
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSingle metal four-electron reduction by U(II) and masked “U(II)” compounds†
Dieuwertje K.
Modder
 a,
Chad T.
Palumbo
a,
Iskander
Douair
b,
Rosario
Scopelliti
a,
Laurent
Maron
b and
Marinella
Mazzanti
*a
a,
Chad T.
Palumbo
a,
Iskander
Douair
b,
Rosario
Scopelliti
a,
Laurent
Maron
b and
Marinella
Mazzanti
*a
aInstitut des Sciences et Ingénierie Chimiques, Ecole Polytechnique Fédérale de Lausanne (EPFL), CH-1015, Lausanne, Switzerland. E-mail: marinella.mazzanti@epfl.ch
bLPCNO, Université de Toulouse, INSA Toulouse, Toulouse, 31077, France
First published on 22nd March 2021
Abstract
The redox chemistry of uranium is dominated by single electron transfer reactions while single metal four-electron transfers remain unknown in f-element chemistry. Here we show that the oxo bridged diuranium(III) complex [K(2.2.2-cryptand)]2[{((Me3Si)2N)3U}2(μ-O)], 1, effects the two-electron reduction of diphenylacetylene and the four-electron reduction of azobenzene through a masked U(II) intermediate affording a stable metallacyclopropene complex of uranium(IV), [K(2.2.2-cryptand)][U(η2-C2Ph2){N(SiMe3)2}3], 3, and a bis(imido)uranium(VI) complex [K(2.2.2-cryptand)][U(NPh)2{N(SiMe3)2}3], 4, respectively. The same reactivity is observed for the previously reported U(II) complex [K(2.2.2-cryptand)][U{N(SiMe3)2}3], 2. Computational studies indicate that the four-electron reduction of azobenzene occurs at a single U(II) centre via two consecutive two-electron transfers and involves the formation of a U(IV) hydrazide intermediate. The isolation of the cis-hydrazide intermediate [K(2.2.2-cryptand)][U(N2Ph2){N(SiMe3)2}3], 5, corroborated the mechanism proposed for the formation of the U(VI) bis(imido) complex. The reduction of azobenzene by U(II) provided the first example of a “clear-cut” single metal four-electron transfer in f-element chemistry.
Introduction
The redox chemistry of uranium is dominated by single electron transfer reactions,1 but examples of U(III) to U(V) electron transfer oxidations are increasingly observed.1a,2a–i Notably, dinitrogen reduction to a hydrazido ligand by a diuranium(III) complex was recently reported to involve a two-electron transfer from each metal centre.3 Single centre U(IV)/U(VI) two-electron transfer reactions have also been reported with oxidative atom transfer agents.4 Redox reactivity of uranium compounds involving the transfer of more than two electrons has so far only been achieved by combining the uranium centre with redox active ligands capable of storing electrons and rendering them available to oxidising agents.5,6 Notably, several uranium-redox active ligand systems have been reported to effect the four-electron reductive cleavage of azobenzene.5c,d,h,6b,c,7–11 In particular, uranium(III)/(IV)-redox active ligands systems such as hydride,5e,7 tetraphenylborate,5d,8 bridging arene,9 bipyridine,6c phosphinidene,10 and metallacyclopropene11 complexes reduce azobenzene to yield U(VI) bis(imido) complexes. Some of these complexes also effect the multielectron reduction of alkynes to yield uranium metallacycles.5d,6c,7,9,10a,11a,b The redox active ligands either remain bound to the metal during the electron transfer5h or are expelled as byproducts.5c–e,6b–d,9The four-electron reductive cleavage of azobenzene was first achieved by C. Burns by using multiple equivalents of the U(III) complex [(C5Me5)2UIIICl(NaCl)] to yield a mixture of U(VI) bis(imido) and U(IV) species.12 The proposed mechanism involves two-electron transfer processes and the comproportionation of a U(V) intermediate, but the possibility of a U(II) intermediate cannot be completely ruled out. Complete cleavage of azobenzene by a single metal centre has so far been reported only for W(II),13 and four-electron transfer by a single metal centre remains unknown in f-element chemistry.
Uranium(II) compounds could potentially effect four-electron transfer, but only very few examples of uranium in the +2 oxidation state have been reported so far14 and their reactivity remains practically unexplored.15 Indeed, the very low redox potential of the isolated U(II) complexes renders it difficult to control their reactivity, which can result in competitive one electron transfer reactions rather than multielectron transfer reactions.10b Conversely, complexes associating uranium in the +3 or +4 oxidation state to redox active ligands have proven effective in performing multielectron transfer reactions with a wide variety of substrates.5
Recently, we reported the synthesis of the diuranium(III) oxo complex [K(2.2.2-cryptand)]2[{((Me3Si)2N)3U}2(μ-O)], 1 (ref. 16) and showed that complex 1 provides a convenient precursor for studying the reactivity of U(II). Indeed, when complex 1 is reacted with N-heterocycles, it releases a “U(II)” synthon that effects one- or two-electron reductions depending on the N-heterocycle.16 We also found that the isolated U(II) complex [K(2.2.2-cryptand)][U{N(SiMe3)2}3], 2, previously reported by the Evans group14f showed similar behaviour but its reactivity turned out to be more complicated to control due to its low stability in solution.
Here we report the reactivity of complexes 1 and 2 with diphenylacetylene and azobenzene. Complex 1 effects the two-electron reduction of diphenylacetylene to yield a stable uranium metallacyclopropene complex through the delivery of a masked U(II). A similar reactivity is also observed for the U(II) complex [K(2.2.2-cryptand)][U{N(SiMe3)2}3], 2.
Moreover, we show that both the masked U(II) in 1 and the U(II) complex 2 reductively cleave azobenzene to yield a U(VI) bis(imido) complex, providing the first example of a “clear-cut” single metal four-electron reduction process in f-element chemistry. Computational studies show that in both cases the reaction proceeds at a single metal centre through the formation of a U(IV) hydrazido intermediate that could also be isolated at −40 °C.
Results and discussion
Synthesis and structural characterisation
The diuranium(III) oxo complex [K(2.2.2-cryptand)]2[{((Me3Si)2N)3U}2(μ-O)], 1 (ref. 16) reacts immediately with 1 equiv. of diphenylacetylene at −80 °C in THF. The 1H NMR spectrum at −80 °C showed full conversion of complex 1 and the appearance of signals corresponding to the previously reported U(IV) oxo complex, [K(2.2.2-cryptand)][U(O){N(SiMe3)2}3]. After removal of the U(IV) oxo complex, the metallocyclopropene complex [K(2.2.2-cryptand)][U(η2-C2Ph2){N(SiMe3)2}3], 3, was isolated in 25% yield (Scheme 1). The conversion, as measured by 1H NMR (using TMS2O as internal standard), is >65%, and the low yield is due to the partial solubility of 3 in toluene used to remove the U(IV) oxo by-product. The products of the reaction are consistent with the release of a “U(II)” synthon and a U(IV) oxo complex upon addition of diphenylacetylene. A closely related U(III) “disproportionation reaction” to afford a U(IV) species and a “U(II)” synthon was reported in an early study by Marks and co-workers for the trinuclear U(III) complex {U(C5Me5)2(μ-Cl)}3 upon reaction with diphenylacetylene.5b | ||
| Scheme 1 Reactivity of 1 and 2 with diphenylacetylene yielding the metallacyclopropene complex 3. | ||
To further corroborate that complex 1 releases a “U(II)” synthon upon reaction with diphenylacetylene, we also explored the reactivity of the previously reported U(II) complex [K(2.2.2-cryptand)][U{N(SiMe3)2}3], 2.14f Because, in our hands, previous attempts to carry out reactivity studies on isolated complex 2 had resulted in intractable mixtures of compounds,16 the reactivity was performed on THF solutions of 2 generated in situ at −80 °C, as previously described,14f by reduction of [U{N(SiMe3)2}3]17 with KC8 in presence of 2.2.2-cryptand. The reaction of in situ generated 2 with 1 equiv. of diphenylacetylene also produced complex 3 in an isolated yield of 37%.
1H NMR studies showed that complex 3 is stable in solid state and in solution at −40 °C but decomposes slowly in THF solution at room temperature.
In the solid-state structure of complex 3, as determined by X-ray diffraction studies (Fig. 1), the uranium centre is bound by three anionic N(SiMe3)2 ligands and a bidentate dianionic PhCCPh. The C–C bond length of 1.342(5) Å is similar to those found in the three reported uranium metallacyclopropene complexes (1.338(11) Å to 1.341(3) Å)10a,11b,18 and is in line with a double bond (1.331 Å). The U–C bond lengths of 2.345(3) Å and 2.352(3) Å and the value of the C1–U1–C2 angle of 33.22(11)° are comparable to those reported in the uranium metallacyclopropene complexes (U–C = 2.303(3) to 2.350(9) Å), and C1–U1–C2 = (33.3(3)°, 33.7(1)°). Overall, the structural parameters are consistent with the assignment of 3 as a U(IV) metallacyclopropene complex.
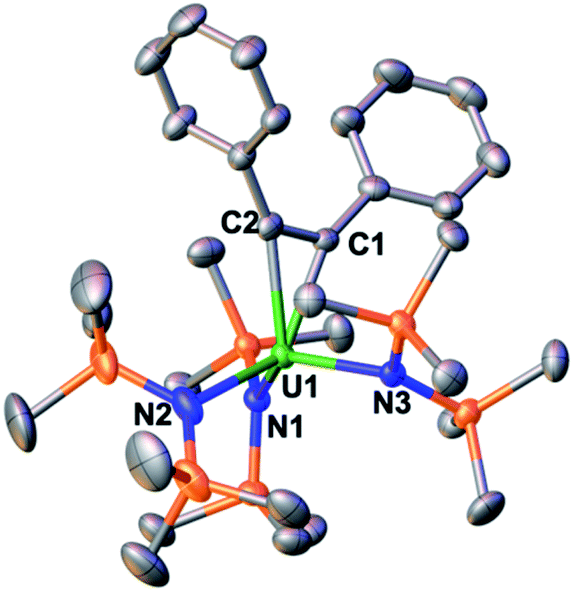 | ||
| Fig. 1 Molecular structure of the [U(η2-C2Ph2){N(SiMe3)2}3]− component of 3. Hydrogen atoms and the [K(2.2.2-cryptand)]+ counterion were omitted for clarity. Selected bond lengths (Å) and angles (°): U1–(Namide)avg 2.34(2), U1–C1 2.345(3), U1–C2 2.352(3), C1–C2 1.342(5), C1–U1–C2 33.22(11). | ||
Actinide metallacyclopropene compounds have shown interesting reactivity with heterounsaturated molecules but remain rare and essentially limited to cyclopentadienyl derivatives.10a,11b,19 Notably, the first example of a stable uranium metallacyclopropene, the cyclopentadienyl derivative [(η5-C5Me5)2U{η2-C2(SiMe3)2}], was isolated only recently.11b Thus, the reactions of 1 and 2 with diphenylacetylene provide access to a new class of uranium metallacyclopropene complexes.
In view of the observed ability of complexes 1 and 2 to effect multielectron transfer, we decided to investigate the reduction of azobenzene, a potential four-electron oxidising agent. Upon addition of a red THF solution of azobenzene to dark purple crystals of complex 1 at −80 °C, the solution immediately turned dark brown and the signals assigned to the terminal oxo complex [K(2.2.2-cryptand)][U(O){N(SiMe3)2}3] were observed in the 1H NMR spectrum. After raising the temperature to 25 °C, the 1H NMR spectrum showed additional signals at 6.97, 5.51, 5.44, and 0.46 ppm, assigned to the trans bis(imido) complex [K(2.2.2-cryptand)][U(NPh)2{N(SiMe3)2}3], 4 (Scheme 2). Single crystals of 4 were grown by slow diffusion of hexane at −40 °C into a concentrated THF solution. Washing with toluene removed the terminal U(IV) oxo complex [K(2.2.2-cryptand)][U(O){N(SiMe3)2}3] and allowed the isolation of clean complex 4 in 69% yield. Thus, following addition of azobenzene to 1, the products of the reaction are consistent with the release of a U(IV) oxo and a “U(II)” synthon. The “U(II)” synthon effects the four-electron reduction of azobenzene to yield the bis(imido) complex.
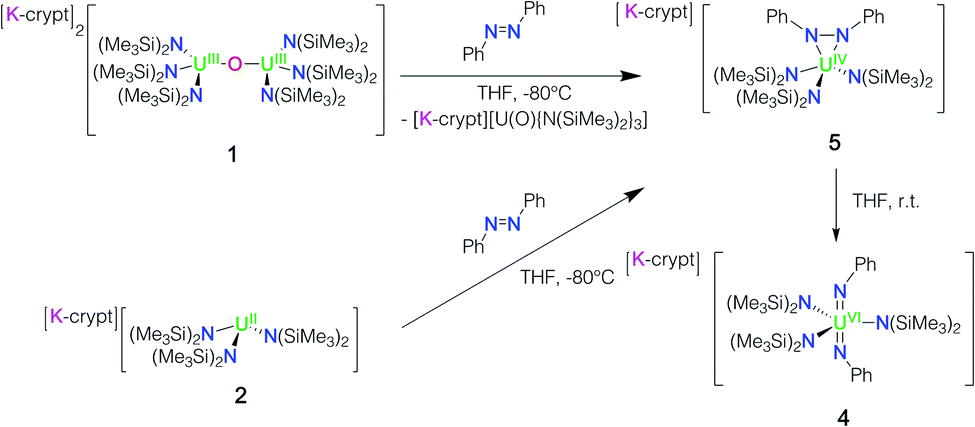 | ||
| Scheme 2 Reactivity of 1 and 2 with azobenzene yielding the bis(imido) complex 4 through the hydrazido intermediate complex 5. | ||
Furthermore, the reaction at −80 °C of the freshly in situ prepared U(II) complex 2 with azobenzene also led to the isolation of the four-electron reduction product, the bis(imido) complex 4 in 37% yield. These results confirm that complex 1 reacts as a U(II) synthon and that both complexes 1 and 2 are able to effect a single-metal four-electron reduction.
In the solid-state structure of 4 (Fig. 2), the uranium centre is five-coordinate and bound by three anionic N(SiMe3)2 ligands and two dianionic imido ligands with a trigonal bipyramidal geometry. The U–Nimido bond lengths of 1.929(8) Å and 1.935(8) Å are similar to those reported in other U(VI) trans bis(imido) complexes (1.840(4) to 1.992(5) Å).5i,12b,20 In addition, the mean U–Namide bond length of 2.34(2) Å is close to the one reported for the analogous uranyl complex, [K(2.2.2-cryptand)][U(O)2{N(SiMe3)2}3] (2.323(2) Å).21 The U![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N-Ph angles (162.5(6) and 165.0(6)°) deviate significantly from linearity as found in other bis(imido) U(VI) complexes12b,22 and suggest partial localization of the pi lone pair on the imido nitrogen (supported by the bonding analysis in ESI†). Trans bis(imido) U(VI) complexes have attracted considerable interest in uranium chemistry because of their fundamental insight in the study of U–L multiple bonds, their isoelectronic structure with the ubiquitous uranyl(VI) cation and their potential application in small molecule activation and catalysis.1a,5i,20,23
N-Ph angles (162.5(6) and 165.0(6)°) deviate significantly from linearity as found in other bis(imido) U(VI) complexes12b,22 and suggest partial localization of the pi lone pair on the imido nitrogen (supported by the bonding analysis in ESI†). Trans bis(imido) U(VI) complexes have attracted considerable interest in uranium chemistry because of their fundamental insight in the study of U–L multiple bonds, their isoelectronic structure with the ubiquitous uranyl(VI) cation and their potential application in small molecule activation and catalysis.1a,5i,20,23
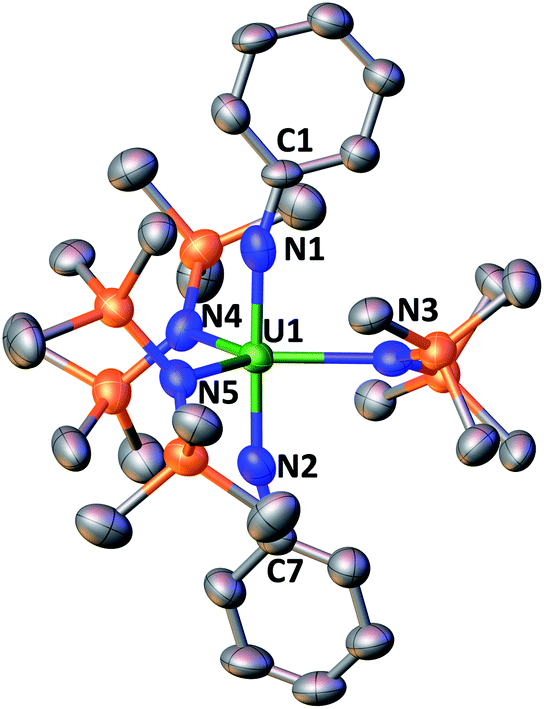 | ||
| Fig. 2 Molecular structure of the [U(NPh)2{N(SiMe3)2}3]− component of 4. Hydrogen atoms and the [K(2.2.2-cryptand)]+ counterion have been omitted for clarity. Selected bond lengths (Å) and angles (°): U1–(Namide)avg 2.34(2), U1–N1 1.929(8), U1–N2 1.935(8), N1–U1–N2 178.8(3), N1–U1–(Namide)avg 89.6(5), N2–U1–(Namide)avg 90.5(9). | ||
Complete cleavage of azobenzene by uranium(III) compounds was achieved in the past by using several equivalents of the U(III) complex2h,12a or by coupling the U(III) centres to redox active ligands.5a,5d The mononuclear U(III) complexes [(C5Me5)2U][(μ-Ph)2BPh2]5d and [(C5Me5)3U]5d and the dinuclear complexes {[(C5Me5)2U]2(μ-C6H6)}24 and {(C5Me5)U[μ-C5Me3(CH2)2](μ-H)2U(C5Me5)2 (ref. 5a) were reported to effect the cleavage of 1 or 2 equiv. of azobenzene by transferring four or eight electrons, respectively. In these systems, it was suggested that the multielectron reductive chemistry proceeded by combining metal-centred three-electron U(III)/U(VI) transfers and ligand based one-electron transfers. Alternatively, the reduction of azobenzene by complexes 1 and 2 proceeds via an unprecedented U(II)/U(VI) four-electron transfer that, so far, was only observed in transition metal chemistry.
Uranium(IV) hydrazido complexes have long been invoked as intermediates in the four-electron reduction of azobenzene.2h,4a,12a Recently, a U(IV) hydrazido species was isolated from the reaction of a U(III)-benzyl complex supported by bulky hydrotris(3,5-dimethylpyrazolyl)borate ligands with azobenzene, but further reduction of the N–N bond to yield the U(VI) bis(imido) species was not observed even at high temperature.6b
In order to gain more insight into the mechanism of the four-electron reduction of azobenzene by complexes 1 and 2, we performed computational studies (see the following section) and pursued the isolation of the probable intermediates by conducting crystallisation experiments at low temperature. X-ray quality crystals of the two-electron reduction intermediate [K(2.2.2-cryptand)][U(N2Ph2){N(SiMe3)2}3], 5, could be isolated from the reaction mixture of 1 and 2 with azobenzene in THF by slow diffusion of hexane at −40 °C. When the THF solution of 5 is left at room temperature, the complex immediately starts to convert into the U(VI) bis(imido) 4 and after 2 h is completely converted into 4.
In the solid-state structure of 5 (Fig. 3), the uranium centre is five coordinate and bound by three N(SiMe3)2 ligands and one bidentate N2Ph22− ligand. A significant elongation of the diazo moiety (N–N = 1.43(2) Å) is observed with respect to neutral cis-azobenzene (1.251 Å) and can be compared to that found in lanthanide complexes25 of the N2Ph22− ligand and in the only other reported U(IV) cis-hydrazido complex (N–N= 1.440(3) Å).6b The N–N elongation is also larger than what is found in mono-reduced azobenzene (1.331(17) Å)26 and in the U(IV) complex [((SiMe2NPh)3-tacn)U(η2-N2Ph2)] (1.353(4) Å) containing a singly reduced azobenzene.27 Furthermore, the U–Nhydrazido bond lengths of 2.271(14) and 2.229(13) Å are similar to those observed in the previously reported U(IV) cis-hydrazide complex (2.2254(15) Å).6b
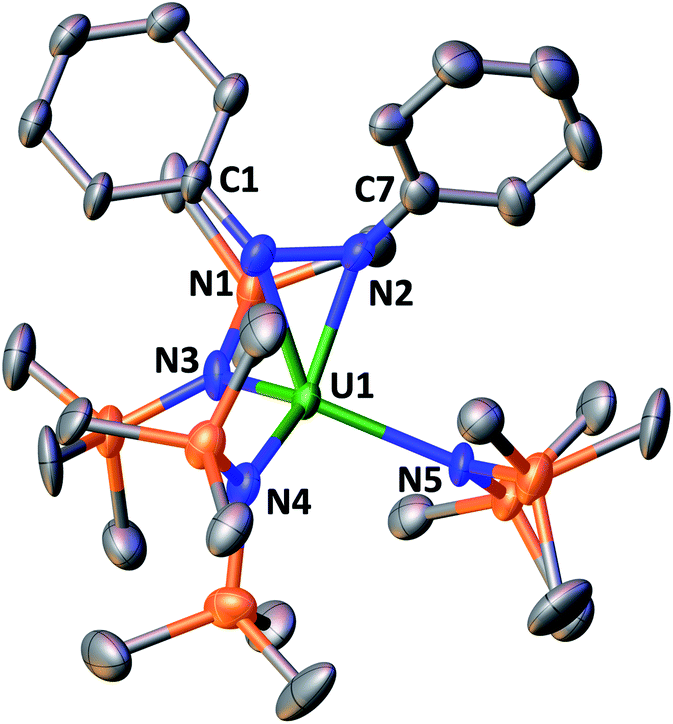 | ||
| Fig. 3 Molecular structure of the [U(N2Ph2){N(SiMe3)2}3]− component of 5. Hydrogen atoms and the [K(2.2.2-cryptand)]+ counterion have been omitted for clarity. Selected bond lengths (Å) and angles (°): U1–(Namide)avg 2.34(2), U1–N1 2.271(14), U1–N2 2.229(13), N1–N2 1.43(2), N1–U1–N2 37.1(5). | ||
The isolation, at low temperature, of complex 5 which readily transforms into the bis(imido) 4 at room temperature provides final evidence for the mechanism involving a step-wise four-electron U(II)/U(VI) redox process at a single metal centre.
Computational studies
The mechanism of the reaction of 2 with azobenzene was further investigated by computational methods (DFT, B3PW91 functional) and shows that the reduction proceeds through a U(IV) intermediate where the two-electron reduced azobenzene, (N2Ph22−), binds the metal centre in a bidentate fashion (Fig. 4). The associated barrier of 20.3 kcal mol−1 is in line with a kinetically accessible reaction. The final U(VI) imido complex is then formed in an exothermic process (9.5 kcal mol−1 from the U(IV) intermediate) via the transfer of two additional electrons in a kinetically favourable process (22.3 kcal mol−1).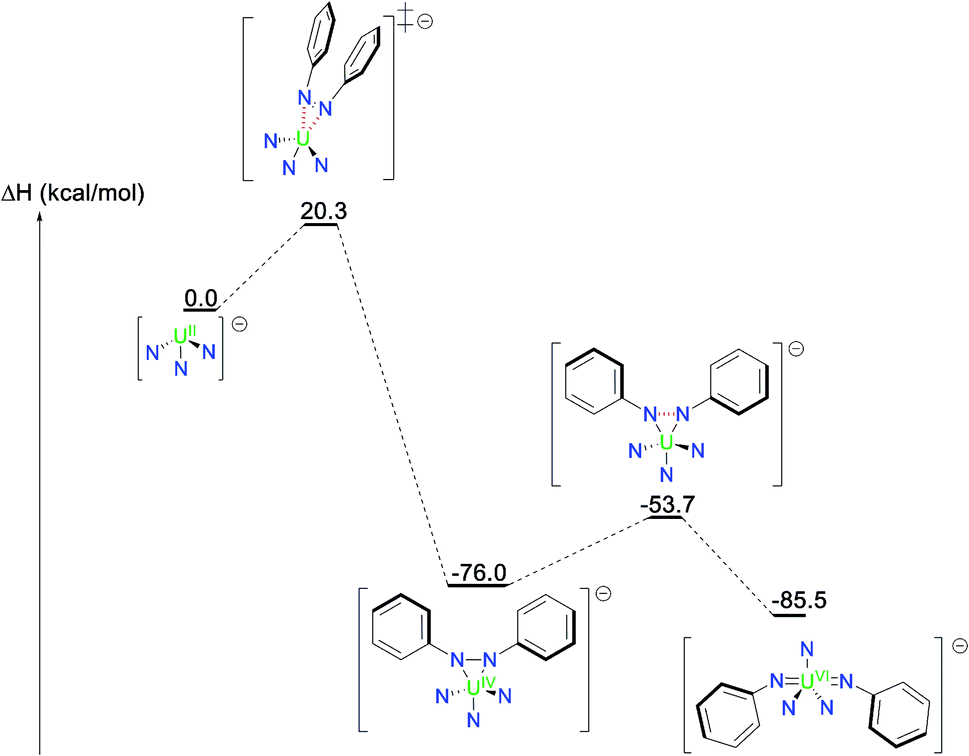 | ||
| Fig. 4 Computed enthalpy profile (in kcal mol−1) for the formation of 4 from the reaction of complex 2 with azobenzene. | ||
Addition of azobenzene to 1 also results in a two-electron reduction that yields the same U(IV) intermediate and the U(IV) terminal oxo complex whose formations are exothermic by 90.5 kcal mol−1 (Fig. 5). The unpaired spin density in the U(IV)/(N2Ph22−) intermediate (2.2) as well as the HOMO of the system (see Fig. S13 in ESI†) highlight the two electrons reduction of the azobenzene ligand. The final U(VI) bis(imido) complex is also in this case formed in an exothermic process (9.5 kcal mol−1 from the U(IV) intermediate) via the transfer of two additional electrons in a kinetically favourable process (22.3 kcal mol−1).
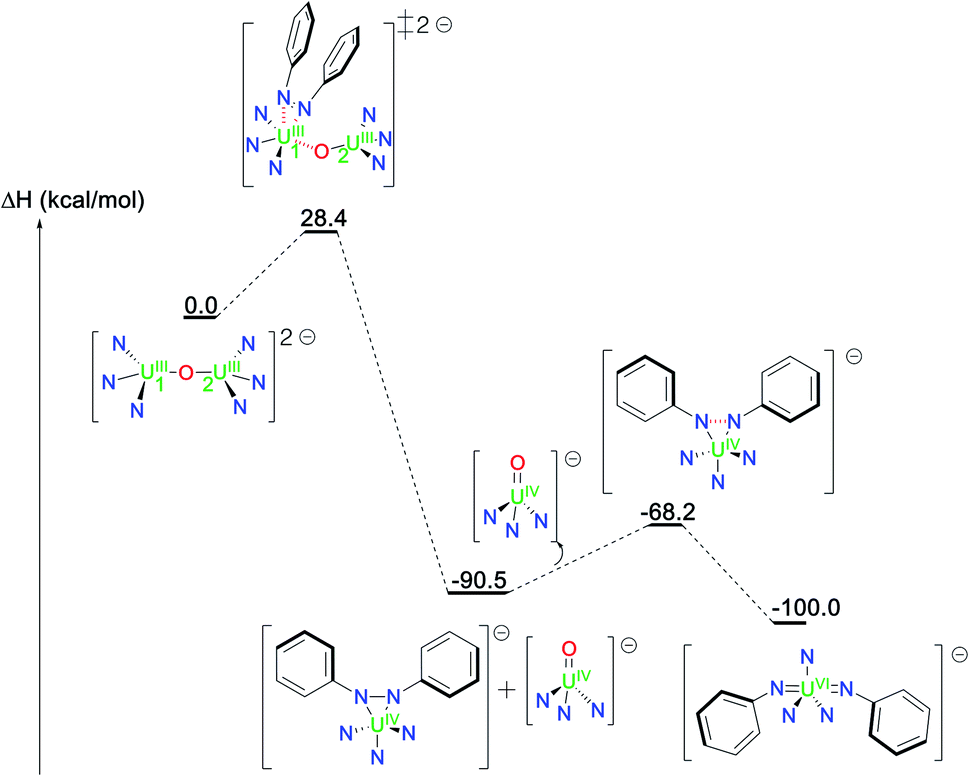 | ||
| Fig. 5 Computed enthalpy profile (in kcal mol−1) for the formation of 4 from the reaction of complex 1 with azobenzene. | ||
Conclusions
In conclusion, we showed that the addition of diphenylacetylene and azobenzene to 1, results in the cleavage of a U–O bond, release of a U(IV) oxo complex and two-electron and four-electron reduction, respectively, of the substrate by a putative U(II) intermediate. The previously reported U(II) complex 2 showed the same reactivity, providing the first example of a two-electron oxidative-addition reaction across the uranium(II/IV) redox couple. Computational studies indicate that the four-electron reduction of azobenzene occurs at a single U(II) centre via two consecutive two-electron transfer reactions and involves the formation of a U(IV) hydrazide intermediate. The isolation of the cis-hydrazide intermediate 5 corroborated the mechanism proposed for the formation of the U(VI) bis(imido) complex 4. The reduction of azobenzene by U(II) provided the first example of a “clear-cut” single metal four-electron transfer in f-elements chemistry.Author contributions
D. K. M. carried out the synthetic experiments; D. K. M. and C. T. P. analysed the experimental data; R. S. carried out the X-ray single crystal structure analyses; I. D. and L. M. carried out and analysed the computational data. M. M. originated the central idea, coordinated the work, and analysed the experimental data. M. M., D. K. M. and C. T. P. wrote the manuscript with input from all co-authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge support from the Swiss National Science Foundation grant number 200021_178793. We thank F. Fadaei-Tirani for important contributions to the X-ray single crystal structure analyses. LM is a member of the Institut Universitaire de France. CAlmip is acknowledged for a generous grant of computing time.Notes and references
- (a) T. W. Hayton, Chem. Commun., 2013, 49, 2956–2973 RSC; (b) S. T. Liddle, Angew. Chem., Int. Ed., 2015, 54, 8604–8641 CrossRef CAS PubMed; (c) M. A. Boreen and J. Arnold, Dalton Trans., 2020, 49, 15124–15138 RSC.
- (a) S. Fortier, N. Kaltsoyannis, G. Wu and T. W. Hayton, J. Am. Chem. Soc., 2011, 133, 14224–14227 CrossRef CAS PubMed; (b) D. M. King, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Science, 2012, 337, 717–720 CrossRef CAS PubMed; (c) C. Camp, J. Pecaut and M. Mazzanti, J. Am. Chem. Soc., 2013, 135, 12101–12111 CrossRef CAS PubMed; (d) C. Camp, M. A. Antunes, G. Garcia, I. Ciofini, I. C. Santos, J. Pecaut, M. Almeida, J. Marçalo and M. Mazzanti, Chem. Sci., 2014, 5, 841–846 RSC; (e) O. Cooper, C. Camp, J. Pécaut, C. E. Kefalidis, L. Maron, S. Gambarelli and M. Mazzanti, J. Am. Chem. Soc., 2014, 136, 6716–6723 CrossRef CAS PubMed; (f) D. P. Halter, F. W. Heinemann, J. Bachmann and K. Meyer, Nature, 2016, 530, 317–321 CrossRef CAS PubMed; (g) N. Tsoureas, A. F. R. Kilpatrick, C. J. Inman and F. G. N. Cloke, Chem. Sci., 2016, 7, 4624–4632 RSC; (h) B. M. Gardner, C. E. Kefalidis, E. Lu, D. Patel, E. J. L. McInnes, F. Tuna, A. J. Wooles, L. Maron and S. T. Liddle, Nat. Commun., 2017, 8, 1898 CrossRef PubMed; (i) N. S. Settineri, A. A. Shiau and J. Arnold, Chem. Commun., 2018, 54, 10913–10916 RSC.
- M. Falcone, L. Chatelain, R. Scopelliti, I. Zivkovic and M. Mazzanti, Nature, 2017, 547, 332–335 CrossRef CAS PubMed.
- (a) D. S. J. Arney and C. J. Burns, J. Am. Chem. Soc., 1995, 117, 9448–9460 CrossRef CAS; (b) J. L. Kiplinger, D. E. Morris, B. L. Scott and C. J. Burns, Chem. Commun., 2002, 30–31 RSC; (c) J. L. Brown, S. Fortier, G. Wu, N. Kaltsoyannis and T. W. Hayton, J. Am. Chem. Soc., 2013, 135, 5352–5355 CrossRef CAS PubMed; (d) A. J. Lewis, P. J. Carroll and E. J. Schelter, J. Am. Chem. Soc., 2013, 135, 13185–13192 CrossRef CAS PubMed; (e) E. Lu, O. J. Cooper, J. McMaster, F. Tuna, E. J. L. McInnes, W. Lewis, A. J. Blake and S. T. Liddle, Angew. Chem., Int. Ed., 2014, 53, 6696–6700 CrossRef CAS PubMed; (f) N. T. Rice, K. McCabe, J. Bacsa, L. Maron and H. S. La Pierre, J. Am. Chem. Soc., 2020, 142, 7368–7373 CrossRef CAS PubMed; (g) R. J. Ward, P. Rungthanaphatsophon, I. del Rosal, S. P. Kelley, L. Maron and J. R. Walensky, Chem. Sci., 2020, 11, 5830–5835 RSC.
- (a) J. M. Manriquez, P. J. Fagan, T. J. Marks, S. H. Vollmer, C. S. Day and V. W. Day, J. Am. Chem. Soc., 1979, 101, 5075–5078 CrossRef CAS; (b) P. J. Fagan, J. M. Manriquez, T. J. Marks, C. S. Day, S. H. Vollmer and V. W. Day, Organometallics, 1982, 1, 170–180 CrossRef CAS; (c) P. L. Diaconescu, P. L. Arnold, T. A. Baker, D. J. Mindiola and C. C. Cummins, J. Am. Chem. Soc., 2000, 122, 6108–6109 CrossRef CAS; (d) W. J. Evans, S. A. Kozimor and J. W. Ziller, Chem. Commun., 2005, 4681–4683 RSC; (e) W. J. Evans, K. A. Miller, S. A. Kozimor, J. W. Ziller, A. G. DiPasquale and A. L. Rheingold, Organometallics, 2007, 26, 3568–3576 CrossRef CAS; (f) W. J. Evans, E. Montalvo, S. A. Kozimor and K. A. Miller, J. Am. Chem. Soc., 2008, 130, 12258–12259 CrossRef CAS PubMed; (g) C. Camp, V. Mougel, P. Horeglad, J. Pecaut and M. Mazzanti, J. Am. Chem. Soc., 2010, 132, 17374–17377 CrossRef CAS PubMed; (h) D. P. Cladis, J. J. Kiernicki, P. E. Fanwick and S. C. Bart, Chem. Commun., 2013, 49, 4169–4171 RSC; (i) N. H. Anderson, S. O. Odoh, Y. Yao, U. J. Williams, B. A. Schaefer, J. J. Kiernicki, A. J. Lewis, M. D. Goshert, P. E. Fanwick, E. J. Schelter, J. R. Walensky, L. Gagliardi and S. C. Bart, Nat. Chem., 2014, 6, 919–926 CrossRef CAS PubMed; (j) J. J. Kiernicki, P. E. Fanwick and S. C. Bart, Chem. Commun., 2014, 50, 8189–8192 RSC; (k) P. L. Diaconescu and C. C. Cummins, Dalton Trans., 2015, 44, 2676–2683 RSC; (l) N. Jori, M. Falcone, R. Scopelliti and M. Mazzanti, Organometallics, 2020, 39, 1590–1601 CrossRef CAS.
- (a) G. F. Zi, L. Jia, E. L. Werkema, M. D. Walter, J. P. Gottfriedsen and R. A. Andersen, Organometallics, 2005, 24, 4251–4264 CrossRef CAS; (b) J. J. Kiernicki, R. F. Higgins, S. J. Kraft, M. Zeller, M. P. Shores and S. C. Bart, Inorg. Chem., 2016, 55, 11854–11866 CrossRef CAS PubMed; (c) L. Zhang, C. Zhang, G. Hou, G. Zi and M. D. Walter, Organometallics, 2017, 36, 1179–1187 CrossRef CAS; (d) P. Rungthanaphatsophon, C. L. Barnes, S. P. Kelley and J. R. Walensky, Dalton Trans., 2018, 47, 8189–8192 RSC.
- J. K. Pagano, J. M. Dorhout, K. R. Czerwinski, D. E. Morris, B. L. Scott, R. Waterman and J. L. Kiplinger, Organometallics, 2016, 35, 617–620 CrossRef CAS.
- W. J. Evans, K. A. Miller, W. R. Hillman and J. W. Ziller, J. Organomet. Chem., 2007, 692, 3649–3654 CrossRef CAS.
- W. J. Evans, C. A. Traina and J. W. Ziller, J. Am. Chem. Soc., 2009, 131, 17473–17481 CrossRef CAS PubMed.
- (a) D. Wang, W. Ding, G. Hou, G. Zi and M. D. Walter, Chem. – Eur. J., 2020, 26, 16888–16899 CrossRef CAS PubMed; (b) D. Wang, G. Hou, G. Zi and M. D. Walter, Organometallics, 2021, 40, 383–396 CrossRef CAS.
- (a) L. Zhang, G. Hou, G. Zi, W. Ding and M. D. Walter, J. Am. Chem. Soc., 2016, 138, 5130–5142 CrossRef CAS PubMed; (b) M. D. Walter, D. Wang, W. Ding, G. Hou and G. Zi, Chem. – Eur. J., 2021 DOI:10.1002/chem.202100089.
- (a) B. P. Warner, B. L. Scott and C. J. Burns, Angew. Chem., Int. Ed., 1998, 37, 959–960 CrossRef CAS; (b) L. Maria, I. C. Santos, V. R. Sousa and J. Marçalo, Inorg. Chem., 2015, 54, 9115–9126 CrossRef CAS PubMed; (c) L. Maria, N. A. G. Bandeira, J. Marçalo, I. C. Santos and J. K. Gibson, Chem. Commun., 2020, 56, 431–434 RSC.
- F. Maseras, M. A. Lockwood, O. Eisenstein and I. P. Rothwell, J. Am. Chem. Soc., 1998, 120, 6598–6602 CrossRef CAS.
- (a) M. R. MacDonald, M. E. Fieser, J. E. Bates, J. W. Ziller, F. Furche and W. J. Evans, J. Am. Chem. Soc., 2013, 135, 13310–13313 CrossRef CAS PubMed; (b) H. S. La Pierre, A. Scheurer, F. W. Heinemann, W. Hieringer and K. Meyer, Angew. Chem., Int. Ed., 2014, 53, 7158–7162 CrossRef CAS PubMed; (c) B. S. Billow, B. N. Livesay, C. C. Mokhtarzadeh, J. McCracken, M. P. Shores, J. M. Boncella and A. L. Odom, J. Am. Chem. Soc., 2018, 140, 17369–17373 CrossRef CAS PubMed; (d) D. N. Huh, C. J. Windorff, J. W. Ziller and W. J. Evans, Chem. Commun., 2018, 54, 10272–10275 RSC; (e) D. N. Huh, J. W. Ziller and W. J. Evans, Inorg. Chem., 2018, 57, 11809–11814 CrossRef CAS; (f) A. J. Ryan, M. A. Angadol, J. W. Ziller and W. J. Evans, Chem. Commun., 2019, 55, 2325–2327 RSC; (g) F. S. Guo, N. Tsoureas, G. Z. Huang, M. L. Tong, A. Mansikkamaki and R. A. Layfield, Angew. Chem., Int. Ed., 2020, 59, 2299–2303 CrossRef CAS PubMed.
- (a) C. J. Windorff, M. R. MacDonald, K. R. Meihaus, J. W. Ziller, J. R. Long and W. J. Evans, Chem. – Eur. J., 2016, 22, 772–782 CrossRef CAS PubMed; (b) S. A. Moehring and W. J. Evans, Chem. – Eur. J., 2020, 26, 1530–1534 CrossRef CAS PubMed.
- D. K. Modder, C. T. Palumbo, I. Douair, F. Fadaei-Tirani, L. Maron and M. Mazzanti, Angew. Chem., Int. Ed., 2021, 60, 3737–3744 CrossRef CAS PubMed.
- R. A. Andersen, Inorg. Chem., 1979, 18, 1507–1509 CrossRef CAS.
- J. K. Pagano, B. L. Scott, D. E. Morris and J. L. Kiplinger, Inorg. Chim. Acta, 2018, 482, 347–352 CrossRef CAS.
- (a) B. Fang, W. Ren, G. Hou, G. Zi, D. C. Fang, L. Maron and M. D. Walter, J. Am. Chem. Soc., 2014, 136, 17249–17261 CrossRef CAS; (b) B. Fang, L. Zhang, G. Hou, G. Zi, D. C. Fang and M. D. Walter, Chem. Sci., 2015, 6, 4897–4906 RSC; (c) L. Zhang, B. Fang, G. Hou, L. Ai, W. Ding, M. D. Walter and G. Zi, Dalton Trans., 2016, 45, 16441–16452 RSC; (d) L. Zhang, G. Hou, G. Zi, W. Ding and M. D. Walter, Dalton Trans., 2017, 46, 3716–3728 RSC; (e) G. Zi, Chem. Commun., 2018, 54, 7412–7430 RSC.
- (a) E. Barnea and M. S. Eisen, Coord. Chem. Rev., 2006, 250, 855–899 CrossRef CAS; (b) T. W. Hayton, J. M. Boncella, B. L. Scott, E. R. Batista and P. J. Hay, J. Am. Chem. Soc., 2006, 128, 10549–10559 CrossRef CAS PubMed.
- P. J. Cobb, D. J. Moulding, F. Ortu, S. Randall, A. J. Wooles, L. S. Natrajan and S. T. Liddle, Inorg. Chem., 2018, 57, 6571–6583 CrossRef CAS PubMed.
- N. C. Tomson, N. H. Anderson, A. M. Tondreau, B. L. Scott and J. M. Boncella, Dalton Trans., 2019, 48, 10865–10873 RSC.
- T. W. Hayton, J. M. Boncella, B. L. Scott, P. D. Palmer, E. R. Batista and P. J. Hay, Science, 2005, 310, 1941–1943 CrossRef CAS PubMed.
- W. J. Evans, S. A. Kozimor, J. W. Ziller and N. Kaltsoyannis, J. Am. Chem. Soc., 2004, 126, 14533–14547 CrossRef CAS PubMed.
- (a) W. J. Evans, D. K. Drummond, L. R. Chamberlain, R. J. Doedens, S. G. Bott, H. M. Zhang and J. L. Atwood, J. Am. Chem. Soc., 1988, 110, 4983–4994 CrossRef CAS; (b) A. R. Willauer, A. M. Dabrowska, R. Scopelliti and M. Mazzanti, Chem. Commun., 2020, 56, 8936–8939 RSC.
- J. Andrez, J. Pecaut, P.-A. Bayle and M. Mazzanti, Angew. Chem., Int. Ed., 2014, 53, 10448–10452 CrossRef CAS PubMed.
- M. A. Antunes, J. T. Coutinho, I. C. Santos, J. Marçalo, M. Almeida, J. J. Baldovi, L. C. J. Pereira, A. Gaita-Ariño and E. Coronado, Chem. – Eur. J., 2015, 21, 17817–17826 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary NMR, IR, and magnetic and crystallographic data. CCDC 2039849–2039851. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/D1SC00668A |
| This journal is © The Royal Society of Chemistry 2021 |