DOI:
10.1039/D1RA07231E
(Paper)
RSC Adv., 2021,
11, 37684-37699
C(acyl)–C(sp2) and C(sp2)–C(sp2) Suzuki–Miyaura cross-coupling reactions using nitrile-functionalized NHC palladium complexes†
Received
28th September 2021
, Accepted 8th November 2021
First published on 23rd November 2021
Abstract
Application of N-heterocyclic carbene (NHC) palladium complexes has been successful for the modulation of C–C coupling reactions. For this purpose, a series of azolium salts (1a–f) including benzothiazolium, benzimidazolium, and imidazolium, bearing a CN-substituted benzyl moiety, and their (NHC)2PdBr2 (2a–c) and PEPPSI-type palladium (3b–f) complexes have been systematically prepared to catalyse acylative Suzuki–Miyaura coupling reaction of acyl chlorides with arylboronic acids to form benzophenone derivatives in the presence of potassium carbonate as a base and to catalyse the traditional Suzuki–Miyaura coupling reaction of bromobenzene with arylboronic acids to form biaryls. All the synthesized compounds were fully characterized by Fourier Transform Infrared (FTIR), and 1H and 13C NMR spectroscopies. X-ray diffraction studies on single crystals of 3c, 3e and 3f prove the square planar geometry. Scanning Electron Microscopy (SEM), energy dispersive X-ray spectroscopy (EDS), metal mapping analyses and thermal gravimetric analysis (TGA) were performed to get further insights into the mechanism of the Suzuki–Miyaura cross coupling reactions. Mechanistic studies have revealed that the stability and coordination of the complexes by the CN group are achieved by the removal of pyridine from the complex in catalytic cycles. The presence of the CN group in the (NHC)Pd complexes significantly increased the catalytic activities for both reactions.
Introduction
The modification of organic compounds such as natural compounds, pharmaceuticals, and novel materials can be achieved through the formation of carbon–carbon bonds. Carbon–carbon bond formation is the most useful transformation for chemists in the preparation of organic compounds.1 Palladium-catalyzed coupling reactions of aryl, alkyl halides or acyl chlorides with organoborons are the easiest and safest way to access C(sp2)–C(sp2) or C(acyl)–C(sp2) bonds and to prepare biaryls or ketones.2 Many synthetic methods have been developed for the synthesis of ketones in general due to their formation in various natural products, agrochemicals, fine chemicals, and pharmaceutical products.3 The palladium-catalyzed acylative Suzuki–Miyaura cross coupling reaction has gained great attention due to its easy accessibility, and use of widely functionalized, partially low toxicity, heat and moisture resistance of organoborons. Besides, this method can prevent the restrictions of the regioselectivity limitation, the requirement of the directional group (carbonylative), the use of the para position (conventional Friedel–Crafts acylation), the use of toxic carbon monoxide, and the formation of biaryl by-products (carboxylative Suzuki–Miyaura cross-coupling), CH activation conditions (oxidation of benzyl alcohols and addition of organometallic reagents to carboxylic derivatives or nitriles), and incompatibility with electron-deficient groups.4 Due to these advantages, recently, palladium-mediated cross-coupling reactions between arylboronic acid and an activated derivative of an acyl chloride has been reported to be effective in synthesizing functionalized ketones.5
The palladium-catalyzed traditional Suzuki–Miyaura cross-coupling reaction of organoboron reagents with aryl halides to form biaryl derivatives has emerged over the past two decades as an extremely powerful tool in organic synthesis.3c-d The reaction conditions for Suzuki–Miyaura couplings involve the use of homogeneous palladium catalysts containing phosphine ligands7 or N-heterocyclic carbene (NHC) ligands.6 The first NHC was discovered by Wanzlick et al. in 1968, and in 1991, it was isolated and structure determined by Arduengo et al.8 Carbenes from many NHC ligands containing other donor functions (such as imidazole, benzimidazole, imidazoline, and 1,2,4-triazole) can affect metal complexes with enhanced stability and catalytic activity.9 On the other hand, hetero-bidentate ligands are hybrid ligands that combine the properties of two different donor atoms. These ligands affect the equilibrium because both the metal ion and the chelating agent are sensitive to changes in the catalytic system. Pd complexes with hybrid NHC ligands (with S–C, O–C, N–C, or P–C coordination mode) are well known as generally good catalysts for C–C bond formation reactions.10 They control metal ions by blocking the reactive sites of the metal ion and preventing them from entering into their normal (and in many cases undesirable) reactions. As coordination flexibility in hybrid ligands can have a profound effect on catalytic performance,11 we were interested in effect of NHC structure and nitrile group on catalytic activities. So, the azolium salts (benzothiazolium, benzimidazolium, imidazolium) containing 2-nitrile substituted benzyl groups and not nitrile group were prepared. Their palladium(II) complexes were synthesized and their catalytic activities were also tested in the direct arylation of acyl chlorides and Suzuki–Miyaura coupling reaction. The catalytic activities of the palladium(II) complexes varied with different NHC skeletons which is the most important way to modify.
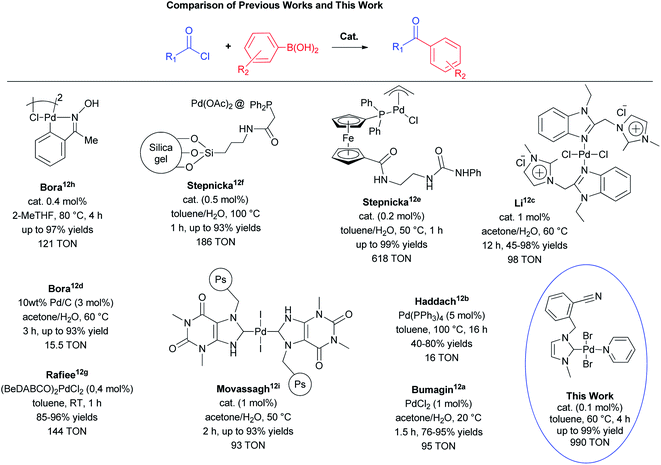
This study primarily focused on the acylative Suzuki–Miyaura cross-coupling reaction. The different strategies for the acylative Suzuki–Miyaura cross-coupling reaction of benzoyl chloride with phenylboronic acid are illustrated in Fig. 1. In early works, Bumagin et al. and Haddach et al. evolved palladium-catalyzed cross-coupling of arylboronic acids with acyl chlorides in 1999.12a,b Li et al. developed an imidazolium chloride tagged Pd(II) complex for the reaction but high catalyst loading and long reaction time was needed.12c Bora et al. reported two different procedure and one was required high catalyst loading,12d while the other one was reached 97% yield in 4 hours with 0.4 mol% catalyst.12h Štěpnička et al. published immobilized palladium catalysts12f and Pd(II) complex with phosphinoferrocene ligands bearing extended polar amidourea pendants12e for the acylative Suzuki–Miyaura coupling reaction. They reached 618 TON value with the Pd(II) complex with phosphinoferrocene ligand. Rafiee et al. developed catalytic system using 1-benzyl-4-aza-1-azoniabicyclo[2.2.2]octane chloride and PdCl2.12g Movassagh et al. reported polystyrene-supported NHC–Pd(II) complex and their heterogeneous catalyst could be reused up to four times.12i In this context, we have prepared nitrile-functionalized NHC precursors and their Pd(II) complexes as a precatalysts to investigate whether (NHC)2PdBr2 and PEPPSI type Pd complexes would be also useful for the acylative Suzuki–Miyaura cross-coupling reaction of acyl chlorides with phenylboronic acids to give biaryl ketones. Also, the performance of the Pd(II) complexes in the traditional Suzuki–Miyaura cross-coupling reaction was evaluated. Both reactions were carried out under mild conditions.
 |
| | Fig. 1 Strategies for the acylative Suzuki–Miyaura cross coupling reaction. | |
 |
| | Scheme 1 Synthesis route to the NHC precursors 1a–c. | |
Results and discussion
Synthesis of the azolium salts and Pd(II) complexes
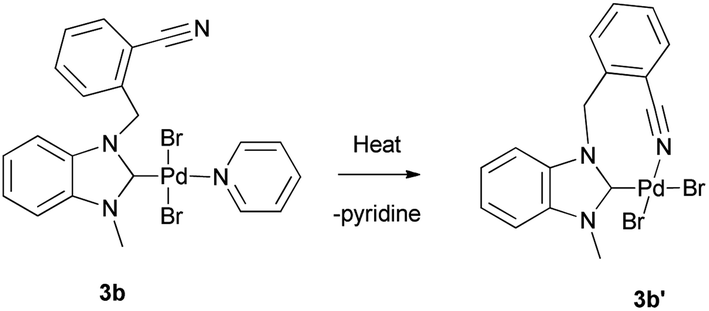
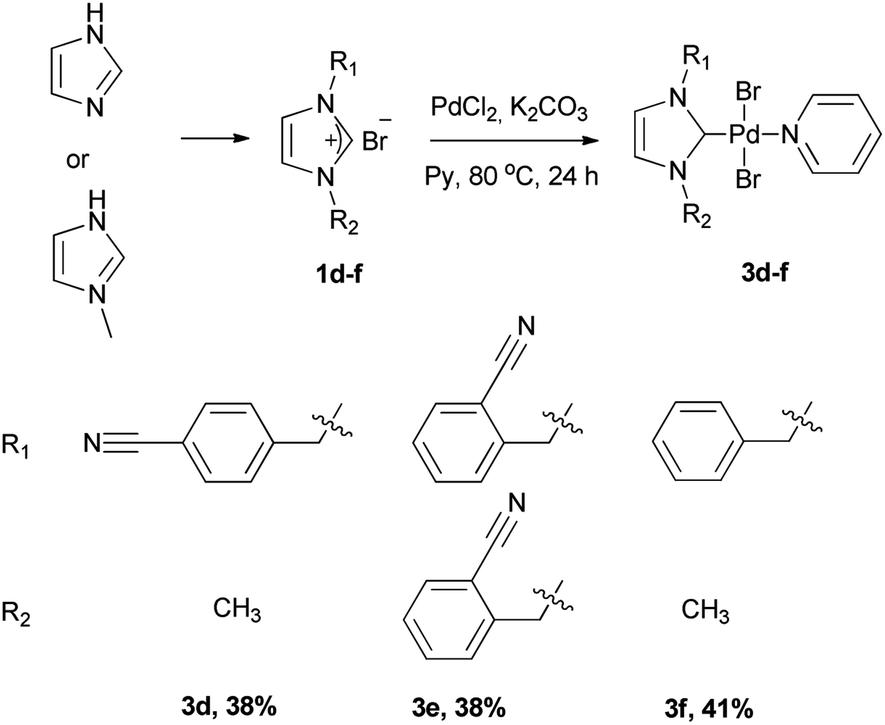
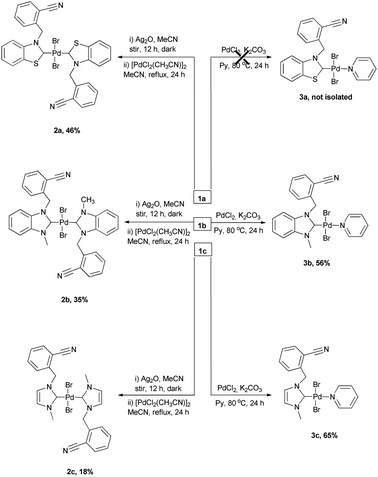
Electronic σ-donor properties of NHCs are significantly affected by the presense of the heterocyclic skeleton.13 The most preferred heterocyclic skeletons in catalyst chemistry are imidazole, benzimidazole and benzothiazole. To understand the effect of heterocyclic skeleton structure (benzothiazole, benzimidazole, and imidazole) on Pd(II) complex in the Suzuki–Miyaura cross-coupling reactions, a series of azolium salts bearing 2-nitrile substituted benzyl group and their palladium complexes were prepared. The azolium salts 1a–c were prepared in one-pot reaction consisting of the alkylation of benzothiazole, 1-methyl-benzimidazole and 1-methyl-imidazole, by 2-(bromomethyl)benzonitrile in toluene for 24 h (Scheme 1). The new azolium salts (1a–c) were obtained as white solids in 81, 85, 89% yields, respectively and they displayed good solubility in polar solvents. They were characterized by 1H and 13C NMR spectroscopy. The NMR spectrums of 1a–c were agreed with the proposed structures: C2–H resonance at δ = 10.80–9.33 ppm as a sharp singlet in the 1H NMR spectrums and these downfield signals demonstrate the formation of azolium salts. Also, the chemical shift of C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N sp hybrid carbon atoms of 1a–c were detected in the 13C NMR spectrums at δ 117.5, 117.4, and 117.0 ppm, respectively. The synthesis route to Pd(II) complexes (2a–c, 3a–c) were summarized in Scheme 2. The new (NHC)2PdBr2 complexes (2a–c) were synthesized in two-step sequence consisting of the transmetalation from the in situ formed NHC–Ag species.14 The PEPPSI type Pd(II) complexes 3b, 3c were obtained deprotonation of 1b, 1c with K2CO3 in pyridine, respectively. Unfortunately, 3a could not be isolated. The chelate complex 3b′ was obtained by heating of 3b to remove the pyridine ligand (Scheme 3). To understand the important of the position, the number of CN, and the absence of CN on NHC–Pd(II) complexes in catalytic activities, imidazolium salts (1d–f) were prepared. The complexes (3d–f) were prepared according to procedure 3b–c (Scheme 4). All the novel Pd complexes were obtained in high yields as air- and moisture-stable pale-yellow solids and were characterized by NMR and infrared spectroscopies. In the 1H-NMR spectra of 2a–c, the CH2 protons of 2a demonstrated a singlet peak, whereas the CH2 protons of 2b and 2c exhibited multiplet peaks. This difference in 2b and 2c may be due to an intermolecular interaction of hydrogen. The Pd(II) complexes (3b–f) displayed characteristic signals Pd–Ccarbene at δ = 164.8, and 154.3, 152.4, 152.6 and 152.5 ppm, respectively. The complexes exhibited almost identical FT–IR spectra indicating their similar structure. Due to the tensile vibrations of nitrile functionality (CN), a medium-density sharp band was observed at 2221–2227 cm−1. The presence of the –C
N sp hybrid carbon atoms of 1a–c were detected in the 13C NMR spectrums at δ 117.5, 117.4, and 117.0 ppm, respectively. The synthesis route to Pd(II) complexes (2a–c, 3a–c) were summarized in Scheme 2. The new (NHC)2PdBr2 complexes (2a–c) were synthesized in two-step sequence consisting of the transmetalation from the in situ formed NHC–Ag species.14 The PEPPSI type Pd(II) complexes 3b, 3c were obtained deprotonation of 1b, 1c with K2CO3 in pyridine, respectively. Unfortunately, 3a could not be isolated. The chelate complex 3b′ was obtained by heating of 3b to remove the pyridine ligand (Scheme 3). To understand the important of the position, the number of CN, and the absence of CN on NHC–Pd(II) complexes in catalytic activities, imidazolium salts (1d–f) were prepared. The complexes (3d–f) were prepared according to procedure 3b–c (Scheme 4). All the novel Pd complexes were obtained in high yields as air- and moisture-stable pale-yellow solids and were characterized by NMR and infrared spectroscopies. In the 1H-NMR spectra of 2a–c, the CH2 protons of 2a demonstrated a singlet peak, whereas the CH2 protons of 2b and 2c exhibited multiplet peaks. This difference in 2b and 2c may be due to an intermolecular interaction of hydrogen. The Pd(II) complexes (3b–f) displayed characteristic signals Pd–Ccarbene at δ = 164.8, and 154.3, 152.4, 152.6 and 152.5 ppm, respectively. The complexes exhibited almost identical FT–IR spectra indicating their similar structure. Due to the tensile vibrations of nitrile functionality (CN), a medium-density sharp band was observed at 2221–2227 cm−1. The presence of the –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N– group in the complexes was verified with the observation of ν(CN) bands between 1640 and 1539 cm−1.
N– group in the complexes was verified with the observation of ν(CN) bands between 1640 and 1539 cm−1.
 |
| | Scheme 2 Synthesis route to the NHC–Pd(II) complexes 2a–c and 3a–c. | |
 |
| | Scheme 3 Preparation of the NHC–Pd(II) chelate complex 3b′. | |
 |
| | Scheme 4 Synthesis route to the NHC–Pd(II) complexes 3d–f. | |
The crystal and molecular structure of 3c, 3e and 3f complexes
The crystal structures of three similar PEPPSI type Pd(II) complexes 3c, 3e, and 3f were determined using the X-ray single-crystal diffraction technique. Asymmetric unit of 3e contains a half molecule and the complete molecular structure is generated by the implementation of the crystallographic two-fold rotation whose axis bisects imidazole and pyridine rings as shown in Fig. 3. Asymmetric units of 3c and 3f are shown in Fig. 2 and 3, respectively. Selected bond lengths and angles are given in the figure captions for each compound. The coordination plane consisting of Pd, Ccarbene, Npyridine and two Br atoms is slightly distorted from square-planar geometry for all three compounds. These planes are almost planar in 3e and 3c. Deviation of Ccarbene from the mean plane through NHC ring in complex 3f is −0.109(2) Å. Due to positional disorder, the most prominent deviation from the coordination plane in complex 3f is observed for Br4A with a value of 0.553(4) Å. Some of Br–Pd–Br and Ccarbene–Pd–Npyridine bond angles are reliably distorted. Deviations from their linearity can be clearly observed for C1–Pd1–N1 with an angle of 173.4(2)° in 3f and Br–Pd–Br bond angles of 172.36(3)° and 174.26(3)° in 3e and 3f, respectively. The other bond angles in the coordination plane are almost perpendicular so as to adopt square planar geometry and in the range of 86.18(2)–93.82(2)° for all three complexes. Bond lengths around the metal center and N–Ccarbene bond lengths are similar to those reported for analogous: Pd–Br: 2.4159(17) Å, 2.4576(16) Å, Pd–Npyridine: 2.095(9) Å, Pd–Ccarbene: 1.959(10) Å, N–Ccarbene 1.335(13) Å, 1.355(14) Å.14 For of each complex, dihedral angles between the mean planes of coordination plane, carbene and pyridine rings, which are denoted by A, B and C respectively, are given in the figure captions.
 |
| | Fig. 2 Molecular structure of complex 3c with displacements ellipsoids plotted at the 30% probability level. Selected bond lengths (Å) and angles (°): Pd1–C1 1.946(12), Pd1–N1 2.113(10), Pd1–Br1 2.389(2), Pd1–Br2 2.393(2), N2–C1 1.357(14), N3–C1 1.346(15), Br1–Pd1–Br2 175.58(7), C1–Pd1–N1 178.4(5), N1–Pd1–Br1 91.6(3), N1–Pd1–Br2 91.8(3), C1–Pd1–Br1 88.6(3), C1–Pd1– Br2 87.9(3), N3–C1–N2 103.9(1). Dihedral angles: A/B 86.73(19)°, A/C 51.97(17)°, B/C 36.02(15)°. | |
 |
| | Fig. 3 Molecular structure of 3e (top) with displacements ellipsoids plotted at the 30% probability level. Symmetry related atoms are labeled with superscript i. Symmetry code: (i) 1 − x, y, 0.5 − z. Selected bond lengths (Å) and angles (°): Pd1–C1 1.946(6), Pd1–N1 2.117(6), Pd1–Br1 2.4465(5), N2–C1 1.342(4), Br1–Pd1–Br1i 172.36(3), C1–Pd1–N1 180.0, C1–Pd1–Br1i 86.18(2), N1–Pd1–Br1 93.82(2), N2–C1–N2i 106.6(5). Dihedral angles: A/B 76.97(19)°, A/C 44.5(3)°, B/C 58.53(19)°. The asymmetric unit of 3f (bottom) showing the atom-labelling scheme. Hydrogen atoms are omitted for clarity. Displacements ellipsoids are drawn at the 30% probability level. Selected bond lengths (Å) and angles (°): Pd1—C1 1.948(6), Pd2—C17 1.963(6), Pd1—N1 2.090(5), Pd2—N4 2.101(5), Pd1—Br1 2.4265(8), Pd1—Br2 2.4308(8), Pd2—Br3 2.4028(9), Pd2—Br4A 2.490(7), Pd2—Br4B 2.388(3), N3—C1 1.337(7), N2—C1 1.333(6), N5—C17 1.336(7), N6—C17 1.342(7), Br1—Pd1—Br2 174.26(3), C1—Pd1—N1 173.4(2), C17—Pd2—N4 176.0(2), C1—Pd1—Br1 86.72(17), C1—Pd1—Br2 89.51(16), N1—Pd1—Br1 91.60(13), N1—Pd1—Br2 92.63(13), N4—Pd2—Br3 92.78(14), C17—Pd2—Br3 91.13(17), C17—Pd2—Br4B 87.91(18), N4—Pd2—Br4B 88.36(14), N2—C1—N3 105.3(5), N5—C17—N6 104.7(5). Dihedral angles in molecule including Pd1: A/B 77.07(17)°, A/C 79.0(2)°, B/C 10.77(17)°. | |
Suzuki–Miyaura cross coupling reactions
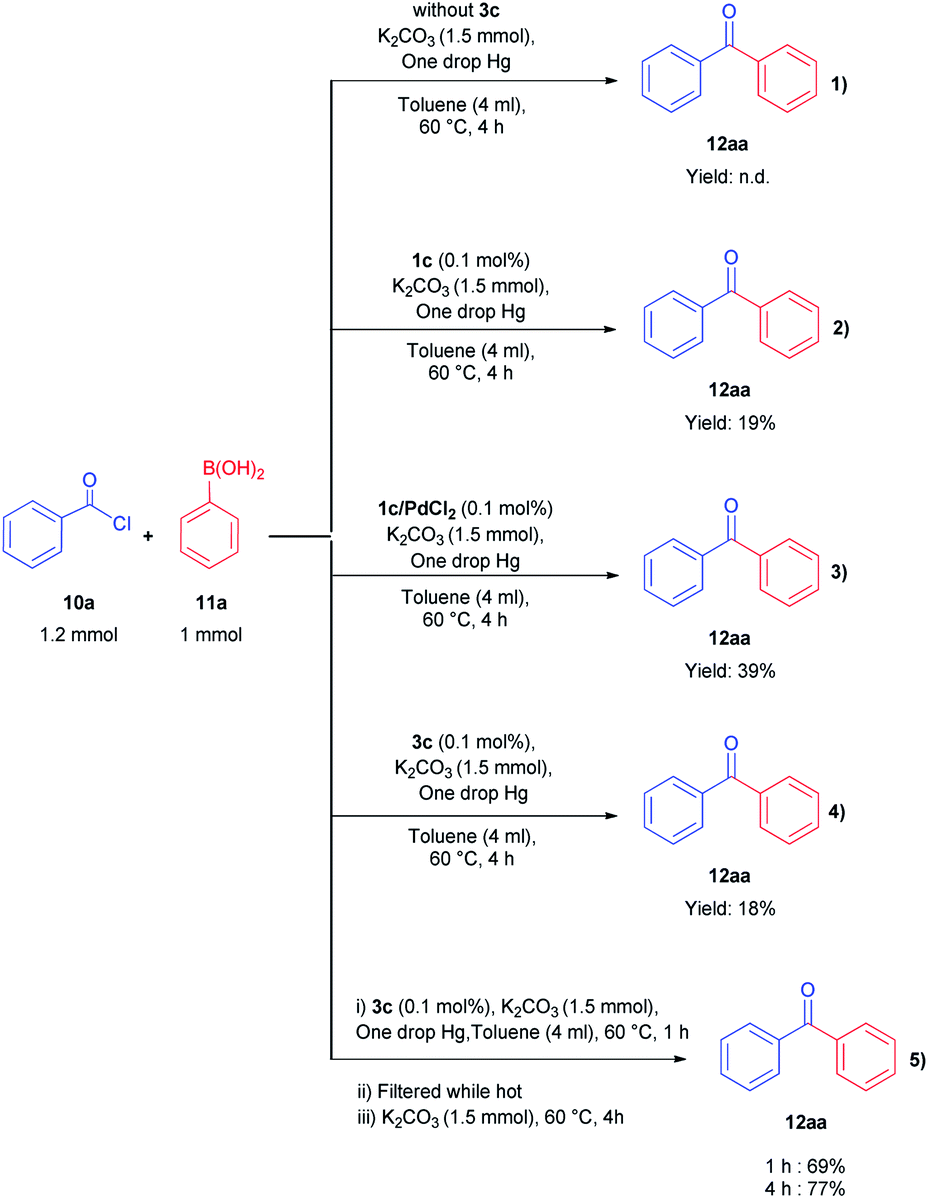
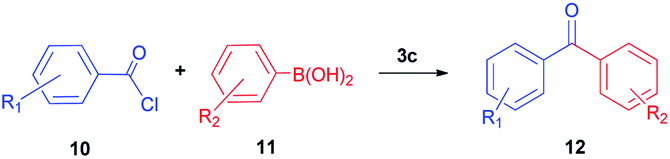
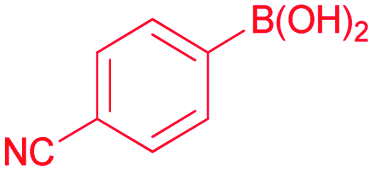
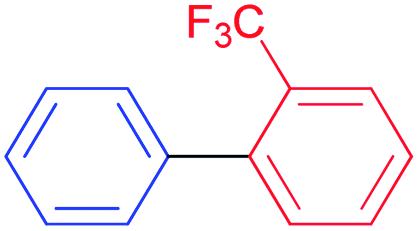
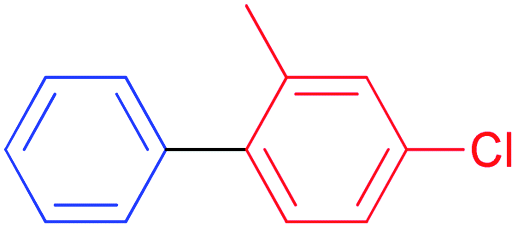
S–M cross-coupling reactions provide additional benefits in terms of designing safer processes. For instance, aromatic ketones can also be prepared effectively by cross-coupling reaction of arylboronic acid with acyl chloride. In the present study, the coupling reaction of phenylboronic acid and benzoyl chloride was selected as a model reaction to test the potential of NHC–Pd(II) complexes 2a–c and 3b–f, and the results are shown in Table 1. The reaction was performed in the presence of 2a–c and 3b–f as the catalyst and K2CO3 (1.5 mmol) in toluene (4 mL) at 60 °C under argon atmosphere for 4 h (Table 1, entries 1–10). Generally, PEPPSI type NHC–Pd(II) complexes 3b–f displayed higher catalytic activity than (NHC)2PdBr2 complexes 2a–c. The (NHC)2PdBr2 complexes 2b–c were demonstrated moderate conversions (Table 1, entries 1–3). The catalytic conversions of NHC–Pd(II) complexes (3b and 3c) were 88, and 96%, respectively (Table 1, entries 4 and 5). The chelate complex 3b′ had similar activity according to the non-chelated complex 3b (Table 1, entry 6). When the effect of the position and number of the CN group in the catalyst structure on the catalytic activity was examined, significant differences were observed. Complex 3d bearing 4-nitrile substituted benzyl group gave 69% yield (Table 1, entry 7). The increased number of nitrile groups in the catalyst did not cause a significant change in the catalytic activity (Table 1, entry 8). The complex 3e showed similar activity to the complex 3c. This showed that at least one CN group at position 2 in the ligand structure of the catalyst was sufficient for high activity. The complex 3f without CN group gave the lowest catalytic activity (Table 1, entry 8). The complex 3c exhibited better activity due to the high solubility in toluene when compared with the counterparts and reached 990 TON value. Unsatisfactorily, decreasing the catalyst loading to 0.05 mol% concluded 81% yield (Table 1, entry 11). In other reports, the authors mentioned that unidentified structures were detected in some catalytic reactions.11 We also observed the undesired product and we found that this occurs when using aqueous solvents. In aqueous solvents, the benzoyl chloride converts the benzoic acid and thus directly affects the reaction efficiency. Eventually, this undesired product formation was minimized in the catalytic reaction with toluene (Table 1, entries 12–14). Reducing or increasing the temperature did not affect the conversion well (Table 1, entries 15 and 16). Replacing K2CO3 with KOH, NEt3, NaOH, or NaHCO3 resulted in a decrease in the conversion, but Cs2CO3 resulted in a good yield (Table 1, entries 17–21). However, K2CO3 was selected as a base for further studies due to its commercial and green advantages over Cs2CO3. Also, an inert atmosphere was required for the reaction (Table 1, entry 23). We also carried out control experiments to understand the progress of the reaction (Scheme 5). Control experiments without catalyst displayed no activity in the absence of the catalyst (Scheme 5, entry 1). In addition, the ligand has minimal effect to the activity without metal center (Scheme 5, entry 2). According to the in situ experiment, the yield of the reaction was reduced (Scheme 5, entry 3). In order to determine the activity of the acylative Suzuki–Miyaura coupling reaction, mercury poisoning and hot filtration experiments were performed for precatalyst 3c using a one drop Hg (Scheme 5, entries 4 and 5). The mercury test showed a remarkable decrease in the conversion and only a 18% product formation was observed after 4 h. This result showed that the Pd nanoparticles were deactivated and the catalytic system took place in the heterogeneous nature. With the optimal reaction conditions determined, various arylboronic acids and benzoyl chlorides were investigated (Table 2). The reactions of benzoyl chloride with phenylboronic acid derivatives bearing 4-methyl, 2-methyl, and 2,4-dimethyl produced the corresponding products 4-methylbenzophenone, 2- methylbenzophenone, and 2,4-dimethylbenzophenone with yields of 88–94% (Table 2, entries 2–4). Accordingly, the reaction of phenylboronic acids bearing 4-Cl-2-CH3, 2-F-4-OCF3, 4-CN, and 4-CF3 substituents with benzoyl chloride gave the desired products 4-chloro-2-methylbenzophenone, 2-fluoro-4-triflouromethoxybenzophenone, 4-cyanobenzophenone, and 4- trifluoromethylbenzophenone in 92–98% yields (Table 2, entries 5–8). We also explored the reactions between 4-nitrobenzoyl chloride and arylboronic acids. Arylboronic acids consisting of different substituents, 4-Cl-2-CH3, 4-CF3, and 4-Br, gave the corresponding products 4-chloro-2-methyl-4′-nitrobenzophenone, 4-trifluoromethyl-4′-nitrobenzophenone, and 4-bromo-4′-nitrobenzophenone with good yields of 93, 97 and 89%, respectively (Table 2, entries 9–11). Lastly, the reaction of 4- nitrobenzoyl chloride with phenylboronic acid resulted in 99% yield (Table 2, entry 12).
Table 1 Optimization of reaction conditions for acylative Suzuki–Miyaura cross-coupling reaction of benzoyl chloride with phenylboronic acida

|
| Entry |
Cat. |
Base |
Solvent |
Yield (%) |
| Reaction conditions: benzoyl chloride (1.2 mmol), phenylboronic acid (1.0 mmol), base (1.5 mmol), solvent (4.0 mL), cat. 0.1 mol%, 60 °C, 4.0 h. Cat. 0.5 mol%. Cat. 0.05 mol%. T = 110 °C. Ambient temperature. Toluene (2.0 mL). Under air atmosphere. Isolated yields. |
| 1 |
2a |
K2CO3 |
Toluene |
64 |
| 2 |
2b |
K2CO3 |
Toluene |
72 |
| 3 |
2c |
K2CO3 |
Toluene |
81 |
| 4 |
3b |
K2CO3 |
Toluene |
88 |
| 5 |
3c |
K2CO3 |
Toluene |
96 |
| 6 |
3b′ |
K2CO3 |
Toluene |
86 |
| 7 |
3d |
K2CO3 |
Toluene |
69 |
| 8 |
3e |
K2CO3 |
Toluene |
85 |
| 9 |
3f |
K2CO3 |
Toluene |
58 |
| 10b |
3c |
K2CO3 |
Toluene |
97 |
| 11c |
3c |
K2CO3 |
Toluene |
81 |
| 12 |
3c |
K2CO3 |
DCM |
87 |
| 13 |
3c |
K2CO3 |
Acetonitrile |
44 |
| 14 |
3c |
K2CO3 |
Acetone |
53 |
| 15d |
3c |
K2CO3 |
Toluene |
61 |
| 16e |
3c |
K2CO3 |
Toluene |
25 |
| 17 |
3c |
Cs2CO3 |
Toluene |
99 |
| 18 |
3c |
KOH |
Toluene |
89 |
| 19 |
3c |
NEt3 |
Toluene |
30 |
| 20 |
3c |
NaOH |
Toluene |
85 |
| 21 |
3c |
NaHCO3 |
Toluene |
50 |
| 22f |
3c |
K2CO3 |
Toluene |
89 |
| 23g |
3c |
K2CO3 |
Toluene |
60 |
 |
| | Scheme 5 Control experiments; (1) without catalyst, (2) only ligand, (3) in situ (4) mercury poisoning and (5) hot filtration for acylative Suzuki–Miyaura cross-coupling reaction. Isolated yields. | |
Table 2 Acylative Suzuki–Miyaura cross-coupling reaction of acyl chlorides with arylboronic acidsa
For a practical availability, gram-scale reaction was performed using benzoyl chloride (10a) with phenylboronic acid (11a), and benzophenone (12aa) was obtained in an 88% isolated yield (Scheme 6).
 |
| | Scheme 6 Gram-scale synthesis of 12aaa. a Reaction conditions: 10a (1.68 g, 12.0 mmol), 11a (1.22 g, 10.0 mmol), 3c (0.1 mol%), K2CO3 (0.2 mg, 1.5 mmol), toluene (12.0 mL), 60 °C, 4 h, under argon atmosphere. Isolated yield. | |
Nolan and Szostak developed [Pd(IPr)(μ-Cl)Cl]2 catalyst for the acylative Suzuki–Miyaura cross-coupling reaction of aryl esters with phenylboronic acids.35a They revealed that the catalyst is the most reactive precatalysts among the other catalyst in the literature for the reaction. Nolan and co-workers also reported Suzuki–Miyaura cross-coupling reactions of amides mediated by [Pd(NHC)(allyl)Cl] precatalysts to form biaryls.35b
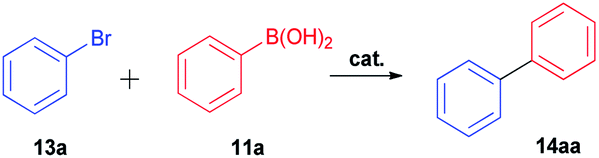
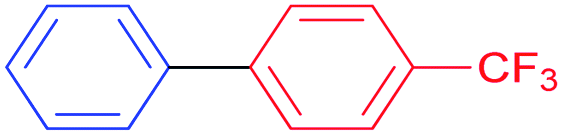
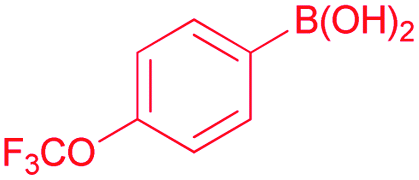
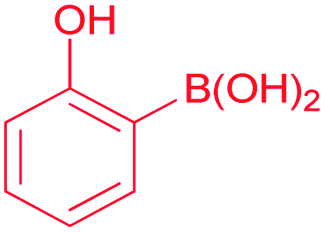
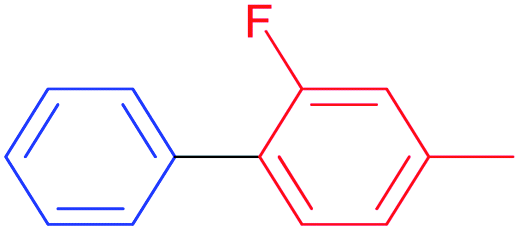
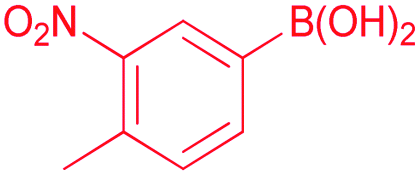
Also, we explored the catalytic activity of NHC–Pd(II) complexes to test their potential in the Suzuki–Miyaura coupling reaction of bromobenzene with phenylboronic acids (Table 3). The coupling reaction of bromobenzene and phenylboronic acid was selected as a benchmark reaction. The reaction was performed in the presence of 2a–c and 3b–f as the catalyst and KOH (0.5 mmol) in H2O/2-propanol (2 mL) at 82 °C open to the air for 0.5 h (Table 3). All the synthesized catalysts (2a–c, 3b–f) were tested in the reaction of bromobenzene with phenylboronic acid (Table 3, entries 1–9). Except for the complex 3f, there was no significant difference between catalysts (Table 3, entry 9). The catalytic result of the complex 3f demonstrated that the effect of the nitrile group was evident in the catalytic reaction. 3c was decided as a catalyst for the Suzuki–Miyaura cross-coupling reaction of bromobenzene with phenylboronic acid. Reducing the catalyst loading also gave the satisfactory yield (Table 3, entry 10). The use of only water or 2-propanol as solvent negatively affected the conversion (Table 3, entries 11 and 12). No product formation was observed in the absence of the catalyst (Table 3, entry 13). The formation of biphenyl was reduced in the presence of different bases such as NaHCO3, NaOH, KOtBu, and K2CO3, while the emulative yield was observed with Cs2CO3 (Table 3, entries 14–18). Decreasing the temperature resulted in lower conversions (Table 3, entries 19 and 20). Assessment of the substrate scope of the reaction of various phenylboronic acids with bromobenzene, 4-bromotoluene, and 4-bromoanisole was performed under the optimized reaction conditions (Table 4). Numerous arylboronic acids bearing electron-withdrawing or electron-donating substituents at the para-position such as 4-CH3, 4-tert-butyl, 4-OH, 4-CN, 4-F, 4-CF3, and 4-OCF3 were converted into the corresponding biphenyls namely 4-methyl-biphenyl, 4-(tert-butyl)-biphenyl, 4-hydroxy-biphenyl, 4-cyano-biphenyl, 4-fluorobiphenyl, 4-(trifluoromethyl)-biphenyl, and 4-(trifluoromethoxy)-biphenyl in high yields (Table 4, entries 2–8). But, ortho-substituted phenylboronic acids like 2-OH, 2-Me and 2-CF3 gave the desired products 2-methyl-biphenyl, 4-hydroxy-biphenyl, and 2-(trifluoromethyl)-biphenyl with a average isolated yields (Table 4, entries 9–11). The double-substituted phenylboronic acids such as 4-Cl-2-Me, 2,4-dimethyl, 4-F-3-Me, 2-F-4-Me, and 4-Me-3-NO2 resulted in good yields in the coupling reactions with the products of 4-chloro-2-methyl-biphenyl, 2,4-dimethyl-biphenyl, 4-fluoro-3-methyl-biphenyl, 2-fluoro-4-methyl-biphenyl, and 4-methyl-3-nitro-biphenyl (Table 4, entries 12–16). Furthermore, different aryl bromides were also examined. Aryl bromides consisting of electron-withdrawing substituents, 4-CH3 and 4- OCH3, gave the corresponding products 4-methyl-biphenyl and 4-methoxy-biphenyl with good yields of 77 and 70%, respectively (Table 4, entries 17, 18). For a practical availability, gram-scale reaction was performed using bromobenzene (13a) with phenylboronic acid (11a), and biphenyl was obtained in an 86% isolated yield (Scheme 7).
Table 3 Suzuki–Miyaura cross-coupling reaction of bromobenzene with phenylboronic acida

|
| Entry |
Cat. |
Base |
Solvent |
Yield (%) |
| Reaction conditions: bromobenzene (1.0 mmol), phenylboronic acid (1.0 mmol), KOH (0.5 mmol), cat. (0.2 mol%), 82 °C, 2 mL solvent or 0.5 mL 2-propanol + 1.5 mL H2O, 0.5 h, under air. Cat. 0.1 mol%. 60 °C. 24 °C. Isolated yields. |
| 1 |
2a |
KOH |
H2O/2-propanol |
80 |
| 2 |
2b |
KOH |
H2O/2-propanol |
71 |
| 3 |
2c |
KOH |
H2O/2-propanol |
78 |
| 4 |
3b |
KOH |
H2O/2-propanol |
90 |
| 5 |
3b′ |
KOH |
H2O/2-propanol |
88 |
| 6 |
3c |
KOH |
H2O/2-propanol |
94 |
| 7 |
3d |
KOH |
H2O/2-propanol |
79 |
| 8 |
3e |
KOH |
H2O/2-propanol |
89 |
| 9 |
3f |
KOH |
H2O/2-propanol |
53 |
| 10b |
3c |
KOH |
H2O/2-propanol |
85 |
| 11 |
3c |
KOH |
2-Propanol |
68 |
| 12 |
3c |
KOH |
H2O |
72 |
| 13 |
— |
KOH |
H2O/2-propanol |
2 |
| 14 |
3c |
NaHCO3 |
H2O/2-propanol |
45 |
| 15 |
3c |
Cs2CO3 |
H2O/2-propanol |
81 |
| 16 |
3c |
NaOH |
H2O/2-propanol |
64 |
| 17 |
3c |
KOtBu |
H2O/2-propanol |
62 |
| 18 |
3c |
K2CO3 |
H2O/2-propanol |
73 |
| 19c |
3c |
KOH |
H2O/2-propanol |
68 |
| 20d |
3c |
KOH |
H2O/2-propanol |
9 |
Table 4 Suzuki–Miyaura cross-coupling reaction of aryl bromides with arylboronic acidsa
 |
| | Scheme 7 Gram-scale synthesis of 14aaa. a Reaction conditions: 13a (1.57 g, 10.0 mmol), 11a (1.22 g, 10.0 mmol), 3c (0.1 mol%), KOH (28 mg, 0.5 mmol), 0.5 mL 2-propanol + 1.5 mL H2O, 82 °C, under air atmosphere. Isolated yield. | |
Kajetanowicz et al. developed catalysts possessing quinone moieties in the pyridine ligand for the Suzuki–Miyaura cross-coupling reaction of phenylboronic acid with 3,5-dimethoxybromobenzene.36 They drew attention to the similarity of the steric and electronic properties of the Organ's Pd–PEPPSI catalysts.
Scanning Electron Microscopy (SEM), energy dispersive X-ray spectroscopy (EDS) and metal mapping analyses were performed to get further insights into the mechanism of the Suzuki–Miyaura cross coupling reactions (Fig. 4 and 5). Au was coated on the surface of the Pd nanoparticles to prevent the specimen from charging during analysis. The black precipitates were collected by filtration at the end of the catalytic reactions and washed with plenty of water. The Pd nanoparticles appeared as cluster with the sizes ranging from 45 to 60 nm for both reaction. EDS and metal mapping images of the black precipitates clearly proven the presence of Pd atom (Fig. 5). Also, the images displayed N and C atoms which are most probably attributed to the NHC ligand. The other atoms like K and O are impurities remaining from the salts (KOH or K2CO3).
 |
| | Fig. 4 SEM images of Pd nanoparticles for acylative Suzuki–Miyaura coupling reaction of benzoyl chloride with phenylboronic acid (top) and Suzuki–Miyaura cross-coupling reaction of phenylboronic acid with bromobenzene (bottom). | |
 |
| | Fig. 5 EDS and metal mapping images of Pd nanoparticles for acylative Suzuki–Miyaura coupling reaction of benzoyl chloride with phenylboronic acid (top) and Suzuki–Miyaura cross-coupling reaction of phenylboronic acid with bromobenzene (bottom). | |
The possible mechanisms of the Suzuki–Miyaura coupling reactions are summarized in Scheme 8. Both reaction mechanisms conducted through the classical Suzuki–Miyaura C–C coupling reaction mechanism, which includes the preactivation, oxidative addition, transmetalation, and reductive elimination, respectively. Pre-activation occurs with the effect of base, solvent or temperature, and the pre-activation of the catalyst is the state with the least energy profile.32 The steric hindrance from NHC ligands plays an important role in the activation process of the catalyst.35b The leaving group (halogen) and ligands are more crucial than metal center in the preactivation. Since bromine is an easily leaving group, the preactivation process is faster.35c However, there may be slight differences in the mechanism due to optimization conditions.16 In the cross-coupling reaction, the possibility of attachment of the hydroxyl group to the palladium center is high due to the use of hydroxyl-containing base (KOH) and water as solvent. In our last study, we supported this attachment with the computational study.15 But, in the acylative cross-coupling reaction, as we have stated before, there must not be any water in the reaction conditions to avoid by-product formation, which reduces the possibility of Pd–OH bond formation. Of course, the formation of Pd–CO3 interaction as an intermediate is expected due to K2CO3.33 The main purpose of the base in Suzuki–Miyaura cross-coupling reactions is to break the Pd–halide bond by increasing the reactivity of boronic acid in the transmetalation step.34
 |
| | Scheme 8 Proposed mechanisms of Suzuki–Miyaura acylative-coupling reaction and cross-coupling reaction with 3c. | |
We supported removal of the pyridine group during the catalytic cycle by thermogravimetric analysis (TGA) (ESI†). The TGA spectrum of the complex 3c showed a band around 210 °C which is attributed to the pyridine group.27 This band did not appear in the TGA spectrum of the nanoparticle of 3c, indicating the absence of pyridine in the nanostructure (Fig. S60 and S61†). In addition, Dyson et al. reported that the nitrile group has two important roles, which are forming a protective group around the nanoparticle and helping to stabilize the active Pd(II) catalyst.28 Both TGA spectrum displayed a band around 305 °C which are attributed to the NHC ligand. This demonstrated that the NHC ligand remains stable in the Pd nanoparticles. Also, the FT-IR spectrum of the Pd nanoparticles depicted the presence of the nitrile group (Fig. S62†).
NHC ligands with electron-rich and steric groups can make subtle changes to the electronic and steric effect of the palladium center.29 The advantages of the electronic and steric effects arising from NHC ligands to the complex are as follows: remarkable activity in the catalytic process, formation of stable complexes against air and moisture, uniquely strong σ-donor feature, wide range scope.30 Pd–PEPPSI–IPr complexes have also been good alternatives to classical Pd–PEPPSI–NHC complexes in C–C and C–N bond formation reactions.31 The steric affects of IPr ligands would improve the efficiency of reductive elimination step. NHC ligands are an influencing factor in determining which step in the cycle is rate limiting.31c Bulky and flexible NHC ligands such as IPr can overcome several limitations of cross-coupling reactions.31d The most important classes of the PEPPSI type Pd catalysts containing NHC ligand was developed by Organ.37 Our Pd-PEPPSI catalysts with nitrile moiety offer comparable results to conventional catalysts in the literature for Suzuki–Miyaura cross-coupling reactions.
Conclusions
In conclusion, a series of CN-containing azolium salts and their palladium complexes were synthesized as the model compounds to systematically study the effect of CN substituents on the both Suzuki–Miyaura cross-coupling reactions. (NHC)2PdBr2 (2a–c) and PEPPSI type (NHC)Pd(II) (3b–f) complexes were used to give C(acyl)–C(sp2) and C(sp2)–C(sp2) Suzuki–Miyaura cross-coupling reactions to yield the corresponding carbonyl compounds and biphenyls, respectively. By introducing the CN substituents onto the phenylene ring of NHC in the Pd complexes, both activity and stability of the resulting nanocatalysts are greatly enhanced. The complex (3c) which has imidazole as the NHC skeleton, and 2-CN benzyl and methyl as the side groups generated higher activity in both catalytic reactions. The SEM analysis, EDS and metal mapping analysis, mercury drop tests and poison experiments of the catalytic solution showed that (NHC)Pd was involved in catalysis. The synthesis of transition metal NHC complexes containing CN group and their different catalytic applications are currently under investigation.
Experimental
X-ray crystallography
X-ray single-crystal diffraction data for complexes 3e, 3f and 3c were collected at 294(2) K on a Rigaku-Oxford Xcalibur diffractometer with an Eos-CCD detector using graphite-monochromated MoKα radiation (λ = 0.71073 Å). Details of the data collection conditions and refinement processes are summarized in Table S1, as ESI†. For each compound, data reduction and analytical absorption corrections were performed using CrysAlisPro 1.171.40.67a software.17 The structure was solved with the ShelXT18 solution program using dual methods and by using Olex2 (ref. 19) as the graphical interface. The model was refined with ShelXL2018/3 (ref. 20) using full matrix least squares minimization on F2. All non-hydrogen atoms were refined as isotropically. Hydrogen atom positions were calculated geometrically and refined using the riding model, fixing the aromatic C–H distances at 0.93 Å, methylene C–H distances at 0.97 Å, methyl C–H distances at 0.96 Å. Uiso(H) values were set to 1.2 Ueq (1.5 Ueq for the methyl group) of the parent atom. Atom Br4 in complex 3f is disordered over two sets of sites, with a refined occupancy ratio of 0.661(13)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.339(13). CCDC 2071690, 2071692, 2071693 contain the supplementary crystallographic data for 3e, 3f and 3c, respectively.
:0.339(13). CCDC 2071690, 2071692, 2071693 contain the supplementary crystallographic data for 3e, 3f and 3c, respectively.
Materials
Unless otherwise noted, all operations were performed without taking precautions to exclude air and moisture. The glass equipment was heated under vacuum in order to remove oxygen and moisture and then they were filled with argon. Starting compounds and reagents were obtained from Merck, Fluka, Alfa Aesar and Acros Organics; pyridine, PdCl2 were obtained from Alfa Aesar and dichloromethane, acetonitrile, ethanol, diethyl ether, toluene, pyridine were obtained from Merck and Ridel de Haen. Catalytic reactions for Suzuki–Miyaura cross-coupling reactions were carried out under argon gas on Carousel 12 Plus Reaction Station system. 1H and 13C NMR spectra were recorded on a Varian AS 400 Mercury instrument. FTIR spectra were recorded on a PerkinElmer Spectrum 100 series. Scanning Electron Microscopy (SEM), energy dispersive X-ray spectroscopy (EDS) and metal mapping analyses were collected on a Thermo Scientific Apreo S.
Synthesis of compound 1a. Benzothiazole (1.00 g, 7.39 mmol) was dissolved in 5 mL of toluene and 2-bromobenzonitrile (1.45 g, 7.39 mmol) was added to boil under reflux for 24 h. The precipitate is then filtered off and washed with diethyl ether. Dried under vacuum to yield. Yield = 81%, 1.98 g. 1H NMR (400 MHz, DMSO-d6): δ 10.8 (s, 1H, S–CH–N), 8.59 (m, 1H, J = 2 Hz, Ar-H), 8.17 (m, 1H, Ar-H), 7.99 (d, J = 7.6 Hz, 1H, Ar-H), 7.86 (m, 2H, Ar-H), 7.69 (t, J = 8 Hz, 1H, Ar-H), 7.67 (t, J = 7.6 Hz, 1H, Ar-H), 7.31 (d, J = 8 Hz, 1H, Ar-H), 6.40 (s, 2H, N–CH2). 13C NMR (100 MHz, DMSO-d6): δ 167.6, 140.7, 136.5, 134.5, 134.4, 132.2, 130.1, 128.9, 126.1, 117.5, 111.1, 54.1. IR, νmax (cm−1) (CH2Cl2): 3367, 3091, 3061, 3021, 2978, 2939, 2897, 2221, 1987, 1682, 1594, 1577, 1493, 1462, 1448, 1426, 1293, 1209, 1175, 1094, 1080, 1032, 900, 834, 792, 774, 760, 751, 730, 643, 597, 581, 512, 452, 425, 404.
Synthesis of compound 1b. Prepared according to procedure 1a using 1-methylbenzimidazole (1.00 g, 7.56 mmol) and 2-bromobenzonitrile (1.48 g, 7.56 mmol). Yield = 85%, 2.12 g. 1H NMR (400 MHz, CDCl3): δ 9.84 (s, 1H, NCHN), 8.08 (d, J = 8 Hz, 1H, Ar-H), 7.98 (d, J = 7.6 Hz, 1H, Ar-H), 7.88 (d, J = 8 Hz, 1H, Ar-H), 7.69 (m, 3H, Ar-H), 7.59 (t, J = 7.6 Hz, 1H, Ar-H), 7.45 (d, J = 7.6 Hz, 1H, Ar-H), 6.03 (s, 2H, N–CH2), 4.13 (s, 3H, N–CH3). 13C NMR (100 MHz, CDCl3): δ 144.0, 137.5, 134.4, 134.2, 132.4, 131.4, 129.9, 129.4, 127.4, 127.2, 117.4, 114.4, 113.9, 111.3, 48.6, 33.9. IR, νmax (cm−1) (CH2Cl2): 3810, 3430, 3137, 3096, 3006, 2979, 2925, 2904, 2843, 2772, 2652, 2584, 2431, 2223, 1967, 1920, 1873, 1807, 1784, 1729, 1696, 1610, 1599, 1570, 1485, 1444, 1366, 1345, 1292, 1277, 1216, 1209, 1165, 1128, 1088, 1047, 1028, 1008, 989, 964, 906, 847, 787, 774, 759, 746, 686, 609, 582, 559, 448, 420.
Synthesis of compound 1c. Prepared according to procedure 1a using 1-methylimidazole (1.00 g, 12.0 mmol) and 2-bromobenzonitrile (2.38 g, 12.0 mmol). Yield = 89%, 3.02 g. 1H NMR (400 MHz, CDCl3): δ 10.2 (s, 1H, NCHN), 7.92 (d, J = 7.6 Hz, 1H, Ar-H), 7.71 (s, 1H, Ar-H), 7.59 (m, 2H, Ar-H), 7.51 (s, 1H, NCHCHN), 7.43 (t, J = 7.6 Hz, 1H, NCHCHN), 5.80 (s, 2H, N–CH2), 4.00 (s, 3H, N–CH3). 13C NMR (100 MHz, CDCl3): δ 182.2, 137.4, 136.5, 134.3, 133.4, 131.1, 130.1, 124.3, 122.3, 117, 0, 112.0, 50.8, 37.0. IR, νmax (cm−1) (CH2Cl2): 3487, 3136, 3084, 3030, 2950, 2919, 2885, 2843, 2407, 2227, 2000, 1740, 1657, 1599, 1572, 1561, 1490, 1451, 1366, 1339, 1298, 1285, 1212, 1169, 1162, 1095, 1082, 962, 887, 866, 824, 775, 652, 622, 557, 477, 447, 404.
Synthesis of compound 1d. Prepared according to procedure 1a using 1-methylimidazole (1.00 g, 12.0 mmol) and 4-bromobenzonitrile (2.38 g, 12.0 mmol). Yield = 59%, 2.00 g. 1H NMR (400 MHz, DMSO-d6): δ 9.33 (s, 1H, NCHN), 7.89 (d, J = 7.6 Hz, 2H, Ar-H), 7.83 (s, 1H, NCHCHN), 7.76 (s, 1H, NCHCHN), 7.60 (d, J = 8 Hz, 2H, Ar-H), 5.57 (s, 2H, N–CH2), 3.86 (s, 3H, N–CH3). 13C NMR (100 MHz, DMSO-d6): δ 166.6, 140.4, 138.8, 133.4, 132.3, 130.2, 129.5, 128.9, 126.0, 118.8, 117.7, 112.1, 54.9. IR, νmax (cm−1) (CH2Cl2): 3418, 3060, 2965, 2910, 2221, 1599, 1510, 1483, 1454, 1379, 1357, 1314, 1284, 1270, 1207, 1141, 1116, 1053, 1029, 982, 913, 793, 762, 713, 674, 604, 552, 450, 432.
Synthesis of compound 1e. Imidazole (1.00 g, 14.68 mmol) was dissolved in 5 mL ethanol and mixed with NaOH for 30 min. 2-Bromobenzonitrile (5.76 g, 29.36 mmol) was then added to the mixture and boiled under reflux for 24 hours. The precipitate is then filtered and washed with diethyl ether. It was dried under vacuum. Yield = 72%, 4.02 g. 1H NMR (400 MHz, DMSO-d6): δ 9.41 (s, 1H, NCHN), 7.95 (d, J = 10.8 Hz, 2H, Ar-H), 7.87 (d, J = 1.6 Hz, 2H, Ar-H), 7.78 (t, J = 5.2 Hz, 2H, Ar-H), 7.61 (t, J = 7.6 Hz, 2H, Ar-H), 7.48 (d, J = 7.6 Hz, 2H, NCHCHN), 5.74 (s, 4H, N–CH2). 13C NMR (100 MHz, DMSO-d6): δ 138.3, 137.9, 134.5, 134.1, 130.1, 129.7, 123.9, 117.3, 111.3, 50.9. IR, νmax (cm−1) (CH2Cl2): 3792, 3414, 3168, 3129, 3044, 3022, 2861, 2222, 1756, 1643, 1617, 1599, 1566, 1487, 1438, 1348, 1303, 1288, 1213, 1182, 1170, 1095, 1045, 1024, 978, 953, 883, 858, 819, 765, 700, 638, 557, 467, 465.
Synthesis of compound 1f. Prepared according to procedure 1a using 1-methylimidazole (1.00 g, 12.0 mmol) and benzoyl bromide (2.25 g, 12.0 mmol). Yield = 73%, 3.23 g. 1H NMR (400 MHz, DMSO-d6): δ 9.41 (s, 1H, NCHN), 7.85 (d, J = 1.6 Hz, 1H, NCHCHN), 7.75 (d, J = 1.2 Hz, 1H, NCHCHN), 7.45 (d, J = 6.4 Hz, 2H, Ar-H), 7.40 (d, J = 7.2 Hz, 2H, Ar-H), 7.35 (m, 1H, Ar-H), 5.47 (s, 2H, N–CH2), 3.86 (s, 3H, N–CH3). 13C NMR (100 MHz, DMSO-d6): δ 137.0, 135.4, 129.4, 129.1, 128.8, 124.4, 122.7, 52.1, 36.4. IR, νmax (cm−1) (CH2Cl2): 3418, 3148, 3090, 2063, 1626, 1573, 1562, 1497, 1456, 1427, 1388, 1362, 1335, 1208, 1161, 1108, 1083, 1029, 1002, 853, 822, 722, 699, 662, 622, 466.
Synthesis of complex 2a. In balloon, 1a (0.50 g, 1.51 mmol) and Ag2O (0.18 g, 0.76 mmol) under argon gas was suspend in acetonitrile (5 mL) and stirred at room temperature for 12 h protected from light. Then, [PdCl2(CH3CN)]2 (0.19 g, 0.76 mmol) was added over and the reflux 24 h. The solid residue remained was washed with diethyl ether. Then dried under vacuum. Yield = 46%, 0.53 g. 1H NMR (400 MHz, CDCl3): δ 7.71 (d, J = 7.2 Hz, 2H, Ar-H), 7.51 (t, J = 8 Hz, 2H, Ar-H), 7.46 (d, J = 8 Hz, 2H, Ar-H), 7.39 (t, J = 7.6 Hz, 2H, Ar-H), 7.24 (m, 4H, Ar-H), 7.17 (t, J = 7.6 Hz, 2H, Ar-H), 6.93 (d, J = 7.6 Hz, 2H, Ar-H), 5.38 (s, 4H, N–CH2). 13C NMR (100 MHz, CDCl3): δ 170.3, 138.8, 136.3, 133.6, 133.1, 128.4, 127.3, 126.7, 123.8, 122.8, 122.5, 117.1, 111.2, 110.9, 43.9. IR, νmax (cm−1) (CH2Cl2): 3791, 3663, 3347, 3068, 2930, 2224, 2190, 1954, 1905, 1767, 1692, 1588, 1561, 1483, 1471, 1447, 1424, 1358, 1352, 1325, 1305, 1285, 1260, 1205, 1197, 1183, 1092, 1044, 1019, 979, 957, 931, 884, 851, 809, 766, 751, 717, 693, 660, 622, 582, 557, 545, 502, 482, 464, 443, 429.
Synthesis of complex 2b. Prepared according to procedure 2a using 1b (0.50 g, 1.52 mmol), Ag2O (0.18 g, 0.76 mmol) and [PdCl2(CH3CN)]2 (0.19 g, 0.76 mmol). Yield = 35%, 0.40 g. 1H NMR (400 MHz, DMSO-d6): δ 8.01–7.96 (dd, J = 7.6 Hz, 2H, Ar-H), 7.84–7.76 (dd, J = 8 Hz, 2H, Ar-H), 7.44 (m, 10H, Ar-H), 7.17 (m, 2H, Ar-H), 6.36–6.32, 6.12–6.08 (dd, J = 16.8 MHz, 2H, N–CH2), 6.42 (s, 2H, N–CH2), 4.31 (d, J = 8.8 Hz, 6H, N–CH3). 13C NMR (100 Hz, DMSO-d6): δ 139.4, 139.2, 134.6, 134.5, 133.9, 133.9, 133.8, 133.7, 129.4, 129.2, 128.8, 128.6, 128.3, 124.8, 124.7, 124.3, 124.2, 118.5, 117.8, 117.7, 112.3, 111.9, 111.6, 111.4, 111.2, 111.1, 49.8, 49.6, 35.6, 35.4. IR, νmax (cm−1) (CH2Cl2): 3436, 2920, 2294, 2224, 1737, 1599, 1484, 1452, 1413, 1355, 1211, 1107, 1030, 986, 824, 798, 762, 559, 447.
Synthesis of complex 2c. Prepared according to procedure 2a using 1c (0.50 g, 1.80 mmol), Ag2O (0.21 g, 0.90 mmol) and [PdCl2(CH3CN)]2 (0.23 g, 0.90 mmol). Yield = 18%, 0.25 g. 1H NMR (400 MHz, DMSO-d6): δ 7.95–7.91 (dd, J = 7.6 Hz, 2H, Ar-H), 7.67 (t, J = 7.6 Hz, 2H, Ar-H), 7.51 (m, 6H, Ar-H), 7.35–7.29 (dd, J = 8 Hz, 2H, Ar-H), 5.88–5.84, 5.77–5.73 (dd, J = 16.0 Hz, 2H, N–CH2), 5.83 (s, 2H, N–CH2), 3.99 (d, J = 9.6 Hz, 6H, N–CH3). 13C NMR (100 MHz, DMSO-d6): δ 179.3, 139.9, 134.0, 133.7, 129.5, 129.0, 124.9, 123.9, 117.7, 111.3, 51.7, 37.9. IR, νmax (cm−1) (CH2Cl2): 3317, 3105, 2926, 2222, 1676, 1602, 1571, 1470, 1244, 1218, 1199, 1135, 1079, 943, 762, 732, 689, 657, 458.
Synthesis of complex 3b. In balloon, 1b (0.50 g, 1.52 mmol) under argon gas was pyridine (3 mL) dissolved. Then, PdCl2 (0.27 g, 1.52 mmol) and K2CO3 (1.05 g, 7.62 mmol) was added and refluxed overnight. The solid residue remained was washed with diethyl ether. Then dried under vacuum. Yield = 56%, 0.51 g. 1H NMR (400 MHz, CDCl3): δ 9.04 (d, J = 1.6 Hz, 2H, Py-H), 7.78 (m, 2H, Py-H), 7.71 (d, J = 8.8 Hz, 1H, Py-H), 7.50 (t, J = 8 Hz, 1H, Ar-H), 7.42 (m, 2H, Ar-H), 7.37 (m, 2H, Ar-H), 7.31 (t, J = 8 Hz, 1H, Ar-H), 7.20 (m, 1H, Ar-H), 7.11 (m, 1H, Ar-H), 6.36 (s, 2H, N–CH2), 4.40 (s, 3H, N–CH3). 13C NMR (100 MHz, CDCl3): δ 164.8, 152.6, 151.9, 138.5, 138.1, 135.4, 133.9, 133.4, 132.9, 129.5, 128.6, 124.6, 123.6, 117.3, 111.4, 110.7, 110.3, 50.4, 35.5. IR, νmax (cm−1) (CH2Cl2): 3381, 3059, 2926, 2223, 1692, 1603, 1539, 1499, 1446, 1403, 1348, 1295, 1253, 1190, 1153, 1126, 1098, 1070, 1011, 919, 803, 760, 691, 668, 558, 439.
Complex 3b′. Yield = 32%, 0.41 g. 1H NMR (400 MHz, CDCl3): δ 7.69 (d, J = 7.2 Hz, 1H, Ar-H), 7.50 (t, J = 7.2 Hz, 1H, Ar-H), 7.37 (t, J = 7.6 Hz, 2H, Ar-H), 7.12 (t, J = 7.6 Hz, 1H, Ar-H), 7.03 (m, 2H, Ar-H), 7.93 (d, J = 7.6 Hz, 1H, Ar-H), 5.31 (s, 2H, N–CH2), 3.49 (s, 3H, N–CH3). 13C NMR (100 MHz, CDCl3): δ 154.5, 153.3, 140.1, 133.4, 132.9, 130.1, 128.6, 128.3, 128.2, 124.9, 121.8, 121.6, 117.3, 111.3, 108.1, 107.7, 42.7, 29.7, 27.4. IR, νmax (cm−1) (CH2Cl2): 3527, 3444, 3379, 3059, 2926, 2222, 1691, 1601, 1541, 1499, 1401, 1341, 1295, 1254, 1210, 1191, 1154, 1100, 1068, 1013, 920, 796, 761, 744, 692, 658, 442.
Synthesis of complex 3c. Prepared according to procedure 3b using 1c (0.50 g, 1.79 mmol), PdCl2 (0.32 g, 1.79 mmol) and K2CO3 (1.24 g, 8.99 mmol). Yield = 65%, 0.63 g. 1H NMR (400 MHz, CDCl3): δ 9.00–8.99 (dd, J = 1.6 Hz, 2H, Py-H), 7.94 (d, J = 7.6 Hz, 1H, Ar-H), 7.75 (m, 2H, Py-H), 7.60 (dt, J = 7.6 Hz, 1H, Py-H), 7.45 (dt, J = 7.6 Hz, 1H, Ar-H), 7.33 (m, 2H, Ar-H), 6.97–6.94 (dd, J = 2 Hz, 2H, N–CHCH–N), 5.97 (s, 2H, N–CH2), 4.14 (s, 3H, N–CH3). 13C NMR (100 MHz, CDCl3): δ 154.3, 152.5, 150.1, 139.0, 138.4, 137.9, 133.4, 132.9, 130.9, 128.9, 124.9, 124.7, 123.9, 122.2, 112.2, 52.4, 38.7. IR, νmax (cm−1) (CH2Cl2): 3437, 3161, 3127, 3104, 2944, 2219, 1706, 1640, 1603, 1567, 1484, 1468, 1445, 1415, 1366, 1327, 1293, 1280, 1247, 1211, 1193, 1148, 1068, 1046, 1018, 943, 795, 767, 731, 684, 643, 552, 447, 431.
Synthesis of complex 3d. Prepared according to procedure 3b using 1d (0.50 g, 1.79 mmol), PdCl2 (0.32 g, 1.79 mmol) and K2CO3 (1.24 g, 8.99 mmol). Yield = 38%, 0.36 g. 1H NMR (400 MHz, DMSO-d6): δ 8.79 (d, J = 5.6 Hz, 2H, Py-H), 7.95 (d, J = 5.6 Hz, 1H, Py-H), 7.85 (d, J = 8 Hz, 2H, Ar-H), 7.66 (d, J = 7.6 Hz, 2H, Py-H), 7.51 (s, 2H, N–CHCH–N), 7.44 (s, 1H, Ar-H), 7.31 (d, J = 3.2 Hz, 1H, Ar-H), 5.80 (d, J = 9.6 Hz, 2H, N–CH2), 4.01 (t, J = 8 Hz, 3H, N–CH3). 13C NMR (100 MHz, DMSO-d6): δ 152.4, 151.9, 132.8, 129.8, 129.7, 129.6, 125.4, 122.9, 119.2, 111.1, 53.3, 38.1. IR, νmax (cm−1) (CH2Cl2): 3736, 3159, 3123, 3104, 2224, 1702, 1602, 1572, 1503, 1485, 1469, 1447, 1407, 1358, 1292, 1237, 1214, 1183, 1151, 1126, 1071, 1016, 944, 862, 838, 824, 768, 756, 735, 692, 682, 651, 552, 491, 468, 442, 416.
Synthesis of complex 3e. Prepared according to procedure 3b using 1e (0.50 g, 1.32 mmol), PdCl2 (0.23 g, 1.32 mmol) and K2CO3 (0.91 g, 6.61 mmol). Yield = 38%, 0.24 g. 1H NMR (400 MHz, CDCl3): δ 8.95 (t, J = 1.6 Hz, 2H, Py-H), 7.98 (d, J = 8 Hz, 2H, Ar-H), 7.76 (t, J = 1.6 Hz, 1H, Py-H), 7.73 (d, J = 8 Hz, 2H, Ar-H), 7.63 (dt, J = 7.6 Hz, 2H, Py-H), 7.47 (m, 2H, Py-H), 7.33 (m, 2H, Ar-H), 7.01 (s, 2H, N–CHCH–N), 6.02 (s, 4H, N–CH2). 13C NMR (100 MHz, CDCl3): δ 152.6, 152.3, 151.9, 151.2, 138.9, 138.8, 138.7, 138.2, 138.1, 138.0, 133.6, 133.5, 133.0, 132.9, 131.0, 130.9, 130.8, 129.1, 124.6, 124.5, 123.1, 123.0, 122.9, 117.3, 112.3, 112.2, 112.0, 52.8, 52.6, 52.4. IR, νmax (cm−1) (CH2Cl2): 3437, 3155, 3104, 2930, 2218, 1695, 1603, 1570, 1482, 1446, 1407, 1377, 1351, 1289, 1263, 1242, 1216, 1156, 1094, 1068, 1046, 1016, 966, 948, 848, 832, 795, 770, 756, 695, 684, 643, 558, 445.
Synthesis of complex 3f. Prepared according to procedure 3b using 1f (0.50 g, 1.98 mmol), PdCl2 (0.35 g, 1.98 mmol) and K2CO3 (1.37 g, 9.92 mmol). Yield = 41%, 0.42 g. 1H NMR (400 MHz, CDCl3): δ 9.04 (t, J = 4.8 Hz, 2H, Py-H), 7.75 (m, 1H, Py-H), 7.91 (d, J = 8 Hz, 2H, Py-H), 7.36 (m, 5H, Ar-H), 6.87 (d, J = 2 Hz, 1H, N–CHCH–N), 6.68 (d, J = 2 Hz, 1H, N–CHCH–N), 5.77 (s, 2H, N–CH2), 4.12 (s, 3H, N–CH3). 13C NMR (100 MHz, CDCl3): δ 152.5, 148.4, 137.9, 135.2, 129.1, 128.9, 128.5, 124.5, 123.6, 121.3, 54.9, 38.4. IR, νmax (cm−1) (CH2Cl2): 3437, 3162, 3129, 3109, 3063, 3027, 3002, 2928, 1920, 1856, 1815, 1598, 1569, 1495, 1486, 1467, 1446, 1421, 1402, 1317, 1229, 1193, 1153, 1121, 1077, 1069, 1044, 1032, 1013, 982, 952, 835, 819, 778, 762, 732, 695, 686, 662, 641, 610, 574, 462, 445.
General procedure for the acylative Suzuki–Miyaura cross-coupling reaction of arylboronic acids with benzoyl chloride
A Radley's tube was charged with benzoyl chloride (1.2 mmol), phenylboronic acid (1.0 mmol), K2CO3 (1.5 mmol) and catalyst (0.1 mol%) in toluene (4 mL). The mixture was stirred to 60 °C for 4 h under an argon atmosphere. The reaction mixture was cooled to room temperature and filtered. The conversion was analyzed by 1H-NMR and 13C-NMR spectroscopy.
General procedure for the Suzuki–Miyaura cross-coupling reaction
A Radley's tube was charged with aryl halide (1.0 mmol), phenylboronic acid (1.0 mmol), KOH (0.5 mmol) and catalyst (0.2 mol%) in 2-propanol–H2O mixture (2 mL, 0.5:1.5). The mixture was stirred to 82 °C for 30 min. under air atmosphere. The reaction mixture was cooled to room temperature and filtered. The conversion was analyzed by 1H-NMR and 13C-NMR spectroscopy.
Suzuki–Miyaura cross coupling products15,20–26
Benzophenone (Table 2, entry 1). 1H NMR (400 MHz, CDCl3): δ 7.81 (d, J = 7.2 Hz, 2H, Ar-H), 7.59 (t, J = 7.6 Hz, 1H, Ar-H), 7.48 (t, J = 7.6 Hz, 2H, Ar-H). 13C NMR (100 MHz, CDCl3): δ 196.7, 137.6, 132.4, 130.0, 128.3.
4-Methylbenzophenone (Table 2, entry 2). 1H NMR (400 MHz, CDCl3): δ 7.78 (m, 2H, Ar-H), 7.72 (d, J = 8.4 Hz, 2H, Ar-H), 7.55 (m, 1H, Ar-H), 7.47 (t, J = 7.2 Hz, 2H, Ar-H), 7.28 (d, J = 8 Hz, 2H, Ar-H), 2.44 (s, 3H, CH3). 13C NMR (100 MHz, CDCl3): δ 196.5, 143.3, 137.9, 134.8, 132.2, 130.3, 129.9, 128.9, 128.2, 21.7.
2-Methylbenzophenone (Table 2, entry 3). 1H NMR (400 MHz, CDCl3): δ 8.17 (d, J = 7.2 Hz, 2H, Ar-H), 7.81 (d, J = 6.8 Hz, 1H, Ar-H), 7.67 (t, J = 8.8 Hz, 1H, Ar-H), 7.58 (t, J = 7.6 Hz, 1H, Ar-H), 7.53 (t, J = 8 Hz, 1H, Ar-H), 7.44 (m, 2H, Ar-H), 7.28 (m, 1H, Ar-H), 2.34 (s, 3H, CH3). 13C NMR (100 MHz, CDCl3): δ 198.6, 162.8, 138.6, 136.7, 134.6, 133.2, 131.0, 130.6, 130.1, 128.9, 128.5, 125.2, 19.9.
2,4-Dimethylbenzophenone (Table 2, entry 4). 1H NMR (400 MHz, CDCl3): δ 8.17–8.16 (dd, J = 6.4 Hz, 1H, Ar-H), 7.79 (d, J = 7.2 Hz, 1H, Ar-H), 7.68 (t, J = 8.8 Hz, 1H, Ar-H), 7.55 (m, 2H, Ar-H), 7.44 (t, J = 8 Hz, 1H, Ar-H), 7.06 (t, J = 8 Hz, 1H, Ar-H), 2.38 (s, 3H, CH3), 2.34 (s, 3H, CH3). 13C NMR (100 MHz, CDCl3): δ 198.6, 140.7, 138.2, 137.3, 135.5, 133.8, 132.8, 131.9, 130.1, 129.3, 128.5, 128.3, 125.8, 21.4, 20.1.
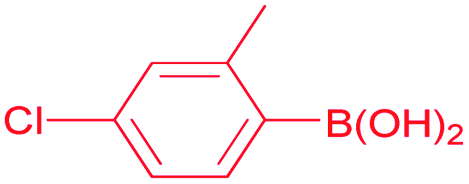
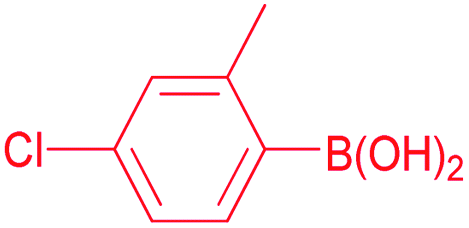
4-Chloro-2-methylbenzophenone (Table 2, entry 5). 1H NMR (400 MHz, CDCl3): δ 7.78 (m, 2H, Ar-H), 7.59 (m, 1H, Ar-H), 7.46 (m, 2H, Ar-H), 7.27 (m, 3H, Ar-H), 2.32 (s, 3H, CH3). 13C NMR (100 MHz, CDCl3): δ 197.5, 139.1, 137.4, 136.8, 136.1, 133.4, 131.0, 130.0, 128.6, 125.4, 19.9.
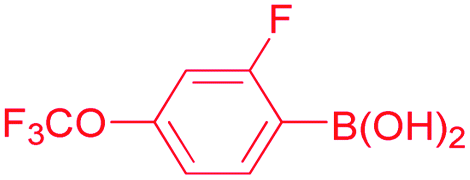
2-Fluoro, 4-methoxybenzophenone (Table 2, entry 6). 1H NMR (400 MHz, CDCl3): δ 7.77–7.75 (dd, J = 6.8 Hz, 2H, Ar-H), 7.70 (d, J = 6.8 Hz, 1H, Ar-H), 7.58 (m, 2H, Ar-H), 7.47 (m, 2H, Ar-H), 7.08 (t, J = 8 Hz, 1H, Ar-H), 2.32 (s, 3H, CH3). 13C NMR (100 MHz, CDCl3): δ 195.6, 165.3, 162.7, 137.6, 133.8, 132.4, 129.9, 128.3, 125.3, 125.1, 115.0, 14.6. 19F NMR (376.2 MHz, CDCl3): δ −110.13 (m, F).
4-Cyanobenzophenone (Table 2, entry 7). 1H NMR (400 MHz, CDCl3): δ 7.87 (m, 2H, Ar-H), 7.79 (m, 4H, Ar-H), 7.65 (m, 1H, Ar-H), 7.52 (m, 2H, Ar-H). 13C NMR (100 MHz, CDCl3): δ 195.0, 141.2, 136.3, 133.3, 132.9, 132.2, 130.2, 130.0, 128.6, 127.9, 117.9, 115.6.
4-(Trifluoromethyl)benzophenone (Table 2, entry 8). 1H NMR (400 MHz, CDCl3): δ 7.90 (m, 2H, Ar-H), 7.81 (m, 2H, Ar-H), 7.76–7.75 (dd, J = 8.8 Hz, 2H, Ar-H), 7.63 (m, 1H, Ar-H), 7.51 (m, 2H, Ar-H). 13C NMR (100 MHz, CDCl3): δ 195.5, 140.7, 140.6, 136.7, 133.1, 130.1, 130.0, 128.5, 125.4, 125.3. 19F NMR (376.2 MHz, CDCl3): δ −63.02 (s, CF3).
4-Chloro-2-methyl-4′-nitrobenzophenone (Table 2, entry 9). 1H NMR (400 MHz, CDCl3): δ 8.30 (d, J = 6.8 Hz, 2H, Ar-H), 7.91 (d, J = 8.8 Hz, 2H, Ar-H), 7.34 (s, 1H, Ar-H), 2.37 (s, 3H, CH3). 13C NMR (100 MHz, CDCl3): δ 195.5, 150.3, 142.5, 139.9, 137.4, 135.2, 131.6, 130.8, 130.6, 125.7, 123.7, 20.2.
4-Trifluoromethyl-4′-nitrobenzophenone (Table 2, entry 10). 1H NMR (400 MHz, CDCl3): δ 8.35 (d, J = 8.8 Hz, 2H, Ar-H), 7.93 (d, J = 8.8 Hz, 2H, Ar-H), 7.87 (d, J = 8.8 Hz, 2H, Ar-H), 7.35 (d, J = 8.4 Hz, 2H, Ar-H). 13C NMR (100 MHz, CDCl3): δ 193.2, 152.8, 149.9, 142.3, 134.4, 132.1, 130.6, 123.7, 120.5. 19F NMR (376.2 MHz, CDCl3): δ −57.62 (s, CF3).
4-Bromo-4′-nitrobenzophenone (Table 2, entry 11). 1H NMR (400 MHz, CDCl3): δ 8.35 (m, 2H, Ar-H), 7.93 (m, 2H, Ar-H), 7.69 (d, J = 2.8 Hz, 2H, Ar-H), 7.62–7.61 (dd, J = 2.4 Hz, 1H, Ar-H), 7.52–7.50 (dd, J = 2.4 Hz, 1H, Ar-H). 13C NMR (100 MHz, CDCl3): δ 193.7, 142.3, 134.9, 132.2, 132.0, 131.5, 130.8, 130.6, 128.8, 127.1, 123.6.
4′-Nitrobenzophenone (Table 2, entry 12). 1H NMR (400 MHz, CDCl3): δ 8.33 (d, J = 8.8 Hz, 2H, Ar-H), 7.93 (d, J = 8.8 Hz, 2H, Ar-H), 7.80 (m, 2H, Ar-H), 7.65 (m, 1H, Ar-H), 7.52 (t, J = 8 Hz, 2H, Ar-H). 13C NMR (100 MHz, CDCl3): δ 194.8, 149.8, 142.9, 136.3, 133.4, 130.7, 130.1, 128.7, 123.5.
Biphenyl [29] (Table 4, entry 1). 1H NMR (400 MHz, CDCl3): δ 7.62 (d, J = 8.4 Hz, 4H, Ar-H), 7.46 (t, J = 7.6 Hz, 4H, Ar-H), 7.37 (t, J = 8 Hz, 2H, Ar-H). 13C NMR (100 MHz, CDCl3): δ 141.3, 128.8, 127.3, 127.2.
4-Methylbiphenyl (Table 4, entry 2). 1H NMR (400 MHz, CDCl3): δ 7.63 (d, J = 8.4 Hz, 2H, Ar-H), 7.52 (d, J = 8 Hz, 2H, Ar-H), 7.47 (t, J = 7.6 Hz, 2H, Ar-H), 7.36 (m, 1H, Ar-H), 7.29 (d, J = 8 Hz, 2H, Ar-H), 2.44 (s, 3H, CH3). 13C NMR (100 MHz, CDCl3): δ 141.2, 138.4, 137.0, 129.5, 128.7, 127.0, 126.9, 21.1.
4-Tert-butylbiphenyl (Table 4, entry 3). 1H NMR (400 MHz, CDCl3): δ 7.62 (d, J = 7.6 Hz, 2H, Ar-H), 7.57 (d, J = 8.4 Hz, 2H, Ar-H), 7.47 (m, 4H, Ar-H), 7.35 (t, J = 8 Hz, 1H, Ar-H), 1.39 (s, 9H, C(CH3)3). 13C NMR (100 MHz, CDCl3): δ 150.3, 141.1, 138.4, 128.7, 127.1, 127.0, 126.8, 125.7, 34.9, 31.4.
4-Hydroxybiphenyl (Table 4, entry 4). 1H NMR (400 MHz, DMSO-d6): δ 9.53 (s, 1H, OH), 7. 54 (d, J = 8.4 Hz, 2H, Ar-H), 7.46 (d, J = 8.4 Hz, 2H, Ar-H), 7.38 (t, J = 7.2 Hz, 1H, Ar-H), 6.84 (d, J = 8.8 Hz, 2H, Ar-H). 13C NMR (100 MHz, DMSO-d6): δ 157.9, 140.7, 131.4, 129.2, 128.2, 126.8, 126.4, 116.2.
4-Cyanobiphenyl (Table 4, entry 5). 1H NMR (400 MHz, CDCl3): δ 7.69 (m, 4H, Ar-H), 7.59 (m, 2H, Ar-H), 7.49 (m, 2H, Ar-H), 7.44 (m, 1H, Ar-H). 13C NMR (100 MHz, CDCl3): δ 145.6, 139.1, 132.9, 129.1, 128.7, 127.7, 127.2, 118.9, 110.9.
4-Fluorobiphenyl (Table 4, entry 6). 1H NMR (400 MHz, CDCl3): δ 7.57 (m, 4H, Ar-H), 7. 46 (t, J = 8 Hz, 2H, Ar-H), 7. 37 (t, J = 8.8 Hz, 1H, Ar-H), 7.15 (m, 2H, Ar-H). 13C NMR (100 MHz, CDCl3): δ 163.7, 161.3, 140.3, 137.4, 128.8, 127.7, 127.6, 127.3, 127.0, 115.7, 115.5. 19F NMR (376.2 MHz, CDCl3): δ −115.82 (m, F).
4-(Trifluoromethyl)biphenyl (Table 4, entry 7). 1H NMR (400 MHz, CDCl3): δ 7.71 (s, 4H, Ar-H), 7.62 (d, J = 7.2 Hz, 2H, Ar-H), 7.49 (t, J = 6.8 Hz, 2H, Ar-H), 7.43 (d, J = 7.6 Hz, 1H, Ar-H). 13C NMR (100 MHz, CDCl3): δ 139.8, 128.9, 128.2, 127.4, 127.3, 125.8, 125.7, 125. 7, 125.6. 19F NMR (376.2 MHz, CDCl3): δ −62.40 (s, CF3).
4-(Trifluoromethoxy)biphenyl (Table 4, entry 8). 1H NMR (400 MHz, CDCl3): δ 7.58 (m, 4H, Ar-H), 7. 46 (t, J = 7.2 Hz, 2H, Ar-H), 7.38 (t, J = 8 Hz, 1H, Ar-H), 7.28 (t, J = 8.8 Hz, 2H, Ar-H). 13C NMR (100 MHz, CDCl3): δ 148.7, 139.9, 139.8, 128.9, 128.4, 127.7, 127.1, 121.8, 121.2, 119.3. 19F NMR (376.2 MHz, CDCl3): δ −57.83 (s, OCF3).
2-Hydroxybiphenyl (Table 4, entry 9). 1H NMR (400 MHz, CDCl3): δ 7.50 (m, 4H, Ar-H), 7.41 (m, 1H, Ar-H), 7.28 (m, 2H, Ar-H), 7.01 (t, J = 7.6 Hz, 2H, Ar-H), 5.21 (s, 1H, OH). 13C NMR (100 MHz, CDCl3): δ 152.4, 137.1, 130.3, 129.3, 129.2, 129.1, 128.1, 127.9, 120.9, 115.8.
2-Methylbiphenyl (Table 4, entry 10). 1H NMR (400 MHz, CDCl3): δ 7.43 (m, 2H, Ar-H), 7.53 (m, 3H, Ar-H), 7.28 (m, 2H, Ar-H), 7.25 (m, 2H, Ar-H), 2.29 (s, 3H, CH3). 13C NMR (100 MHz, CDCl3): δ 141.9, 141.9, 135.3, 130.3, 129.8, 129.2, 128.1, 127.2, 126.7, 125.7, 20.5.
2-(Trifluoromethyl)biphenyl (Table 4, entry 11). 1H NMR (400 MHz, CDCl3): δ 7.75 (d, J = 7.2 Hz, 1H, Ar-H), 7.59 (m, 1H, Ar-H), 7.47 (t, J = 8 Hz, 1H, Ar-H), 7.41 (d, J = 5.2 Hz, 3H, Ar-H), 7.34 (m, 3H, Ar-H). 13C NMR (100 MHz, CDCl3): δ 139.8, 132.0, 131.2, 128.9, 128.7, 127.7, 127.9, 127.3, 127.2, 126.0, 125.9. 19F NMR (376.2 MHz, CDCl3): δ −56.86 (s, CF3).
4-Chloro-2-methylbiphenyl (Table 4, entry 12). 1H NMR (400 MHz, CDCl3): δ 7.42 (t, J = 7.2 Hz, 2H, Ar-H), 7.36 (m, 1H, Ar-H), 7.29 (d, J = 8 Hz, 3H, Ar-H), 7.23 (d, J = 7.6 Hz, 1H, Ar-H), 7.16 (d, J = 8.4 Hz, 1H, Ar-H), 2.26 (s, 3H, CH3). 13C NMR (100 MHz, CDCl3): δ 140.8, 140.4, 137.3, 132.9, 131.0, 130.1, 129.1, 128.2, 127.1, 125.9, 20.4.
2,4-Methylbiphenyl (Table 4, entry 13). 1H NMR (400 MHz, CDCl3): δ 7.42 (m, 2H, Ar-H), 7. 35 (t, J = 5.6 Hz, 3H, Ar-H), 7.12 (m, 3H, Ar-H), 2.39 (s, 3H, CH3), 2.27 (s, 3H, CH3). 13C NMR (100 MHz, CDCl3): δ 141.9, 139.1, 136.9, 135.1, 131.1, 129.8, 129.3, 128.0, 126.6, 126.5, 21.1, 20.4.
4-Fluoro-3-methylbiphenyl (Table 4, entry 14). 1H NMR (400 MHz, CDCl3): δ 7.55 (d, J = 8.4 Hz, 2H, Ar-H), 7.43 (t, J = 7.6 Hz, 3H, Ar-H), 7.37 (m, 3H, Ar-H), 7.07 (t, J = 8.8 Hz, 1H, Ar-H), 2.35 (s, 3H, CH3). 13C NMR (100 MHz, CDCl3): δ 162.3, 159.8, 140.5, 130.3, 130.2, 128.8, 127.2, 127.1, 127.0, 126.0, 125.9, 125.1, 124.9, 115.1, 14.7, 14.7. 19F NMR (376.2 MHz, CDCl3): δ −120.18 (m, F).
2-Fluoro-4-methylbiphenyl (Table 4, entry 15). 1H NMR (400 MHz, CDCl3): δ 7.55 (d, J = 8.4 Hz, 2H, Ar-H), 7.45 (t, J = 7.6 Hz, 3H, Ar-H), 7.39 (m, 2H, Ar-H), 7.08 (t, J = 8.8 Hz, 1H, Ar-H), 2.36 (s, 3H, CH3). 13C NMR (100 MHz, CDCl3): δ 130.3, 130.2, 128.7, 127.1, 126.9, 125.9, 125.8, 115.3, 115.1, 14.7. 19F NMR (376.2 MHz, CDCl3): δ −120.17 (m, F).
4-Methyl-3-nitrobiphenyl (Table 4, entry 16). 1H NMR (400 MHz, CDCl3): δ 8.21 (d, J = 1.6 Hz, 1H, Ar-H), 7.73 (d, J = 8 Hz, 1H, Ar-H), 7.60 (d, J = 8 Hz, 2H, Ar-H), 7.48 (t, J = 7.2 Hz, 2H, Ar-H), 7.41 (m, 2H, Ar-H), 2.64 (s, 3H, CH3). 13C NMR (100 MHz, CDCl3): δ 140.2, 138.4, 133.6, 133.2, 132.2, 131.3, 129.1, 128.3, 126.9, 122.9, 20.1.
4′-Methylbiphenyl (Table 4, entry 17). 1H NMR (400 MHz, CDCl3): δ 7.61 (d, J = 8 Hz, 2H, Ar-H), 7.53 (d, J = 8 Hz, 2H, Ar-H), 7.45 (t, J = 7.6 Hz, 2H, Ar-H), 7.35 (t, J = 7.6 Hz, 1H, Ar-H), 7.28 (d, J = 8 Hz, 2H, Ar-H), 2.42 (s, 3H, CH3). 13C NMR (100 MHz, CDCl3): δ 141.2, 138.4, 137.0, 129.5, 128.7, 127.0, 127.0, 21.1.
4′-Methoxybiphenyl (Table 4, entry 18). 1H NMR (400 MHz, CDCl3): δ 7.56 (m, 4H, Ar-H), 7.43 (t, J = 7.6 Hz, 2H, Ar-H), 7.32 (t, J = 7.6 Hz, 1H, Ar-H), 7.00 (m, 2H, Ar-H), 3.87 (s, 3H, CH3). 13C NMR (100 MHz, CDCl3): δ 159.2, 140.9, 133.8, 128.8, 128.2, 126.8, 126.7, 114.2, 55.4.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Financial support from Ege University (Project FYL-2018-20197) is gratefully acknowledged. The authors acknowledge Dokuz Eylul University for using of the Rigaku Oxford Xcalibur Eos Diffractometer (purchased under University Research Grant No. 2010.KB.FEN.13). Dr Senthil Rethinam acknowledges the funding support granted by the 2232-International Fellowship for Outstanding Researcher Program of TUBITAK (Project No: 118C350).
Notes and references
- K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4442–4489 CrossRef CAS PubMed.
- L. Yin and J. Liebscher, Chem. Rev., 2007, 107, 133–137 CrossRef CAS PubMed.
-
(a) B. M. Baughman, E. Stennett, R. E. Lipner, A. C. Rudawsky and S. J. Schidtke, J. Phys. Chem. A, 2009, 113, 8011 CrossRef CAS PubMed;
(b) C. Torborg and M. Beller, Adv. Synth. Catal., 2009, 351, 3027–3043 CrossRef CAS;
(c) C. M. So and F. Y. Kwong, Chem. Soc. Rev., 2011, 40, 4963 RSC;
(d) I. P. Beletskaya, F. Alonso and V. Tyurin, Coord. Chem. Rev., 2019, 385, 137–173 CrossRef CAS.
- M. Blangetti, H. Rosso, C. Prandi, A. Deagostino and P. Venturello, Molecules, 2013, 18, 1188 CrossRef CAS PubMed.
- J. Buchspies and M. Szostak, Catalysts, 2019, 9, 53 CrossRef.
-
(a) C. Fliedel, A. Labande, E. Manoury and R. Poli, Coord. Chem. Rev., 2019, 394, 65–103 CrossRef CAS;
(b) G. C. Fortman and S. P. Nolan, Chem. Soc. Rev., 2011, 40, 5151–5516 RSC.
-
(a) C. Fliedel, A. Ghisolfi and P. Braunstein, Chem. Rev., 2016, 116(16), 9237–9304 CrossRef CAS PubMed;
(b) N. T. S. Phan, M. V. D. Sluys and C. W. Jones, Adv. Synth. Catal., 2006, 348(6), 609–679 CrossRef CAS;
(c) Y. C. Wong, K. Parthasarathy and C. H. Cheng, Org. Lett., 2010, 12, 1736 CrossRef CAS PubMed.
-
(a) H. W. Wanzlick and H. J. Schonherr, Angew. Chem., Int. Ed. Engl., 1968, 7, 141–143 CrossRef CAS;
(b) A. J. Arduengo, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361–363 CrossRef CAS.
- H. V. Huynh, The Organometallic Chemistry of N-Heterocyclic Carbenes, John Wiley & Sons, Ltd, Chichester, UK, 2017 Search PubMed.
- A. Rühling, K. Schaepe, L. Rakers, B. Vonhören, P. Tegeder, B. J. Ravoo and F. Glorius, Angew. Chem., Int. Ed., 2016, 55, 5856–5860 CrossRef PubMed.
- L. Jiang, F. Shan, Z. Li and D. Zhao, Molecules, 2012, 17, 12121–12139 CrossRef CAS PubMed.
-
(a) N. A. Bumagin and D. N. Korolev, Tetrahedron Lett., 1999, 40, 3057–3060 CrossRef CAS;
(b) M. Haddach and J. R. McCarthy, Tetrahedron Lett., 1999, 40, 3109–3112 CrossRef CAS;
(c) L. Zhang, J. Wu, L. Shi, C. Xia and F. Li, Tetrahedron Lett., 2011, 52, 3897–3901 CrossRef CAS;
(d) M. Mondal and U. Bora, Appl. Organomet. Chem., 2018, 28, 354–358 CrossRef;
(e) H. Solařová, I. Císařová and P. Štěpnička, Organometallics, 2014, 33, 4131–4147 CrossRef;
(f) M. Semler and P. Štěpnička, Catal. Today, 2015, 243, 128–133 CrossRef CAS;
(g) F. Rafiee and A. R. Hajipour, Appl. Organomet. Chem., 2015, 29, 181–184 CrossRef CAS;
(h) M. Mondal and U. Bora, New J. Chem., 2016, 40, 3119–3123 RSC;
(i) B. Movassagh, F. Hajizadeh and E. Mohammadi, Appl. Organomet. Chem., 2018, 32, e3982 CrossRef.
-
(a) J.-Y. Chen, S.-C. Chen, Y.-J. Tang, C.-Y. Mou and F.-Y. Tsai, J. Mol. Catal. A: Chem., 2009, 88, 307 Search PubMed;
(b) D. J. Nelson and S. P. Nolan, Chem. Soc. Rev., 2013, 42, 6723 RSC;
(c) H. Türkmen and B. Çetinkaya, J. Organomet. Chem., 2006, 691, 3749–3759 CrossRef;
(d) E. Gacal, S. Denizaltı, A. Kınal, A. G. Gökçe and H. Türkmen, Tetrahedron, 2018, 74, 6829–6838 CrossRef CAS;
(e) H. Türkmen, R. Can and B. Çetinkaya, Dalton Trans., 2009, 7039–7044 RSC.
- H. M. J. Wang and I. J. B. Lin, Organometallics, 1998, 17, 972–975 CrossRef CAS.
- M. Sevim, S. B. Kavukcu, A. Kınal, O. Şahin and H. Türkmen, Dalton Trans., 2020, 49, 16903–16915 RSC.
- C. F. R. A. C. Lima, A. S. M. C. Rodrigues, V. L. M. Silva, A. M. S. Silva and L. M. N. B. F. Santos, ChemCatChem, 2014, 6(5), 1291–1302 CAS.
- CrysAlisPro Software System, Rigaku Oxford Diffraction, 2019.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- Y. C. Wong, K. Parthasarathy and C. H. Cheng, Org. Lett., 2010, 12, 1736 CrossRef CAS PubMed.
- J.-Y. Chen, S.-C. Chen, Y.-J. Tang, C.-Y. Mou and F.-Y. Tsai, J. Mol. Catal. A: Chem., 2009, 88, 307 Search PubMed.
- M. Li, C. Wang and H. Ge, Org. Lett., 2011, 13, 2062 CrossRef CAS PubMed.
- D. Xing, B. Guan, G. Cai, Z. Fang, L. Yang and Z. Shi, Org. Lett., 2006, 8, 693 CrossRef CAS PubMed.
- W. J. Zhou, K. H. Wang and J. X. Wang, J. Org. Chem., 2009, 74, 5599 CrossRef CAS PubMed.
- L. Liu, Y. Zhang and B. Xin, J. Org. Chem., 2006, 71, 3994 CrossRef CAS PubMed.
- L. Liu, Y. Zhang and Y. Wang, J. Org. Chem., 2005, 70, 6122 CrossRef CAS PubMed.
-
(a) S. Çakır, G. Türkmen and H. Türkmen, Appl. Organomet. Chem., 2018, 32, e3969 CrossRef;
(b) S. Çakır and H. Türkmen, Appl. Organomet. Chem., 2020, 34, e5499 CrossRef.
- Z. Fei, D. Zhao, D. Pieraccini, W. H. Ang, T. J. Geldbach, R. Scopelliti, C. Chiappe and P. J. Dyson, Organometallics, 2007, 26, 1588–1598 CrossRef CAS.
-
(a) F. E. Hahn and M. C. Jahnke, Angew. Chem., Int. Ed., 2008, 47, 3122–3172 CrossRef CAS PubMed;
(b) S. Díez-González, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612–3676 CrossRef PubMed;
(c) T. Dröge and F. Glorius, Angew. Chem., Int. Ed., 2010, 49, 6940–6952 CrossRef PubMed;
(d) Z. Jin, L.-L. Qiu, Y.-Q. Li, H.-B. Song and J.-X. Fang, Organometallics, 2010, 29, 6578–6586 CrossRef CAS;
(e) G. C. Fortman and S. P. Nolan, Chem. Soc. Rev., 2011, 40, 5151–5169 RSC.
-
(a) R. A. Kelly III, H. Clavier, S. Giudice, N. M. Scott, E. D. Stevens, J. Bordner, I. Samardjiev, C. D. Hoff, L. Cavallo and S. P. Nolan, Organometallics, 2008, 27, 202–210 CrossRef;
(b) D. J. Nelson and S. P. Nolan, Chem. Soc. Rev., 2013, 42, 6723–6753 RSC.
-
(a) M. G. Organ, M. AbdelHadi, S. Avola, I. Dubovyk, N. Hadei, E. A. B. Kantchev, C. J. O'Brien, M. Sayah and C. Valente, Chem.–Eur. J., 2008, 14, 2443–2452 CrossRef CAS PubMed;
(b) L. Xu and Y. Shi, J. Org. Chem., 2008, 73, 749–751 CrossRef CAS PubMed;
(c) K. H. Hoi, S. Çalimsiz, R. D. J. Froese, A. C. Hopkinson and M. G. Organ, Chem.–Eur. J., 2011, 17, 3086–3090 CrossRef CAS PubMed;
(d) C. Valente, S. Çalimsiz, K. H. Hoi, D. Mallik, M. Sayah and M. G. Organ, Angew. Chem., Int. Ed., 2012, 51, 3314–3332 CrossRef CAS PubMed;
(e) A. Chartoire, X. Frogneux, A. Boreux, A. M. Z. Slawin and S. P. Nolan, Organometallics, 2012, 31, 6947–6951 CrossRef CAS;
(f) J.-S. Ouyang, S. Liu, B. Pen, Y. Zhang, H. Liang, B. Chen, X. He, W. T. K. Chan, A. S. C. Chan, T.-Y. Sun, Y.-D. Wu and L. Qiu, ACS Catal., 2021, 11, 9252–9261 CrossRef CAS.
-
(a) C. Yang, L. Zhang, C. Lu, S. Zhou, X. Li, Y. Li, Y. Yang, Y. Li, Z. Liu, J. Yang, K. N. Houk, F. Mo and X. Guo, Nat. Nanotechnol., 2021, 16, 1214–1223 CrossRef CAS PubMed;
(b) N. Marion and S. P. Nolan, Acc. Chem. Res., 2008, 41, 1440–1449 CrossRef CAS PubMed.
- G. Li, P. Lei, M. Szostak, E. Casals-Cruañas, A. Poater, L. Cavallo and S. P. Nolan, ChemCatChem, 2018, 10, 3096–3106 CrossRef CAS.
- C. F. R. A. C. Lima, A. S. M. C. Rodrigues, V. L. M. Silva, A. M. S. Silva and L. M. N. B. F. Santos, ChemCatChem, 2014, 6, 1291–1302 CrossRef CAS.
-
(a) S. Yang, T. Zhou, A. Poater, L. Cavallo, S. P. Nolan and M. Szostak, Catal. Sci. Technol., 2021, 11, 3189–3197 RSC;
(b) G. Li, P. Lei, M. Szostak, E. Casals-Cruañas, A. Poater, L. Cavallo and S. P. Nolan, ChemCatChem, 2018, 10, 3096–3106 CrossRef CAS;
(c) G. M. Meconi, S. V. C. Vummaleti, J. A. Luque-Urrutia, P. Belanzoni, S. P. Nolan, H. Jacobsen, L. Cavallo, M. Solà and A. Poater, Organometallics, 2017, 36, 2088–2095 CrossRef CAS.
- R. Gajda, A. Poater, A. Brotons-Rufes, S. Planer, K. Woźniak, K. Grela and A. Kajetanowicz, Arkivoc, 2021, 138–156 Search PubMed.
-
(a) M.
G. Organ, G. A. Chass, D.-C. Fang, A. C. Hopkinson and C. Valente, Synthesis, 2008, 2008, 2776–2797 CrossRef;
(b) C. Valente, M. Pompeo, M. Sayah and M. G. Organ, Org. Process Res. Dev., 2014, 18, 180–190 CrossRef CAS;
(c) R. D. J. Froese, C. Lombardi, M. Pompeo, R. P. Rucker and M. G. Organ, Acc. Chem. Res., 2017, 50, 2244–2253 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Serdar Batıkan Kavukcu
a,
Serdar Batıkan Kavukcu