Fig. S10†), which further suggested the generation of 1O2 inside the cells via activation of OTE-BN, leading to the breakage of lysosomes.To gain deep insight into the influence that Ru-OTE has on the integrity of lysosomes, acridine orange (AO) staining was subsequently performed.46,47 Generally, AO emits intense red fluorescence in acidic lysosomes, while it generates green fluorescence in cytoplasm and nuclei. As shown in Fig. 3e and S8b,† when cells were incubated with Ru-OTE and dyed with AO, a large number of red dots in the cytoplasm and green fluorescence in the nuclei were observed, validating the existence of intact lysosomes. After CO activation, there were fewer red dots, while the red dots disappeared after light activation, implying the effective disruption of the lysosomal structures in MDA-MB-231. Moreover, Fig. S11† shows that there were fewer red dots in the cells incubated with OTE-BN and these even completely disappeared after light irradiation because of the production of 1O2. The existence of red dots in the cells incubated with Ru-H2O in the presence or absence of light irradiation confirmed the integrity of the lysosomes (Fig. S8b†). According to the results, it can be concluded that CO/light activates Ru-OTE and triggers the release of the OTE-BN and Ru(II) species. OTE-BN specifically accumulates in lysosomes and physically breaks lysosomes, and the generated 1O2 completely destroys lysosome structures and leads to the uncontrolled release of lysosomal proteases, thus inducing cell apoptosis.14,48
NIR two-photon activatable PDT
To further expand the bioapplications of the CO/light dual-activatable Ru-OTE system for anticancer therapeutics and increase the tissue penetration depth of the used light, a two-photon laser (λ = 800 nm) was applied for NIR light activatable synergistic PDT. As shown in Fig. 4a, in the Ru-OTE treated MDA-MB-231 cell group, the two-photon fluorescence signal was clearly observed in the selected area after two-photon laser (λ = 800 nm) irradiation, while no signal was detected in the unirradiated region, confirming that OTE-BN was successfully two-photon-activated followed by its release in a precise optically-controlled manner. The cells without Ru-OTE treatment did not emit blue fluorescence neither in the presence nor absence of laser irradiation. Importantly, the morphology of the cells after selective laser irradiation was indeed destroyed, implying the NIR PDT potential of Ru-OTE. Furthermore, the two-photon PDT of Ru-OTE was evaluated by live/dead cell staining after two-photon laser (λ = 800 nm) irradiation. As shown in Fig. 4b, the cells incubated with Ru-OTE without laser irradiation and only irradiated with the laser for 120 s were almost kept alive. However, Ru-OTE treated cells upon laser irradiation for 30, 60 and 120 s gradually died and all the cells in the selected region were killed after 120 s of laser irradiation, confirming that the Ru-OTE system could be directly activated by a two-photon laser for efficient PDT of cancer cells. | ||
| Fig. 4 NIR two-photon laser activatable Ru-OTE for PDT of cancer cells. (a) Two-photon laser activatable and fluorescence images of MDA-MB-231 cells treated with Ru-OTE (4 μM) in a selected area. Scale bar: 100 μm. (b) Live/dead staining of MDA-MB-231 cells treated with Ru-OTE (4 μM) for 6 h and then irradiated using a two-photon laser (λ = 800 nm) for 0, 30, 60, and 120 s, respectively. The laser irradiation area was 600 μm × 600 μm. The fluorescence images of Calcein AM and PI were collected at 500–540 nm (λex: 488 nm) and 570–620 nm (λex: 559 nm), respectively. Scale bars: 200 μm. | ||
In vivo anticancer therapy
In vivo tumor suppression experiments were carried out to further evaluate the validity of Ru-OTE based on a MDA-MB-231 breast tumor model. The tumor-bearing mice were divided into three groups: PBS as a control, Ru-OTE, and Ru-OTE with white light irradiation. As shown in Fig. 5a, mice in the Ru-OTE group exhibited fractional tumor suppression due to the partial release of OTE-BN from Ru-OTE under CO activation. Expectedly, the Ru-OTE with light irradiation (10 mw cm−2) group demonstrated significant hindrance of tumor growth, in which the tumor volume was suppressed greatly over 14 days. The average tumor weight in the Ru-OTE + light group was much lighter than that in the other two groups (Fig. 5c). The superior antitumor effect of Ru-OTE in the presence of light was mainly due to synergistic PDT and chemotherapy by CO/light dual-activation. Representative photos of the mice and tumors extracted from the mice also confirmed these results (Fig. 5d and e). Moreover, the body weight was monitored for these groups to estimate toxicity. As shown in Fig. 5b, no obvious fluctuations in body weight were observed for all of the groups, indicating the negligible side effects of Ru-OTE and white light irradiation. To further verify the dramatic therapeutic efficiency, histological hematoxylin and eosin (H&E) staining assays were carried out after 14 days of treatment (Fig. 5f). The results showed large areas of apoptosis and necrosis, indicating that Ru-OTE exposure to light irradiation exhibits remarkable therapeutic effectiveness.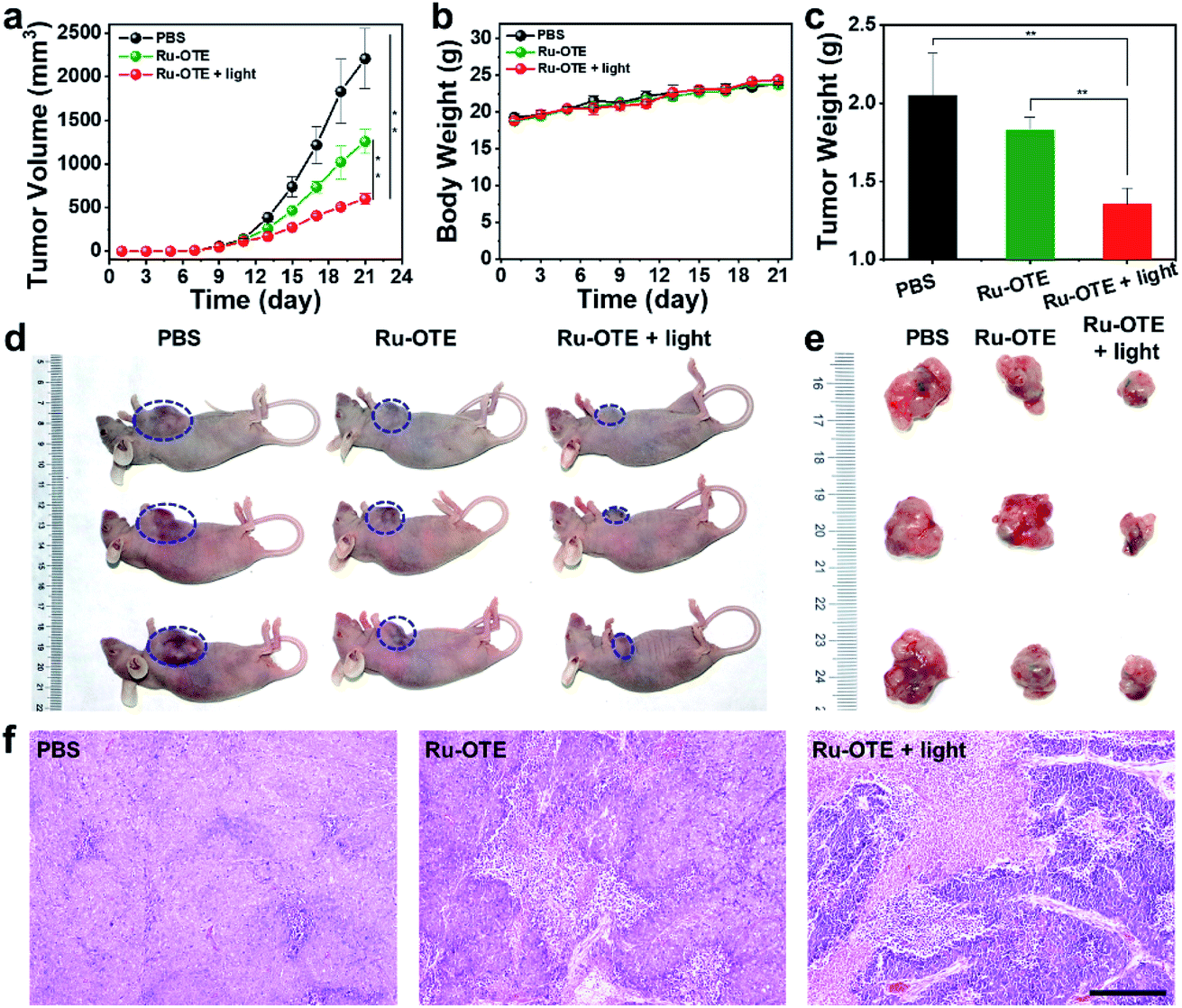 | ||
| Fig. 5 Evaluation of the in vivo anticancer efficiency of the Ru-OTE system in MDA-MB-231 tumor-bearing BALB/c-nude mice. (a) Tumor volume, (b) body weight, and (c) tumor weight variations of the mice with various treatments. n = 4, mean ± SD, *P < 0.05; **P < 0.01; ***P < 0.001. (d) Representative photographs of the mice and (e) tumors extracted from the mice in the various groups at the end of the treatments. (f) Hematoxylin and eosin (H&E)-stained tumor sections from tumor-bearing mice at the end of treatment. Scale bar: 300 μm. | ||
Conclusions
In summary, a CO/light dual-activatable Ru-OTE agent for lysosome-targeted multimodal cancer therapeutics was firstly reported. Upon the dual-triggering of CO and light, Ru-OTE undergoes ligand substitution and releases OTE-BN, followed by dramatic fluorescence recovery for monitoring drug delivery. Multimodal cancer therapeutics was achieved relying on the following models: (1) the released OTE-BN selectively accumulated in the lysosome, physically breaking its integrity. (2) Then, the generated cytotoxic 1O2 causes severe lysosome damage, thus leading to cancer cell death via PDT. (3) The release of the Ru(II) agent also suppressed cancer cell growth as an anticancer metal drug. (4) Importantly, Ru-OTE was directly photo-activated using a two-photon laser (800 nm) for efficient NIR PDT. This study demonstrates that the development of a dual-activatable Ru(II)-conjugated oligomer potential drug provides a new strategy for highly precise and effective subcellular organelle-targeted multimodal cancer therapeutics.Author contributions
M. Y. and Y. L. T. conceived the project. M. Y., H. Z., Z. Q. Z., Q. Y. and Q. F. performed the experiments. Y. L. T. supervised the research. M. Y., H. Z. and Y. L. T. wrote and revised the paper. X. R. D., S. W. and Y. L. T. validate, review and edit the writing. All the authors discussed the results and contributed to the preparation of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grants 21974084 and 21675106), the Innovation Capability Support Program of Shaanxi (Program no. 2021TD-42), and the Fundamental Research Funds for the Central Universities (No. GK201901003, GK202101001). Animal experiments were approved by the China Committee for Research and Animals Ethics in compliance with the law on experimental animals.Notes and references
- F. Bray, J. Ferlay, I. Soerjomataram, R. L. Siegel, L. A. Torre and A. Jemal, Ca-Cancer J. Clin., 2018, 68, 394–424 CrossRef PubMed.
- L. Zeng, P. Gupta, Y. Chen, E. Wang, L. Ji, H. Chao and Z. S. Chen, Chem. Soc. Rev., 2017, 46, 5771–5804 RSC.
- N. Rubio, I. Coupienne, E. Di Valentin, I. Heirman, J. Grooten, J. Piette and P. Agostinis, Autophagy, 2012, 8, 1312–1324 CrossRef CAS PubMed.
- H. Chen, S. Li, M. Wu, K. Huang, C.-S. Lee and B. Liu, Angew. Chem., Int. Ed., 2020, 59, 632–636 CrossRef CAS PubMed.
- P. Agostinis, K. Berg, K. A. Cengel, T. H. Foster, A. W. Girotti, S. O. Gollnick, S. M. Hahn, M. R. Hamblin, A. Juzeniene, D. Kessel, M. Korbelik, J. Moan, P. Mroz, D. Nowis, J. Piette, B. C. Wilson and J. Golab, Ca-Cancer J. Clin., 2011, 61, 250–281 CrossRef PubMed.
- J. P. Celli, B. Q. Spring, I. Rizvi, C. L. Evans, K. S. Samkoe, S. Verma, B. W. Pogue and T. Hasan, Chem. Rev., 2010, 110, 2795–2838 CrossRef CAS PubMed.
- D. E. Dolmans, D. Fukumura and R. K. Jain, Nat. Rev. Cancer, 2003, 3, 380–387 CrossRef CAS PubMed.
- S. Gao, G. Wang, Z. Qin, X. Wang, G. Zhao, Q. Ma and L. Zhu, Biomaterials, 2017, 112, 324–335 CrossRef CAS PubMed.
- J. Sun, K. Du, J. J. Diao, X. T. Cai, F. D. Feng and S. Wang, Angew. Chem., Int. Ed., 2020, 59, 12122–12128 CrossRef CAS PubMed.
- D. Peer, J. M. Karp, S. Hong, O. C. Farokhzad, R. Margalit and R. Langer, Nat. Nanotechnol., 2007, 2, 751–760 CrossRef CAS PubMed.
- G. C. Yu, X. L. Zhao, J. Zhou, Z. W. Mao, X. L. Huang, Z. T. Wang, B. Hua, Y. J. Liu, F. W. Zhang, Z. M. He, O. Jacobson, C. Y. Gao, W. L. Wang, C. Y. Yu, X. Y. Zhu, F. H. Huang and X. Y. Chen, J. Am. Chem. Soc., 2018, 140, 8005–8019 CrossRef CAS PubMed.
- T. Senthilkumar, L. Zhou, Q. Gu, L. Liu, F. Lv and S. Wang, Angew. Chem., Int. Ed., 2018, 57, 13114–13119 CrossRef CAS PubMed.
- V. P. Torchilin, Nat. Rev. Drug Discovery, 2014, 13, 813–827 CrossRef CAS PubMed.
- L. Galluzzi, J. M. Bravo-San Pedro and G. Kroemer, Nat. Cell Biol., 2014, 16, 728–736 CrossRef CAS PubMed.
- C. J. Zhang, Q. L. Hu, G. X. Feng, R. Y. Zhang, Y. Y. Yuan, X. M. Lu and B. Liu, Chem. Sci., 2015, 6, 4580–4586 RSC.
- A. Jhaveri and V. Torchilin, Expert Opin. Drug Delivery, 2016, 13, 49–70 CrossRef CAS PubMed.
- R. Villasenor, Y. Kalaidzidis and M. Zerial, Curr. Opin. Cell Biol., 2016, 39, 53–60 CrossRef CAS PubMed.
- Z. X. Zhou, J. P. Liu, J. J. Huang, T. W. Rees, Y. L. Wang, H. Wang, X. P. Li, H. Chao and P. J. Stang, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 20296–20302 CrossRef CAS PubMed.
- H. Y. Huang, B. L. Yu, P. Y. Zhang, J. J. Huang, Y. Chen, G. Gasser, L. N. Ji and H. Chao, Angew. Chem., Int. Ed., 2015, 54, 14049–14052 CrossRef CAS PubMed.
- W. K. Martins, N. F. Santos, C. D. Rocha, I. O. L. Bacellar, T. M. Tsubone, A. C. Viotto, A. Y. Matsukuma, A. B. D. Abrantes, P. Siani, L. G. Dias and M. S. Baptista, Autophagy, 2019, 15, 259–279 CrossRef CAS PubMed.
- M. R. Gill, P. J. Jarman, S. Halder, M. G. Walker, H. K. Saeed, J. A. Thomas, C. Smythe, K. Ramadan and K. A. Vallis, Chem. Sci., 2018, 9, 841–849 RSC.
- N. Dai, H. Zhao, R. L. Qi, Y. Y. Chen, F. T. Lv, L. B. Liu and S. Wang, Chem.–Eur. J., 2020, 26, 4489–4495 CrossRef CAS PubMed.
- C. Mari, V. Pierroz, S. Ferrari and G. Gasser, Chem. Sci., 2015, 6, 2660–2686 RSC.
- W. Sun, S. Li, B. Haupler, J. Liu, S. Jin, W. Steffen, U. S. Schubert, H. J. Butt, X.-J. Liang and S. Wu, Adv. Mater., 2017, 29, 1603702 CrossRef PubMed.
- J. Liu, Y. Chen, G. Li, P. Zhang, C. Jin, L. Zeng, L. Ji and H. Chao, Biomaterials, 2015, 56, 140–153 CrossRef CAS PubMed.
- L. Kohler, L. Nease, P. Vo, J. Garofolo, D. K. Heidary, R. P. Thummel and E. C. Glazer, Inorg. Chem., 2017, 56, 12214–12223 CrossRef CAS PubMed.
- R. Li, C. Zhang, B. Xie, W. Yu, W. Qiu, H. Cheng and X. Zhang, Biomaterials, 2019, 194, 84–93 CrossRef CAS PubMed.
- J. D. Knoll and C. Turro, Coord. Chem. Rev., 2015, 282, 110–126 CrossRef PubMed.
- X. Zeng, Y. Wang, J. Han, W. Sun, H.-J. Butt, X.-J. Liang and S. Wu, Adv. Mater., 2020, 32, 2004766 CrossRef CAS PubMed.
- B. Wang, B. N. Queenan, S. Wang, K. P. R. Nilsson and G. C. Bazan, Adv. Mater., 2019, 31, 1806701–1806722 CrossRef PubMed.
- B. Wang, M. Wang, A. Mikhailovsky, S. Wang and G. C. Bazan, Angew. Chem., Int. Ed., 2017, 56, 5031–5034 CrossRef CAS PubMed.
- D. G. Whitten, Y. Tang, Z. Zhou, J. Yang, Y. Wang, E. H. Hill, H. C. Pappas, P. L. Donabedian and E. Y. Chi, Langmuir, 2019, 35, 307–325 CrossRef CAS PubMed.
- X. Duan, X. F. Jiang, D. Hu, P. Liu, S. Li, F. Huang, Y. Ma, Q. H. Xu and Y. Cao, Nanoscale, 2018, 11, 185–192 RSC.
- Q. Feng, Z. Q. Zhang, Q. Yuan, M. Yang, C. Zhang and Y. L. Tang, Sens. Actuators, B, 2020, 312, 127981 CrossRef CAS.
- L. Zhou, F. Lv, L. Liu and S. Wang, Acc. Chem. Res., 2019, 52, 3211–3222 CrossRef CAS PubMed.
- Q. Yuan, Y. Zhao, Z. Zhang and Y. Tang, ACS Appl. Mater. Interfaces, 2021, 13, 257–265 CrossRef CAS PubMed.
- C. Torre, A. Toscani, C. Marin-Hernandez, J. A. Robson, M. C. Terencio, A. J. P. White, M. J. Alcaraz, J. Wilton-Ely, R. Martinez-Manez and F. Sancenon, J. Am. Chem. Soc., 2017, 139, 18484–18487 CrossRef CAS PubMed.
- L. Wu and R. Wang, Pharmacol. Rev., 2005, 57, 585–630 CrossRef CAS PubMed.
- J. Karges, J. Li, L. Zeng, H. Chao and G. Gasser, ACS Appl. Mater. Interfaces, 2020, 12, 54433–54444 CrossRef CAS PubMed.
- W. Sun, Y. Wen, R. Thiramanas, M. Chen, J. Han, N. Gong, M. Wagner, S. Jiang, M. S. Meijer, S. Bonnet, H. J. Butt, V. Mailänder, X. J. Liang and S. Wu, Adv. Funct. Mater., 2018, 28, 1804227 CrossRef.
- Z. Deng, P. Gao, L. Yu, B. Ma, Y. You, L. Chan, C. Mei and T. Chen, Biomaterials, 2017, 129, 111–126 CrossRef CAS PubMed.
- G. Feng, Y. Fang, J. Liu, J. Geng, D. Ding and B. Liu, Small, 2017, 13, 1602807 CrossRef PubMed.
- Y. Zhao, Z. Zhang, Z. Lu, H. Wang and Y. Tang, ACS Appl. Mater. Interfaces, 2019, 11, 38467–38474 CrossRef CAS PubMed.
- B. Wang, H. Yuan, Z. Liu, C. Nie, L. Liu, F. Lv, Y. Wang and S. Wang, Adv. Mater., 2014, 26, 5986–5990 CrossRef CAS PubMed.
- B. Wang, S. L. Fronk, Z. D. Rengert, J. Limwongyut and G. C. Bazan, Chem. Mater., 2018, 30, 5836–5840 CrossRef CAS.
- X. Li, R. R. Tao, L. J. Hong, J. Cheng, Q. Jiang, Y. M. Lu, M. H. Liao, W. F. Ye, N. N. Lu, F. Han, Y. Z. Hu and Y. H. Hu, J. Am. Chem. Soc., 2015, 137, 12296–12303 CrossRef CAS PubMed.
- M. Li, T. Xiong, J. Du, R. Tian, M. Xiao, L. Guo, S. Long, J. Fan, W. Sun, K. Shao, X. Song, J. W. Foley and X. Peng, J. Am. Chem. Soc., 2019, 141, 2695–2702 CrossRef CAS PubMed.
- L. He, Y. Li, C. P. Tan, R. R. Ye, M. H. Chen, J. J. Cao, L. N. Ji and Z. W. Mao, Chem. Sci., 2015, 6, 5409–5418 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc01317c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |