
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Difluorination of α-(bromomethyl)styrenes via I(I)/I(III) catalysis: facile access to electrophilic linchpins for drug discovery†
Joel
Häfliger
,
Keith
Livingstone
,
Constantin G.
Daniliuc
and
Ryan
Gilmour
 *
*
Organisch-Chemisches Institut, Westfälische Wilhelms-Universität Münster, Corrensstraße 36, 48149 Münster, Germany. E-mail: ryan.gilmour@uni-muenster.de
First published on 26th March 2021
Abstract
Simple α-(bromomethyl)styrenes can be processed to a variety of 1,1-difluorinated electrophilic building blocks via I(I)/I(III) catalysis. This inexpensive main group catalysis strategy employs p-TolI as an effective organocatalyst when combined with Selectfluor® and simple amine·HF complexes. Modulating Brønsted acidity enables simultaneous geminal and vicinal difluorination to occur, thereby providing a platform to generate multiply fluorinated scaffolds for further downstream derivatization. The method facilitates access to a tetrafluorinated API candidate for the treatment of amyotrophic lateral sclerosis. Preliminary validation of an enantioselective process is disclosed to access α-phenyl-β-difluoro-γ-bromo/chloro esters.
Structural editing with fluorine enables geometric and electronic variation to be explored in functional small molecules whilst mitigating steric drawbacks.1 This expansive approach to manipulate structure–function interplay continues to manifest itself in bio-organic and medicinal chemistry.2 Of the plenum of fluorinated motifs commonly employed, the geminal difluoromethylene group3 has a venerable history.4 This is grounded in the structural as well as electronic ramifications of CH2 → CF2 substitution, as is evident from a comparison of propane and 2,2-difluoropropane (Fig. 1, upper). Salient features include localized charge inversion (C–Hδ+ to C–Fδ−) and a widening of the internal angle from 112° to 115.4°.5 Consequently, geminal difluoromethylene groups feature prominently in the drug discovery repertoire6 to mitigate oxidation and modulate physicochemical parameters. Catalysis-based routes to generate electrophilic linchpins that contain the geminal difluoromethylene unit have thus been intensively pursued, particularly in the realm of main group catalysis.7–9 Motivated by the potential of this motif in contemporary medicinal chemistry, it was envisaged that an I(I)/I(III) catalysis platform could be leveraged to convert simple α-(bromomethyl)styrenes to gem-difluorinated linchpins: the primary C(sp3)–Br motif would facilitate downstream synthetic manipulations (Fig. 1, lower). To that end, p-TolI would function as a catalyst to generate p-TolIF2in situ in the presence of an external oxidant10 and an amine·HF complex. Alkene activation (I) with subsequent bromonium ion formation (II)11 would provide a pre-text for the first C–F bond forming process (III) with regeneration of the catalyst. A subsequent phenonium ion rearrangement12/fluorination sequence (III and IV) would furnish the geminal difluoromethylene group and liberate the desired electrophilic building block.
 | ||
| Fig. 1 The geminal difluoromethylene group: bioisosterism, and catalysis-based access from α-(bromomethyl)styrenes via I(I)/I(III) catalysis. | ||
To validate this conceptual framework, a short process of reaction optimization (1a → 2a) was conducted to assess the influence of solvent, amine·HF ratio (Brønsted acidity)13 and catalyst loading (Table 1). Initial reactions were performed with p-TolI (20 mol%), Selectfluor® (1.5 equiv.) as an oxidant, and CHCl3 as the reaction medium. Variation of the amine![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :HF ratio was conducted to explore the influence of Brønsted acidity on catalysis efficiency (entries 1–4). An optimal ratio of 1:6 was observed enabling the product 2a to be generated in >95% NMR-yield. Although reducing the catalyst loading to 10 and 5 mol% (entries 5 and 6, respectively) led to high levels of efficiency (79% yield with 5 mol%), the remainder of the study was performed with 20 mol% p-TolI. Notably, catalytic vicinal difluorination was not observed at any point during this optimization, in contrast with previous studies from our laboratory.9d,i A solvent screen revealed the importance of chlorinated solvents (entries 7 and 8): in contrast, performing the reaction in ethyl trifluoroacetate (ETFA) and acetonitrile resulted in a reduction in yield (9 and 10). Finally, a control reaction in the absence of p-TolI confirmed that an I(I)/I(III) manifold was operational (entry 11). An expanded optimization table is provided in the ESI.†
:HF ratio was conducted to explore the influence of Brønsted acidity on catalysis efficiency (entries 1–4). An optimal ratio of 1:6 was observed enabling the product 2a to be generated in >95% NMR-yield. Although reducing the catalyst loading to 10 and 5 mol% (entries 5 and 6, respectively) led to high levels of efficiency (79% yield with 5 mol%), the remainder of the study was performed with 20 mol% p-TolI. Notably, catalytic vicinal difluorination was not observed at any point during this optimization, in contrast with previous studies from our laboratory.9d,i A solvent screen revealed the importance of chlorinated solvents (entries 7 and 8): in contrast, performing the reaction in ethyl trifluoroacetate (ETFA) and acetonitrile resulted in a reduction in yield (9 and 10). Finally, a control reaction in the absence of p-TolI confirmed that an I(I)/I(III) manifold was operational (entry 11). An expanded optimization table is provided in the ESI.†
|
|
||||
|---|---|---|---|---|
| Entry | Solvent | Amine/HF | Catalyst loading [mol%] | Yieldb [%] |
|
a Standard reaction conditions: 1a (0.2 mmol), Selectfluor® (1.5 equiv.), amine:HF source (0.5 mL), solvent (0.5 mL), p-TolI, 24 h, rt.
b Determined by 19F NMR using α,α,α-trifluorotoluene as internal standard.
|
||||
| 1 | CHCl3 | 1:4.5 |
20 | 72 |
| 2 | CHCl 3 |
1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :6.0 :6.0 |
20 | >95 |
| 3 | CHCl3 | 1:7.5 |
20 | 94 |
| 4 | CHCl3 | 1:9.23 |
20 | 87 |
| 5 | CHCl3 | 1:6.0 |
10 | 87 |
| 6 | CHCl3 | 1:6.0 |
5 | 79 |
| 7 | DCM | 1:6.0 |
20 | >95 |
| 8 | DCE | 1:6.0 |
20 | 93 |
| 9 | ETFA | 1:6.0 |
20 | 84 |
| 10 | MeCN | 1:6.0 |
20 | 50 |
| 11 | CHCl3 | 1:6.0 |
0 | <5 |

To explore the scope of this geminal difluorination, a series of α-(bromomethyl)styrenes were exposed to the standard reaction conditions (Fig. 2). Gratifyingly, product 2a could be isolated in 80% yield after column chromatography on silica gel. The parent α-(bromomethyl)styrene was smoothly converted to species 2b, as were the p-halogenated systems that furnished 2c and 2d (71 and 79%, respectively). The regioisomeric bromides 2e and 2f (70 and 62%, respectively) were also prepared for completeness to furnish a series of linchpins that can be functionalized at both termini by displacement and cross-coupling protocols (2a, 2e and 2f). Modifying the amine:HF ratio to 1:4.5 provided conditions to generate the tBu derivative 2g in 68% yield.14 Electron deficient aryl derivatives were well tolerated as is demonstrated by the formation of compounds 2h–2k (up to 91%). Disubstitution patterns (2l, 81%), sulfonamides (2m, 75%) and phthalimides (2n, 80%) were also compatible with the standard catalysis conditions. Gratifyingly, compound 2n was crystalline and it was possible to unequivocally establish the structure by X-ray crystallography (Fig. 2, lower).15 The C9–C8–C7 angle was measured to be 112.6° (cf. 115.4° for 2,2-difluoropropane).5 Intriguingly, the C(sp3)–Br bond eclipses the two C–F bonds rather than adopting a conformation in which dipole minimization is satisfied (F1–C8–C9–Br dihedral angle is 56.3°).
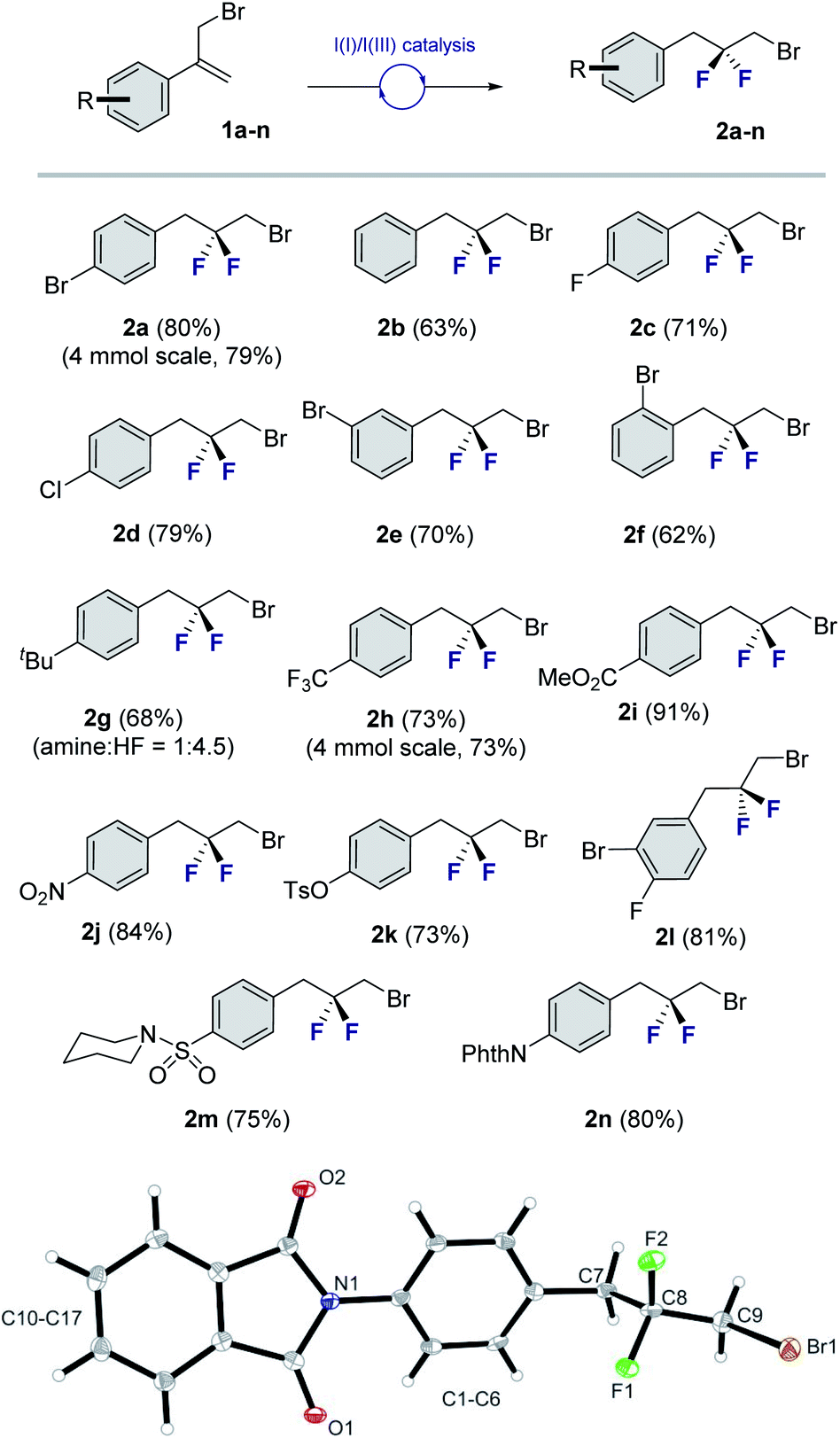 | ||
| Fig. 2 Exploring the scope of the geminal difluorinative rearrangement of α-(bromomethyl)styrenes via I(I)/I(III) catalysis. Isolated yields after column chromatography on silica gel are reported. X-ray crystal structure of compound 2n (CCDC 2055892†). Thermal ellipsoids shown at 50% probability. | ||
Cognizant of the influence of Brønsted acidity on the regioselectivity of I(I)/I(III) catalyzed alkene difluorination,9d the influence of the amine:HF ratio on the fluorination of electronically non-equivalent divinylbenzene derivatives was explored (Fig. 3, top). Initially, compound 3 bearing an α-(trifluoromethyl)styrene motif was exposed to the standard catalysis conditions with a 1:4.5 amine:HF ratio. Exclusive, chemoselective formation of 4 was observed in 79% yield. Simple alteration of the amine:HF ratio to 1:7.5 furnished the tetrafluorinated product 5 bearing both the geminal and vicinal difluoromethylene16 groups (55% yield. 20% of the geminal–geminal product was also isolated. See ESI†). Relocating the electron-withdrawing group (α-CF3 → β-CO2Me) and repeating the reaction with 1:4.5 amine:HF generated the geminal CF2 species 7 in analogy to compound 4. However, increasing the amine:HF ratio to 1:6.0 led exclusively to double geminal difluorination (8, 55%).
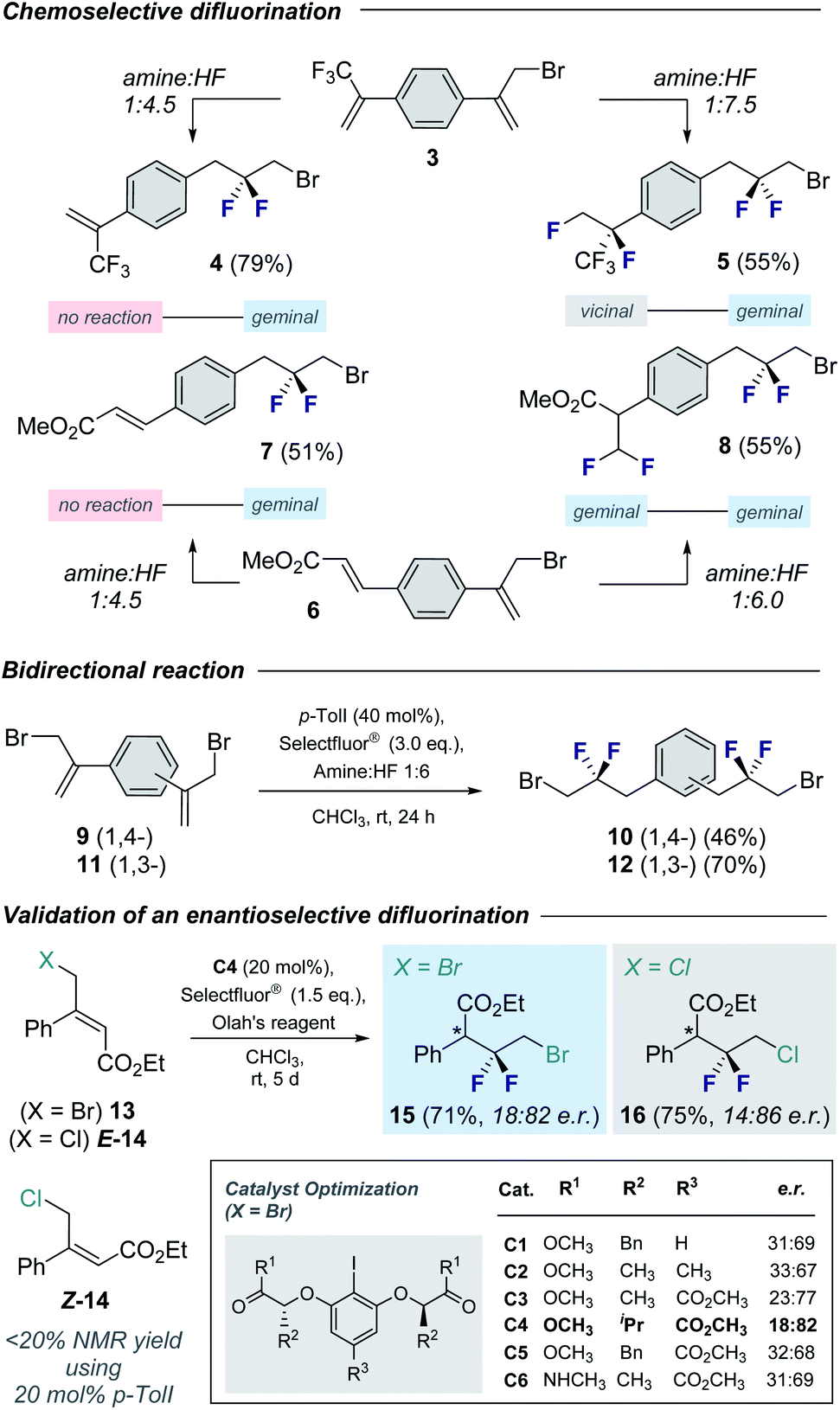 | ||
| Fig. 3 Exploring the synthetic versatility of this platform. (Top) Leveraging Brønsted acidity to achieve chemoselective fluorination. (Centre) Bidirectional functionalization. (Bottom) Preliminary validation of an enantioselective variant. | ||
Similarly, bidirectional geminal difluorination of the divinylbenzene derivatives 9 and 11 was efficient, enabling the synthesis of 10 (46%) and 12 (70%), respectively. This enables facile access to bis-electrophilic fluorinated linchpins for application in materials chemistry.
Preliminary validation of an enantioselective variant8d was achieved using the trisubstituted alkene 13. To that end, a series of C2-symmetric resorcinol-based catalysts were explored (see Fig. 3, inset). This enabled the generation of product 15 in up to 18:82 e.r. and 71% isolated yield. It is interesting to note that this catalysis system was also compatible with the chlorinated substrate E-14. A comparison of geometric isomers revealed a matched-mismatched scenario: whilst E-14 was efficiently converted to 16 (75%, 14:86 e.r.), Z-14 was recalcitrant to rearrangement (<20%).
To demonstrate the synthetic utility of the products, chemoselective functionalization of linchpin 2a was performed to generate 17 (57%) and 18 (87%), respectively (Fig. 4). Finally, this method was leveraged to generate an API for amyotrophic lateral sclerosis. Whereas the reported synthesis17 requires the exposure of α-bromoketone 19 to neat DAST over 7 days,18 compound 2h can be generated using this protocol over a more practical timeframe (24 h) on a 4 mmol scale. This key building block was then processed, via the amine hydrochloride salt 20, to API 21.
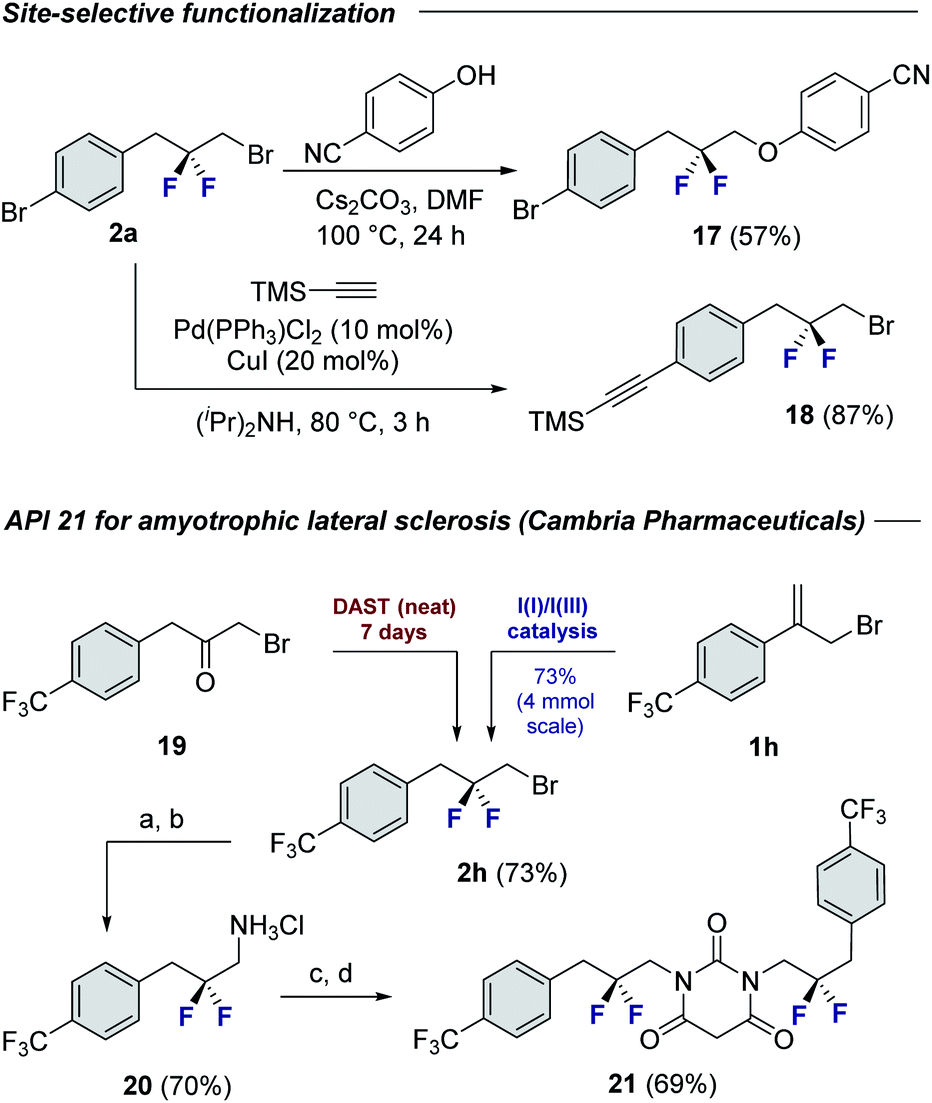 | ||
| Fig. 4 Selected modification of building blocks 2a and 2h. Conditions: (a) NaN3, DMF, 110 °C, 16 h. (b) Pd(OH)2/C (10 mol%), EtOH, 1 M HCl, rt, 24 h; (c) CDI, Et3N, THF, 60 °C, 16 h; (d) malonyl chloride, DCM, 0 °C, 2 h. | ||
Conclusions
In conclusion, an I(I)/I(III) catalysis manifold that facilitates the difluorinative rearrangement of α-(bromomethyl)styrenes is disclosed. In addition to generating electrophiles with a single geminal difluoro motif, bidirectional processes are presented together with simultaneous geminal and vicinal difluorination. Preliminary validation of an enantioselective reaction is demonstrated, to enable the generation of versatile α-phenyl-β-difluoro-γ-bromo/chloro esters. Finally, the transformation has been leveraged to enable the synthesis of an amyotrophic lateral sclerosis drug: this provides an operationally simple alternative to common deoxyfluorinating reagents when preparing gem-difluoro linchpins for contemporary medicinal chemistry.Author contributions
All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Note added after first publication
This article replaces the version published on 31st March 2021. The title contained a typesetting error. The oxidation state change in the title was incorrect and should read I(I)/I(III).Acknowledgements
We acknowledge generous financial support from the WWU Münster and the European Commission (ERC Consolidator Grant, 818949 RECON).Notes and references
- (a) B. E. Smart, J. Fluorine Chem., 2001, 109, 3 CrossRef CAS; (b) D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308 RSC; (c) L. Zimmer, C. Sparr and R. Gilmour, Angew. Chem., Int. Ed., 2011, 50, 11860 CrossRef CAS PubMed; (d) M. Aufiero and R. Gilmour, Acc. Chem. Res., 2018, 51, 1701 CrossRef CAS PubMed.
- (a) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef PubMed; (b) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC.
- Q. A. Huchet, B. Kuhn, B. Wagner, N. A. Kratochwil, H. Fischer, M. Kansy, D. Zimmerli, E. M. Carreira and K. Müller, J. Med. Chem., 2015, 58, 9041 CrossRef CAS PubMed.
- (a) G. M. Blackburn, D. E. Kent and F. Kolkmann, J. Chem. Soc., Perkin Trans. 1, 1984, 1, 1119 RSC; (b) G. M. Blackburn and D. E. Kent, J. Chem. Soc., Chem. Commun., 1981, 511 RSC.
- D. O'Hagan, Y. Wang, M. Skibinski and A. M. Z. Slawin, Pure Appl. Chem., 2012, 84, 1587 Search PubMed.
- (a) N. A. Meanwell, J. Med. Chem., 2011, 54, 2529 CrossRef CAS PubMed; (b) E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med. Chem., 2015, 58, 8315 CrossRef CAS PubMed; (c) I. G. Molnár, C. Thiehoff, M. C. Holland and R. Gilmour, ACS Catal., 2018, 6, 7167 CrossRef.
- For selected reviews on I(III)-mediated fluorination of alkenes, see (a) S. V. Kohlhepp and T. Gulder, Chem. Soc. Rev., 2016, 45, 6270 RSC; (b) A. M. Arnold, A. Ulmer and T. Gulder, Chem.–Eur. J., 2016, 22, 8728 CrossRef CAS PubMed; (c) S. Doobary and A. J. J. Lennox, Synlett, 2010, 31, 1333 Search PubMed.
- For selected examples of 1,1-difluorination, see: (a) S. Hara, J. Nakahigashi, K. Ishi-I, T. Fukuhura and N. Yoneda, Tetrahedron Lett., 1998, 39, 2589 CrossRef CAS; (b) N. O. Ilchenko, B. O. A. Tasch and K. J. Szabó, Angew. Chem., Int. Ed., 2014, 53, 12897 CrossRef CAS PubMed; (c) T. Kitamura, K. Muta and J. Oyamada, J. Org. Chem., 2015, 80, 10431 CrossRef CAS PubMed; (d) S. M. Banik, J. W. Medley and E. N. Jacobsen, Science, 2016, 353, 51 CrossRef CAS PubMed; (e) N. O. Ilchenko and K. J. Szabó, J. Fluorine Chem., 2017, 203, 104 CrossRef CAS; (f) F. Scheidt, J. Neufeld, M. Schäfer, C. Thiehoff and R. Gilmour, Org. Lett., 2018, 20, 8073 CrossRef CAS PubMed; (g) Z. Zhao, L. Racicot and G. K. Murphy, Angew. Chem., Int. Ed., 2017, 56, 11620 CrossRef CAS PubMed; (h) T. Kitamura, K. Yoshida, S. Mizuno, A. Miyake and J. Oyamada, J. Org. Chem., 2018, 83, 14853 CrossRef CAS PubMed; (i) W.-X. Lv, Q. Li, J.-L. Li, Z. Li, E. Lin, D.-H. Tan, Y.-H. Cai, W.-X. Fan and H. Wang, Angew. Chem., Int. Ed., 2018, 57, 16544 CrossRef CAS PubMed.
- For selected examples of 1,2-difluorination, see (a) S. M. Banik, J. W. Medley and E. N. Jacobsen, J. Am. Chem. Soc., 2016, 138, 5000 CrossRef CAS PubMed; (b) I. G. Molnár and R. Gilmour, J. Am. Chem. Soc., 2016, 138, 5004 CrossRef PubMed; (c) M. K. Haj, S. M. Banik and E. N. Jacobsen, Org. Lett., 2019, 21, 4919 CrossRef CAS PubMed; (d) F. Scheidt, M. Schäfer, J. C. Sarie, C. G. Daniliuc, J. J. Molloy and R. Gilmour, Angew. Chem., Int. Ed., 2018, 57, 16431 CrossRef CAS PubMed; (e) N. Erdeljac, G. Kehr, M. Ahlqvist, L. Knerr and R. Gilmour, Chem. Commun., 2018, 54, 12002 RSC; (f) N. Erdeljac, K. Bussmann, A. Schöler, F. K. Hansen and R. Gilmour, ACS Med. Chem. Lett., 2019, 10, 1336 CrossRef CAS PubMed; (g) S. Doobary, A. T. Sedikides, H. P. Caldora, D. L. Poole and A. J. J. Lennox, Angew. Chem., Int. Ed., 2020, 59, 1155 CrossRef CAS PubMed; (h) N. Erdeljac, C. Thiehoff, R. Jumde, C. Daniliuc, S. Hoeppner, A. Faust, A. K. H. Hirsch and R. Gilmour, J. Med. Chem., 2020, 63, 6225 CrossRef CAS PubMed; (i) S. Meyer, J. Häfliger, M. Schäfer, J. J. Molloy, C. G. Daniliuc and R. Gilmour, Angew. Chem., Int. Ed., 2021, 60, 6430 CrossRef CAS PubMed.
- (a) C. Ye, B. Twamley and J. M. Shreeve, Org. Lett., 2005, 7, 3961 CrossRef CAS PubMed; (b) J. C. Sarie, C. Thiehoff, R. J. Mudd, C. Daniliuc, G. Kehr and R. Gilmour, J. Org. Chem., 2017, 82, 11792 CrossRef CAS PubMed.
- For an elegant example of harnessing bromonium ions in I(III)-mediated fluorination see: (a) M. D. Levin, J. M. Ovian, J. A. Read, M. S. Sigman and E. N. Jacobsen, J. Am. Chem. Soc., 2020, 142, 14831 CrossRef CAS PubMedFor an example of the synthesis of β-difluoroalkyl azides via 1,2-azide migration, see: (b) Y. Ning, P. Sivaguru, G. Zanoni, E. A. Anderson and X. Bi, Chem, 2020, 6, 486 CrossRef CAS.
- T. Bykova, N. Al-Maharik, A. M. Z. Slawin and D. O'Hagan, J. Fluorine Chem., 2015, 179, 188 CrossRef CAS.
- J. L. Cotter, L. J. Andrews and R. M. Keefer, J. Am. Chem. Soc., 1962, 84, 793 CrossRef CAS.
- Highly electron rich substrates were avoided due to their known ability to undergo uncatalysed vicinal difluorination with HF and Selectfluor®: G. S. Lal, J. Org. Chem., 1993, 58, 2791 CrossRef CAS.
- CCDC 2055892 contains the ESI crystallographic data for compound 2n.†.
- C. Thiehoff, Y. P. Rey and R. Gilmour, Isr. J. Chem., 2017, 57, 92 CrossRef CAS.
- (a) D. R. Kirsch, R. Benmohamed, A. C. Arvanites, R. I. Morimoto, G. Xia and R. B. Silverman, WO Pat., 2010129665A2, 2010 Search PubMed; (b) G. Xia, R. Benmohamed, R. I. Morimoto, D. R. Kirsch and R. B. Silverman, Bioorg. Med. Chem. Lett., 2014, 24, 5098 CrossRef CAS PubMed.
- P. A. Messina, K. C. Mange and W. J. Middleton, J. Fluorine Chem., 1989, 42, 137 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2055892. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc01132d |
| This journal is © The Royal Society of Chemistry 2021 |