
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An umpolung-enabled copper-catalysed regioselective hydroamination approach to α-amino acids†
Soshi
Nishino
a,
Masahiro
Miura
 b and
Koji
Hirano
*a
b and
Koji
Hirano
*a
aDepartment of Applied Chemistry, Graduate School of Engineering, Osaka University, Suita, Osaka 565-0871, Japan. E-mail: k_hirano@chem.eng.osaka-u.ac.jp
bInnovative Catalysis Science Division, Institute for Open and Transdisciplinary Research Initiatives (ICS-OTRI), Osaka University, Suita, Osaka 565-0871, Japan
First published on 27th July 2021
Abstract
A copper-catalysed regio- and stereoselective hydroamination of acrylates with hydrosilanes and hydroxylamines has been developed to afford the corresponding α-amino acids in good yields. The key to regioselectivity control is the use of hydroxylamine as an umpolung, electrophilic amination reagent. Additionally, a judicious choice of conditions involving the CsOPiv base and DTBM-dppbz ligand of remote steric hindrance enables the otherwise challenging C–N bond formation at the α position to the carbonyl. The point chirality at the β-position is successfully controlled by the Xyl-BINAP or DTBM-SEGPHOS chiral ligand with similarly remote steric bulkiness. The combination with the chiral auxiliary, (−)-8-phenylmenthol, also induces stereoselectivity at the α-position to form the optically active unnatural α-amino acids with two adjacent stereocentres.
Introduction
α-Amino acids are prevalent structural motifs in many biologically active compounds and pharmaceutical agents, especially peptide drugs. In particular, unnatural α-amino acids have received significant attention because when they replace natural α-amino acids in the original drug structure, the potential for activity improvement and the discovery of new functions increases.1 Multicomponent couplings such as the Strecker, Ugi, and Petasis reactions are classical but the most powerful approaches to the aforementioned target structures.2 The catalytic hydrogenation of α-dehydroamino acid derivatives also provides promising access to unnatural α-amino acids.3 Additionally, the decoration of naturally occurring α-amino acids by the C–C bond forming reactions under phase-transfer,4 palladium,5 copper,6 and iridium/copper dual catalysis7 as well as cross-dehydrogenative-coupling (CDC) conditions8 has also been explored. On the other hand, the C–N bond formation at the α position to the carbonyl in carboxylic acid derivatives can also be a good alternative (Scheme 1). Vedejs reported the KO-t-Bu-mediated direct α-amination of phenylacetates with the electrophilic amination reagent (4-MeOC6H4)2(O)PO–NH2 (Scheme 1a).9 Shi also revealed that the related α-amination was possible with di-tert-butyldiaziridinone in the presence of copper salts.10 MacMillan developed the CuBr2/O2-catalysed ideal C–H/N–H coupling of phenylacetates and free NH amines through the transient formation of α-bromo esters.11 The copper-catalysed electrophilic amination of ketene silyl acetals with hydroxylamines12 or chloroamines13 can also access α-amino acids and was independently developed by our group and the Miura/Murakami research group (Scheme 1b). Kiyokawa and Minakata recently designed (diarylmethylene)amino-substituted hypervalent iodine(III) reagents and succeeded in the amination of the same ketene silyl acetals under catalyst-free conditions.14 Yazaki and Ohshima achieved the copper-catalysed direct α-amination of acylpyrazoles as the carboxylic acid surrogates with the PhI = NTs nitrenoid source (Scheme 1c).15 Wasa16 and Sawamura/Shimizu17 groups independently developed boron-based catalyst systems for the α-amination of esters, amides, and free carboxylic acids with azodicarboxylates (Scheme 1d). Despite these certain advances, there are still several drawbacks; the substrates are limited to the relatively acidic C–H of phenylacetates (Scheme 1a); the preactivation of carboxylic acids by strong bases/silyl halides is inevitable (Scheme 1b); condensation with a special pyrazole-based directing group is necessary (Scheme 1c); the initial product is the hydrazine derivative, and thus the resultant N–N bond should be reductively cleaved to obtain the targeted α-amino acids (Scheme 1d). Moreover, the highly stereocontrolled process remains largely elusive. Thus, the concise and efficient synthesis of α-amino acids based on C–N bond formation is still a formidable challenge.18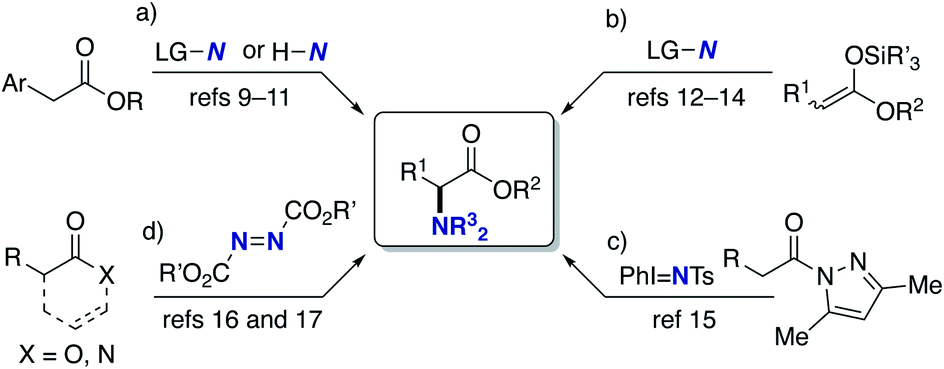 | ||
| Scheme 1 C–N bond forming approaches to α-amino acids. (a) Amination of phenylacetates, (b) amination of ketene silyl acetals, (c) amination of acylpyrazoles, and (d) amination with azo reagents. LG = leaving group, Ts = p-toluenesulfonyl. | ||
Meanwhile, the catalytic hydroamination of readily available α,β-unsaturated carboxylic acid derivatives such as acrylic acids also seems to be an attractive approach to amino acids. However, because of the innate polarity of α,β-unsaturated carbonyls and amines, the nucleophilic amino group generally adds at the electrophilic β-position, thus delivering the β-amino acids selectively (Scheme 2a).19 Therefore, in spite of its potential, α-amino acids are generally difficult to prepare by conventional hydroamination reactions. Herein, we report a copper-catalysed regioselective hydroamination of α,β-unsaturated esters with hydrosilanes and hydroxylamines to form α-amino acid derivatives with high regioselectivity (Scheme 2b). The key to successful regioselectivity control is the introduction of a polarity inversion concept, that is an umpolung strategy;20 the hydrosilane and hydroxylamine work as the nucleophilic hydrogen (hydride) and amino electrophile, respectively, to induce the desired α-amination selectivity. The judicious choice of the CsOPiv base and bisphosphine ligands of remote steric hindrance enables the otherwise challenging C–N bond formation at the α position to the carbonyl. The asymmetric induction at the β position is also possible by a similarly bulky Xyl-BINAP or DTBM-SEGPHOS chiral ligand. Moreover, the point chirality at the α-position is also successfully controlled by using an (−)-8-phenylmenthol auxiliary. Thus, unnatural α-amino acids with adjacent two stereocentres are obtained with high enantioselectivity. The detailed optimization studies, substrate scope, application to conjugation with bioactive amines, and preliminary mechanistic studies are disclosed herein.
 | ||
| Scheme 2 Hydroamination approaches to amino acids. (a) Usual hydroamination and (b) umpolung hydroamination. Bz = benzoate, Piv = tert-butylcarbonyl. | ||
Results and discussion
The blueprint for the regioselective hydroamination of acrylates is based on the recent advances of copper-catalysed net hydroamination of alkenes with hydrosilanes and hydroxylamines, which was originally and independently developed by our group21 and the Buchwald research group.22 Our working hypothesis is shown in Scheme 3. A copper hydride species A is initially formed from the starting copper salt CuX2 and hydrosilane Si–H with the assistance of an external base. The acrylate 1 undergoes the regioselective 1,4-addition with the LnCu–H A to afford the O-bound copper enolate B, where the regioselectivity is controlled by the innate electronic bias of acrylate 1 toward the nucleophilic copper hydride.23 Subsequent electrophilic amination24 with the O-benzoylhydroxylamine 2 delivers the desired α-amino acid derivative 3.25 The concurrently formed copper benzoate C is converted back to the catalytically active copper hydride Avia direct metathesis with the hydrosilane Si–H or base-assisted stepwise ligand exchange. If the appropriate chiral ligand (L) is employed, enantioselectivity is induced in the 1,4-addition step (A to B) to control the point chirality at the β-position. The additional point chirality at the α-position can also be controlled by the proximal chirality at the β-position (substrate control), the chirality of the ligand on copper (catalyst control), or both, giving optically active α-amino acids with two adjacent stereocentres. However, there is a considerable challenge associated with the aforementioned reaction design; Guo and Buchwald recently reported the related attempt of the copper-catalysed regioselective hydroamination of cinnamates with 1,2-benzisoxazole as the electrophilic amino source (Scheme 4).26a While the (S,S)-Ph–BPE ligand successfully gave the corresponding β-amino acid derivative, the α-amino acid was not obtained at all, and instead the simply reduced byproduct 4 was observed exclusively. Around the same time, the research group of Xiong, Guan, and Zhang also reported the related hydroamination of cinnamates, in which the corresponding α-amino acids were detected but as the minor product.26b Thus, the development of suitable reaction conditions for the suppression of the undesired protonation from the copper enolate B is the most important but challenging issue (B to 4). An additional potent side reaction is the reductive N–O bond cleavage of 2 with the copper hydride A, just consuming the electrophilic amination reagent (A and 2 to 2-H). Namely, the copper hydride A should react with the unsaturated ester 1 preferably over the hydroxylamine 2. | ||
| Scheme 3 Working hypothesis. | ||
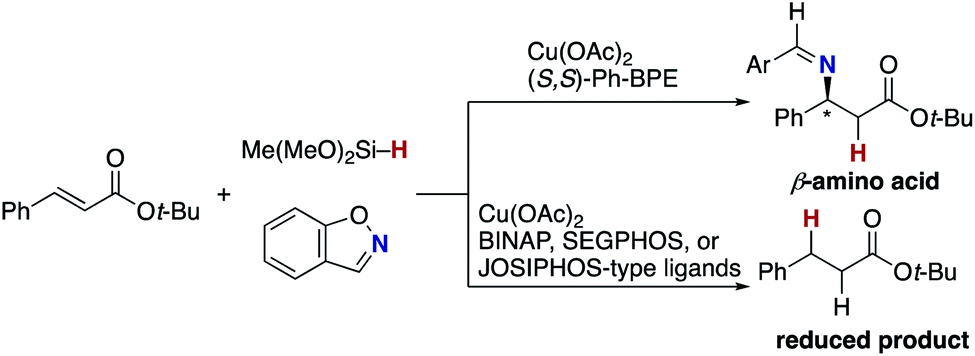 | ||
| Scheme 4 Attempt to synthesize α-amino acids by Guo and Buchwald. | ||
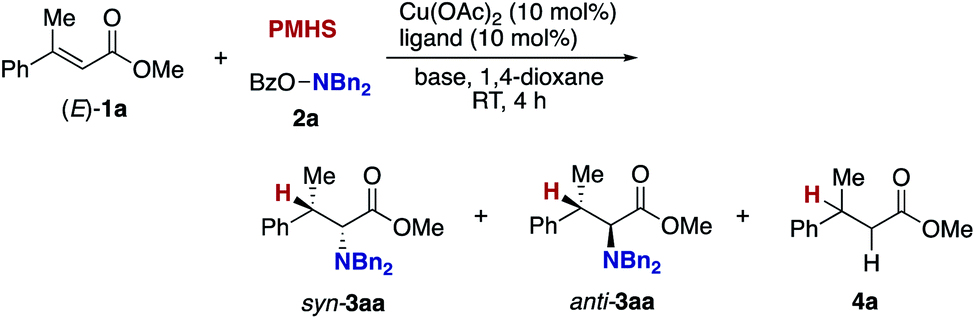
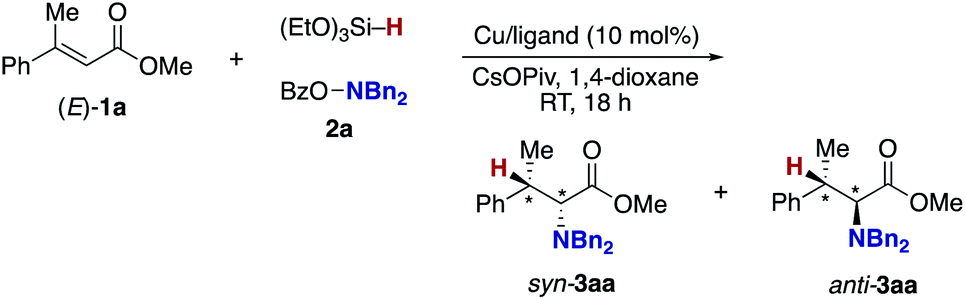
Our optimization studies commenced with (E)-β-methylcinnamate (E)-1a and O-benzoly-N,N-dibenzylhydroxylamine (2a) to identify the suitable ligand, base, and solvent in the presence of Cu(OAc)2 and polymethylhydrosiloxane (PMHS; Table 1). The initial attempt to apply our previous optimal conditions for the styrene hydroamination (CF3-dppbz (dppbz = 1,2-bis(diphenylphosphino)benzene) ligand and the LiO-t-Bu base)21a remained unsuccessful; similar to the aforementioned result in Scheme 4, just saturated ester 4a was observed (entry 1). Additional screening of ligands and solvents was also performed, but the use of LiO-t-Bu only afforded the simply reduced product regardless of other reaction parameters. Thus, we then switched attention to conditions including the bulky dppbz-type ligand, namely, DTBM-dppbz,27 the CsOAc base, and 1,4-dioxane solvent, which were uniquely effective for the related hydroamination of 1-trifluoromethylalkenes.21f Gratifyingly, the desired β-methyl-α-amino acid derivative 3aa![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 28 was formed in 62% 1H NMR yield (syn/anti = 44:56) albeit with the concomitant formation of 4a in 43% (entry 2). Prompted by this preliminary but intriguing result, several acetate-type bases were investigated (entries 3–7), with CsOPiv proving to be best in terms of efficiency and diastereoselectivity. As a general trend, the yield increased with increasing the size of the counter cation (Cs > K > Na). Other cesium bases such as Cs2CO3 and CsF showed lower performance (entries 8 and 9). The reaction also proceeded even in the absence of external bases, but the yield of 3aa largely dropped (entry 10). Even with the DTBM-dppbz ligand, the Li- or NaO-t-Bu bases totally shut down the formation of 3aa (entries 11 and 12). We next examined the effect of hydrosilanes; some alkoxy-substituted hydrosilanes provided the desired 3aa, with (EtO)3SiH giving the α-amino acid 3aa in a comparable yield with a slightly better syn/anti ratio (entry 13). As far as we tested, the reaction was less dependent on the copper catalyst precursor, but Cu(OAc)2·H2O slightly improved the yield (entry 14). On the other hand, the substituent on the aromatic ring in the dppbz-type ligand was critical; the parent dppbz and ortho-substituted o-Me-dppbz resulted in much less productivity (entries 15 and 16) whereas para- and meta-substituted dppbzs gave the α-amino acid 3aa in moderate to good yields, except for the electron-withdrawing p-CF3-dppbz (entries 17–22). The observed better performance of dppbzs of remote steric hindrance can be associated with the attractive London dispersion,29 which accelerates the C–N bond formation (B to C and 3 in Scheme 3) as well as the addition step (A to B in Scheme 3). The yield of 3aa further increased at a higher reaction concentration (entry 23). The final investigation of reaction stoichiometry revealed that the use of 2a as the limiting agent was the best, and the desired 3aa was isolated in 92% yield with 42:58 syn/anti ratio (entry 24). Additional observations are to be noted; other bidentate/monodentate phosphine and NHC ligands mainly formed the reduced 4a or recovered the starting 1a intact; the solvent screening uncovered that 3aa was formed uniquely in 1,4-dioxane; no conversion was observed in the absence of any copper salts or ancillary ligands; other leaving groups on the nitrogen were tested, but OBz was identified to be the best in terms of reactivity and availability (see the ESI† for more detailed optimization studies).
28 was formed in 62% 1H NMR yield (syn/anti = 44:56) albeit with the concomitant formation of 4a in 43% (entry 2). Prompted by this preliminary but intriguing result, several acetate-type bases were investigated (entries 3–7), with CsOPiv proving to be best in terms of efficiency and diastereoselectivity. As a general trend, the yield increased with increasing the size of the counter cation (Cs > K > Na). Other cesium bases such as Cs2CO3 and CsF showed lower performance (entries 8 and 9). The reaction also proceeded even in the absence of external bases, but the yield of 3aa largely dropped (entry 10). Even with the DTBM-dppbz ligand, the Li- or NaO-t-Bu bases totally shut down the formation of 3aa (entries 11 and 12). We next examined the effect of hydrosilanes; some alkoxy-substituted hydrosilanes provided the desired 3aa, with (EtO)3SiH giving the α-amino acid 3aa in a comparable yield with a slightly better syn/anti ratio (entry 13). As far as we tested, the reaction was less dependent on the copper catalyst precursor, but Cu(OAc)2·H2O slightly improved the yield (entry 14). On the other hand, the substituent on the aromatic ring in the dppbz-type ligand was critical; the parent dppbz and ortho-substituted o-Me-dppbz resulted in much less productivity (entries 15 and 16) whereas para- and meta-substituted dppbzs gave the α-amino acid 3aa in moderate to good yields, except for the electron-withdrawing p-CF3-dppbz (entries 17–22). The observed better performance of dppbzs of remote steric hindrance can be associated with the attractive London dispersion,29 which accelerates the C–N bond formation (B to C and 3 in Scheme 3) as well as the addition step (A to B in Scheme 3). The yield of 3aa further increased at a higher reaction concentration (entry 23). The final investigation of reaction stoichiometry revealed that the use of 2a as the limiting agent was the best, and the desired 3aa was isolated in 92% yield with 42:58 syn/anti ratio (entry 24). Additional observations are to be noted; other bidentate/monodentate phosphine and NHC ligands mainly formed the reduced 4a or recovered the starting 1a intact; the solvent screening uncovered that 3aa was formed uniquely in 1,4-dioxane; no conversion was observed in the absence of any copper salts or ancillary ligands; other leaving groups on the nitrogen were tested, but OBz was identified to be the best in terms of reactivity and availability (see the ESI† for more detailed optimization studies).
a
|
|
||||
|---|---|---|---|---|
| Entry | Ligand | Base | Yield of 3aa (%), syn/antib | Yield of 4ac (%) |
| a Conditions: Cu(OAc)2 (0.025 mmol), ligand (0.025 mmol), (E)-1a (0.25 mmol), 2a (0.38 mmol), PMHS (0.75 mmol based on Si–H), base (0.75 mmol), solvent (1.5 mL), RT, 4 h, N2. b Estimated by 1H NMR based on 0.25 mmol with CH2Br2 as the internal standard. The syn/anti ratio is determined in the crude mixture. Isolated yield is in parentheses. c Estimated by 1H NMR based on 0.25 mmol with CH2Br2 as the internal standard. d With (EtO)3SiH instead of PMHS. e With Cu(OAc)2·H2O instead of anhydrous Cu(OAc)2. f In 1,4-dioxane (1.0 mL). g With 1a (0.50 mmol) and 2a (0.25 mmol). | ||||
| 1 | CF3-dppbz | LiO-t-Bu | 0, — | 9 |
| 2 | DTBM-dppbz | CsOAc | 62, 44:56 |
43 |
| 3 | DTBM-dppbz | KOAc | 38, 47:53 |
57 |
| 4 | DTBM-dppbz | NaOAc | 26, 50:50 |
63 |
| 5 | DTBM-dppbz | CsOPiv | 71, 42:58 |
29 |
| 6 | DTBM-dppbz | KOPiv | 70, 44:56 |
30 |
| 7 | DTBM-dppbz | NaOPiv | 0, — | 0 |
| 8 | DTBM-dppbz | Cs2CO3 | 30, 37:67 |
59 |
| 9 | DTBM-dppbz | CsF | 34, 44:56 |
64 |
| 10 | DTBM-dppbz | None | 22, 32:68 |
55 |
| 11 | DTBM-dppbz | LiO-t-Bu | 0, — | 51 |
| 12 | DTBM-dppbz | NaO-t-Bu | 0, — | 49 |
| 13d | DTBM-dppbz | CsOPiv | 71, 38:62 |
30 |
| 14d,e | DTBM-dppbz | CsOPiv | 77, 42:58 |
27 |
| 15d,e | dppbz | CsOPiv | 10, 40:60 |
67 |
| 16d,e | o-Me-dppbz | CsOPiv | 0, — | 0 |
| 17d,e | MeO-dppbz | CsOPiv | 21, 48:52 |
80 |
| 18d,e | p-CF3-dppbz | CsOPiv | 0, — | 0 |
| 19d,e | p-t-Bu-dppbz | CsOPiv | 32, 41:59 |
73 |
| 20d,e | CF3-dppbz | CsOPiv | 46, 41:59 |
55 |
| 21d,e | t-Bu-dppbz | CsOPiv | 47, 38:62 |
53 |
| 22d,e | TMS-dppbz | CsOPiv | 26, 42:58 |
65 |
| 23d,e,f | DTBM-dppbz | CsOPiv | 82, 44:56 |
19 |
| 24d,e,f,g | DTBM-dppbz | CsOPiv | 99, 38:62 (92, 42:58) |
84 |
|
||||
With optimal conditions in hand (entry 24 in Table 1), we next examined the substrate scope of the umpolung-enabled regioselective hydroamination of acrylates toward α-amino acids (Scheme 5). The reaction was compatible with the electron-donating methoxy, electron-withdrawing trifluoromethyl, and chloro groups at the para position of the phenyl ring in the model substrate 1a to form the corresponding β-methylphenylalanine derivatives 3ba–da in good yields. The methylenedioxy substituent was also tolerated (3ea), while the ortho-substitution was somewhat detrimental to the reaction (3fa). The substrates that bear higher fused naphthalene and heteroaromatic benzofuran, thiophene, and pyridine all worked well to deliver the targeted α-amino acids 3ga–ja in 77–95% yields. Additionally, the biologically interesting β-methyltryptophan303ka was accessible. The copper catalysis accommodated some other bulkier alkyl substituents at the β-position; the ethyl-, chloropropyl-, and cyclopropyl-substituted cinnamates underwent the regioselective hydroamination smoothly (3la–na). Moreover, the cyclic systems could also be employed (3oa and 3pa). The successful conversion of β,β-dialkyl-substituted acrylates to deliver the corresponding isoleucine derivatives 3qa and 3ra in synthetically useful yields is particularly notable. The β,β-diaryl substitution pattern was also viable (3sa–ua) albeit with somewhat lower efficiency. The ester, boryl, and silyl functionalities at the β-position were amenable to the regioselective hydroamination, and the biologically interesting aspartic acid 3va, β-boryl-α-amino acid 3wa,31 and β-silyl-α-amino acid 3xa32 were obtained in acceptable yields. Uniquely in the case of 3wa and 3xa, the syn-isomer was mainly formed because of the intramolecular oxygen to boron or silicon coordination in the O-bound copper enolate intermediate (B in Scheme 3).33 On the other hand, the reaction of the simple cinnamate and crotonate also proceeded without any difficulties to form 3ya and 3za in high yields. Different from the work by Guo and Buchwald,26 the regioisomeric β-amino acid was not detected at all in the hydroamination of cinnamate 1y. The α,β,γ,δ-unsaturated sorbate was also applicable, and the corresponding 1,4-adduct 3Aa was formed exclusively.
 | ||
| Scheme 5 Scope of copper-catalysed regioselective hydroamination of acrylates 1 with O-benzoylhydroxylamines 2 for synthesis of α-amino acids 3. Conditions: Cu(OAc)2·H2O (0.015 mmol), DTBM-dppbz (0.015 mmol), 1 (0.30 mmol), 2 (0.15 mmol), (EtO)3Si–H (0.45 mmol), CsOPiv (0.45 mmol), 1,4-dioxane (0.60 mL), RT, 4 h, N2. Isolated yields are shown. a On a 0.25 mmol scale. b On a 1.0 mmol scale. | ||
Several acyclic and cyclic O-benzoylhydroxylamines 2 underwent copper-catalysed hydroamination; N-benzyl-N-methylamine, N,N-diallylamine, piperidine, morpholine, thiomorpholine, and piperazine all were adopted in the reaction to afford the corresponding α-amino acids 3ab–ae, 3bf, and 3ag in good to excellent yields. Additionally it is worth noting that (1) the reaction could also be conducted on a 1.0 mmol scale (3aa); (2) when the (Z)-type substrate was employed, the yield was slightly lower, but the same syn/anti ratio was observed (3aa), thus supporting the intermediacy of the common O-bound copper enolate (B in Scheme 3).34
The aforementioned success prompted us to explore enantioselective conditions by the judicious choice of ancillary chiral ligands (Table 2). Given the positive effects of the DTBM substituent observed in Table 1, we first investigated (R)-DTBM-BINAP, -SEGPHOS, and -MeO-BIPHEP in conjunction with a Cu(OAc)2·H2O catalyst. The DTBM-SEGPHOS ligand promoted the reaction to form 3aa in 60% isolated yield with 42:56 syn/anti ratio and 99:1 e.r. for each diastereomer (entry 2), while no conversion occurred in the presence of DTBM-BINAP and -MeO-BIPHEP (entries 1 and 3). Intriguingly, the relatively small (R)-DM-SEGPHOS also showed high enantioselectivity (98:2 e.r.) albeit with somewhat lower yield of 3aa (entry 4). On the other hand, the parent (R)-SEGPHOS largely decreased the yield (entry 5). Inspired by the comparable performance of DM-SEGPHOS, (R)-Xyl-BINAP and parent (R)-BINAP were also tested (entries 6 and 7). Gratifyingly, the better isolated yield and similarly high enantioselectivity were obtained with the (R)-Xyl-BINAP ligand (81% yield and 97:3 e.r.; entry 6). Additional screening of copper salts revealed that the combination of CuCl/(R)-DTBM-SEGPHOS resulted in better conversion than that of Cu(OAc)2·H2O/(R)-DTBM-SEGPHOS (entries 8 vs. 1) whereas in the case of (R)-Xyl-BINAP, CuCl showed slightly lower activity than Cu(OAc)2·H2O (entries 9 vs. 6). On the basis of the above optimization studies, conditions with Cu(OAc)2·H2O and (R)-Xyl-BINAP were identified to be the best from the viewpoints of catalytic activity and enantioselectivity (entry 6).35 The syn/anti ratio was low, but both isomers could be separated to each other by chromatographic purification. The relative and absolute configurations were assigned by comparison of 1H NMR spectra and specific rotation with the reported values after the derivatization. Given the high enantioselectivity also in the reduced byproduct 4a, the point chirality at the β-position was well controlled but not at the α-position (see the ESI† for details).
a
|
|
||||
|---|---|---|---|---|
| Entry | Cu/ligand | Yield of 3aa (%), syn/antib | e.r.c | |
| syn | anti | |||
| a Conditions: Cu (0.015 mmol), ligand (0.015 mmol), (E)-1a (0.30 mmol), 2a (0.15 mmol), (EtO)3Si–H (0.45 mmol), CsOPiv (0.45 mmol), 1,4-dioxane (0.60 mL), RT, 18 h, N2. b Isolated yields are shown. The syn/anti ratio is determined in the crude mixture. c The enantiomeric ratios (e.r.) were determined by HPLC analysis on a chiral stationary phase. d 1H NMR yield. e The isolated yields of syn-3aa and anti-3aa after the separation. f 4 h. n.d. = not determined. | ||||
| 1 | Cu(OAc)2·H2O/(R)-DTBM-BINAP | 0, — | — | — |
| 2 | Cu(OAc)2·H2O/(R)-DTBM-SEGPHOS | 60, 44:56 |
99:1 |
99:1 |
| 3 | Cu(OAc)2·H2O/(R)-DTBM-MeO-BIPHEP | 0, — | — | — |
| 4 | Cu(OAc)2·H2O/(R)-DM-SEGPHOS | 42, 44:56 |
98:2 |
98:2 |
| 5 | Cu(OAc)2·H2O/(R)-SEGPHOS | 18,d 44:56 |
n.d. | n.d. |
| 6 | Cu(OAc)2·H2O/(R)-Xyl-BINAP | 81, 43:57 (31, 41)e |
97:3 |
97:3 |
| 7 | Cu(OAc)2·H2O/(R)-BINAP | 23, 44:56 |
94:6 |
94:6 |
| 8 | Cu(OAc)2·H2O/(R)-DTBM-SEGPHOS | 73, 42:58 |
96:4 |
96:4 |
| 9f | CuCl/(R)-Xyl-BINAP | 65, 43:57 |
97:3 |
97:3 |
|
||||
Under conditions of entry 6 in Table 2, a variety of β-methylcinnamates underwent regio- and enantioselective hydroamination to form the corresponding β-methylphenylalanine derivatives 3ba–ga in good yields with 92:8–98:2 e.r. (Scheme 6). Similarly under the nonenantioselective conditions, heteroaromatic substituents were also compatible to deliver the hydroaminated products with high enantioselectivity, except for the benzofuran substrate (3ha–ka). The asymmetric catalysis accommodated several other alkyl substituents at the β-position (3la–na and 3pa) as well as the β,β-dialkyl substitution (3qa and 3ra). In particular, both diastereomers of isoleucine derivative 3qa were obtained with high enantiopurity. In the reaction of β,β-diaryl-substituted acrylates, the yield was somewhat lower, but the high enantiomeric ratio still remained (3sa–ua). Moreover, the β-boryl- and β-silyl-α-amino acids 3wa and 3xa were successfully synthesized in enantioenriched forms. In contrast, the simple methyl cinnamate formed the completely racemic product 3ya, thus suggesting almost no control of point chirality at the α-position by the catalyst. As the amino sources, not only the acyclic but also cyclic hydroxylamines were readily and stereoselectively coupled with the β-methylcinnamates to afford the corresponding optically active α-amino acids 3ab–ae, 3bf, and 3ag with high enantiomeric ratios (96:4–99:1 e.r.). In some cases (3qa, 3sa, and 3ta), the combination of CuCl/(R)-DTBM-SEGPHOS instead of Cu(OAc)2·H2O/(R)-Xyl-BINAP showed better performance from the viewpoints of efficiency and enantioselectivity. The enantioselective reaction could also be performed on a 1.0 mmol scale (3aa), indicating the good reliability and reproducibility of the asymmetric copper catalysis.
 | ||
| Scheme 6 Scope and limitation of copper-catalysed regio- and enantioselective hydroamination of acrylates 1 with O-benzoylhydroxylamines 2 for asymmetric synthesis of α-amino acids 3. Conditions: Cu(OAc)2·H2O (0.015 mmol), (R)-Xyl-BINAP (0.015 mmol), 1 (0.30 mmol), 2 (0.15 mmol), (EtO)3Si–H (0.45 mmol), CsOPiv (0.45 mmol), 1,4-dioxane (0.60 mL), RT, 18 h, N2. Isolated yields are shown. a On a 1.0 mmol scale. b With CuCl/(R)-DTBM-SEGPHOS instead of Cu(OAc)2·H2O/(R)-Xyl-BINAP. | ||
On the other hand, the stereoisomeric (Z)-1a was converted to the opposite enantiomers with a moderate enantiomeric ratio (3aa), supporting that the enantioface selection in the reaction of the copper hydride and acrylate 1 (A to B in Scheme 3) occurs in the 1,4-addition manner rather than the 1,2-insertion.36
The newly developed asymmetric copper catalysis was applicable to the derivatization of several biologically active alkylamines (Fig. 1). Nortriptyline and maprotiline, antidepressant drugs, were conjugated with the β-methylcinnamate 1a and 1b under the (R)-Xyl-BINAP-ligated asymmetric copper catalysis to form the corresponding α-amino acids 3ah and 3bi, respectively, with high enantioselectivity. In a similar manner, desloratadine, an antihistamine agent, was successfully modified with 1b, where the heterocyclic pyridine and aryl-Cl moiety were tolerated (3bj). Moreover, the chiral amines including duloxetine (antidepressant and anticonvulsant), paroxetine, and sertraline (selective serotonin reuptake inhibitors) were viable substrates, giving the highly functionalized α-amino acids 3ak, 3al, and 3bm with acceptable diastereomeric ratios (d.r.). Notably, the α-chiral amine, sertraline, showed a significant match/mismatch phenomenon and resulted in almost no formation of the aminated product in the presence of (S)-Xyl-BINAP, while the reaction of paroxetine that bears the chiral centres at the remote positions proceeded smoothly even with (S)-Xyl-BINAP to furnish the product with the opposite stereoselectivity.
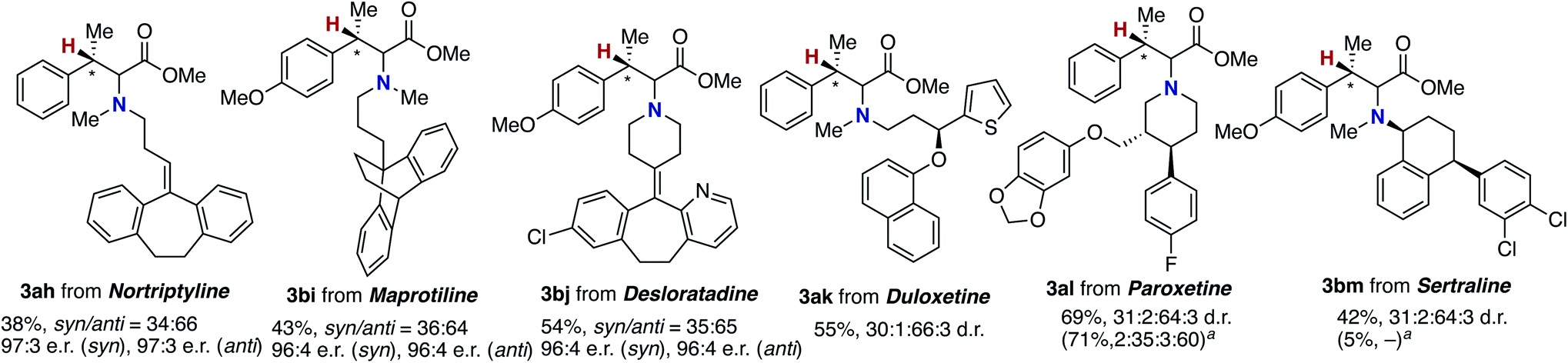 | ||
| Fig. 1 Modification of biologically active amines by conjugation with acrylate 1a through coper-catalysed enantioselective hydroamination. For conditions, see the footnote in Scheme 6. a With (S)-Xyl-BINAP instead of (R)-Xyl-BINAP. | ||
As mentioned above, the Cu/Xyl-BINAP catalyst system successfully controlled the point chirality at the β-position but not at the α-position. Given the intermediacy of the O-bound copper enolate (B in Scheme 3), the steric and electronic effects of the alcohol moiety in 1 (R3 of B in Scheme 3) can affect the stereochemical outcome. Accordingly, several alkyl and aryl esters were prepared and subjected to the enantioselective conditions (Scheme 7a). Unfortunately, the ethyl, tert-butyl, and diphenylmethyl esters showed negligible effects on the stereoselectivity, and the corresponding α-amino acids were formed with low to moderate diastereomeric ratios similar to the methyl ester model substrate (3Ba–Davs.3aa). The additionally coordinating pyridyl ester did not undergo hydroamination at all (3Ea). We then switched attention to the use of chiral alcohol, namely, the chiral auxiliary for improvement of the diastereoselectivity. Pleasingly, the L-(−)-menthol skeleton was found to be the promising candidate to increase the diastereomeric ratio to 25:75 (3Fa), only when combined with (S)-Xyl-BINAP. The value of d.r. was further improved to 9:91 with the assistance of readily prepared (−)-8-phenylmenthol37 (3Ga). The achiral DTBM-dppbz resulted in poor reactivity and moderate stereoselectivity, thus suggesting the necessity of double asymmetric induction arising from the chiral ligand and auxiliary. Treatment with LiAlH4 readily converted 3Ga to the corresponding chiral 1,2-aminoalcohol, and the major anti-5 could be isolated in a pure form with recovery of the chiral auxiliary (Scheme 7b). Subsequent protecting group exchange on nitrogen and TEMPO oxidation afforded the Boc-protected β-methylphenylalanine anti-6 with >99:1 d.r. and >99:1 e.r. (see the ESI† for detailed stereochemical assignment). Given the absolute configuration of anti-6, the Si-face of copper enolate is efficiently blocked by the bulky PhMe2C group, and the hydroxylamine 2a selectively approaches from the Re-face to produce the observed stereoisomer. The Xyl-BINAP/8-phenylmenthol double asymmetric induction strategy was applicable to the cyclic amine (3Gd) as well as other β-alkyl-substituted cinnamate derivatives (3Ha–Ja) to furnish the corresponding α-amino acids with good diastereoselectivity (>10:90; Scheme 7c). The β,β-dialky and -diaryl substrates (3Ka and 3La) could also be employed albeit with slightly reduced stereoselectivity. On the other hand, the simple crotonate resulted in moderate stereochemical induction (3Ma). Even with the achiral DTBM-dppbz, the β-monosubstituted 1M gave 3Ma with the same 31:69 d.r. (data not shown), thus suggesting that the point chirality at the α-position is controlled just by the steric repulsion between the 8-phenylmenthol auxiliary and the substituent at the β-position without influence of the phosphine ligand on copper.
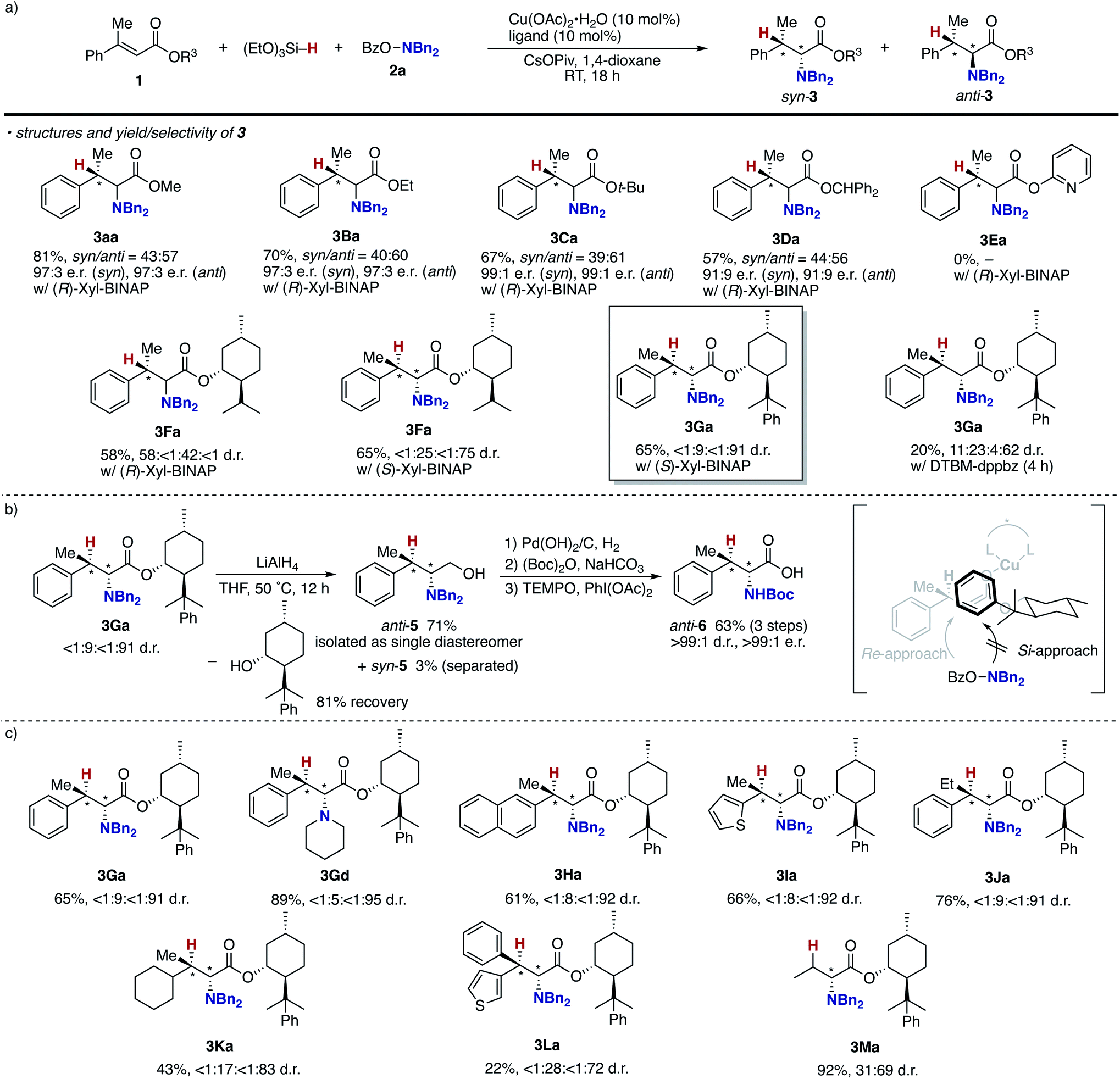 | ||
| Scheme 7 Attempts to control point chirality at the α-position. (a) Identification of alkyl groups in acrylate 1, (b) removal of auxiliary and additional transformations, and (c) substrate scope. | ||
Finally mechanistic studies were implemented. In Scheme 3, we propose C–N bond formation by the reaction of the hydroxylamine 2 and copper enolate B directly formed through the 1,4-addition of the copper hydride A to the acrylate 1, but there are two other potential pathways: one is the stepwise conjugate reduction/enolization/electrophilic amination (Scheme 8a) and another is the hydrosilylation/transmetalation from Si to Cu/electrophilic amination (Scheme 8b). To investigate these possibilities, we performed some control experiments. When the independently prepared simply reduced 4a was subjected to reaction conditions including the copper catalyst, base, and 2a, no aminated product 3aa was observed with 4a left intact (Scheme 9a). The additional use of (EtO)3SiH also gave no 3aa, thus excluding the possibility demonstrated in Scheme 8a. The intermediacy of the ketene silyl acetal (Scheme 8b) was also examined by the following experiments; the copper-catalysed reaction of 1a with (EtO)3SiH in the absence of 2a (4 h) was followed by the addition of D2O to afford the deuterated 4a-d in 99% yield with 82% D content (Scheme 9b).38 This result suggests in situ formation of the ketene silyl acetal. However, the quenching with the hydroxylamine 2a instead of D2O only formed the protonated 4a without any detectable amount of the aminated product 3aa (Scheme 9c). Thus, the ketene silyl acetal can be generated under the optimal catalytic conditions but as nonproductive species, just en route to the protonated byproduct; under optimal conditions, the transmetalation from Si to Cu in the ketene silyl acetal might be unfavored.39 Actually, also under catalytic optimal conditions, the D2O quenching afforded the partial but a significant amount of deuterated 4a-d along with the aminated product 3aa (Scheme 9d). On the basis of the findings in Scheme 9, the originally proposed direct electrophilic amination of the firstly generated copper enolate with hydroxylamines is most favourable.
 | ||
| Scheme 8 Two other possible pathways in C–N bond formation. (a) Conjugate reduction/enolization/electrophilic amination pathway and (b) hydrosilylation/transmetalation/electrophilic amination pathway. | ||
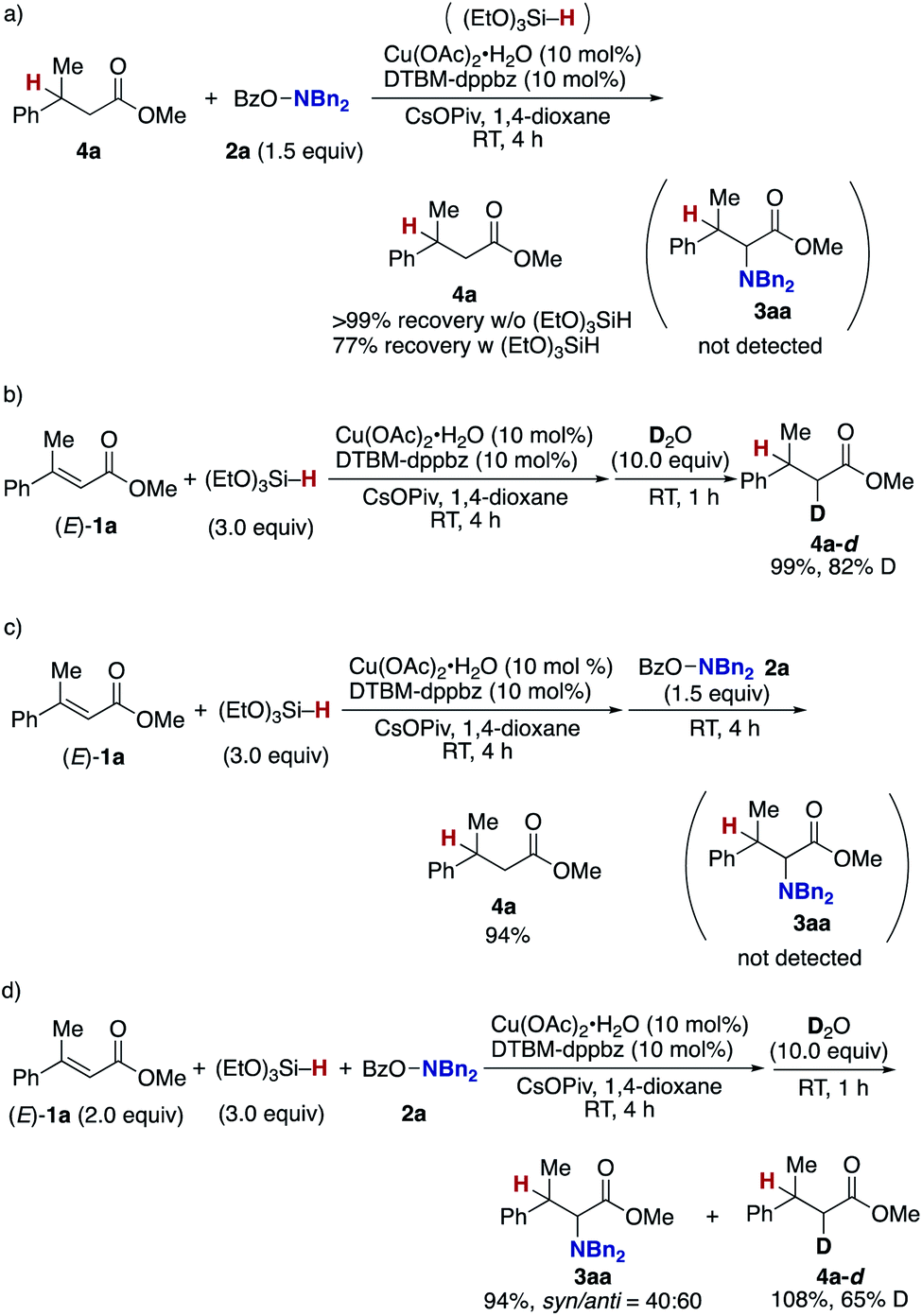 | ||
| Scheme 9 Control experiments. (a) Possibility of 4a as intermediate and (b)–(d) possibilities of ketene silyl acetal as intermediate. | ||
To gain insight into the origin of the positive effects of the CsOPiv base (Table 1, entries 5 vs. 10), we then investigated the amination/protonation selectivity under silane-free, stoichiometric conditions on CuH in the absence and presence of CsOPiv (Scheme 10); upon treatment of unsaturated ester 1N with Stryker's reagent, [(PPh3)CuH]6,40 and the hydroxylamine 2a, the corresponding α-amino acid derivative 3Na was formed in 21% yield along with 34% of the simply reduced 4N. The addition of CsOPiv was almost negligible, delivering 3Na and 4N in similar 15 and 47% yields, respectively. Thus, acceleration of the C–N bond forming step (B to C and 3 in Scheme 3) by the action of CsOPiv is unlikely. On the other hand, LiO-t-Bu totally shut down the formation of 3Na, which is consistent with the optimization studies in entries 1 and 11 of Table 1.
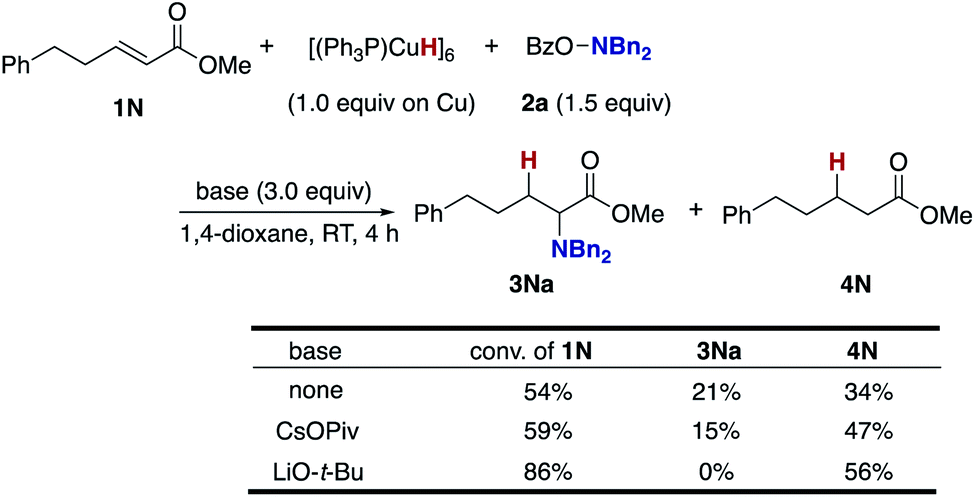 | ||
| Scheme 10 Effects of external bases under silane-free, stoichiometric conditions with [(Ph3P)CuH]6. | ||
The aforementioned results prompted us to check the dependence of the hydroxylamine decomposition side pathway (A and 2 to 2-H in Scheme 3)22e,f on the external base (Scheme 11). Under the external-base free conditions, 80% of the hydroxylamine 2a was decomposed within 4 h. In sharp contrast, the rate of decomposition dramatically decreased in the presence of CsOPiv, and 72% of 2a was retained after 4 h. These outcomes suggest that the CsOPiv base suppresses the competitive N–O bond cleavage by the CuH species to keep the concentration of the hydroxylamine higher and increase the amination product over the protonation byproduct.41 Other bases also suppressed the decomposition to some extent, except for strongly basic LiO-t-Bu, but with CsOPiv proving to be best.
 | ||
| Scheme 11 Effects of external bases in the decomposition of hydroxylamine 2a. | ||
Conclusions
We have developed an umpolung-enabled copper-catalysed regioselective hydroamination of α,β-unsaturated esters with hydrosilanes and hydroxylamines to deliver the corresponding α-amino acid derivatives. The judicious choice of the CsOPiv external base and supporting ligand with remote steric bulkiness promotes the otherwise challenging C–N bond formation at the α position to the carbonyl. The asymmetric induction at the β-position is possible by using the suitable chiral Xyl-BINAP or DTBM-SEGPHOS bisphosphine ligand. Moreover, combined with the 8-phenylmenthol chiral auxiliary, the point chirality at the α-position can also be controlled, giving optically active unnatural α-amino acids with two adjacent stereocentres. Asymmetric copper catalysis is also applied to the conjugation of α-amino acids with biologically active complex amines. Some mechanistic experiments suggest the pivotal role of the copper enolate in the C–N forming step and the unique effect of CsOPiv to suppress the competitive but nonproductive decomposition pathway of the hydroxylamines. The obtained results can provide a new repertoire of C–N bond formation approaches to unnatural and complicated chiral α-amino acid derivatives. More detailed mechanistic studies and further development of related copper catalysis for more complicated and densely functionalized α-amino acids are ongoing in our laboratory and will be reported in due course.Data availability
All experimental procedures and spectroscopic data can be found in the ESI.†Author contributions
S. N. and K. H. conceived the idea. S. N. performed all experiments including condition optimizations and exploring the scope. K. H. supervised the project. M. M. supported other authors to perform the project well. All the authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI Grant no. JP 17H06092 (Grant-in-Aid for Specially Promoted Research) to MM and 18K19078 (Grant-in-Aid for Challenging Research (Exploratory)) to KH. KH also acknowledges Toyota Riken Scholar for financial support. We thank Mr Tatsuaki Takata (Osaka University) for his initial experimental assistance.Notes and references
- (a) S. V. Bhat, B. A. Nagasampagi and M. Sivakumar, Chemistry of Natural Products, Springer, 2005, pp. 317–393 Search PubMed; (b) L. Weber, Curr. Med. Chem., 2002, 9, 2085 CrossRef CAS PubMed; (c) C. Hulme and V. Gore, Curr. Med. Chem., 2003, 10, 51 CrossRef CAS PubMed; (d) L. Wang and P. G. Schultz, Angew. Chem., Int. Ed., 2005, 44, 34 CrossRef CAS PubMed; (e) Y. S. Tsantrizos, Acc. Chem. Res., 2008, 41, 1252 CrossRef CAS PubMed; (f) A. A. Vinogradov, Y. Yin and H. Suga, J. Am. Chem. Soc., 2019, 141, 4167 CrossRef CAS PubMed; (g) M. Muttenthaler, G. F. King, D. J. Adams and P. F. Alewood, Nat. Rev. Drug Discovery, 2021, 20, 309 CrossRef CAS PubMed.
- (a) A. Domling and I. Ugi, Angew. Chem., Int. Ed., 2000, 39, 3168 CrossRef CAS; (b) H. Gröger, Chem. Rev., 2003, 103, 2795 CrossRef PubMed; (c) A. Domling, Chem. Rev., 2006, 106, 17 CrossRef PubMed; (d) N. R. Candeias, F. Montalbano, P. M. S. D. Cal and P. M. P. Gois, Chem. Rev., 2010, 110, 6169 CrossRef CAS PubMed; (e) J. Wang, X. Liu and X. Feng, Chem. Rev., 2011, 111, 6947 CrossRef CAS PubMed.
- Accounts and reviews: (a) W. S. Knowles, Acc. Chem. Res., 1983, 16, 106 CrossRef CAS; (b) W. Tang and X. Zhang, Chem. Rev., 2003, 103, 3029 CrossRef CAS PubMed; (c) C. S. Shultz and S. W. Krska, Acc. Chem. Res., 2007, 40, 1320 CrossRef CAS PubMed ; selected examples:; (d) M. J. Burk, M. F. Gross and J. P. Martinez, J. Am. Chem. Soc., 1995, 117, 9375 CrossRef CAS; (e) C. Molinaro, J. P. Scott, M. Shevlin, C. Wise, A. Ménard, A. Gibb, E. M. Junker and D. Lieberman, J. Am. Chem. Soc., 2015, 137, 999 CrossRef CAS PubMed.
- (a) K. Maruoka and T. Ooi, Chem. Rev., 2003, 103, 3013 CrossRef CAS PubMed; (b) T. Hashimoto and K. Maruoka, Chem. Rev., 2007, 107, 5656 CrossRef CAS PubMed; (c) S.-s. Jew and H.-g. Park, Chem. Commun., 2009, 7090 RSC; (d) T. Ooi, D. Kato, K. Inamura, K. Ohmatsu and K. Maruoka, Org. Lett., 2007, 9, 3945 CrossRef CAS PubMed.
- (a) S. Lee, N. A. Beare and J. F. Hartwig, J. Am. Chem. Soc., 2001, 123, 8410 CrossRef CAS; (b) O. Gaertzen and S. L. Buchwald, J. Org. Chem., 2002, 67, 465 CrossRef CAS PubMed; (c) M. Hocek, Heterocycles, 2004, 63, 1673 CrossRef CAS.
- Y. Matsumoto, J. Sawamura, Y. Murata, T. Nishikata, R. Yazaki and T. Ohshima, J. Am. Chem. Soc., 2020, 142, 8498 CrossRef CAS PubMed.
- (a) L. Wei, Q. Zhu, S.-M. Xu, X. Chang and C.-J. Wang, J. Am. Chem. Soc., 2018, 140, 1508 CrossRef CAS PubMed; (b) X. Huo, J. Zhang, J. Fu, R. He and W. Zhang, J. Am. Chem. Soc., 2018, 140, 2080 CrossRef CAS.
- (a) L. Zhao and C. J. Li, Angew. Chem., Int. Ed., 2008, 47, 7075 CrossRef CAS PubMed; (b) J. Xie and Z. Z. Huang, Angew. Chem., Int. Ed., 2010, 49, 10181 CrossRef CAS PubMed; (c) G. Zhang, Y. Zhang and R. Wang, Angew. Chem., Int. Ed., 2011, 50, 10429 CrossRef CAS PubMed; (d) S. Zhu and M. Rueping, Chem. Commun., 2012, 48, 11960 RSC; (e) Z.-Q. Wang, M. Hu, X.-C. Huang, L.-B. Gong, Y.-X. Xie and J.-H. Li, J. Org. Chem., 2012, 77, 8705 CrossRef CAS PubMed; (f) X.-W. Gao, Q.-Y. Meng, M. Xiang, B. Chen, K. Feng, C.-H. Tung and L.-Z. Wu, Adv. Synth. Catal., 2013, 355, 2158 CrossRef CAS; (g) X.-W. Gao, Q.-Y. Meng, J.-X. Li, J.-J. Zhong, T. Lei, X.-B. Li, C.-H. Tung and L.-Z. Wu, ACS Catal., 2015, 5, 2391 CrossRef CAS.
- J. A. Smulik and E. Vedejs, Org. Lett., 2003, 5, 4187 CrossRef CAS PubMed.
- B. Zhao, H. Du and Y. Shi, J. Am. Chem. Soc., 2008, 130, 7220 CrossRef CAS PubMed.
- R. W. Evano, J. R. Zbieg, S. Zhu, W. Li and D. W. C. MacMillan, J. Am. Chem. Soc., 2013, 135, 16074 CrossRef.
- N. Matsuda, K. Hirano, T. Satoh and M. Miura, Angew. Chem., Int. Ed., 2012, 51, 11827 CrossRef CAS PubMed.
- T. Miura, M. Morimoto and M. Murakami, Org. Lett., 2012, 14, 5214 CrossRef CAS PubMed.
- K. Kiyokawa, D. Okumatsu and S. Minakata, Angew. Chem., Int. Ed., 2019, 58, 8907 CrossRef CAS PubMed.
- K. Tokumasu, R. Yazaki and T. Ohshima, J. Am. Chem. Soc., 2016, 138, 2664 CrossRef CAS PubMed.
- M. Shang, X. Wang, S. M. Koo, J. Youn, J. Z. Chan, W. Yao, B. T. Hastings and M. Wasa, J. Am. Chem. Soc., 2017, 139, 95 CrossRef CAS PubMed.
- T. Morisawa, M. Sawamura and Y. Shimizu, Org. Lett., 2019, 21, 7466 CrossRef CAS PubMed.
- For the related α-amination reactions of activated 1,3-dicarbonyl compounds, ketones, and aldehydes, see: (a) A. M. R. Smith and K. K. Hii, Chem. Rev., 2011, 111, 1637 CrossRef CAS PubMed; (b) S. Mukherjee, J. W. Yang, S. Hoffmann and B. List, Chem. Rev., 2007, 107, 5471 CrossRef CAS PubMed; (c) P. Melchiorre, M. Marigo, A. Carlone and G. Bartoli, Angew. Chem., Int. Ed., 2008, 47, 6138 CrossRef CAS PubMed; (d) W. Adam, K. J. Roschmann and C. R. Saha-Möller, Eur. J. Org. Chem., 2000, 557 CrossRef CAS; (e) J. L. Liang, X. Q. Yu and C. M. Che, Chem. Commun., 2002, 124 RSC; (f) T. Baumann, M. Bächle and S. Bräse, Org. Lett., 2006, 8, 3797 CrossRef CAS PubMed; (g) M. Tanaka, Y. Kurosaki, T. Washio, M. Anada and S. Hashimoto, Tetrahedron Lett., 2007, 48, 8799 CrossRef CAS; (h) D. M. S. Schietroma, M. R. Monaco, V. Bucalossi, P. E. Walter, P. Gentili and M. Bella, Org. Biomol. Chem., 2012, 10, 4692 RSC; (i) J.-S. Tian, K. W. J. Ng, J.-R. Wong and T.-P. Loh, Angew. Chem., Int. Ed., 2012, 51, 9105 CrossRef CAS PubMed.
- For selected reviews: (a) L.-W. Xu and C.-G. Xia, Eur. J. Org. Chem., 2005, 633 CrossRef CAS; (b) D. Enders, C. Wang and J. X. Liebich, Chem.–Eur. J., 2009, 15, 11058 CrossRef CAS PubMed; (c) P. R. Krishna, A. Sreeshailam and R. Srinivas, Tetrahedron, 2009, 65, 9657 CrossRef CAS; (d) A. Y. Rulev, Russ. Chem. Rev., 2011, 80, 197 CrossRef CAS; (e) J. Wang, P. Li, P. Y. Choy, A. S. C. Chan and F. Y. Kwong, ChemCatChem, 2012, 4, 917 CrossRef CAS; (f) M. G. Vinogradov, O. V. Turova and S. G. Zlotin, Org. Biomol. Chem., 2019, 17, 3670 RSC.
- (a) D. Seebach and E. J. Corey, J. Org. Chem., 1975, 40, 231 CrossRef CAS; (b) D. Seebach, Angew. Chem., Int. Ed. Engl., 1979, 18, 239 CrossRef.
- (a) Y. Miki, K. Hirano, T. Satoh and M. Miura, Angew. Chem., Int. Ed., 2013, 52, 10830 CrossRef CAS PubMed; (b) Y. Miki, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2014, 16, 1498 CrossRef CAS PubMed; (c) D. Nishikawa, K. Hirano and M. Miura, J. Am. Chem. Soc., 2015, 137, 15620 CrossRef CAS PubMed; (d) D. Nishikawa, R. Sakae, Y. Miki, K. Hirano and M. Miura, J. Org. Chem., 2016, 81, 12128 CrossRef CAS PubMed; (e) T. Takata, D. Nishikawa, K. Hirano and M. Miura, Chem.–Eur. J., 2018, 24, 10975 CrossRef CAS PubMed; (f) T. Takata, K. Hirano and M. Miura, Org. Lett., 2019, 21, 4284 CrossRef CAS PubMed; (g) S. Nishino, K. Hirano and M. Miura, Chem.–Eur. J., 2020, 26, 8725 CrossRef CAS PubMed.
- (a) S. Zhu, N. Niljianskul and S. L. Buchwald, J. Am. Chem. Soc., 2013, 135, 15746 CrossRef CAS PubMed; (b) S. Zhu and S. L. Buchwald, J. Am. Chem. Soc., 2014, 136, 15913 CrossRef CAS PubMed; (c) N. Niljianskul, S. Zhu and S. L. Buchwald, Angew. Chem., Int. Ed., 2015, 54, 1638 CrossRef CAS PubMed; (d) S.-L. Shi and S. L. Buchwald, Nat. Chem., 2015, 7, 38 CrossRef CAS PubMed; (e) Y. Yang, S.-L. Shi, D. Niu, P. Liu and S. L. Buchwald, Science, 2015, 349, 62 CrossRef CAS PubMed; (f) D. Niu and S. L. Buchwald, J. Am. Chem. Soc., 2015, 137, 9716 CrossRef CAS PubMed; (g) J. S. Bandar, M. T. Pirnot and S. L. Buchwald, J. Am. Chem. Soc., 2015, 137, 14812 CrossRef CAS PubMed; (h) H. Wang, J. C. Yang and S. L. Buchwald, J. Am. Chem. Soc., 2017, 139, 8428 CrossRef CAS PubMed; (i) Y. Zhou, O. D. Engl, J. S. Bandar, E. D. Chant and S. L. Buchwald, Angew. Chem., Int. Ed., 2018, 57, 6672 CrossRef CAS PubMed.
- For a review on copper hydride species in organic synthesis, see: C. Deutsch, N. Krause and B. H. Lipshutz, Chem. Rev., 2008, 108, 2916 CrossRef CAS PubMed.
- For pioneering work on the electrophilic amination using the hydroxylamines, see: (a) H. Tsutsui, Y. Hayashi and K. Narasaka, Chem. Lett., 1997, 26, 317 CrossRef; (b) A. M. Berman and J. S. Johnson, J. Am. Chem. Soc., 2004, 126, 5680 CrossRef CAS PubMed; (c) S. Liu and L. S. Liebeskind, J. Am. Chem. Soc., 2008, 130, 6918 CrossRef CAS PubMed ; representative reviews:; (d) E. Erdik and M. Ay, Chem. Rev., 1989, 89, 1947 CrossRef CAS; (e) K. Narasaka and M. Kitamura, Eur. J. Org. Chem., 2005, 4505 CrossRef CAS; (f) E. Ciganek, Org. React., 2009, 72, 1 Search PubMed; (g) T. J. Barker and E. R. Jarvo, Synthesis, 2011, 3954 CAS; (h) M. Corpet and C. Gosmini, Synthesis, 2014, 46, 2258 CrossRef CAS; (i) M. T. Pirnot, Y.-M. Wang and S. L. Buchwald, Angew. Chem., Int. Ed., 2016, 55, 48 CrossRef CAS PubMed; (j) X. Dong, Q. Liu, Y. Dong and H. Liu, Chem.–Eur. J., 2017, 23, 2481 CrossRef CAS PubMed.
- For recent computational studies on the electrophilic amination of organocopper species with the hydroxylamine, see: (a) S. Tobisch, Chem.–Eur. J., 2016, 22, 8290 CrossRef CAS PubMed; (b) S. Tobisch, Chem.–Eur. J., 2017, 23, 17800 CrossRef CAS PubMed; (c) S. Tobisch, Chem. Sci., 2017, 8, 4410 RSC.
- (a) S. Guo, J. Zhu and S. L. Buchwald, Angew. Chem., Int. Ed., 2020, 59, 20841 CrossRef CAS PubMed; (b) G. Zhang, Y. Liang, T. Qin, T. Xiong, S. Liu, W. Guan and Q. Zhang, CCS Chem., 2020, 2, 1737 Search PubMed.
- For recent applications of the modified dppbz ligands in catalysis, see: (a) S. Ito, T. Itoh and M. Nakamura, Angew. Chem., Int. Ed., 2011, 50, 454 CrossRef CAS PubMed; (b) T. Hatakeyama, T. Hashimoto, Y. Kondo, Y. Fujiwara, H. Seike, H. Takaya, Y. Tamada, T. Ono and M. Nakamura, J. Am. Chem. Soc., 2010, 132, 10674 CrossRef CAS PubMed; (c) T. Fujihara, A. Sawada, T. Yamaguchi, Y. Tani, J. Terao and Y. Tsuji, Angew. Chem., Int. Ed., 2017, 56, 1539 CrossRef CAS PubMed; (d) D. Nishikawa, K. Hirano and M. Miura, Org. Lett., 2016, 18, 4856 CrossRef CAS PubMed; (e) K. Kato, K. Hirano and M. Miura, Angew. Chem., Int. Ed., 2016, 55, 14400 CrossRef CAS PubMed; (f) K. Kato, K. Hirano and M. Miura, J. Org. Chem., 2017, 82, 10418 CrossRef CAS PubMed; (g) H. Iwamoto, K. Kubota and H. Ito, Chem. Commun., 2016, 52, 5916 RSC.
- For the β-methyl-α-amino acids as the important nonproteinogenic amino acids in the synthesis of a bioactive natural product assembly, see: (a) B. Wilkinson and J. Micklefield, Methods Enzymol., 2009, 458, 353 CAS; (b) N. Sitaram, Curr. Med. Chem., 2006, 13, 679 CrossRef CAS PubMed; (c) J. A. Ma, Angew. Chem., Int. Ed., 2003, 42, 4290 CrossRef CAS PubMed; (d) G. Cardillo, L. Gentilucci and A. Tolomelli, Mini-Rev. Med. Chem., 2006, 6, 293 CrossRef CAS PubMed; (e) M. S. Butler and A. D. Buss, Biochem. Pharmacol., 2006, 71, 919 CrossRef CAS PubMed.
- (a) D. J. Liptrot and P. P. Power, Nat. Rev. Chem., 2017, 1, 00004 CrossRef; (b) G. Lu, R. Y. Liu, Y. Yang, C. Fang, D. S. Lambrecht, S. L. Buchwald and P. Liu, J. Am. Chem. Soc., 2017, 139, 16548 CrossRef CAS PubMed; (c) Y. Xi, B. Su and X. Qi, J. Am. Chem. Soc., 2020, 142, 18213 CrossRef PubMed . For a related interaction between dppbz-type ligands and aromatic substrates in the insertion into the borylcopper species, see:; (d) T. Kang, T. G. Erbay, K. L. Xu, G. M. Gallego, A. Burtea, S. K. Nair, R. L. Patman, R. Zhou, S. C. Sutton, I. J. McAlpine, P. Liu and K. M. Engle, ACS Catal., 2020, 10, 13075 CrossRef CAS PubMed.
- (a) W. Balk-Bindseil, E. Helmke, H. Weyland and H. Laatsch, Liebigs Ann., 1995, 7, 1291 CrossRef; (b) T. Ueda, M. Inada, I. Okamoto, N. Morita and O. Tamura, Org. Lett., 2008, 10, 2043 CrossRef CAS PubMed; (c) S. Takase, N. Shigematsu, I. Shima, I. Uchida, M. Hashimoto, T. Tada, S. Koda and Y. Morimoto, J. Org. Chem., 1987, 52, 3485 CrossRef CAS; (d) O. E. Christian, J. Compton, K. R. Christian, S. L. Mooberry, F. A. Valeriote and P. Crews, J. Nat. Prod., 2005, 68, 1592 CrossRef CAS PubMed; (e) T. H. Chan and R. K. Hill, J. Org. Chem., 1970, 35, 3519 CrossRef CAS PubMed; (f) J. C. Sheehan, D. Mania, S. Nakamura, J. A. Stock and K. Maeda, J. Am. Chem. Soc., 1968, 90, 462 CrossRef CAS PubMed.
- C. Cabrele, T. A. Martinek, O. Reiser and Ł. Berlicki, J. Med. Chem., 2014, 57, 9718 CrossRef CAS PubMed.
- E. Rémond, C. Martin, J. Martinez and F. Cavelier, Chem. Rev., 2016, 116, 11654 CrossRef PubMed.
- J.-B. Xie, S. Lin, S. Qiao and G. Li, Org. Lett., 2016, 18, 3926 CrossRef CAS PubMed.
- Attempts to apply α,β-unsaturated lactones, ketones, amides, and carboxylic acids remained unsuccessful. The secondary amine and benzisoxazole (primary amine surrogate) were also unfavourable to the reaction. See the ESI† for more details.
- See the ESI for more detailed optimization studies for enantioselective conditions.
- C. Wu, G. Yue, C. D.-T. Nielsen, K. Xu, H. Hirao and J. Zhou, J. Am. Chem. Soc., 2016, 138, 742 CrossRef CAS PubMed.
- (a) E. J. Corey and H. E. Ensley, J. Am. Chem. Soc., 1975, 97, 6908 CrossRef CAS PubMed; (b) W. Oppolzer, C. Robbiani and K. Battig, Helv. Chim. Acta, 1980, 63, 2015 CrossRef CAS; (c) O. Ort, Org. Synth., 1987, 65, 203 CrossRef CAS.
- At present, the H source for the α position to the carbonyl is unclear. Even in 1,4-dioxane-d8 under otherwise identical catalytic conditions, the deuterated 4a-d was not formed, and the simple 4a was observed exclusively.
- For related studies, see: S. Bouaouli, K. Spielmann, E. Vrancken, J.-M. Campagne and H. Gérard, Chem.–Eur. J., 2018, 24, 6617 CrossRef CAS PubMed.
- W. S. Mahoney, D. M. Brestensky and J. M. Stryker, J. Am. Chem. Soc., 1988, 110, 291 CrossRef CAS . In the stoichiometric reaction studies, the β-mono-substituted 1N was used because β,β-disubstituted substrates such as 1a underwent no conversion even in the presence of a stoichiometric amount of [(Ph3P)CuH]6..
- At present, we cannot completely exclude the possibility that CsOPiv also accelerates the C–N bond forming step in the case of β,β-disubstituted substrates such as 1a.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc03692k |
| This journal is © The Royal Society of Chemistry 2021 |