
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA novel Ce0.8Fe0.1Zr0.1O2 solid solution with high catalytic activity for hydrogen storage in MgH2
Ying Chenga,
Shuhua Zhoub,
Biqing Shia,
Bing Donga,
Xianbin Jia,
Siqi Lia and
Wei Zhang *b
*b
aDepartment of Environmental Engineering, Hebei University of Environmental Engineering, Qinhuangdao, 066102, PR China
bHebei Key Laboratory of Applied Chemistry, School of Environmental and Chemical Engineering, Yanshan University, Qinhuangdao 066004, PR China. E-mail: zhangweihh@ysu.edu.cn; Tel: +86-010-8387744
First published on 23rd November 2021
Abstract
The effect of the solid solution Ce0.8Fe0.1Zr0.1O2, successfully prepared by a hydrothermal synthesis method, on the hydrogen sorption properties of MgH2 is systemically investigated. The Ce0.8Fe0.1Zr0.1O2-modified MgH2 composite exhibits remarkable hydrogen kinetics properties and thermodynamics behavior compared to those of as-milled MgH2, with a reduction in the initial desorption temperature of approximately 82 K. With respect to the hydrogen kinetics, the Ce0.8Fe0.1Zr0.1O2-added sample could uptake approximately 5.3 wt% H2 at 473 K in 2500 s, whereas only 1.5 wt% hydrogen could be absorbed by pristine MgH2 in the same conditions. Furthermore, about 4.5 wt% of hydrogen could be desorbed by Ce0.8Fe0.1Zr0.1O2-doped MgH2 composite at 623 K, which was 2 wt% higher than the as-milled MgH2 sample over the same period of time. The decomposition apparent activation energy for MgH2–Ce0.8Fe0.1Zr0.1O2 is reduced to 84.3 kJ mol−1, which is about 77 kJ mol−1 lower than that of pristine MgH2. It is believed that the notable improvement in the hydrogen sorption kinetics is due to the in situ-formed active species of CeH2.51 and MgO as well as the abundant oxygen vacancies, which play a vital role in catalyzing the hydrogen sorption performance of MgH2.
1. Introduction
Hydrogen, as a green fuel, is seen as one of the most promising energy carriers due to its high energy density, cost-effectiveness, and non-pollution, which could also efficiently solve the major issues in regard to fossil fuels: urban air pollution and climate change impact.1,2 To make full use of hydrogen energy sources and realize hydrogen economy, many factors, including production, distribution, transportation, and storage need to be considered. The key to the widespread utilization of hydrogen is to develop safe and efficient storage materials for hydrogen. It is generally accepted that solid-state hydrogen storage materials act like a sponge with the ability to absorb and release hydrogen, which make them propitious candidates for hydrogen storage. Among the solid-state hydrogen storage materials, magnesium has received much attention owing to its high hydrogen capacity, abundant availability, and low cost. However, to make MgH2 a benign and viable hydrogen storage material, two critical barriers need to be overcome. It is well known that MgH2 possesses superior stability and relatively high temperatures are needed to decompose the Mg–H bond to release H2.Numerous methods have been optimized to overcome the disadvantages of MgH2 by ball-milling, nano-structuring, alloying, catalyst doping, etc. Among them, ball-milling various metal catalysts with MgH2 is considered an efficient way to significantly improve the hydrogen storage properties of MgH2. One of the well-known ergastic additives is the rare earth material ceria (Ce). A large number of experiments have been launched to study the catalytic effect of Ce-based materials. Ismail et al.3 reported that doping with CeCl3 could significantly reduce the initial decomposition temperature and enhance the sorption kinetics of MgH2. Some research showed that the peak temperature and the decomposition apparent activation energy for the hydrogen desorption of Mg–20Ni–CeO2 decreased to 318.9 °C and 72.7 kJ mol−1 due to the presence of CeO2.4 Leng et al.5 systemically investigated the influence of Ce on TiFe0.9Mn0.1 alloy and found that the addition of Ce significantly enhanced the hydrogen storage properties of TiFe0.9Mn0.1 alloy on account of its high dispersion on the TiFe0.9Mn0.1 matrix.
Fe has also been widely proved to be an effective catalyst that can promote remarkable improvement of the hydrogen absorption and desorption performances of MgH2.6 Chen et al.7 prepared nanosheet Fe through a wet-chemical ball milling method which contributes to the enhancement of the hydrogen storage properties of MgH2. Wang and Yan et al.8 have shown the improved kinetics results of MgH2 catalyzed by FeB/CNTs; the composites could absorb about 6.2 wt% of hydrogen at 150 °C. Song et al.9 prepared MgH2 catalyzed by 10 wt% Fe2O3 through a mechanical milling method. They reported that 10 wt% Fe2O3 shows far better hydrogen/dehydrogen storage behaviors compared to pristine MgH2.
Furthermore, recent studies have demonstrated that oxygen vacancies (Ovac) are greatly important active sites for hydrogen storage which can capture H2 molecules and help the diffusion of H atoms during the hydrogen absorption/desorption processes.10 The enhancement of hydrogen dynamics and thermodynamics properties is mainly ascribed to the abundant oxygen vacancies, as confirmed by Zhou et al.11 An abundance of oxygen vacancies can be achieved by the addition of Zr into CeO2.12,13 Moreover, extensive research has revealed that the density of oxygen vacancies on transition doped Ce–Zr oxides is far better than on single Ce–Zr oxides;14,15 Ovac defects can be produced on the CeO2 surface by Ce3+–Ce4+ transfer owing to the transition metal dopant.
Inspired by these studies, a Ce–Fe–Zr oxide solid solution catalyst with high activity and Ovac defects was designed and the influence of the Ce–Fe–Zr solid solution on the hydrogen storage properties of MgH2 was systemically investigated. Moreover, the catalytic mechanism of the synthesized Ce–Fe–Zr solid solution in MgH2 is explained, which will provide novel strategies for the design of multiple catalysts and enrich the hydrogen storage field.
2. Experimental section
2.1 Synthesis of Ce0.8Fe0.1Zr0.1O2 catalyst
Ce(NO3)3·6H2O (99.9%), ZrOCl2 (99.9%) and Fe(NO3)3·6H2O (99.9%) were purchased from Aladdin Chemical Reagent Co. Ltd and used as reactants without any purification. The above reactants in a mass ratio of 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 were dissolved in a solvent composed of CTAB with NH3·H2O added dropwise into the mixed solution to adjust the pH to 9–11. The mixture was stirred in the reactor for 1–2 hours and then stood at room temperature for 12 hours. After standing, the solution was filtered and washed several times with deionized water, then put into an oven at 60 °C to dry for 12 h. After drying, the sample was calcined in a muffle furnace and raised to the target temperature of 550 °C at a rate of 1 °C min−1. After holding at the target temperature for a period, the solid solution of cerium zirconium was obtained.
:1:1 were dissolved in a solvent composed of CTAB with NH3·H2O added dropwise into the mixed solution to adjust the pH to 9–11. The mixture was stirred in the reactor for 1–2 hours and then stood at room temperature for 12 hours. After standing, the solution was filtered and washed several times with deionized water, then put into an oven at 60 °C to dry for 12 h. After drying, the sample was calcined in a muffle furnace and raised to the target temperature of 550 °C at a rate of 1 °C min−1. After holding at the target temperature for a period, the solid solution of cerium zirconium was obtained.
2.2 Synthesis of MgH2–Ce0.8Fe0.1Zr0.1O2 composites
MgH2 was prepared from commercial Mg powder (98%) through hydrogen combustion. The Mg powder was purchased from Aladdin Chemical Reagent Co. Ltd and hydrogenated at 400 °C at 4 MPa for 10 h. Pure MgH2 was successfully obtained by repeating the above procedure five times.The doped MgH2–Ce0.8Fe0.1Zr0.1O2 sample was fabricated through a mechanical ball-milling method by milling MgH2 and Ce0.8Fe0.1Zr0.1O2 in a mass ratio of 5:1. An increased milling temperature was inhibited by controlling the mill direction, starting in one direction for 15 min, switching to the opposite direction for 15 min and then pausing for 10 min. The ball-to-sample weight ratio was 20:1. To prevent the sample from contacting oxygen and vapor, the operation was conducted in an Ar-filled glove box.
2.3 Characterization
X-ray diffraction (XRD) was conducted on a SmartLab high resolution X-ray diffractometer (Rigaku) with Cu Kα radiation at 40 kV and 40 mA. The scanning speed was 4° min−1 in the range of 10° to 80°. Scanning electron microscopy (SEM) was employed to observe the micro-structure and morphology of the formed sample. The element distribution and active oxygen species on the surface of the catalyst were determined by X-ray photoelectron spectroscopy (XPS).The measurements of the initial desorption temperature and hydrogen absorption/desorption kinetics were performed on a Sieverts-type pressure-composition-temperature (PCT) apparatus (GRINM Co., China). During the initial desorption temperature test, the sample was heated from room temperature to 1000 K at 0.01 MPa with a heating rate of 10 K min−1 in a vacuum chamber. For the hydrogen absorption kinetics tests, the sample was heated to 473 K and 523 K under 3.0 MPa of pressure, while for the hydrogen desorption kinetics tests, the sample was heated to 598 K and 623 K under a pressure of 0.001 MPa. The thermal behaviors of the as-milled MgH2 and Ce0.8Fe0.1Zr0.1O2-catalyzed MgH2 were examined by differential scanning calorimetry (Mettler Toledo, TGA/DSC 1). All the samples were heated under an Ar atmosphere from room temperature to 500 °C at heating rates of 5 K min−1, 10 K min−1, 15 K min−1, and 20 K min−1.
3. Results and discussion
3.1 Characteristics of the prepared Ce0.8Fe0.1Zr0.1O2
The XRD pattern of the synthesized Ce0.8Fe0.1Zr0.1O2 in Fig. 1 obviously shows strong peaks at 2θ = 28.86°, 33.52°, 47.78°, 56.66°, 59.63°, 70.35°, 77.56°, and 78.19° which correspond well to the standard card (PDF# 34-0394). All the strong peaks are shifted to the left compared to the standard card (PDF# 34-0394), indicating the successful synthesis of the solid solution Ce0.8Fe0.1Zr0.1O2 catalyst. | ||
| Fig. 1 XRD pattern of Ce0.8Fe0.1Zr0.1O2 catalyst. | ||
3.2 TPD curves of MgH2–Ce0.8Fe0.1Zr0.1O2 and MgH2 samples
Fig. 2 depicts the temperature programmed desorption (TPD) patterns for the hydrogen desorption process of ball-milled MgH2 and Ce0.8Fe0.1Zr0.1O2-catalyzed MgH2 materials. The ball-milled pristine MgH2 begins to release hydrogen at about 643 K, with a maximum hydrogen capacity of about 6.5 wt% of hydrogen reached by 800 K. Moreover, Ce0.8Fe0.1Zr0.1O2 contributes to the hydrogen desorption of MgH2 in a relatively lower temperature range than that of pure MgH2. An obvious feature seen in Fig. 2 is that Ce0.8Fe0.1Zr0.1O2-catalyzed MgH2 starts to release hydrogen at a lower temperature, approximately 561 K, achieving a full hydrogen capacity of about 5.5 wt% H2 at 690 K. Furthermore, the modified sample completed dehydrogenation at 710 K. In contrast, the saturation of the desorption process for pure MgH2 is at 830 K. According to the TPD curves, the desorption rate and temperature determined for Ce0.8Fe0.1Zr0.1O2-catalyzed MgH2 are superior to those of MgH2 without the catalyst. The onset temperature reduction (82 K) on the doped sample signifies that Ce0.8Fe0.1Zr0.1O2 is a promising catalyst in the MgH2 system which helps lower the desorption temperature and rate. | ||
| Fig. 2 TPD curves of ball-milled MgH2 and Ce0.8Fe0.1Zr0.1O2-catalyzed MgH2 at the heating rate of 10 K min−1. | ||
3.3 Sorption kinetics of MgH2–Ce0.8Fe0.1Zr0.1O2 and MgH2 samples
Further investigation of the catalytic activity of Ce0.8Fe0.1Zr0.1O2 on the hydrogen desorption properties of MgH2 was performed by hydrogen sorption release. Fig. 3 exhibits the hydrogen desorption properties for the milled MgH2 and the modified sample at 598 K and 623 K. It is obvious that the ball-milled MgH2 shows sluggish desorption kinetic properties compared to those with catalyst addition at the investigated temperature. Almost no hydrogen is released by MgH2 at the temperature of 598 K. At this temperature, the Ce0.8Fe0.1Zr0.1O2-doped MgH2 yields a significant increase to 2.5 wt% hydrogen at 598 K within 2000 s. When the temperature shows a sharp shift to 623 K, both the hydrogen desorption properties and desorption rate are highly improved. The Ce0.8Fe0.1Zr0.1O2-added sample results in the liberation of approximately 4.5 wt% hydrogen at 623 K after 2000 s of dehydrogenation, whereas the as-milled MgH2 sample desorbs less than 2.5 wt% hydrogen over the same period. It is evident that Ce0.8Fe0.1Zr0.1O2 plays a significant catalytic role in enhancing the desorption behavior of MgH2.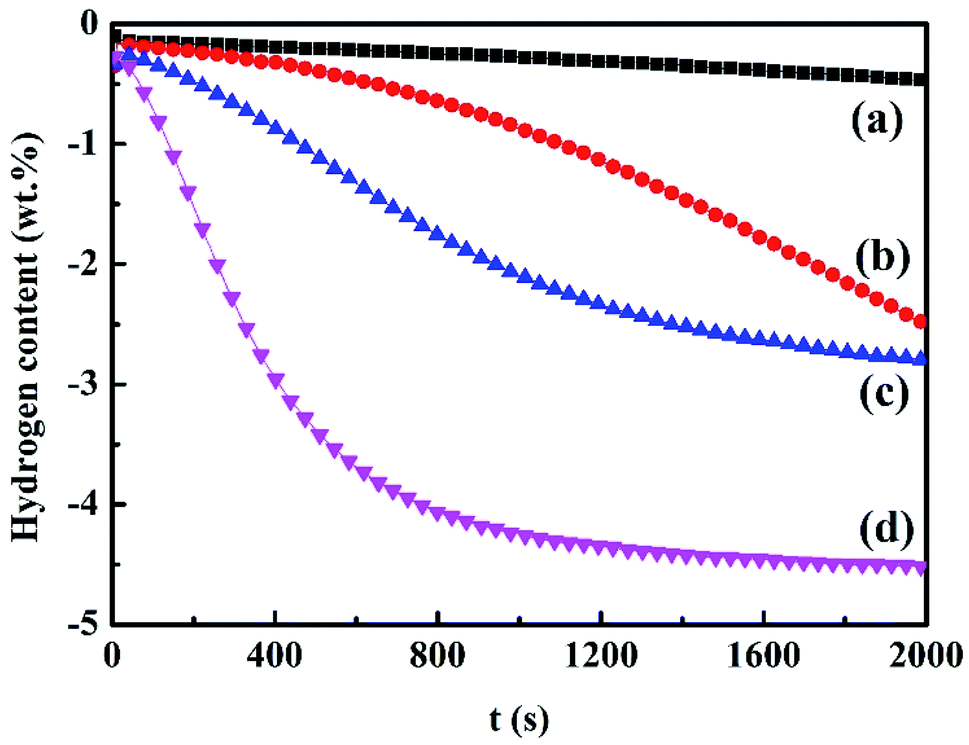 | ||
| Fig. 3 Desorption kinetics curves of ball-milled MgH2 at 598 K (a) and 623 K (b) and Ce0.8Fe0.1Zr0.1O2 catalyzed MgH2 at 598 K (c) and 623 K (d). | ||
The excellent hydrogen release kinetics of MgH2 are related to the energy barrier of the desorption process on MgH2 and the apparent energy (Ea) is a key parameter that reflects the improved desorption kinetics. To determine the Ea for the desorption stage, the Kissinger method was used. The equation is as follows:
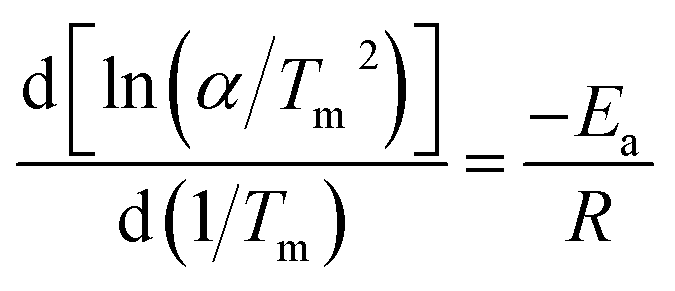 | (1) |
 | ||
| Fig. 4 DSC curves of MgH2 (a) and Ce0.8Fe0.1Zr0.1O2-catalyzed MgH2 (b) at different heating rates; (c) Kissinger's plots for milled MgH2 and MgH2–Ce0.8Fe0.1Zr0.1O2 composite. | ||
To further clarify the effect of the synthesized Ce0.8Fe0.1Zr0.1O2 on the hydrogen sorption performance of pristine MgH2, the isothermal hydrogen absorption was systematically investigated. Fig. 5 collates the findings of the isothermal rehydrogenation kinetics at 473 K and 523 K in the presence of 3.0 MPa hydrogen pressure which demonstrate that the sample of Ce0.8Fe0.1Zr0.1O2 takes in hydrogen faster than pure MgH2 and the MgH2–Ce0.8Fe0.1Zr0.1O2 sample has the superior hydrogen absorption kinetics rate. At 473 K, only 1.5 wt% of hydrogen could be absorbed by pristine MgH2 in 2500 s. For the Ce0.8Fe0.1Zr0.1O2-modified MgH2 sample, the hydrogen absorption capacity is about three times higher than that of pure MgH2, with 5.3 wt% of hydrogen adsorbed. Raising the temperature to 598 K leads to a significant increase in hydrogen capacity as well as in the hydrogen rate compared to those presented by the other sample at the identical temperature. The ball-milled MgH2 presents a slight increase when the temperature rises from 473 K to 523 K, absorbing about 4.4 wt% hydrogen after 2500 s at 523 K. The hydrogen capacity for the modified Ce0.8Fe0.1Zr0.1O2 sample achieves 5.6 wt% hydrogen in the same conditions. The results signify that Ce0.8Fe0.1Zr0.1O2 is a striking catalyst that endows the MgH2–Ce0.8Fe0.1Zr0.1O2 sample with excellent hydrogen absorption behavior. Many researchers have reported a similar phenomenon where increased temperature exhibits a promising effect on the hydrogen storage capacity and hydrogen storage rate of solid-state hydrogen storage materials.23
 | ||
| Fig. 5 Absorption kinetics curves of ball-milled samples at different temperatures under 3.0 MPa H2:MgH2 at 473 K (a) and 523 K (b) and Ce0.8Fe0.1Zr0.1O2-catalyzed MgH2 at 473 K (c) and 523 K (d). | ||
In order to verify the cause of the improvement of the hydrogen absorption kinetics for MgH2 attributed to the addition of Ce0.8Fe0.1Zr0.1O2, the hydrogenation mechanism was investigated by comparing the hydrogen absorption rate curves with the rate equations for MgH2–Ce0.8Fe0.1Zr0.1O2 and MgH2 composites. The Avrami–Erofeev equation (eqn (2)) is usually employed to fit the hydrogenation absorption process and gives strong insight into the nucleation and growth processes.
| α = 1 − exp(−ktm) | (2) |
Here, α is the reacted fraction, k is the rate constant, and m is the order of the reaction. Based on the obtained results, it is evident that there exists a distinct difference between the hydrogen sorption kinetics and the fitting curves result for MgH2. The same phenomenon does not appear in the Ce0.8Fe0.1Zr0.1O2-catalyzed MgH2. It is obvious that for the Ce0.8Fe0.1Zr0.1O2-catalyzed MgH2 composites, the correlation and fitting degree of both curves agree very well. The fitting lines in Fig. 6 depict that the reaction mechanism of MgH2 catalyzed by the Ce0.8Fe0.1Zr0.1O2 composite is subject to nucleation and growth processes. The value of m gives a specific explanation of the rate-controlling step for the hydrogenation diffusion process. It is reported that an m value approaching 0.620 belongs to a one-dimensional diffusion process, whereas a value approximating 1.070 could be ascribed to a three-dimensional diffusional process; a one-dimensional diffusion process is beneficial to hydrogen adsorption.24 According to the fitted curves, the m value for as-milled MgH2 is equal to 0.91, which is close to 1.070, indicating that the hydrogen sorption process for pure MgH2 is a three-dimensional diffusion process. For the MgH2 catalyzed by Ce0.8Fe0.1Zr0.1O2, the value of m (0.36) approaches 0.620, which indicates one-dimensional diffusion. Therefore, the rate-controlling step for the hydrogenation process changes from a three-dimensional diffusion process to a one-dimensional diffusion process due to the addition of Ce0.8Fe0.1Zr0.1O2.
 | ||
| Fig. 6 Fitted hydrogenation kinetic curves of MgH2 and Ce0.8Fe0.1Zr0.1O2-catalyzed MgH2 composite at 523 K under the pressure of 3 MPa. | ||
3.4 XRD analysis
To further investigate the reaction mechanism of the improved hydrogenation/dehydrogenation kinetics and thermodynamics of Ce0.8Fe0.1Zr0.1O2-catalyzed MgH2, XRD was employed to clarify the phase components of MgH2–Ce0.8Fe0.1Zr0.1O2 composites at different stages; the XRD patterns are presented in Fig. 7. At the ball-milled stage in Fig. 7a, the dominant diffraction peak is indexed to MgH2 and some new peaks relating to CeH2.51 could be detected. Additionally, a diffraction peak at 2θ = 42.5° ascribed to MgO could be found. The appearance of MgO may be due to slight oxygen contamination during XRD. The newly formed phase gives an indication that there has been a chemical reaction between Ce0.8Fe0.1Zr0.1O2 and MgH2 during the ball-milling process. After the composites were hydrogenated at 3.0 MPa (Fig. 7b), all the diffraction peaks become sharper and narrower compared to those of the ball-milled stage and the diffraction peaks for MgH2 and CeH2.51 remain. In the XRD pattern of the dehydrogenated sample at 623 K (Fig. 7c), the characteristic diffraction peaks for MgH2 vanish, while those for Mg are obvious, indicating that MgH2 has been largely transformed to Mg during the dehydrogenation process. Fe and Zr cannot be detected in the XRD patterns, due to the small amounts or the formation of solid solution, and the obtained results correspond well to the XRD analysis. It is obvious that the diffraction peaks for CeH2.51 and MgO remain present through the whole hydrogenation/dehydrogenation process. It has been confirmed by numerous studies that CeH2.51 is beneficial to the enhancement of MgH2 hydrogen uptake and release.25 | ||
| Fig. 7 XRD patterns of Ce0.8Fe0.1Zr0.1O2-catalyzed MgH2 at different stages: (a) after ball-milling, (b) hydrogenation at 3.0 MPa and (c) dehydrogenation at 623 K. | ||
3.5 XPS analysis
To explain the reason for the improvement of the hydrogen performance by Ce0.8Fe0.1Zr0.1O2-modified MgH2, the chemical states of key elements which play vital roles in the enhancement by Ce0.8Fe0.1Zr0.1O2 are investigated. The Ce 3d and O 1s spectra for Ce0.8Fe0.1Zr0.1O2 are presented with that of pure CeO2 as a comparison. The O 1s spectrum (Fig. 8A) can be divided into three main peaks: the one centered at 529.5 eV, which is related to lattice oxygen Oα(O2−), and two others centered at 530.4 and 532.8 eV, which are ascribed to surface chemically adsorbed oxygen Oβ(O−, O2−).26 It is obvious that the amount of lattice oxygen Oα on the surface of Ce0.8Fe0.1Zr0.1O2 is higher than that in pure CeO2, indicating that more oxygen vacancies exist in the Fe and Zr doped CeO2. | ||
| Fig. 8 (A) O 1s XPS spectra of (a) CeO2 and (b) Ce0.8Fe0.1Zr0.1O2; (B) Ce 3d XPS spectra of (a) CeO2 and (b) Ce0.8Fe0.1Zr0.1O2. | ||
According to Fig. 8B, the complex spectrum of Ce 3d can be decomposed into eight peaks, labeled as V (882.4 eV), V′ (885.0 eV), V′′ (889.2 eV), V′′′ (898.1 eV), U (900.9 eV), U′ (903.3 eV), U′′ (907.6 eV), and U′′′ (916.7 eV). The split peaks labeled V, V′′, V′′′, U, U′′, and U′′′ are ascribed to Ce4+ species, while the peaks located at V′ and U′ correspond to Ce3+ species.27 The primary chemical valence state on the surface of the sample is Ce3+. It can be clearly seen in Fig. 8B that the surface Ce3+ amount is higher on Ce0.8Fe0.1Zr0.1O2 than on pure CeO2. Huang28 and Weng et al.29 confirmed that the improved catalytic activity could be due to the formation of more Ce3+ accompanied by oxygen defects. According to the obtained results of the XPS analysis, the Fe and Zr doped CeO2 possesses more oxygen vacancies and Ce3+ on the Ce0.8Fe0.1Zr0.1O2 surface.
Based on the XRD and XPS analyses, the catalytic mechanism of Ce0.8Fe0.1Zr0.1O2 that improves the hydrogenation/dehydrogenation properties of MgH2 can be summarized. The generation of CeH2.51 and MgO derived from the reaction between Ce0.8Fe0.1Zr0.1O2 and MgH2 may play a vital role in altering the hydrogenation/dehydrogenation behaviors of MgH2. It has been reported by numerous researchers that the in situ-formed light rare earth hydride CeH2.51 could serve as the real catalyst promoting the hydrogenation/dehydrogenation procedures.30,31 The light rare earth hydrides generate the activation of the surface of MgH2 and lead to more nucleation sites on the substrate of MgH2, also providing H diffusion channels and easily facilitating the hydrogen absorption and desorption processes.32 In addition, the produced MgO also brings a positive catalytic effect for the improvement of MgH2. Ismail et al.33 showed that MgO could work with Fe, Mg–Cu alloy as an active species to catalyze the hydrogen storage properties of MgH2 in CuFe2O4-doped MgH2 composite. MgO also played a crucial role in improving the sorption performance of the MgH2–MgFe2O4 system through the synergetic catalytic effect.34 Also, its high electronegativity promotes the improvement of the kinetics of magnesium-based hydrides.35 Furthermore, the addition of Fe and Zr promotes more oxygen vacancies and Ce3+ on the surface of Ce0.8Fe0.1Zr0.1O2 and multi-valence catalysts and abundant oxygen vacancies have shown great potential in enhancing the de/hydrogenation kinetics of MgH2.11,36 Therefore, the great improvement in the hydrogenation kinetics is ascribed to the in situ-formed active species CeH2.51 and MgO and the abundant oxygen vacancy defects, which may be the major factors boosting the hydrogen sorption performance of MgH2.
4. Conclusions
The solid solution Ce0.8Fe0.1Zr0.1O2 was successfully prepared by hydrothermal synthesis method and exhibited surprisingly high catalytic activity, leading to the MgH2–Ce0.8Fe0.1Zr0.1O2 composite reflecting excellent hydrogen sorption kinetics. The Ce0.8Fe0.1Zr0.1O2-doped MgH2 presented striking hydrogen storage properties as it starts to liberate H2 at 561 K in the dehydrogenation process, 82 K lower than the pristine MgH2. With respect to hydrogen adsorption, the Ce0.8Fe0.1Zr0.1O2-added sample could uptake approximately 5.3 wt% H2 at 473 K, while only 1.5 wt% hydrogen could be absorbed by pristine MgH2 in the same conditions. The dehydrogenation properties of MgH2 are also improved, with a desorption amount of 4.5 wt% hydrogen at 623 K, where the as-milled MgH2 sample desorbs less than 2.5 wt% hydrogen over the same period of time. The Kissinger plot shows that the apparent activation energy for MgH2 is reduced from 161.2 kJ mol−1 to 84.3 kJ mol−1 due to the presence of the solid solution Ce0.8Fe0.1Zr0.1O2-modified sample. It is believed that the great improvement in hydrogen sorption kinetics is due to the in situ-formed active species of CeH2.51 and MgO and the abundant oxygen vacancies, which play vital roles in boosting the hydrogen sorption performance of MgH2.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by Natural Science Foundation of Hebei Province of China (E2019415036; B2021415002); Science and Technology Project of Hebei Education Department (BJ2020043; QN2020142); Hebei University of Environmental Engineering (Top-notch Talents Cultivation Program for Young Science and Technology 2020ZRBJ01); Doctoral Foundation of Hebei University of Environmental Engineering (201805).Notes and references
- Z. Shao, Y. Li, C. Liu, W. Ai, S.-P. Luo and Q. Liu, Nat. Commun., 2020, 11, 591 CrossRef CAS PubMed.
- D. J. Han, S. Kim and E. S. Cho, J. Mater. Chem. A, 2021, 9, 9875–9881 RSC.
- M. Ismail, N. S. Mustafa, N. Juahir and F. A. H. Yap, Mater. Chem. Phys., 2016, 170, 77–82 CrossRef CAS.
- P. Liu, J. J. Lian, H. P. Chen, X. J. Liu, Y. L. Chen, T. H. Zhang, H. Yu, G. J. Lu and S. X. Zhou, Chem. Eng. J., 2020, 385, 9 Search PubMed.
- H. Leng, Z. Yu, J. Yin, Q. Li, Z. Wu and K.-C. Chou, Int. J. Hydrogen Energy, 2017, 42, 23731–23736 CrossRef CAS.
- N. A. Abdul Majid, J. Watanabe and M. Notomi, Int. J. Hydrogen Energy, 2021, 46, 4181–4187 CrossRef CAS.
- L. Zhang, L. Ji, Z. Yao, N. Yan, Z. Sun, X. Yang, X. Zhu, S. Hu and L. Chen, Int. J. Hydrogen Energy, 2019, 44, 21955–21964 CrossRef CAS.
- S. Gao, X. Wang, H. Liu, T. He, Y. Wang, S. Li and M. Yan, J. Power Sources, 2019, 438, 227006–227016 CrossRef CAS.
- M. Y. Song and Y. J. Kwak, Mater. Res. Bull., 2021, 140, 111304 CrossRef CAS.
- X. Lin, S. Li, H. He, Z. Wu, J. Wu, L. Chen, D. Ye and M. Fu, Appl. Catal., B, 2018, 223, 91–102 CrossRef CAS.
- P. Liu, H. Chen, H. Yu, X. Liu, R. Jiang, X. Li and S. Zhou, Int. J. Hydrogen Energy, 2019, 44, 13606–13612 CrossRef CAS.
- J. Xiong, Y. C. Wei, Y. L. Zhang, X. L. Mei, Q. Q. Wu, Z. Zhao, J. Liu, D. Wu and J. M. Li, Catal. Today, 2020, 355, 587–595 CrossRef CAS.
- J. Xiong, X. Mei, J. Liu, Y. Wei, Z. Zhao, Z. Xie and J. Li, Appl. Catal., B, 2019, 251, 247–260 CrossRef CAS.
- S. Ali, L. Chen, F. Yuan, R. Li, T. Zhang, S. u. H. Bakhtiar, X. Leng, X. Niu and Y. Zhu, Appl. Catal., B, 2017, 210, 223–234 CrossRef CAS.
- Y. Cheng, W. Song, J. Liu, H. Zheng, Z. Zhao, C. Xu, Y. Wei and E. J. M. Hensen, ACS Catal., 2017, 7, 3883–3892 CrossRef CAS PubMed.
- X. Zhang, K. Wang, X. Zhang, J. Hu, M. Gao, H. Pan and Y. Liu, Int. J. Energy Res., 2021, 45, 3129–3141 CrossRef CAS.
- N. A. Sazelee, N. H. Idris, M. F. Md Din, N. S. Mustafa, N. A. Ali, M. S. Yahya, F. A. Halim Yap, N. N. Sulaiman and M. Ismail, Int. J. Hydrogen Energy, 2018, 43, 20853–20860 CrossRef CAS.
- L. Dan, L. Hu, H. Wang and M. Zhu, Int. J. Hydrogen Energy, 2019, 44, 29249–29254 CrossRef CAS.
- S. Singh, A. Bhatnagar, V. Shukla, A. K. Vishwakarma, P. K. Soni, S. K. Verma, M. A. Shaz, A. S. K. Sinha and O. N. Srivastava, Int. J. Hydrogen Energy, 2020, 45, 774–786 CrossRef CAS.
- N. A. Ali, N. H. Idris, M. F. M. Din, M. S. Yahya and M. Ismail, J. Alloys Compd., 2019, 796, 279–286 CrossRef CAS.
- D. Pukazhselvan, N. Nasani, P. Correia, E. Carbó-Argibay, G. Otero-Irurueta, D. G. Stroppa and D. P. Fagg, J. Power Sources, 2017, 362, 174–183 CrossRef CAS.
- N. A. Sazelee, N. H. Idris, M. F. Md Din, M. S. Yahya, N. A. Ali and M. Ismail, Results Phys., 2020, 16, 102844 CrossRef.
- L. T. Zhang, Z. L. Cai, X. Q. Zhu, Z. D. Yao, Z. Sun, L. Ji, N. H. Yan, B. B. Xiao and L. X. Chen, J. Alloys Compd., 2019, 805, 295–302 CrossRef CAS.
- J. S. Pedersen, J. Appl. Crystallogr., 1994, 27, 595–608 CrossRef.
- N. S. Mustafa and M. Ismail, J. Alloys Compd., 2017, 695, 2532–2538 CrossRef CAS.
- Y. Cheng, J. Liu, Z. Zhao, W. Song and Y. Wei, J. Hazard. Mater., 2018, 342, 317–325 CrossRef CAS PubMed.
- Y. Cheng, J. Liu, Z. Zhao, W. Song and Y. Wei, Chem. Eng. Sci., 2017, 167, 219–228 CrossRef CAS.
- X. Yuan, H. Ge, X. Liu, X. Wang, W. Chen, W. Dong and F. Huang, J. Alloys Compd., 2016, 688, 613–618 CrossRef CAS.
- J. Fan, X. Wu, X. Wu, Q. Liang, R. Ran and D. Weng, Appl. Catal., B, 2008, 81, 38–48 CrossRef CAS.
- Z. Cao, L. Ouyang, H. Wang, J. Liu, L. Sun and M. Zhu, J. Alloys Compd., 2015, 639, 452–457 CrossRef CAS.
- D. Wu, L. Ouyang, C. Wu, H. Wang, J. Liu, L. Sun and M. Zhu, J. Alloys Compd., 2015, 642, 180–184 CrossRef CAS.
- L. Z. Ouyang, X. S. Yang, M. Zhu, J. W. Liu, H. W. Dong, D. L. Sun, J. Zou and X. D. Yao, J. Phys. Chem. C, 2014, 118, 7808–7820 CrossRef CAS.
- M. Ismail, N. S. Mustafa, N. A. Ali, N. A. Sazelee and M. S. Yahya, Int. J. Hydrogen Energy, 2019, 44, 318–324 CrossRef CAS.
- N. A. Ali, N. H. Idris, M. F. M. Din, N. S. Mustafa, N. A. Sazelee, F. A. H. Yap, N. N. Sulaiman, M. S. Yahya and M. Ismail, RSC Adv., 2018, 8, 15667–15674 RSC.
- J.-R. Ares-Fernández and K.-F. Aguey-Zinsou, Catalysts, 2012, 2, 330–343 CrossRef.
- L. S. Xie, J. S. Li, T. B. Zhang and L. Song, Mater. Charact., 2017, 133, 94–101 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |