
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePreparation and catalytic behavior of antioxidant cassava starch with selenium active sites and hydrophobic microenvironments
Cheng Shia,
Qiugang Huanga,
Ruirui Zhangb,
Xingtang Liangb,
Feng Wangb,
Zijie Liub,
Min Liub,
Huayu Hu*a and
Yanzhen Yin *ab
*ab
aSchool of Chemistry and Chemical Engineering, Guangxi University, Nanning 530004, China. E-mail: 404631785@qq.com
bQinzhou Key Laboratory of Biowaste Resources for Selenium-enriched Functional Utilization, College of Petroleum and Chemical Engineering, Beibu Gulf University, Qinzhou 535011, China. E-mail: yinyanzhen2018@163.com
First published on 14th December 2021
Abstract
The preparation of antioxidant starch with the activity of glutathione peroxidase (GPx) for scavenging free radicals can not only enrich the types of modified starch but also alternate native GPx to overcome its drawbacks. In this work, an antioxidant cassava starch (AO-ca-starch) was prepared by the sequential esterification and selenylation of cassava starch. The process was optimized based on the selenium content. Various characterizations for the AO-ca-starch indicated that the catalytic center of GPx, the selenium, was anchored on the starch. The catalytic activity of AO-ca-starch, a starch-based biomimetic GPx, was about 4.95 × 105 times higher than that of the typical artificial selenoenzyme (diphenyl diselenide, PhSeSePh), and it exhibited a typical saturation kinetic catalytic behavior. The surface changes of the starch during the modification were conducive to the formation of hydrophobic microenvironments, which played an important role in the catalytic reaction of biomimetic GPx due to the binding of the hydrophobic substrates. The match of the catalytic center and the hydrophobic microenvironments was the key factor for maintaining the high catalytic activity of AO-ca-starch. Without cytotoxicity, AO-ca-starch exemplified a new and promising modified starch as a selenium-enriched functional food and antioxidant drug.
Introduction
Modified starch is a carbon-based material with the advantages of a wide variety of sources, low cost, and diverse functions. It is widely used in fields such as food additives and biological materials.1,2 At present, modified starches include etherified starch,3 esterified starch,4 cross-linked starch,5 and grafted starch.6 The octenyl succinic anhydride-modified starch (OSA-starch) is a typical esterified starch that can be legally used in food and medicine.7–9 With the increasing demand for functional foods for health benefits, the development of functional starch has become a prospective research work.Selenium, as an important trace element, has attracted wide attention due to its unique chemical- and bio-activities. Selenium-functionalized materials have been extensively applied as bioactive components in pharmaceutical industries and as functional constituents in materials.10–12 For example, carbon-based materials with anchored selenium revealed high performances in catalysis and anti-oxidation.12–15 Previous studies have shown that many diseases such as inflammation, cancer and Keshan disease are caused by excessive accumulation of free radicals in the human body.16,17 Glutathione peroxidase (GPx, EC 1.11.1.9) is one of the typical selenoenzymes that reduces the excess free radicals and hydroperoxides (ROOH) at the expense of glutathione (GSH) and consequently inhibits oxidative diseases.18,19 However, there are intrinsic vulnerabilities such as limited source, low yield and high cost in the extraction of native GPx. Therefore, it is of great significance to develop GPx mimics for overcoming the drawbacks of native GPx. The development of antioxidant starch with the ability to scavenge free radicals could not only expand the application of modified starch but also provide new functional materials for antioxidant drugs.
Our group and other researchers have devoted considerable efforts to produce a series of biomimetic GPx using small molecules,20,21 polymers,22,23 proteins,24,25 and nanomaterials,26,27 and to expose the relationship between the structure and the function of biomimetic GPx. The previous results indicated that the three important catalytic factors for maintaining the high catalytic activity of GPx are the selenium element as the catalytic center on GPx, the hydrophobic microenvironment composed of hydrophobic amino acid residues such as 150Phe, 148Trp, and 34Leu, and the recognition site for binding substrates via hydrogen bonds between substrates and arginine-based 40Arg, 130Arg, and 167Arg.28,29 In order to construct a biodegradable biomimetic GPx with high catalytic activity, we intended to prepare the biomimetic GPx with modified starch (OSA-starch), resulting in a new antioxidant starch. The OSA-starch prepared from cassava starch revealed an amphiphilic surface and a double bond due to the anchoring of the OSA chain, which make it a promising candidate for the high-performance biomimetic GPx. On the one hand, the double bond in the OSA-starch can be reacted with sodium hydrogen selenide (NaSeH) by nucleophilic addition to offer the starch with –SeH, the catalytic center of GPx. On the other hand, the structure of OSA-starch with an amphiphilic surface provides the possibility to simulate catalytic factors such as the hydrophobic microenvironment and substrate recognition site.
The process for preparing the antioxidant cassava starch (AO-ca-starch) with GPx activity is shown in Scheme 1. The cassava starch was first modified with octenyl succinic anhydride to produce octenyl succinic anhydride modified cassava starch (OSA-ca-starch). The OSA-ca-starch was then reacted with NaSeH under nitrogen to provide the starch with a catalytic center and hydrophobic microenvironments. The catalytic behavior and mechanism of the antioxidant cassava starch were also studied. This work would open a new way for the preparation and application of Se-functionalized starch.
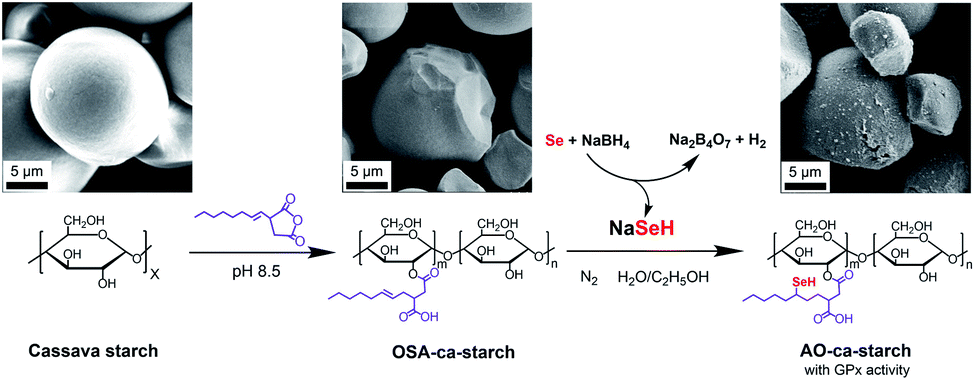 | ||
| Scheme 1 Graphical representation of the preparation of AO-ca-starch using cassava starch as the raw material. | ||
Material and methods
Material
Cassava starch was purchased from Guangxi Nongken Mingyang Biochemical Group Co., Ltd. Octenyl succinic anhydride (OSA), 4-nitrothiophenol (NBT), and cumene hydroperoxide (CUOOH) were purchased from J&K Scientific Ltd. Selenium (Se) and pyrene were commercially acquired from Shanghai Aladdin Bio-Chem Technology Co., Ltd. 3-Carboxy-4-nitrothiophenol (TNB) was prepared according to the previously reported method by Dong et al.30 All chemicals were of analytical grade and used without further purification.Preparation of OSA-ca-starch
Cassava starch (30.0 g) was dispersed in 70 g of water to produce a starch slurry (30%, w/w). OSA (1.05 g, 3.5% of weight of starch) was dissolved in 5 mL of ethanol and then slowly added to the starch slurry. A NaOH solution (3 wt%) was used to adjust the solution pH to be about 8.5. The reaction was performed at 40 °C for 6 h. After that, deionized water and 75% ethanol were used alternately to wash the product until the eluate became neutral. The octenyl succinic anhydride modified cassava starch (OSA-ca-starch) was obtained after vacuum drying at 55 °C for 24 h.Determination of the double bond content of OSA-ca-starch
The degree of substitution (DS) was measured by the titration method as described by He et al.31 Typically, OSA-ca-starch (0.5 g) was first acidified by mixing it with 3 mL of HCl ethanol solution (2.5 M) for 30 min. The suspension was filtered and the residue was washed with ethanol until no chloride ions were detected by a AgNO3 solution (0.1 M). The residue was dispersed again in distilled water (30 mL) and heated in a boiling water bath for 30 min. The resulting mixture was titrated with a NaOH solution (0.1 M), using phenolphthalein as the end-point indicator. The DS and the double bond (−CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH−) content of OSA-ca-starch (n) were calculated using the following equations:
CH−) content of OSA-ca-starch (n) were calculated using the following equations:
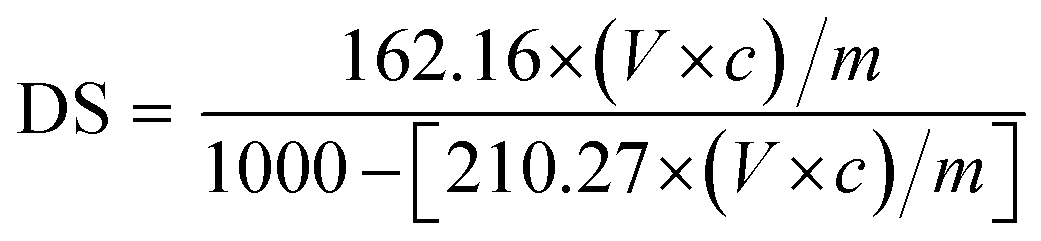 | (1) |
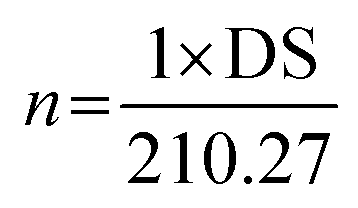 | (2) |
Preparation of AO-ca-starch
The sodium hydrogen selenide (NaSeH) solution was prepared by mixing the selenium powder and sodium borohydride (NaBH4) in distilled water under nitrogen. OSA-ca-starch (5.0 g) was dispersed in ethanol (the volume ratios of ethanol and distilled water were 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3, 6:4, 5:5, 4:6 and 3:7, respectively). Then, an appropriate volume of NaSeH solution (the molar ratios of NaSeH to double bond were 2:1, 4:1, 6:1, 8:1 and 10:1, respectively) was added to the OSA-ca-starch slurry under nitrogen. This reaction system was stirred at different temperatures (20, 30, 40, 50 and 60 °C, respectively) for different durations (2, 4, 6, 8 and 10 h, respectively). After that, the product was filtered and washed in a nitrogen atmosphere. The targeted AO-ca-starch was obtained after vacuum drying at 55 °C for 24 h.
:3, 6:4, 5:5, 4:6 and 3:7, respectively). Then, an appropriate volume of NaSeH solution (the molar ratios of NaSeH to double bond were 2:1, 4:1, 6:1, 8:1 and 10:1, respectively) was added to the OSA-ca-starch slurry under nitrogen. This reaction system was stirred at different temperatures (20, 30, 40, 50 and 60 °C, respectively) for different durations (2, 4, 6, 8 and 10 h, respectively). After that, the product was filtered and washed in a nitrogen atmosphere. The targeted AO-ca-starch was obtained after vacuum drying at 55 °C for 24 h.
Determination of the selenium content
Typically, 0.2 g of starch sample was added into a conical flask containing 10 mL of HNO3–HClO4 mixed acid (VHNO3:VHClO4 = 4:1). This sample was thermally digested at 120, 150 and 180 °C for 1 h, respectively, and then deacidified at about 220 °C. After that, it was reduced with 2.5 mL of HCl solution (6 M) for 6 h. The resulting solution was diluted with a HCl solution (10%, volume fraction), and the selenium concentration was measured using an atomic fluorescence spectrometer (AFS, AFS-390, Haiguang Instrument). All measurements were carried out in triplicate and the average value was presented.
Determination of GPx activity
The catalytic activity of GPx was assayed according to the Hilvert's method,32 using thiophenols (ArSH) and hydroperoxides (ROOH) as substrates using an Ultraviolet-visible Spectrophotometer (UV2600, Shimadzu). First, 700 μL of phosphate buffer (PBS, pH = 7.0, 50 mM), 100 μL of starch sample dispersion and 100 μL of thiophenol substrate solution (1.5 mM) were added into a quartz cuvette (1 mL, L = 1 cm). The mixture in the quartz cuvette was pre-incubated for 1 min at room temperature under ultrasound. The enzymatic reaction was initiated by adding 100 μL of ROOH (2.5 mM). The decrease in absorption at 410 nm (ΔA) was monitored using a Ultraviolet-visible Spectrophotometer (εNBT = 14600 M−1 cm−1, εTNB = 13600 M−1 cm−1, pH = 7.0). The control test was performed using the same method mentioned above, but the starch sample dispersion was replaced with 100 μL of PBS. The initial reaction rate (v0) of the enzymatic reaction, namely, the activity of GPx, was calculated using the following equation:
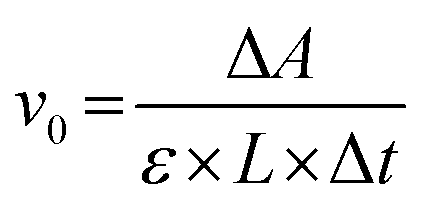 | (3) |
Characterizations
The proton nuclear magnetic resonance (1H NMR) spectra for starch samples were recorded using a Bruker AVANCE III HD 500 MHz NMR spectrometer. The sample was dissolved in deuterium oxide (D2O).The Fourier-transform infrared (FT-IR) spectroscopic measurements for the related samples were performed using a PerkinElmer Frontier FT-IR spectrometer with the attenuated total reflectance (ATR) mode in the region from 500 to 4000 cm−1.
The X-ray diffraction (XRD) patterns for the relevant samples were recorded using a Bruker D8 ADVANCE X-ray diffractometer at 40 mA and 40 kV. The scanning range of diffraction angle (2θ) was from 5 to 50°. Prior to the analysis, the samples were equilibrated at 50 °C for 24 h. The relative crystallinity for the samples was quantitatively estimated according to the previously reported method by Frost et al.33 Briefly, the crystallinity degree was considered to be the ratio of crystalline domain area (the area of the diffraction peaks) to total area of the XRD pattern:
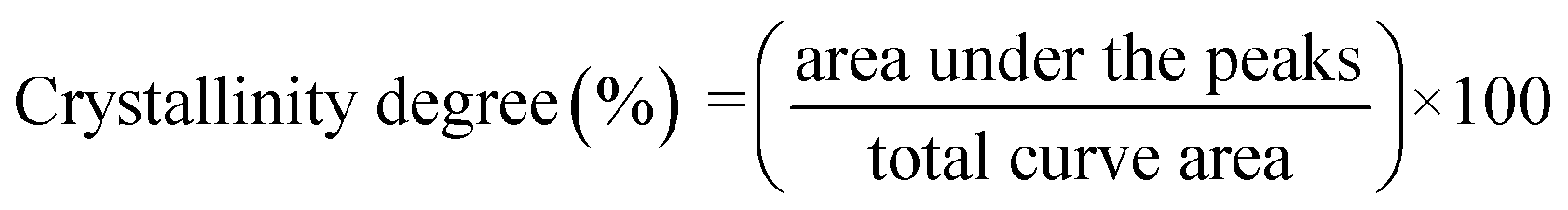 | (4) |
The test of the thermogravimetric analysis was performed using a Mettler-Toledo TGA/DSC1 synchronous thermal analyzer in the temperature range from 30 to 600 °C at a heating rate of 10 °C min−1.
A JEOL FESEM 6700F Scanning Electron Microscope (SEM) was employed to observe the morphology of the relevant samples.
Analysis of the hydrophobicity of starch samples
The hydrophobicity of modified starch sample was analyzed by the pyrene fluorescence probe method. Typically, 100 μL of pyrene solution (5 × 10−4 mol L−1) was added into 10 mL of the starch sample dispersion (2 mg mL−1) followed by ultrasonic treatment for 30 min at room temperature. The fluorescence spectrum of the resulting mixture was recorded using an Agilent Cary Eclipse Fluorospectrophotometer at an excitation wavelength of 334 nm. The intensity ratio (I1/I3) of the peak at λ1 = 372 nm and the peak at λ3 = 383 nm was employed to evaluate the hydrophobic property.34Cytotoxicity test of starch samples
The cytotoxicity of starch samples was examined by the methylthiazol tetrazolium (MTT) assay.35 In the study, human hepatocellular carcinoma cell line HepG2 was obtained from Cell Bank of the Type Culture Collection Committee of the Chinese Academy of Sciences. Briefly, 100 μL of HepG2 cell suspension (2 × 105 cells per mL) was added into each well of a 96-well plate and incubated for 24 h under standard conditions (37 °C, 5% CO2, >90% humidity). After incubation, the substrate was exchanged with 100 μL of starch sample slurry with different concentrations (10, 50, 100, 250, 500, 1000, and 2000 μg mL−1) and a fresh culture medium (control group), respectively. The cell was further incubated for 24 h, and then the culture medium was removed. The resulting cell was washed with PBS. Then, 100 μL of MTT solution (5 mg mL−1) was introduced into each well and then the cells were incubated for another 4 h under light-free conditions. Subsequently, the culture medium was removed and the resulting formazan crystals were fully dissolved in 100 μL of dimethyl sulfoxide in each well. The optical density (OD) at 490 nm was measured using a microplate reader (PerkinElmer Victor X5). The relative cell viability (RCV) was calculated according to the following equation:
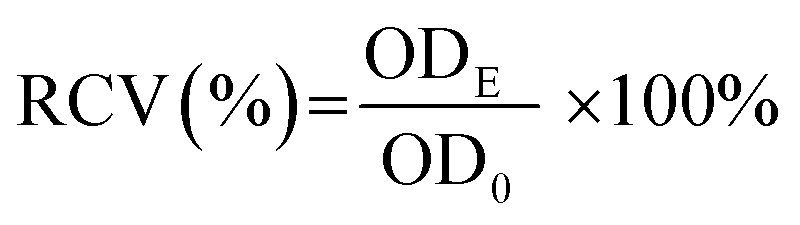 | (5) |
Results and discussion
Preparation and structure characterization of AO-ca-starch
In this work, the widely used cassava starch was used as the raw material to produce the antioxidant starch and mimic the antioxidant catalytic behavior of native GPx. As shown in Scheme 1, the double bond (−CHCH−) group on OSA-ca-starch was reacted with NaSeH by nucleophilic addition to anchor the selenium element (−SeH), the catalytic center of biomimetic GPx, on the surface of the starch. AFS and 1H NMR were used to analyze the selenium content and the changes in functional groups of AO-ca-starch.
The selenium content of the sample was measured by AFS to optimize the reaction conditions. Unsurprisingly, selenium was not detected for the native cassava starch and the OSA-ca-starch. However, the selenium content of AO-ca-starch was changed with the reaction condition including reaction time (A), reaction temperature (B), molar ratio of NaSeH to double bond (–CHCH–) of OSA-ca-starch, and volume ratio of ethanol (D), as shown in Fig. 1A, B, C, and D, respectively.
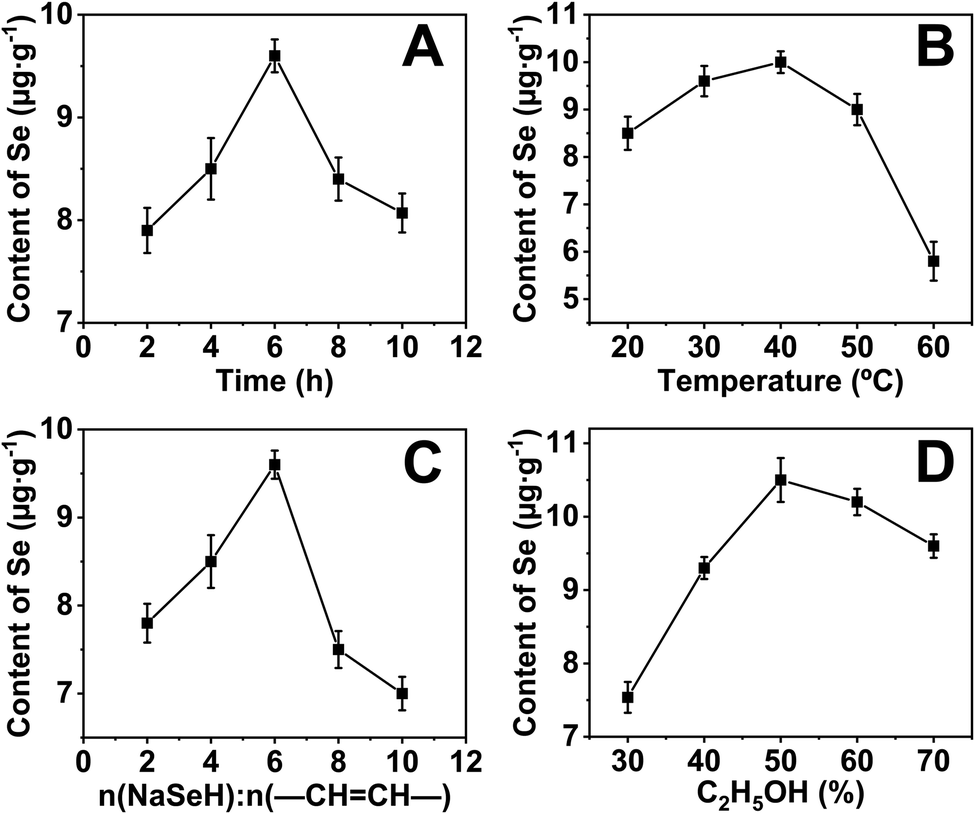 | ||
| Fig. 1 Selenium contents of AO-ca-starch as a function of reaction time (A), reaction temperature (B), molar ratio of NaSeH to double bond (−CHCH−) of OSA-ca-starch (C) and volume ratio of ethanol (D). | ||
In order to investigate the effect of reaction time on the selenium content of AO-ca-starch, the reaction time was set to 2, 4, 6, 8, and 10 h, while other conditions including reaction temperature, molar ratio of NaSeH to double bond, and volume ratio of ethanol were fixed to be 30 °C, 6:1, and 70%, respectively. As shown in Fig. 1A, the selenium content of AO-ca-starch gradually increased with the reaction time at the beginning 6 h. However, the further prolongation of the reaction time resulted in a decrease in the selenium content. A possible explanation was the reversible reaction of esterification between starch and OSA. Therefore, the optimal reaction time for the preparation of AO-ca-starch was 6 h.
For the effect of reaction temperature, a reaction time of 6 h, a volume ratio of ethanol of 70%, and a molar ratio of NaSeH to double bond of 6:1 were chosen. As shown in Fig. 1B, the selenium content of AO-ca-starch slightly rose to a high point as the reaction temperature increased and peaked at 40 °C. The further increase in temperature declined the selenium content. A possible explanation for these results was that the nucleophilic addition reaction was dominant at low temperatures, while the elimination of C–Se predominated the reaction at high temperatures. Therefore, the optimal temperature was 40 °C.
The effect of the molar ratio of NaSeH to double bond (−CHCH−) of OSA-ca-starch on the selenium content of AO-ca-starch was investigated in the system with the conditions of 6 h reaction time, 70% volume ratio of ethanol and 30 °C reaction temperature. The result is shown in Fig. 1C. The selenium content of AO-ca-starch reached a peak with the increase in the molar ratio of NaSeH to double bond from 2:1 to 6:1, and fell when the molar ratio of NaSeH to double bond was further increased. The increase in NaSeH, as a reactant, could promote its contact to the OSA-ca-starch, and consequently enhance the nucleophilic addition reaction. However, the exceeded NaSeH (the molar ratio of NaSeH to double bond was greater than 6:1), a strong alkaline substance, could raise the alkalinity of the reaction system. It might cause the hydrolysis of the ester bond in the starch and strip the OSA chains, thereby reducing the selenium content. Therefore, the optimal molar ratio of NaSeH to double bond was 6:1.
The volume ratio of ethanol to water is another important factor to influence the reaction due to the change in the reaction medium. The effect of volume ratio of ethanol to water on the selenium content of AO-ca-starch was studied in the system with the conditions of 6 h reaction time, 30 °C reaction temperature, and 6:1 molar ratio of NaSeH to the double bond. As shown in Fig. 1D, the selenium content of AO-ca-starch increased with the volume ratio of ethanol and peaked at 50% volume ratio of ethanol; the selenium content decreased with the further promotion of the volume ratio of ethanol. At the low volume ratio of ethanol, the increase in the concentration of ethanol in the reaction medium could decrease the polarity of the reaction medium, which benefited the solubility of OSA chains anchored on the starch granules and facilitated the nucleophilic reactions. However, in the reaction medium with high content of ethanol, the free starch molecular chains on the surface would be re-aggregated due to the low polarity of the medium, which might hinder the exposure of the double bonds for reaction, and consequently, decrease the selenium content of product. Therefore, the optimal volume ratio of ethanol for the ethanol/water system was 50%.
Taken together, the optimal conditions for preparing AO-ca-starch were considered to be 6 h reaction time, 40 °C reaction temperature, 6:1 molar ratio of NaSeH to double bond, and 50% volume ratio of ethanol for the ethanol/water system. The selenium content of AO-ca-starch prepared under this condition was 11.2 μg g−1, and such AO-ca-starch was selected as a biomimetic GPx for studying the catalytic behavior and mechanism.
The 1H NMR spectra of cassava starch, OSA-ca-starch, and AO-ca-starch are shown in Fig. 2. All characteristic peaks were assigned based on the previous report in the literature.36 Signal peaks at about 5.46 ppm (b) and 3.5–4.3 ppm (c), attributed to the protons of –CH2− and –CH− in the starch glucose unit, appeared in both 1H NMR spectra of cassava starch and OSA-ca-starch. Compared to the cassava starch, OSA-ca-starch revealed several new signal peaks at 5.56 ppm (a), and 0.8–2.8 ppm (d), respectively. The signal of a in the 1H NMR spectrum of OSA-ca-starch was attributed to the protons of –CHCH− in the OSA side chain; the signal peaks at d represented the protons of the OSA chain except the double bond.37 These signals at a and d indicated that the OSA chain was anchored on the starch through the esterification reaction. In the 1H NMR spectrum of AO-ca-starch, the exhibition of the signals of b, c and d suggested that the main skeleton structure of OSA-ca-starch remained during modification. However, the peak of a almost disappeared, indicating the consumption of –CHCH− during the reaction. It was found that –CHCH− in the OSA-ca-starch was the only active site for the reaction to NaSeH, a strong nucleophilic reagent. Therefore, it could be speculated that the disappearance of the signal double bond may be caused by the nucleophilic addition reaction between NaSeH and the double bond.
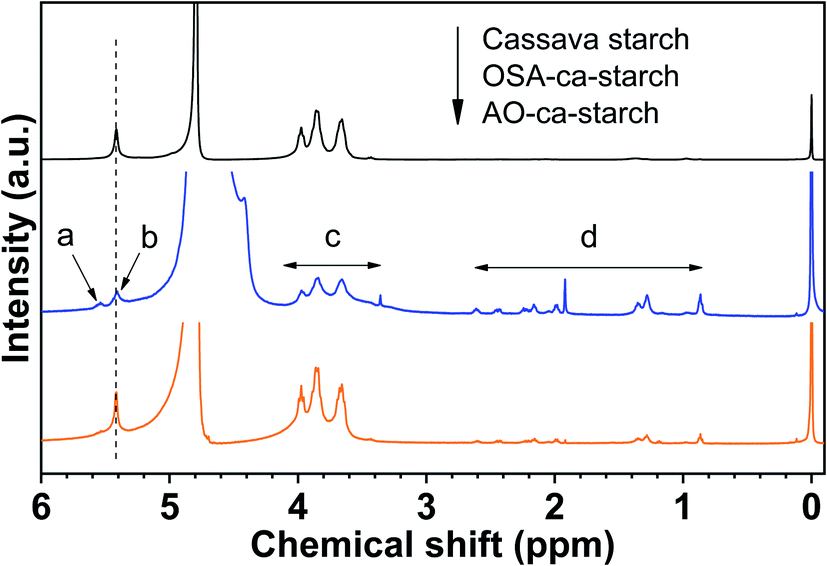 | ||
| Fig. 2 1H NMR spectra of cassava starch, OSA-ca-starch and AO-ca-starch. | ||
The structural changes of the starch during the modification were further characterized by SEM, FT-IR, XRD and TGA.
The SEM images of cassava starch (a), OSA-ca-starch (b) and AO-ca-starch (c) are shown in Fig. 3. Cassava starch revealed a spheroidal or hemispheric structure with a smooth surface. After esterification with OSA, the starch granules maintained their original shape but showed some slight corrosion on the surface. This result is similar to other report for the OSA-starch.38 The AO-ca-starch almost retained the granule structure of the starch with the same size, but exhibited a rough surface. This surface was probably due to the erosion of the strongly alkaline NaSeH solution and/or the aggregation of the anchored OSA chains and some free starch chains during the preparation process. However, the complete structure of AO-ca-starch implied that the nucleophilic addition reaction did not damage the spherical structure of OSA-ca-starch, that is, most of the active sites (−SeH) were anchored on the surface of the starch, which would benefit the exposure of the active sites for catalytic reactions.
 | ||
| Fig. 3 SEM images of cassava starch (a), OSA-ca-starch (b) and AO-ca-starch (c). | ||
The FT-IR spectra of cassava starch, OSA-ca-starch and AO-ca-starch are presented in Fig. 4A. In the spectrum of cassava starch, the peaks at 3300 cm−1, 2930 cm−1 and 1640 cm−1 are assigned to the –OH stretch, –CH stretch and the absorbed water characteristic peak, respectively. The major adsorption bands in the region 1400–1000 cm−1 are attributed to the –CH2 bending vibration, C–O, C–C and C–O–H stretching vibration and C–O–H bending vibration. In comparison to the FT-IR spectrum of cassava starch, the OSA-ca-starch exhibited two new peaks at 1724 and 1572 cm−1, which are assigned to the stretching vibration of the ester substituent group (CO) and the asymmetric stretching vibration of the carboxyl group (−COO−).37 In addition, the intensity of the peak at 1640 cm−1 is decreased by the modification. These results indicated that OSA chains were linked on cassava starch by the reaction between OSA and the –OH of starch, which endowed the starch with hydrophobicity (derived from the OSA chains) and dropped the absorbed water of starch. For the AO-ca-starch, the FT-IR spectrum showed the essentially identical peaks to that of OSA-ca-starch, indicating that the molecular framework of starch was stable during the selenized modification. It is consistent with the 1H NMR results, suggesting that AO-ca-starch was successfully synthesized by the nucleophilic addition and the starch structure was retained during the modification.
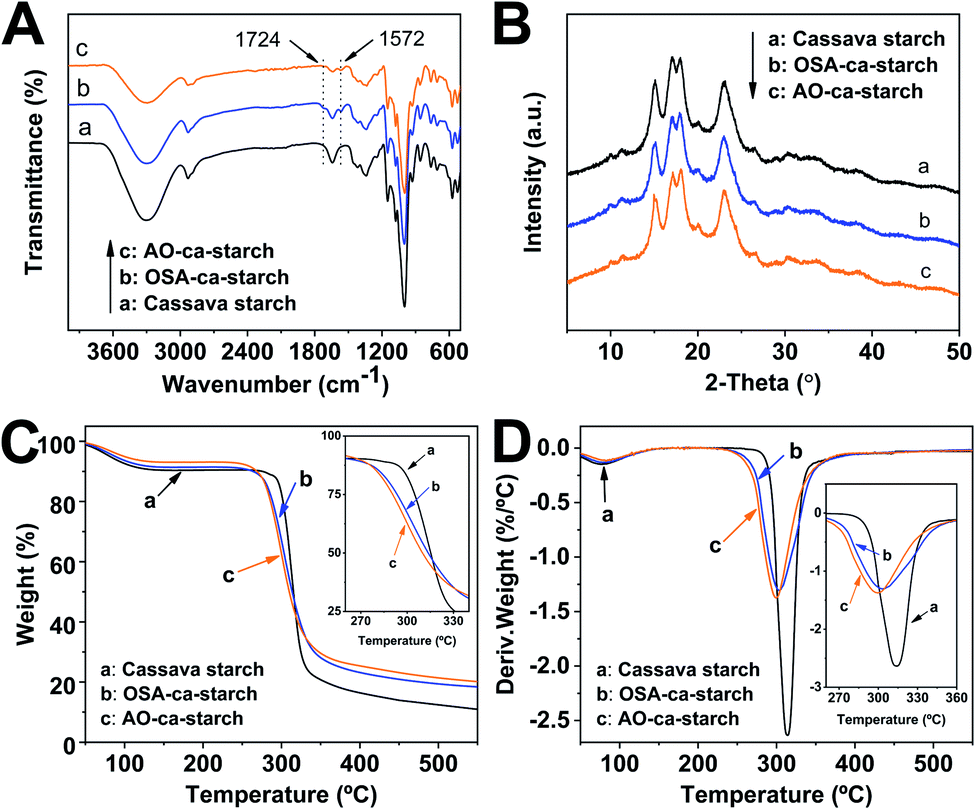 | ||
| Fig. 4 FT-IR spectra (A), XRD patterns (B), TGA curves (C) and DTG curves (D) of cassava starch, OSA-ca-starch and AO-ca-starch. | ||
X-ray diffraction patterns of cassava starch, OSA-ca-starch and AO-ca-starch are shown in Fig. 4B. The XRD patterns of all starch samples consisted of the peak diffraction and the scattering diffraction, designating the non-crystalline region and crystalline region in the starch granules. All starch samples showed a typical A-type diffraction pattern with peaks at 15, 17, 18, and 23°,39 indicating that the modifications did not destroy the crystal structure of starch. The crystallinity degree of OSA-ca-starch (43.5%) was close to that of the cassava starch (44.1%), suggesting that the esterification occurred primarily on the surface and/or in the amorphous region of native cassava starch. However, the crystallinity degree of AO-ca-starch was reduced to 38.1%. Due to the nucleophilic addition occurring in the OSA molecular chain, which did not affect the crystallinity of starch, the mild reduction of the crystallinity degree might be caused by the strong alkaline environment.40
In order to further study the changes in starch during the modification, thermogravimetric analysis (TGA) was used to analyze the thermal decomposition behavior of starch. The curves of TGA and the corresponding derivative thermogravimetric analysis (DTG) for the cassava starch, OSA-ca-starch, and AO-ca-starch are presented in Fig. 4C and D, respectively. The thermal decomposition for all starch samples could be mainly divided into two stages in the DTG curves.41 The first stage between 50 and 150 °C was contributed to the physical dehydration; the second stage from 250 to 350 °C was ascribed to the decomposition of the polymer chain of starch. As shown in Fig. 4C, the weight loss of the dehydration for AO-ca-starch, OSA-ca-starch and cassava starch was 6.9, 8.6 and 9.5%, respectively. Compared with the cassava starch, the OSA-ca-starch showed a lower weight loss, which might be due to the anchoring of the hydrophobic OSA chains on the surface of starch to suppress its water absorption. For the AO-ca-starch, the selenized modification was performed in the alkali-alcohol mixture, which could dehydrate the starch, resulting in a lower water content in comparison to the OSA-ca-starch. These results were in accordance with the decrease in the peak intensity at 2800–3500 cm−1 for the relevant samples (Fig. 4A). As shown in Fig. 4D, The DTG peaks for the polymer chain decomposition of OSA-ca-starch and AO-ca-starch were located at about 304 and 300 °C, respectively, which are lower than that of cassava starch (315 °C). This was probably due to that the modification mildly corroded the surface of starch, which benefited the heat transfer and consequently accelerated the decomposition of the polymer chain. Even so, the modified starch, with a starting decomposition temperature higher than 250 °C, maintained a favorable thermostability.
Antioxidant catalytic behavior and catalytic mechanism of AO-ca-starch
The catalytic activity (v0, μM min−1) of native GPx and biomimetic GPx was used as the anti-oxidative indicator and tested using the method established by Hilvert et al.32 As shown in Scheme 2, the anti-oxidation catalytic rate (v0, μM min−1) of AO-ca-starch was measured using the thiophenol (ArSH) substrate and hydroperoxide (ROOH) substrate as the dual substrate, in which the thiophenol substrate was 3-carboxy-4-nitrothiophenol (TNB) or 4-nitrothiophenol (NBT), while the hydroperoxide substrate was cumene hydroperoxide (CUOOH) or hydrogen peroxide (H2O2). For the thiophenol substrates, the additional carboxyl group in TNB increases its hydrophilicity in comparison to the NBT. For the hydroperoxide substrates, CUOOH with a p-cumyl group reveals more hydrophobicity than that of H2O2. In order to test v0, one molecule catalytic center (Se-monomer) in the AO-ca-starch was regarded as one active site. The v0 values for reducing ROOH (CUOOH or H2O2, 250 μM) by ArSH (TNB or NBT, 150 μM) in the presence of AO-ca-starch at pH 7.0 (50 mM PBS) are shown in Table 1. Intuitively, the v0 value was changed in the system with different substrates. It is speculated that the difference in v0 for AO-ca-starch in different substrate systems was caused by the various binding abilities between AO-ca-starch and substrates. The related mechanism would be studied later. In addition, AO-ca-starch showed a significantly high antioxidant activity. For example, the v0 value for AO-ca-starch in the system containing NBT and CUOOH was 11.79 ± 0.45 μM min−1, which is almost 4.95 × 105 times of the value of PhSeSePh. Meanwhile, the v0 value for AO-ca-starch in the system containing TNB and H2O2 was greater than that of the micellar GPx mimic, a micellar catalyst that was considered to be an excellent GPx mimic with high substrate recognition ability and catalytic activity.42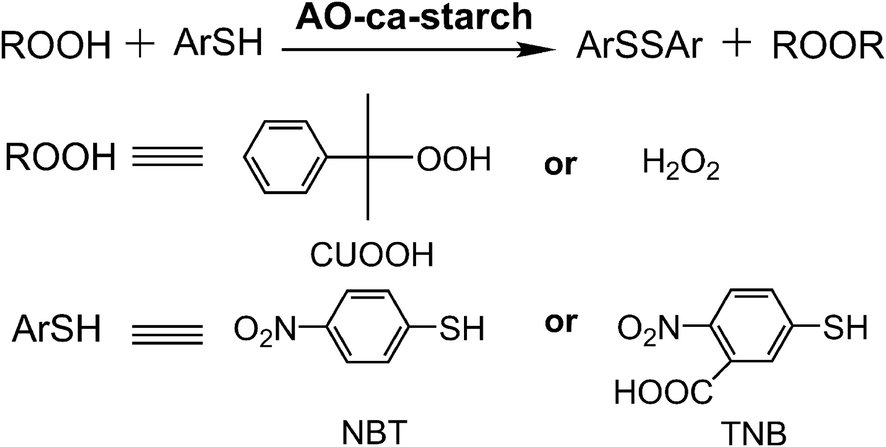 | ||
| Scheme 2 Determination of GPx activity of AO-ca-starch for using ArSH (TNB or NBT) and ROOH (CUOOH or H2O2) as substrates. | ||
| Catalyst | ArSH | ROOH | v0a (μM min−1) |
|---|---|---|---|
| a The v0 was calculated based on 1.0 μM selenium monomer.b No detected. | |||
| PhSeSePh | NBT | CUOOH | 2.38 × 10−5 |
| Micellar catalyst | TNB | CUOOH | 2.25 ± 0.24 |
| OSA-ca-starch | NBT | CUOOH | NDb |
| AO-ca-starch | TNB | CUOOH | 9.26 ± 0.37 |
| AO-ca-starch | TNB | H2O2 | 6.32 ± 0.31 |
| AO-ca-starch | NBT | CUOOH | 11.79 ± 0.45 |
| AO-ca-starch | NBT | H2O2 | 7.74 ± 0.33 |
In order to further investigate the catalytic behavior, the concentration of TNB or NBT was fixed to be 150 μM, the v0 values of AO-ca-starch at different reaction systems containing TNB or NBT and ROOH (CUOOH or H2O2) with different concentrations were measured. As shown in Fig. 5, for all reaction systems, the v0 value of AO-ca-starch showed a similar variation trend to the ROOH (CUOOH or H2O2) concentration increase, that is, the v0 value increased with the increase in ROOH concentration and then reached the equilibrium. These profiles of v0 against ROOH concentrations for AO-ca-starch in the four systems are similar to the real catalytic behavior of native GPx and show a typical saturation kinetic.42,43 The plots of the reciprocal of the v0 value per Se-monomer group ([E]0/v0) against the reciprocal of the ROOH concentration (1/[COOH] or 1/[H2O2]) are presented in Fig. 6. These double-reciprocal plots are regarded as the Lineweaver–Burk plots, which are parallel to each other at different concentrations of ArSH, suggesting that the catalytic mechanism of AO-ca-starch toward the reaction between ArSH and ROOH is similar to native GPx, following the “ping-pong” catalytic mechanism.30,44
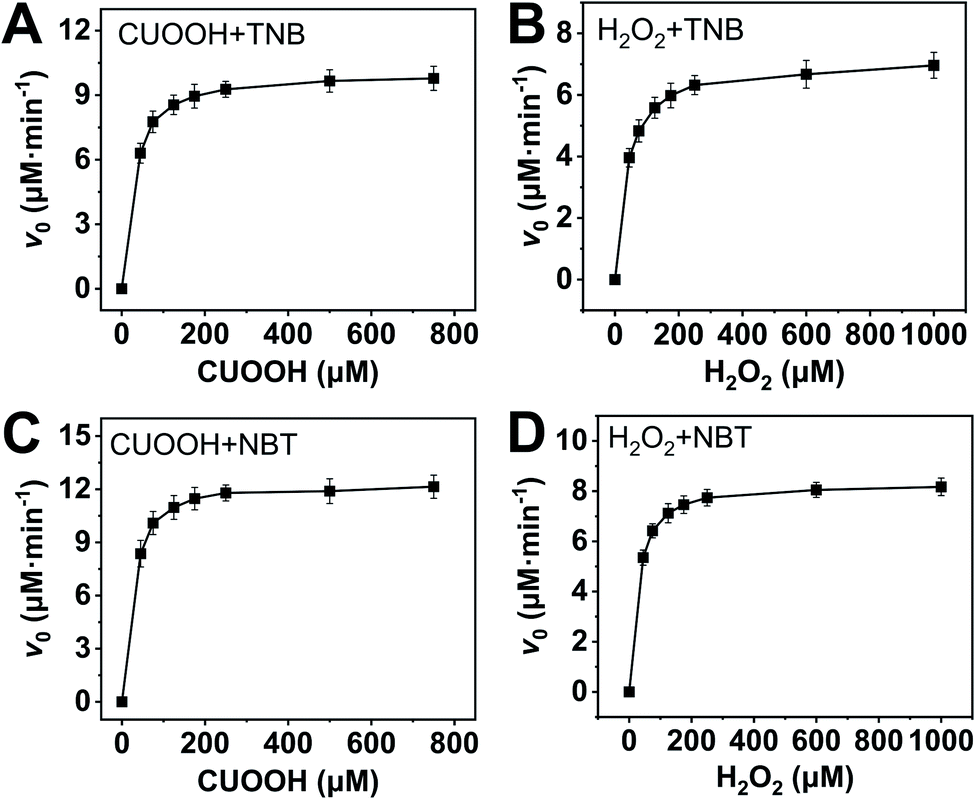 | ||
| Fig. 5 Plots of v0 against ROOH with different concentrations at the system containing ArSH (150 μM): CUOOH + TNB (A); H2O2 + TNB (B); CUOOH + NBT (C); H2O2 + NBT (D). | ||
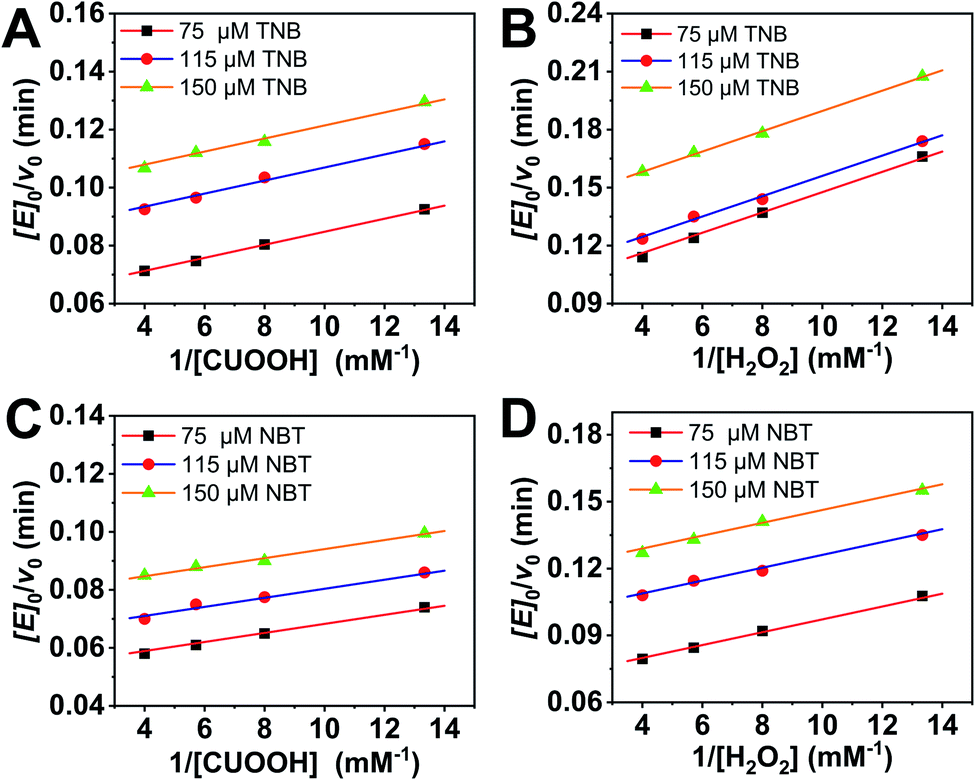 | ||
| Fig. 6 Double reciprocal plots of catalytic rate versus hydroperoxide concentration for AO-ca-starch at ArSH concentrations of 75, 115 and 150 μM: TNB + CUOOH (A); TNB + H2O2 (B), NBT + CUOOH (C), and NBT + H2O2 (D). | ||
The double-reciprocal plots derived from the ArSH concentration of 150 μM were selected to calculate the catalytic kinetic constants including the maximum reaction rate (vmax, μM min−1), the reaction constant (Kcat, min−1), Michaelis–Menten constant (Km, μM), and the catalytic efficiency (Kcat/Km, M−1 min−1). In general, Km represents the concentration of the substrate when the reaction rate is half the maximum reaction rate, reflecting the binding strength between the substrate and the enzyme.30,44,45 As shown in Table 2, the values of Km(CUOOH) are smaller than that of Km(H2O2) in both reaction systems where TNB and NBT were employed, suggesting the higher affinity of AO-ca-starch toward CUOOH in comparison to H2O2. In addition, the AO-ca-starch revealed a higher vmax and Kcat/Km when catalyzing the reaction of CUOOH and ArSH than that the reaction was conducted by H2O2 and ArSH, indicating that the reaction efficiency of AO-ca-starch toward CUOOH reduction was higher than that of the H2O2 reduction. In these reaction systems, the ROOH (CUOOH and H2O2) was the only difference. Therefore, the structural difference of ROOH was the main cause of the changes of the kinetic constants. The CUOOH is more hydrophobic than H2O2 due to the bearing of the p-cumyl group, which exhibited a higher reducing capacity under the catalysis of AO-ca-starch. Therefore, it could be concluded that AO-ca-starch is more suitable for capturing the hydrophobic substrates for the catalytic reaction.
| Reaction | vmax (μM min−1) | Kcat (min−1) | Km(ROOH) (μM) | Kcat/Km (×105 M−1 min−1) |
|---|---|---|---|---|
| TNB + CUOOH | 10.11 | 10.11 | 22.75 | 4.44 |
| TNB + H2O2 | 7.29 | 7.29 | 38.29 | 1.90 |
| NBT + CUOOH | 12.73 | 12.73 | 19.87 | 6.41 |
| NBT + H2O2 | 8.51 | 8.51 | 24.50 | 3.47 |
This result was further testified by changing the reductive substrates (ArSH). NBT revealed a higher hydrophobicity in comparison to TNB due to the absence of the carboxyl group. However, compared with TNB, the catalytic reaction with NBT as the reductive substrate showed a lower Km(ROOH) and a higher Kcat/Km in both systems containing CUOOH and H2O2 (Table 2). Appreciably, the reaction between two hydrophobic substrates (NBT and CUOOH) catalyzed by AO-ca-starch revealed the highest vmax, Kcat, and Kcat/Km, and the smallest Km compared to other reactions, whereas the AO-ca-starch-catalyzed reaction of TNB and H2O2 displayed the lowest vmax, Kcat, and Kcat/Km, but the highest Km. These results are in good agreement with the v0 value of AO-ca-starch in different reaction systems (Table 1). The AO-ca-starch revealed a higher v0 in the reaction system with more hydrophobic substrates. For example, the v0 value for the AO-ca-starch-catalyzed reaction of NBT and CUOOH is the maximum one among the four kinds of substrate combinations (Table 1).
According to the previous reports in the literature,23,43,46,47 the hydrophobic microenvironments in the GPx mimic and the active sites play an important role in offering the high catalytic activity. The anchoring of the hydrophobic OSA chains on the starch surface provided the hydrophobic microenvironments for gathering the hydrophobic substrates via hydrophobic interaction, which would benefit the catalytic reaction. Generally, the rate of the spontaneous reaction between ArSH and ROOH is in the order v0(H2O2) > v0(CUOOH).48 However, the v0 value for AO-ca-starch conducted in the system containing CUOOH was greater than that in the system containing H2O2 (Table 1). This result suggested that the hydrophobic microenvironment played an important role in maintaining the high catalytic activity of AO-ca-starch. However, the OSA-ca-starch, without −SeH (catalytic center), could not catalyze the reaction between ROOH and ArSH (Table 1). Therefore, –SeH was the key factor for the catalytic reaction, while the hydrophobic microenvironments in AO-ca-starch enhanced the catalytic activity. In order to testify the formation of hydrophobic microenvironments in AO-ca-starch, the pyrene fluorescent probe method was used.
Pyrene is a commonly used fluorescent probe for testing the hydrophobicity of materials as its molecular fluorescence emission spectrum is very sensitive to the polarity of the environment.49,50 The hydrophobic pyrene molecules can be assembled in the hydrophobic microenvironments of the materials, resulting in a decrease in fluorescence intensity ratio (I1/I3) of the peak at λ1 = 372 nm and the peak at λ3 = 383 nm.51 Therefore, the value of I1/I3 can be used to characterize the hydrophobic microenvironments of the targeted materials, that is, the smaller the I1/I3, the stronger the hydrophobicity of the material. The fluorescence spectra of pyrene in the aqueous solution and pyrene in dispersions of AO-ca-starch, OSA-ca-starch and cassava starch are shown in Fig. 7. The concentration of pyrene for each sample was 5 × 10−6 mol L−1. The value of I1/I3 for the pyrene/OSA-ca-starch mixture was 1.54, which is smaller than that of pyrene/starch mixture (1.75), indicating that the anchoring of the hydrophobic long-chains of OSA offered the OSA-ca-starch with some hydrophobic microenvironments. This result further indicated the successful esterification of starch with OSA. The nucleophilic addition reaction between NaSeH and OSA-ca-starch would consume –CHCH− in the OSA chains, and consequently decrease the hydrophobicity of the OSA-ca-starch. However, the value of I1/I3 for the pyrene/AO-ca-starch mixture (1.46) is slightly smaller than that of the pyrene/OSA-ca-starch mixture, suggesting more hydrophobic microenvironments were formed on the AO-ca-starch. A possible explanation is that the aggregation of the starch molecular segments and OSA chains caused a rough surface, which increased its hydrophobicity. The nucleophilic addition was conducted in the medium of ethanol and water (1:1, v/v), in which ethanol dissolved the anchored OSA chains and water dissolved some external molecular of starch. These dissolved molecular chains with high freedom could be re-aggregated during the drying, resulting in a rough surface (Fig. 3c). In a word, these results of pyrene fluorescent probe tests indicated that the hydrophobic microenvironment was formed in AO-ca-starch. Such hydrophobic microenvironment was conducive to gather hydrophobic substrates, resulting in a high catalytic activity and catalytic efficiency.
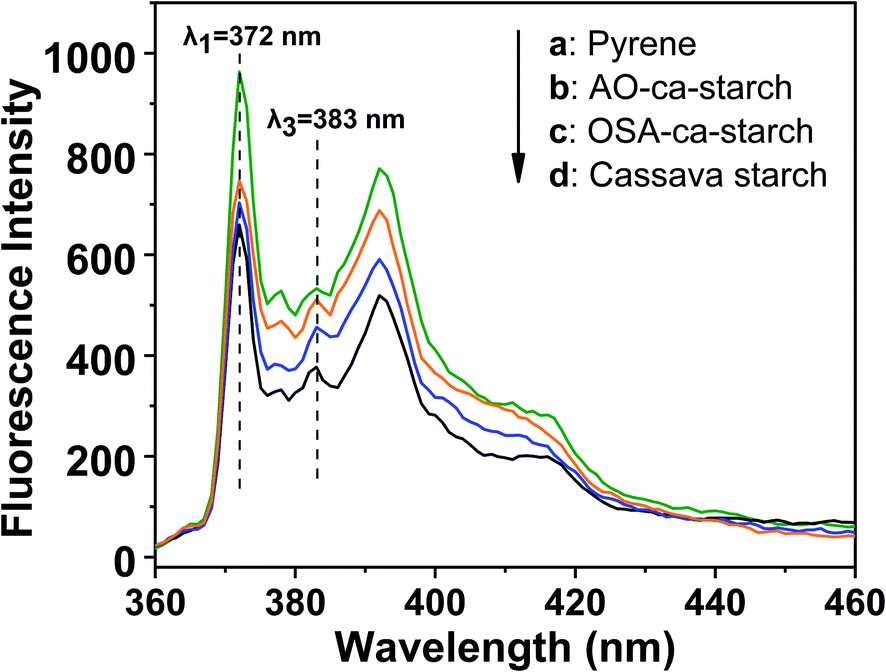 | ||
| Fig. 7 Fluorescence spectra of pyrene in solution of pyrene (a) and pyrene in dispersions of AO-ca-starch (b), OSA-ca-starch (c) and cassava starch (d). | ||
Overall, the nucleophilic addition of OSA-ca-starch with NaSeH offered the resulting starch with active sites (–SeH) and hydrophobic microenvironments, which endowed the starch with favorable antioxidant property.
Cytotoxicity of AO-ca-starch
The cytotoxicity of the native cassava starch and AO-ca-starch was measured by the MTT assay, which tested the cell growth of tumor cells HepG2 in a starch concentration-dependent manner. As shown in Fig. 8, the minimum cell viability is greater than 92% when exposed to cassava starch in the concentration ranging from 10 to 2000 μg mL−1, suggesting the negligible cytotoxicity of cassava starch. For the AO-ca-starch, the cell viability is higher than 95% at a low concentration, while at a high concentration (>200 μg mL−1), the cell viability is greater than 100%. For example, the cell viability was 108.2% and 107.7% as the cells were incubated in the media containing 1000 μg mL−1 and 2000 μg mL−1 AO-ca-starch, respectively. A likely explanation is that the AO-ca-starch with anti-oxidation property facilitated the propagation of cell. Overall, the AO-ca-starch is not toxic to cells.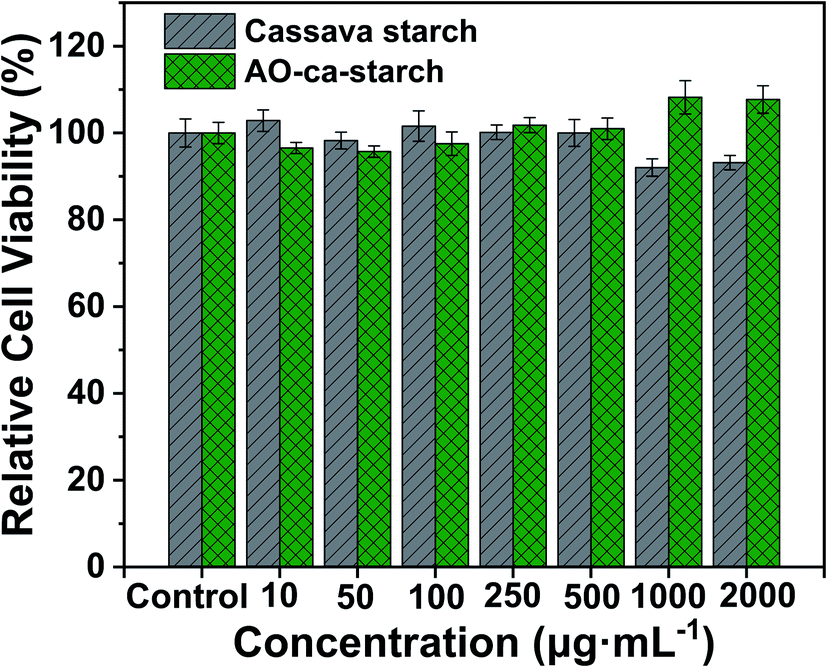 | ||
| Fig. 8 Relative cell viability as a function of concentrations of cassava starch and AO-ca-starch. | ||
Conclusions
In this work, a modified starch (AO-ca-starch) with a favorable antioxidant property was prepared to mimic GPx. The cassava starch was first modified with OSA and then reacted with NaSeH by nucleophilic addition to innovatively produce the antioxidant starch with active sites (–SeH) and hydrophobic microenvironments on the surface. The optimal conditions for the preparation of the antioxidant starch were a reaction time of 6 h, a reaction temperature of 50 °C, a molar ratio of NaSeH to double bonds of 6:1, and an ethanol volume ratio of 50%. The selenium content of AO-ca-starch prepared under the above-mentioned conditions was 11.2 μg g−1. The crystalline structure and particle size of starch did not change significantly during the modifications. With a typical saturated kinetic catalytic behavior, this AO-ca-starch revealed a catalytic activity of about 11.79 μM min−1, which is 4.95 × 105 times that of PhSeSePh and 4.12 times of the micellar catalyst. The combination of the active sites and hydrophobic microenvironments was the main contribution of the high catalytic activity. The AO-ca-starch was not toxic to cells. This work may open a new way for the functional application of selenium-enriched starch as antioxidant food and drugs.
Author contributions
Conceptualization, C. S. and Q. H.; methodology, C. S. and Q. H.; validation, C. S., Q. H. and R. Z.; investigation, C. S., F. W. and M. L.; resources, M. L. and Z. L.; writing – original draft preparation, C. S.; writing – review & editing, C. S. and X. L.; visualization, C. S.; supervision, X. L. and Y. Y.; project administration, Y. Y. and H. H.; funding acquisition, Y. Y. and R. Z. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from National Natural Science Foundation of China (51663020), Natural Science Foundation for Distinguished Young Scholars of Guangxi Province (2017GXNSFFA198007) and Project of Guangxi Colleges and Universities for the Promotion of Foundation Ability of Young Teachers (2019KY0469). The authors also acknowledge Guangxi Colleges and Universities Innovation Research Team.Notes and references
- M. A. V. T. Garcia, C. F. Garcia and A. A. G. Faraco, Starch/Staerke, 2020, 72, 1900270 CrossRef CAS.
- M. A. Labelle, P. Ispas Szabo and M. A. Mateescu, Starch/Staerke, 2020, 72, 2000002 CrossRef CAS.
- H. J. Li, T. Cai, B. Yuan, R. H. Li, H. Yang and A. M. Li, Ind. Eng. Chem. Res., 2014, 54, 59–67 CrossRef.
- N. N. Shah, C. Vishwasrao, R. S. Singhal and L. Ananthanarayan, Food Hydrocolloids, 2016, 55, 179–188 CrossRef CAS.
- A. J. Cole, A. E. David, J. Wang, C. J. Galbán, H. L. Hill and V. C. Yang, Biomaterials, 2011, 32, 2183–2193 CrossRef CAS PubMed.
- A. P. Zhang, Z. Zhang, F. H. Shi, J. X. Ding, C. S. Xiao, X. L. Zhuang, C. L. He, L. Chen and X. S. Chen, Soft Matter, 2013, 9, 2224 RSC.
- L. Altuna, M. L. Herrera and M. L. Foresti, Food Hydrocolloids, 2018, 80, 97–110 CrossRef CAS.
- T. Dapčević Hadnađev, M. Hadnađev, M. Pojić, S. Rakita and V. Krstonošić, J. Food Eng., 2015, 167, 133–138 CrossRef.
- X. Y. Wang, X. X. Li, L. Chen, F. W. Xie, L. Yu and B. Li, Food Chem., 2011, 126, 1218–1225 CrossRef CAS.
- X. R. Xiao, C. Guan, J. Xu, W. J. Fu and L. Yu, Green Chem., 2021, 23, 4647–4655 RSC.
- X. Y. Chen, J. F. Mao, C. Liu, C. Chen, H. E. Cao and L. Yu, Chin. Chem. Lett., 2020, 31, 3205–3208 CrossRef CAS.
- X. R. Xiao, Z. F. Shao and L. Yu, Chin. Chem. Lett., 2021, 32, 2933–2938 CrossRef CAS.
- M. X. Liu, X. L. Zhang, S. N. Chu, Y. Y. Ge, T. Huang, Y. H. Liu and L. Yu, Chin. Chem. Lett., 2021 DOI:10.1016/j.cclet.2021.05.061.
- X. Y. Mao, P. Z. Li, T. Li, M. M. Zhao, C. Chen, J. Liu, Z. Q. Wang and L. Yu, Chin. Chem. Lett., 2020, 31, 3276–3278 CrossRef CAS.
- H. E. Cao, R. N. Ma, S. N. Chu, J. Q. Xi, L. Yu and R. Guo, Chin. Chem. Lett., 2021, 32, 2761–2764 CrossRef CAS.
- G. M. Luo, X. J. Ren, J. Q. Liu, Y. Mu and J. C. Shen, Curr. Med. Chem., 2003, 10, 1151–1183 CrossRef CAS PubMed.
- H. Sies, Exp. Physiol., 1997, 82, 291–295 CrossRef CAS PubMed.
- O. M. Ighodaro and O. A. Akinloye, Alex. J. Med., 2018, 54, 287–293 Search PubMed.
- H. J. Yu, J. Q. Liu, A. Böck, J. Li, G. M. Luo and J. C. Shen, J. Biol. Chem., 2005, 280, 11930–11935 CrossRef CAS.
- S. Thangamani, H. E. Eldesouky, H. Mohammad, P. E. Pascuzzi, L. Avramova, T. R. Hazbun and M. N. Seleem, BBA-Gen. Subjects, 2017, 1861, 3002–3010 CrossRef CAS PubMed.
- V. P. Singh, J. F. Poon, R. J. Butcher, X. Lu, G. Mestres, M. K. Ott and L. Engman, J. Org. Chem., 2015, 80, 7385–7395 CrossRef CAS PubMed.
- S. F. Jiao, R. R. Zhang, Y. Z. Yin, S. M. Zhong, Z. J. Liu, Y. Y. Zheng, X. X. Hu, X. T. Liang and Z. Q. Huang, RSC Adv., 2019, 9, 28814–28822 RSC.
- Y. Z. Yin, S. F. Jiao, R. R. Zhang, X. X. Hu, Z. F. Shi and Z. Q. Huang, Soft Matter, 2015, 11, 5301–5312 RSC.
- S. Y. Li, W. J. Xu, S. N. Chu, N. Ma, S. D. Liu, X. M. Li, T. T. Wang, X. J. Jiang, F. Li, Y. J. Li, D. M. Zhang, Q. Luo and J. Q. Liu, Chem.–Eur. J., 2019, 25, 10350–10358 CrossRef CAS PubMed.
- D. Su, D. L. You, X. J. Ren, G. M. Luo, Y. Mu, G. L. Yan, Y. Xue and J. C. Shen, Biochem. Biophys. Res. Commun., 2001, 285, 702–707 CrossRef CAS PubMed.
- A. A. Vernekar, D. Sinha, S. Srivastava, P. U. Paramasivam, P. D Silva and G. Mugesh, Nat. Commun., 2014, 5, 1–13 Search PubMed.
- Y. Tang, L. P. Zhou, J. X. Li, Q. Luo, X. Huang, P. Wu, Y. G. Wang, J. Xu, J. C. Shen and J. Q. Liu, Angew. Chem., Int. Ed., 2010, 49, 3920–3924 CrossRef CAS PubMed.
- O. Epp, R. Ladenstein and A. Wendel, Eur. J. Biochem., 1983, 133, 51–69 CrossRef CAS PubMed.
- J. Rotruck, A. Pope, H. Ganther, A. Swanson, D. Hafeman and W. Hoekstra, Nutr. Rev., 1973, 179, 588–591 CAS.
- Z. Y. Dong, J. Q. Liu, S. Z. Mao, X. Huang, B. Yang, X. J. Ren, G. M. Luo and J. C. Shen, J. Am. Chem. Soc., 2004, 126, 16395–16404 CrossRef CAS PubMed.
- G. Q. He, X. Y. Song, H. Ruan and F. Chen, J. Agric. Food Chem., 2006, 54, 2775–2779 CrossRef CAS PubMed.
- Z. P. Wu and D. Hilvert, J. Am. Chem. Soc., 1990, 112, 5647–5648 CrossRef CAS.
- K. Frost, D. Kaminski, G. Kirwan, E. Lascaris and R. Shanks, Carbohydr. Polym., 2009, 78, 543–548 CrossRef CAS.
- J. Matsui, M. Mitsuishi and T. Miyashita, Macromolecules, 1999, 32, 381–386 CrossRef CAS.
- A. Drabczyk, S. Kudłacik-Kramarczyk, M. Głąb, M. Kędzierska, A. Jaromin, D. Mierzwiński and B. Tyliszczak, Materials, 2020, 13, 3073 CrossRef CAS PubMed.
- Y. J. Bai, Y. C. Shi, A. Herrera and O. Prakash, Carbohydr. Polym., 2011, 83, 407–413 CrossRef CAS.
- F. Ye, M. Miao, C. Huang, K. Y. Lu, B. Jiang and T. Zhang, J. Agric. Food Chem., 2014, 62, 11696–11705 CrossRef CAS PubMed.
- N. F. Zainal Abiddin, A. Yusoff and N. Ahmad, Food Hydrocolloids, 2018, 75, 138–146 CrossRef CAS.
- P. S. Hornung, L. Do Prado Cordoba, S. R. Da Silveira Lazzarotto, E. Schnitzler, M. Lazzarotto and R. H. Ribani, J. Therm. Anal. Calorim., 2017, 127, 1869–1877 CrossRef CAS.
- R. H. Lin, Y. Y. Fan, T. Liu, H. Yang, L. J. Ma, X. J. Huang and Y. Liu, Starch/Staerke, 2019, 72, 1900049 CrossRef.
- X. X. Liu, Y. F. Wang, L. Yu, Z. Tong, L. Chen, H. S. Liu and X. X. Li, Starch/Staerke, 2013, 65, 48–60 CrossRef CAS.
- X. Huang, Z. Y. Dong, J. Q. Liu, S. Z. Mao, J. Y. Xu, G. M. Luo and J. C. Shen, Langmuir, 2007, 23, 1518–1522 CrossRef CAS PubMed.
- X. Huang, Y. Z. Yin, Y. Tang, X. L. Bai, Z. M. Zhang, J. Y. Xu, J. Q. Liu and J. C. Shen, Soft Matter, 2009, 5, 1905 RSC.
- S. W. Lv, X. G. Wang, Y. Mu, T. Z. Zang, Y. T. Ji, J. Q. Liu, J. C. Shen and G. M. Luo, FEBS J., 2007, 274, 3846–3854 CrossRef CAS PubMed.
- Z. Y. Dong, X. Huang, S. Z. Mao, K. Liang, J. Q. Liu, G. M. Luo and J. C. Shen, Chem.–Eur. J., 2006, 12, 3575–3579 CrossRef CAS PubMed.
- H. X. Zou, H. C. Sun, L. Wang, L. L. Zhao, J. X. Li, Z. Y. Dong, Q. Luo, J. Y. Xu and J. Q. Liu, Soft Matter, 2016, 12, 1192–1199 RSC.
- L. Wang, H. X. Zou, Z. Y. Dong, L. P. Zhou, J. X. Li, Q. Luo, J. Y. Zhu, J. Y. Xu and J. Q. Liu, Langmuir, 2014, 30, 4013–4018 CrossRef CAS PubMed.
- Y. Z. Yin, L. Wang, H. Y. Jin, C. Y. Lv, S. J. Yu, X. Huang, Q. Luo, J. Y. Xu and J. Q. Liu, Soft Matter, 2011, 7, 2521 RSC.
- F. Mravec, M. Pekař and V. Velebný, Colloid Polym. Sci., 2008, 286, 1681–1685 CrossRef CAS.
- K. Kalyanasundaram and J. K. Thomas, J. Am. Chem. Soc., 1977, 7, 2039–2044 CrossRef.
- O. E. Philippova, E. V. Volkov, N. L. Sitnikova, A. R. Khokhlov, J. Desbrieres and M. Rinaudo, Biomacromolecules, 2001, 2, 483–490 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |