
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRegioselective difunctionalization of pyridines via 3,4-pyridynes†
Benjamin
Heinz‡
a,
Dimitrije
Djukanovic‡
a,
Paolo
Filipponi
b,
Benjamin
Martin
b,
Konstantin
Karaghiosoff
a and
Paul
Knochel
 *a
*a
aLudwig-Maximilians-Universität München, Department Chemie, Butenandtstraße 5-13, 81377 Munich, Germany. E-mail: paul.knochel@cup.uni-muenchen.de
bNovartis Pharma AG, Chemical Development, Fabrikstraße, 4002 Basel, Switzerland. E-mail: benjamin.martin@novartis.com
First published on 25th March 2021
Abstract
A new regioselective 3,4-difunctionalization of 3-chloropyridines via 3,4-pyridyne intermediates is reported. Regioselective lithiation of 3-chloro-2-ethoxypyridine and a related 2-thio-derivative followed by treatment with aryl- and alkylmagnesium halides as well as magnesium thiolates at −78 °C produced 3,4-pyridynes during heating to 75 °C. Regioselective addition of the Grignard moiety in position 4 followed by an electrophilic quench in position 3 led to various 2,3,4-trisubstituted pyridines. This method was adapted into a continuous flow set-up. As an application, we have prepared a key intermediate for (±)-paroxetine.
Introduction
Pyridines are important heterocycles present in numerous biologically relevant molecules.1 As a consequence, many synthetic methods have been developed for the functionalization of such N-heterocycles.2 Especially, the regioselective metalation of pyridines has been broadly used.3 Also, highly unsaturated intermediates such as pyridynes (analogs of arynes4,5) offer a unique possibility of adjacent regioselective double functionalization.6However, pyridyne chemistry is relatively unexplored. The generation of pyridynes often requires elaborated precursors and their further reactions with nucleophiles were of limited scope and complicated by a lack of regioselectivity.7 Garg showed, that the introduction of a substituent in position 2 significantly improved the regioselectivity of the nucleophilic addition using the aryne distortion model.8 However, the starting material preparation was lengthy and the reactivity of such 3,4-pyridynes was limited to cycloadditions and aminations.8 To overcome some of the previously described drawbacks, we have chosen, according to Hegarty,9 to use readily prepared 3-chloro-2-ethoxypyridine (1) as a precursor of 3,4-pyridyne, achieving an effective synthesis of highly decorated pyridines.
Thus, the regioselective lithiation of the pyridine 1 with n-BuLi will afford the 4-lithiated pyridine 2.10 Transmetalation with an organomagnesium halide RMgBr·LiCl of type 3 will produce mixed diorganomagnesiums of type 4. At elevated temperatures, an elimination should proceed leading to the 3,4-pyridyne 5. After the regioselective addition of RMgX (RMgBr·LiCl or RMgCl), affording magnesiated pyridines of type 6, quenching with various electrophiles (E–X) should produce the desired polyfunctional pyridines of type 7 (Scheme 1).
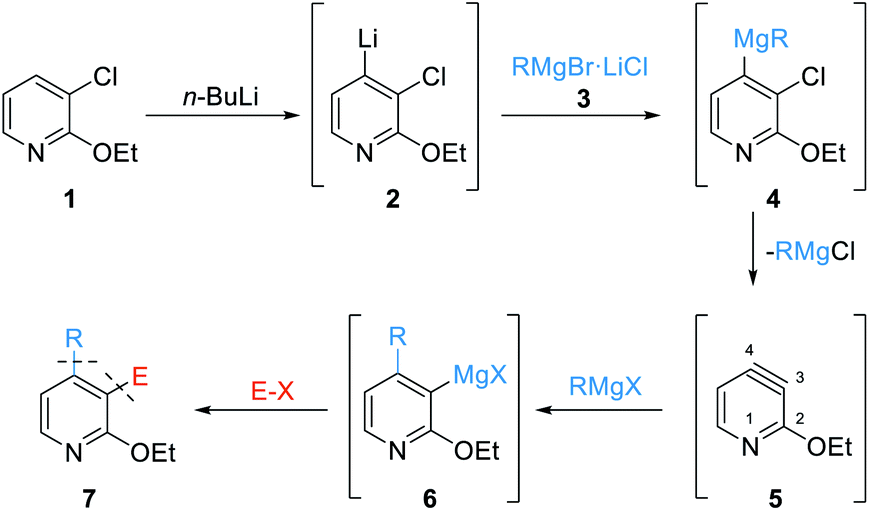 | ||
| Scheme 1 General reaction sequence towards difunctionalized pyridines of type 7 starting from 3-chloro-2-ethoxypyridine (1) via the 3,4-pyridyne intermediate 5. | ||
Results and discussion
Herein, we report the successful outcome of this synthetic approach. In preliminary experiments, we have treated 1 with n-BuLi in THF at −78 °C and found that a fully regioselective lithiation towards 2 was achieved within 2 h. Addition of 4-anisylmagnesium bromide (AnMgBr·LiCl, 3a) at −78 °C, which was prepared by the reaction of 4-anisyl bromide with magnesium turnings in the presence of LiCl,11 led tentatively to the mixed diorganomagnesium reagent 4. The optimum for the elimination of 4 providing the corresponding pyridyne occurred at 75 °C in a sealed tube with 1 h reaction time.12 Also, the use of 2 equivalents of 4-anisylmagnesium bromide (An-MgBr) was required to achieve a high yield.13 The regioselectivity of the organomagnesium addition to the pyridyne intermediate was explained by a coordinating effect of the ethoxy group at position C2. Therefore, the stabilized 3-magnesiated species 6 gave regioselectively the pure 4-arylated pyridine 7a (64% yield) after quenching the reaction mixture with sat. aq. NH4Cl. This procedure was quite general and a range of arylmagnesium bromides added regioselectively at position 4. After aqueous workup, the 4-arylated pyridines 7b–g were obtained in 41–61% yield.14 Additionally, the use of alkylmagnesium halides (5.0 equiv.) provided the desired pyridines 7h–i, bearing an alkyl substituent at position 4, in 56–58% yield (Scheme 2). The missing material was a result of the unstability of the pyridine intermediate which may oligomerize or polymerize.15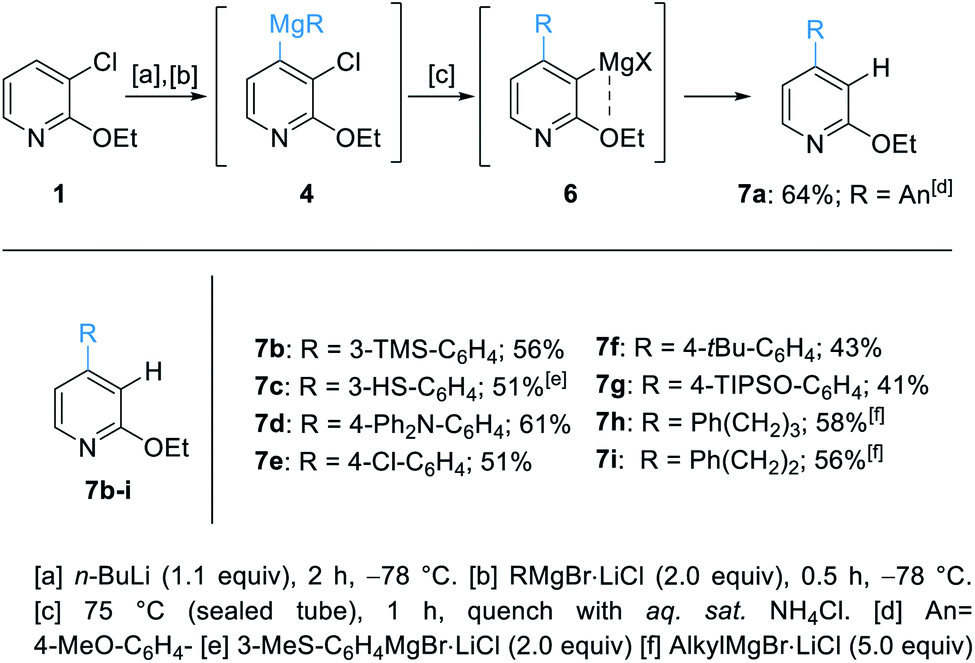 | ||
| Scheme 2 Regioselective lithiation of 3-chloro-2-ethoxypyridine (1) and subsequent transmetalation to diorganomagnesium reagents of type 4 leading to 4-arylated pyridines of type 7via the 3,4-pyridyne 5. | ||
With these optimized conditions, we have trapped the newly generated pyridylmagnesium halides of type 6 with various electrophiles leading to 3,4-difunctionalized pyridines of type 7 (Scheme 3).
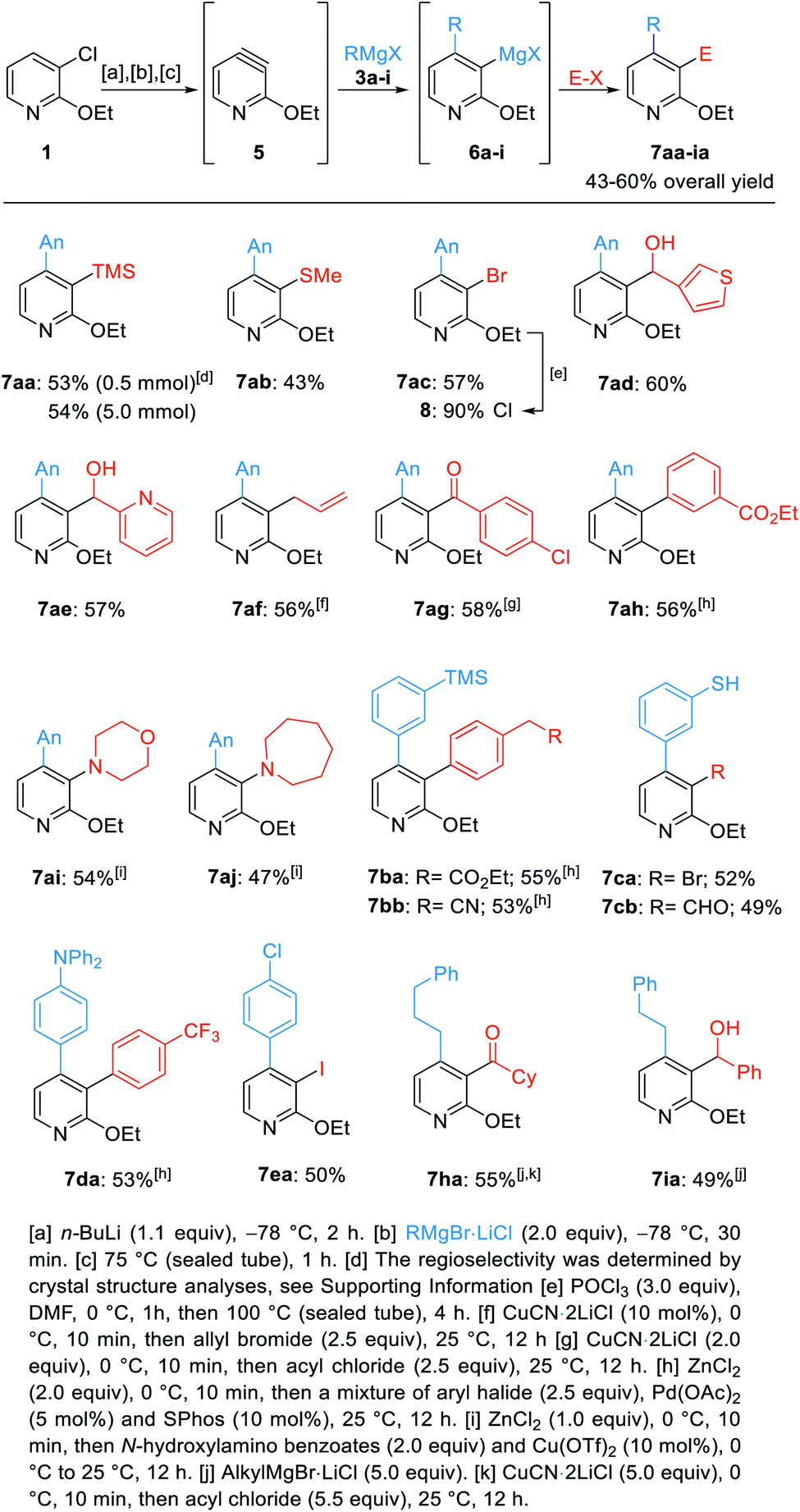 | ||
| Scheme 3 Regioselective difunctionalization of 3-chloro-2-ethoxypyridine (1) towards the pyridines 7aa–7iavia the 3,4-pyridyne 5. | ||
As previously described, we treated 1 with n-BuLi followed by addition of AnMgBr (3a, An = 4-MeO-C6H4). Heating to 75 °C in a sealed tube for 1 h led to the pyridyne formation. Regioselective addition of 3a afforded the 3-pyridylmagnesium species 6a. Addition of TMSCl (2.5 equiv., 25 °C, 12 h) gave the 3,4-difunctionalized pyridine 7aa in an overall yield of 53% (0.5 mmol scale, Scheme 3). A scale-up to 5 mmol provided a similar yield (54%). Thiolation with S-methyl methanesulfonothioate and bromination with (CCl2Br)2 led to the thioether 7ab in 43% yield and the 3-halogenated pyridine 7ac in 57% yield. A further derivatization of the previously prepared trisubstituted pyridines (7ac) was made. This pyridine was treated with POCl3 and DMF leading to the trisubstituted 2-chloropyridine 8 in 90% yield.16 Quenching the pyridylmagnesium bromide 6a with both, electron-rich and electron-poor heterocyclic aldehydes afforded the benzylic alcohols 7ad and 7ae in 57–60% yield. Copper(I)-catalyzed allylation gave the pyridine 7af in 56% yield. Additionally, copper(I)-mediated acylation with 4-chlorobenzoyl chloride led to the biaryl ketone 7ag in 58% yield. Negishi cross-coupling17 of the magnesium species 6a, after transmetalation with ZnCl2, with ethyl 3-bromobenzoate in the presence of 5 mol% Pd(OAc)2 and 10 mol% SPhos18 led to the 3,4-bis-arylated pyridine 7ah in 56% yield. A Cu-catalyzed electrophilic amination with N-hydroxylamino benzoates afforded the 3-aminated pyridines 7ai and 7aj in 47–54% yield.19
Next, cross-coupling reactions of 4-(3-(trimethylsilyl)phenyl)-3-pyridylmagnesium bromide (6b) led to the highly functionalized pyridines 7ba and 7bb in 53–55% isolated yield. Using the 3-pyridylmagnesium compound 6c gave the thiols 7ca and 7cb in 49–52% yield by quenching with either (CCl2Br)2 or DMF.14 A cross-coupling of 6d with 4-iodobenzotrifluoride gave the expected trisubstituted pyridine 7da in 53% yield. Iodolysis of 3-magnesiated pyridine 6e gave the iodinated pyridine 7ea in 50% yield.
Finally, the 4-alkylated pyridylmagnesium species 6h and 6i were further functionalized either by acylation affording the ketone 7ha in 55% yield or quench with benzaldehyde leading to the alcohol 7ia in 49% overall yield.
In order to extend the scope of these difunctionalizations, we investigated the use of magnesium thiolates. These nucleophiles proved to be excellent reaction partners and the treatment of the lithiated pyridine 2 with RSMgX·LiCl20 gave regioselectively, after heating to 75 °C for 1 h in a sealed tube, 4-thiolated pyridines such as 10a–b in 69–72% yield (Scheme 4). Further quenching of the intermediate 9a with various electrophiles such as TMSCl, DMF, benzaldehyde and benzophenone gave the 2,3,4-trifunctionalized pyridines 10aa–ad in 50–71% yield. Additionally, the treatment of 9a with ethyl 2-formylbenzoate produced the phthalide 10ae in 71% yield. Further, a palladium-catalyzed cross-coupling on the sterically demanding substrate 9b gave, after 3,4-pyridyne formation, the pyridine 10ba in 67% isolated yield.
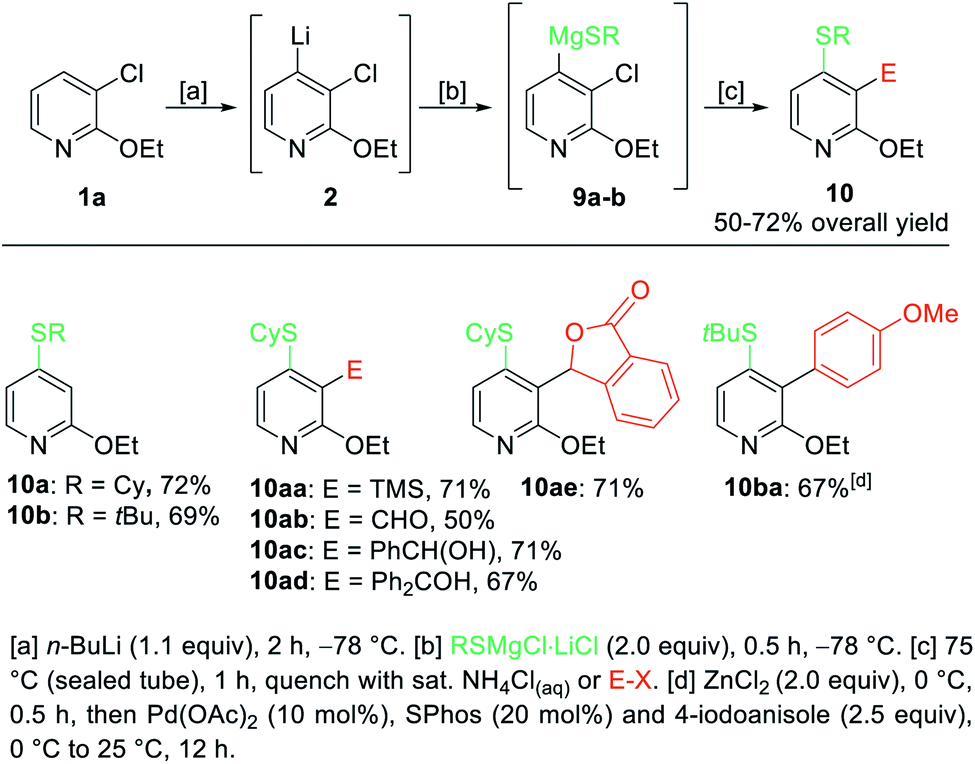 | ||
| Scheme 4 Selective lithiation of 3-chloro-2-ethoxypyridine (1) and subsequent transmetalation towards the diorganomagnesium reagents of type 9 leading to 4-thiolated pyridines of type 10. | ||
The performance of multi-step organometallic reactions in a continuous flow set-up using microreactors often proved to be advantageous, allowing a much better reaction control than in batch with milder and more selective reactions conditions.21 Thus, we examined the arylation of 2-isopropylthio-3-chloropyridine (11) and subsequent trapping with electrophiles.22 In a calibration experiment in batch, we treated 11 with n-BuLi (−60 °C, 10 min) followed by the addition of AnMgBr (3a, 6.0 equiv., −60 °C, 0.5 h, then 75 °C, 1 h) leading to the desired 2,4-disubstituted pyridine 12a in 56% yield.
We transposed this reaction sequence into a continuous flow set-up as shown in Scheme 5. Mixing n-BuLi with the starting material 11 afforded the lithiated species 13 after 5 min residence time at −60 °C. Adding various arylmagnesium bromides of type 3via a third pump gave the transient magnesium species of type 14 which were heated to 75 °C for 10 min. This very reproducible procedure provided the stable 3-pyridylmagnesium species 15 which was directly pumped into a round bottom flask containing various electrophiles. Quenching with sat. aq. NH4Cl gave the pyridine 12a in 57% isolated yield. Alternatively, iodination (with I2), silylation (with TMSCl), allylation (with allyl bromide) and formylation (with DMF) gave the expected 2,3,4-trisubstituted pyridines 12b–f in 49–53% overall yield. For this multi-step procedure, the flow set-up proved to be advantageous for its high reproducibility, good temperature control and its potential for easy scale up for eventual industrial applications.
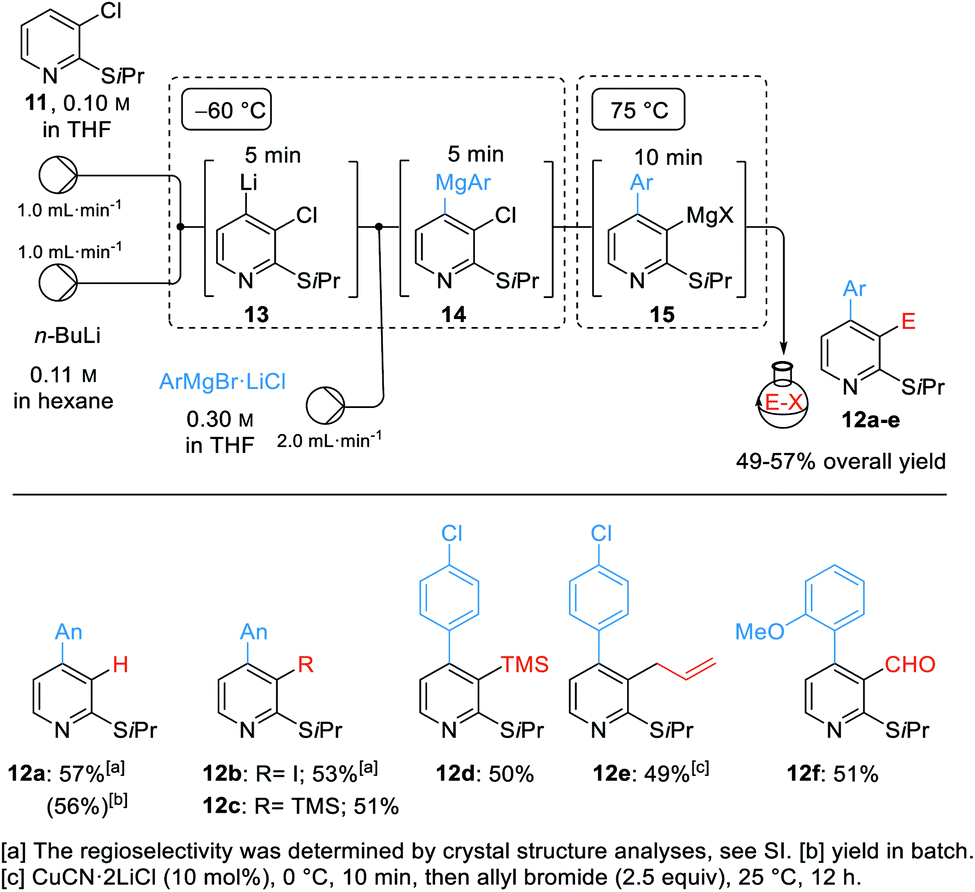 | ||
| Scheme 5 Continuous flow set-up for the difunctionalization of 3-chloro-2-(isopropylthio)pyridine 11via pyridyne formation and subsequent quenching of the generated 3-pyridylmagnesium reagent 15 with various electrophiles. | ||
Next, we applied this method for the preparation of a key intermediate of the synthesis of (±)-paroxetine (16).23 Thus, starting from 3-chloro-2-ethoxypyridine (1), we have prepared the trisubstituted pyridine (7ja) on a 5 mmol scale with 53% overall yield. Treatment of 7ja with neat benzyl bromide at 120 °C for 30 h afforded the N-benzylated pyridone 17 in 80% yield.24 Then, a selective hydrogenation of the pyridone 17 using H2 gas and Pd/C gave the desired trans-substituted piperidone 18 in 50% yield (Scheme 6).25
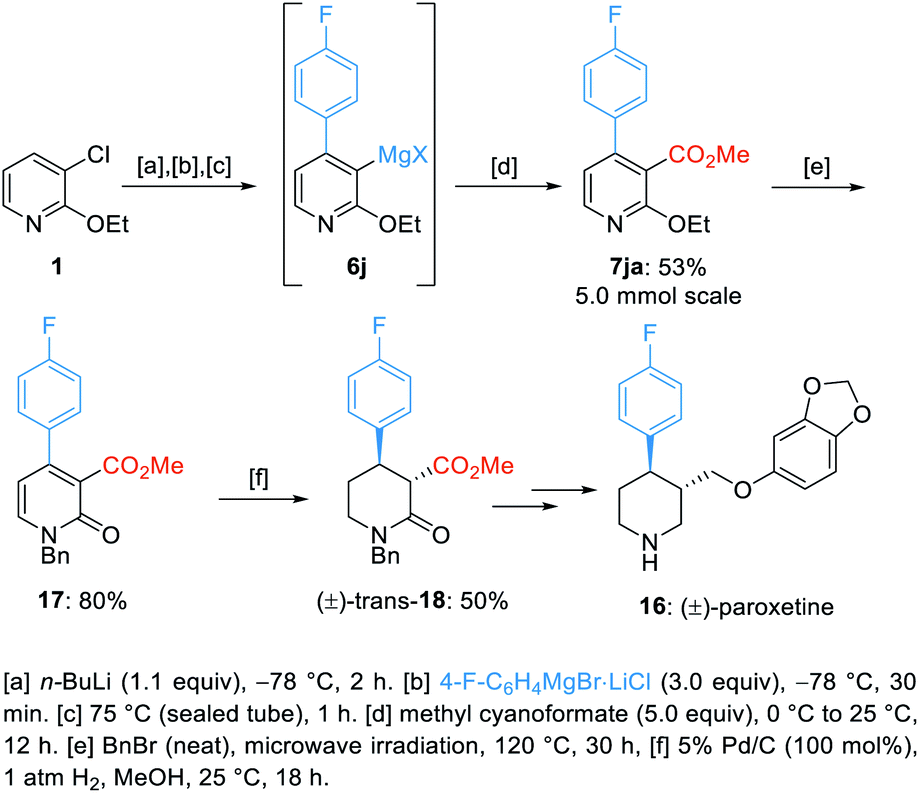 | ||
| Scheme 6 Route towards the key intermediate 18 of the synthesis of (±)-paroxetine (16). | ||
Conclusions
In conclusion, we have developed a new regioselective 3,4-difunctionalization of 3-chloropyridines via 2-alkoxy or 2-alkylthiol 3,4-pyridyne intermediates using as nucleophiles aryl- and alkylmagnesium halides as well as magnesium alkythiolates. The resulting 3-pyridylmagnesium species were quenched with various electrophiles, producing polyfunctional 2,3,4-substituted pyridines.In addition, this methodology was converted into a convenient continuous flow set-up. As an application, we have prepared a key intermediate for the synthesis of the antidepressant (±)-paroxetine 16.
Author contributions
B. H. and D. D. performed the experiments and analysed the data. P. F. and B. M. discussed the results. K. K. measured and analyzed the crystal structures. P. K. conceived the project and wrote the paper. B. H., D. D., P. F. and B. M. contributed to the editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
B. Heinz thanks the Novartis Pharma AG for the fellowship. We thank Albermarle (Hoechst, Germany) and BASF for the generous gift of chemicals and Vapourtec for technical support. Helpful discussions with Dr B. Schenkel, Dr F. Mandrelli and Dr S. Mostarda (Novartis Pharma) are gratefully acknowledged.Notes and references
-
(a) V. Bisai and R. Sarpong, Org. Lett., 2010, 12, 2551–2553 CrossRef CAS PubMed
; (b) D. F. Fischer and R. Sarpong, J. Am. Chem. Soc., 2010, 132, 5926–5927 CrossRef CAS PubMed
-
(a) L. C. Campeau, S. Rousseaux and K. Fagnou, J. Am. Chem. Soc., 2005, 127, 18020–18021 CrossRef CAS PubMed
-
(a) D. L. Comins and M. O. Killpack, J. Org. Chem., 1990, 55, 69–73 CrossRef CAS
- For aryne chemistry starting with direct lithiation, see:
(a) K. H. Reuter and W. J. Scott, J. Org. Chem., 1993, 58, 4722–4726 CrossRef CAS
- For other methods to generate arynes, see:
(a) E. R. Biehl, S. M. Smith and P. C. Reeves, J. Org. Chem., 1971, 36, 1841–1842 CrossRef CAS
-
(a) G. W. Gribble and M. G. Saulnier, Heterocycles, 1993, 35, 151–169 CrossRef CAS
-
(a) M. Tsukazaki and V. Snieckus, Heterocycles, 1992, 33, 533–536 CrossRef CAS
-
(a) A. E. Goetz and N. K. Garg, Nat. Chem., 2013, 5, 54–60 CrossRef CAS PubMed
- S. J. Connon and A. F. Hegarty, J. Chem. Soc., Perkin Trans. 1, 2000, 1245–1249 RSC
-
(a) S. Choppin, P. Gros and Y. Fort, Eur. J. Org. Chem., 2001, 2001, 603–606 CrossRef
- F. M. Piller, P. Appukkuttan, A. Gavryushin, M. Helm and P. Knochel, Angew. Chem., Int. Ed., 2008, 47, 6802–6806 CrossRef CAS PubMed
- For the optimization of the reaction conditions, see ESI.†.
- For the optimization of the stoichiometry of nucleophiles, see ESI.†.
- We have observed that by using of 3-methylthiophenyl-magnesium bromide the resulting product 6g bears a free thiol group. We assumed that a magnesiation of the methylthio group takes place during the reaction, resulting into a magnesium carbenoid which decomposed under the reaction conditions (75 °C).
-
(a) W. E. Bachmann and H. T. Clarke, J. Am. Chem. Soc., 1927, 49, 2089–2098 CrossRef CAS
-
(a) T. R. Kasturi, H. R. Y. Jois and L. Mathew, Synthesis, 1984, 9, 743–746 CrossRef
-
(a) G. Manolikakes, C. M. Hernandez, M. A. Schade, A. Metzger and P. Knochel, J. Org. Chem., 2008, 73, 8422–8436 CrossRef CAS PubMed
-
(a) T. E. Barder, S. D. Walker, J. R. Martinelli and S. L. Buchwald, J. Am. Chem. Soc., 2005, 127, 4685–4696 CrossRef CAS
-
(a) A. M. Berman and J. S. Johnson, J. Am. Chem. Soc., 2004, 126, 5680–5681 CrossRef CAS
- B. Heinz, M. Balkenhohl and P. Knochel, Synthesis, 2019, 51, 4452–4462 CrossRef CAS
-
(a) Z. He and T. F. Jamison, Angew. Chem., Int. Ed., 2014, 53, 3353–3357 CrossRef CAS
- The continuous flow set-up was not applicable for the reaction starting from 3-chloro-2-ethoxypyridine (1) due to longer reaction times and pressure limitations. For a detailed continuous flow/batch comparison, see ESI.†.
-
(a) C. De Risi, G. Fanton, G. P. Pollini, C. Trapella, F. Valente and V. Zanirato, Tetrahedron: Asymmetry, 2008, 19, 131–155 CrossRef CAS
- W. R. Bowman and C. F. Bridge, Synth. Commun., 1999, 29, 4051–4059 CrossRef CAS
-
(a) S. Maris and N. Castagnoli, J. Org. Chem., 1996, 61(1), 309–313 CrossRef
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2057612, 2057613 and 2057614. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc01208h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |