
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotoresponsive, switchable, pressure-sensitive adhesives: influence of UV intensity and hydrocarbon chain length of low molecular weight azobenzene compounds†
Tae-Hyung Lee a,
Gi-Yeon Hana,
Mo-Beom Yia,
Jae-Ho Shina and
Hyun-Joong Kim*ab
a,
Gi-Yeon Hana,
Mo-Beom Yia,
Jae-Ho Shina and
Hyun-Joong Kim*ab
aLaboratory of Adhesion and Bio-Composites, Program in Environmental Materials Science, Department of Agriculture, Forestry and Bioresources, Seoul National University, 1 Gwanak-ro, Gwanak-gu, Seoul 08826, Republic of Korea. E-mail: hjokim@snu.ac.kr
bResearch Institute of Agriculture and Life Sciences, Seoul National University, 1 Gwanak-ro, Gwanak-gu, Seoul 08826, Republic of Korea
First published on 22nd November 2021
Abstract
Unlike traditional adhesives with a fixed adhesive force, switchable adhesives, which have an adhesive force that can be adjusted by external stimuli, are specifically designed to be released according to user demand, or to enable the transfer of fine electronic devices. Previously developed switchable adhesives have limitations such as a slow switching rate, narrow adhesion modulation range, or the lack of reusability. Thus, we fabricated switchable pressure-sensitive adhesives (PSAs) that can overcome these limitations. The adhesive force of each switchable PSA, which comprises an azobenzene-containing acrylic polymer and low molecular weight compounds, was designed to be activated/deactivated via ultraviolet (UV) and visible light irradiation. The adhesive force and UV intensity required for the switch were found to be dependent on the aliphatic chain length of the compound. The adhesive force of the SP-C10, i.e., a switchable PSA containing a azobenzene compound with an aliphatic chain of 10 hydrocarbons, increased to 3.5 N from nearly zero in response to only 30 s of low-level (25 mW cm−2) UV irradiation. Additionally, SP-C10 did not lose its adhesive force even after 30 cycles of repeated adhesion switching. The mechanism of adhesion switching influenced by UV intensity and the structure of low molecular weight azobenzene compounds are also reported.
Introduction
An adhesive is a material used to attach two substrates; it is generally known as a material that is applied in a liquid state and cured into a solid through the use of moisture, heat, and/or light, forming an adhesive bond. Pressure-sensitive adhesives (PSAs) are adhesives that maintain their stickiness under the service temperature conditions, and can form an adhesive bond without being cured.1 To achieve adhesion without a liquid-to-solid phase transition, a PSA has to have a Tg that is 25–45 °C lower than service temperature,2 and the appropriate viscoelastic properties.3 Tg is a characteristic of amorphous polymers, and is the temperature at which molecular segments comprising several molecules begin to move. The wetting of an adhesive on the substrate is essential for adhesion. The relatively low PSA Tg allows them to be wetted at the service temperature and form an adhesive bond in the absence of a phase transition. Chang published a study on the viscoelastic window of PSAs.3 They were found to have specific storage modulus (G′) and loss modulus (G′′) ranges, and to have characteristics and applications that can be determined by G′ and G′′.Thus, when the Tg and viscoelastic properties of a PSA are determined by the chemical composition or film fabrication process, the adhesive force is fixed under constant environmental conditions such as temperature and humidity. General adhesives maintain their adhesive force after being cured. However, there is growing interest in adhesives that can be released from a substrate upon user demand, thereby enabling multi-functionality. This type of adhesive, i.e., switchable adhesives, is attached to the substrate with a strong force, and can be easily detached in response to external stimuli such as heat, light, water, and/or electromagnetic force.4
Heat is the most common stimulus for adhesion switching. Because the mechanical properties of the base polymer are temperature-dependent, the adhesive strength can be changed by heating or cooling it. Alternatively, an excessive temperature change is necessary to control the adhesive strength of general adhesives. In the case of conventional PSAs, a significant change in adhesive force can be achieved by applying a temperature that is at least 30–40 °C lower or higher than Tg.5 As such, liquid crystal polymers or liquid crystal elastomers have been used to reduce the temperature range required for adhesion switching.6–9 Adhesion switching is induced by a change in the mechanical or chemical properties of the comprising materials.4 Thus, owing to their better molecular mobility as compared to dried materials, many researchers have opted to use hydrogel systems to obtain the desired switchable adhesion characteristics. The adhesion properties of hydrogels can be controlled by modulating the molecular conformation or reversible bonding phases, particularly heat-, pH-, and solvent-dependent host–guest interactions or coordination with metal ions.10–15 The frequency dependence of elastomer stamp16 or cross-linking by UV light17–19 can be applied in the electronics manufacturing process to enable rapid adhesion switching that does not damage the substrate. There are also switchable adhesives that utilize shape-memory polymers,20 solvent wettability,21 and magneto-rheological materials.22,23 However, the applicability of switchable adhesives that require heat, solvent, or hydrated conditions is limited. In addition, irreversibly cross-linked adhesives cannot be reused; other methods also have limitations, such as a slow switching speed or narrow adhesive force modulation range.
Thus, we have studied photo-sensitive materials with rapid adhesion switching characteristics and a wide adhesive force modulation range in the dried state. Some compounds have photo-reversible bonds in the dried state.24 A photo-reversible cycloaddition reaction of anthracene moiety has been applied in studies to realize re-workable adhesives.25–27 Although these compounds with reversible reactions form stable bonded structures, they require long-term stimulation for bond transition. We have applied an azobenzene moiety to fabricate the photoresponsive adhesive. The azobenzene group isomerizes from the trans form to the cis form in response to UV radiation, and returns to the trans form under visible light. Although trans-azobenzene has a planar symmetric structure, cis-azobenzene has a three-dimensionally bent asymmetric structure.28 For this reason, the intermolecular arrangement and polarity of the azobenzene moiety can be converted by photoisomerization. Some studies have been conducted on switchable adhesives that incorporate an azobenzene moiety as an acrylic polymer side chain.29–34 However, these adhesives have limitations in terms of repeated usage, because separation tends to occur during the solid-to-liquid phase transition. In this study, we prepared PSAs that are capable of adhesion switching without solid/liquid phase transitioning; they comprise mixtures of a copolymer of butyl acrylate and azobenzene-containing acrylate, and low molecular weight compounds containing an azobenzene moiety. The isomerization of azobenzene moiety is influenced by its substituent, polymer containing azobenzene groups, and the environment of the surrounding matrix by electronic and steric effects.35–38 We studied the influence of substituent's steric effect on photoisomerization of azobenzene moiety and adhesion switching by varying the hydrocarbon chain length of the substituent. Our novel switchable PSA is triggered by UV and visible light irradiation, and is capable of repeated adhesion switching without any loss of adhesive force. It was found to be able to obtain a large adhesive force even at low UV intensity, and under the condition of a short exposure time of 30 s. Here, we also report on the mechanism of adhesion switching and influence of the UV intensity on the switchable PSAs.
Experimental
Materials
The following were purchased from Sigma-Aldrich: 4-phenylazophenol (98%), 1-chlorohexane (99%), 1-chlorodecane (98%), dibutyltin dilaurate (DBTDL, 95%), and butyl acrylate (>99%). Potassium carbonate (>99%), potassium iodide (>99.5%), 6-chloro-1-hexanol (>96%), 1-chlorotetradecane (>98%), and 1-chlorooctadecane (>98%) were purchased from Tokyo Chemical Industry Co., Ltd. The following were purchased from Samchun Chemicals Co., Ltd.: 2,2′-azobisisobutyronitrile (98%), 2-butanone (MEK, 99.5%), N,N-dimethylformamide (DMF, 99.5%), tetrahydrofuran (THF, 99.9%), methanol (99.5%), n-hexane (96%), acetone (99.5%), and ethyl acetate (EA, 99.5%). Lastly, 2-isocyanatoethyl acrylate (Karenz AOI, Showa Denko) was used as the isocyanate-containing acrylic monomer.Synthesis and polymerization
Preparation of switchable PSA
After dissolving the azo-polymer in THF, 12 mol% of one of the four azo-compounds was added to the polymer solution. The content of the azo-compound was calculated in consideration of the number of butyl acrylate and azo-acrylate molecules in the azo-polymer. The azo-compounds were dissolved in their respective polymer solution by using a vortex mixer. After using a 120 μm coating applicator to cast each mixture on a corona-treated polyethylene terephthalate (PET) film (50 μm), the films were dried at 100 °C for 20 min in a convection oven. After being cooled, the dried films were irradiated with UV light (365 nm LED lamp, 125 mW cm−2, 30 s) and visible light (50 W white LED lamp, 30 s). The thicknesses of the switchable PSAs were 6–7 μm, as measured by using a digital micrometer (S-Mike_Pro, Sylvac). The lap shear test specimen was prepared by applying the same method, using a silicone-release PET film (50 μm) instead of the corona-treated PET film. The PSA samples were named SP-6, SP-10, SP-14, and SP-18, according to the type of azo-compound (Table 1).| Azo-polymer | THF | Azo-C6 | Azo-C10 | Azo-C14 | Azo-C18 | |
|---|---|---|---|---|---|---|
| Azo-polymer | 0.1 g (88 mol% of butyl and azobenzene pendant groups) | 1 ml | — | — | — | — |
| SP-C6 | 25.1 mg (12 mol%) | — | — | — | ||
| SP-C10 | — | 30.1 mg (12 mol%) | — | — | ||
| SP-C14 | — | — | 35.1 mg (12 mol%) | — | ||
| SP-C18 | — | — | — | 40.1 mg (12 mol%) |
Characterization
A 400 MHz NMR spectrometer (JNM-ECX400, JEOL) was used to record the 1H-NMR spectra. The operating temperature was room temperature, and tetramethylsilane (δ = 0 ppm) was used as a reference to determine the chemical shift. The ratio of azobenzene groups present in the azo-polymer was calculated based on its 1H-NMR spectrum, which was obtained after it was washed with methanol (Fig. S8†). The ratio was calculated based on the bandsrespectively corresponding to the hydrogen of the aromatic group of azobenzene, and the CH3 at the end of butyl acrylate, as described below.
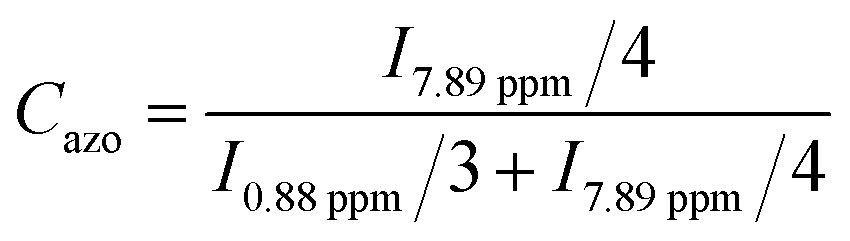 | (1) |
The adhesive forces of the switchable PSAs were evaluated by applying a probe tack test. The tack forces were measured by operating a texture analyzer (TA.XT plus, Stable Micro Systems) and a 500 N load cell at 25 °C (RH: 50 ± 10%). A cylindrical 5 mm-diameter stainless-steel probe was used. The probe was in contact with each PSA specimen for 1 s, applying a force of 100 gf; the contact speed was 0.2 mm s−1. After the probe was detached from the specimen at a speed of 10 mm s−1, the maximum force was taken as the probe tack value. The average of five measurements was used for each specimen.
The azobenzene moiety photoisomerization was monitored by using a UV/Vis spectrometer (UV-1601PC, Shimadzu) to record the UV/Vis absorption spectrum; the scan range was 300–600 nm. To measure each switchable PSA cast on the PET film, the UV/Vis absorption of the 50 μm-thick corona-treated PET film was applied as the spectrum baseline.
A lap shear test was conducted to measure the shear modulus of each switchable PSA. The PSA cast on the release film was transferred to a poly(methyl methacrylate) (PMMA) substrate with dimensions of 6 mm × 20 mm × 1 mm (width × length × thickness) after UV irradiation (125 mW cm−2, 30 s). The adhesive area was 6 mm × 20 mm (width × length). After another PMMA substrate was attached, visible light was applied for 30 s. Two pieces of PMMA with dimensions of 6 mm × 6 mm × 1 mm were attached to each end of the substrate with an instant adhesive. The shear stress was measured by using dynamic mechanical analysis (DMA, Q800, TA Instruments). The test was conducted in strain ramp mode at 25 °C; the test speed was 1% s−1. This test was applied three times for each specimen; the average value was used.
A drop-shape analyzer (DSA 100, KRÜSS) was used to measure the water contact angle of each PSA surface at 25 °C (RH: 50 ± 10%). The contact angle was measured 10 s after 5 μl of water droplets were dropped onto the surface. The average of five measurements was used for each specimen.
The Tg of the switchable PSAs and phase-transition temperatures of the azo-compounds were measured by loading each PSA (2–3 mg) onto a sample pan (Tzero Pan, 901683.901, TA Instruments) and applying differential scanning calorimetry (DSC, Q200, TA Instruments). After being cooled to −50 °C at 30 °C min−1, the temperature of each specimen was maintained for 2 min. The DSC curve was recorded as the specimen was heated to 50 °C at a rate of 5 °C min−1. A photothermal DSC method was used to measure the Tg of each UV-irradiated switchable PSA. Although the cooling and heating rates were the same, each specimen was irradiated with UV light during the cooling and isothermal steps. The heating step was carried out under dark conditions. A spot UV curing system (S2000-XLA, OmniCure) with a 320–500 nm filter was used for UV irradiation. The UV intensity at specimen surface was 75 mW cm−2. The azo-compounds (1–2 mg) were loaded onto the Tzero Pan. After each specimen was heated to 150 °C at 30 °C min−1 and subjected to a 3 min isothermal process, the data were recorded as it was cooled to 0 °C and heated to 200 °C at 10 °C min−1.
Results and discussion
Adhesion switching characteristics of switchable PSAs
The chemical structures of the azo-polymer and azo-compounds are shown in Fig. 1. The switchable PSAs, as fabricated with a mixture of azo-polymer and azo-compound, were expected to have a structure in which the azo-compound was dispersed in the azo-polymer matrix. It was also expected to have different molecular arrangements, depending on the length of the hydrocarbon chain of the azo-compounds. Because PSAs must have a low Tg to have adhesion properties at the service temperature, the acrylic copolymer was polymerized with 90 mol% of butyl acrylate with a low Tg (approximately −53 °C), and 10 mol% of azo-acrylate (Fig. 1a). The content of the azobenzene moiety contained in the polymer chain was determined from the 1H-NMR spectrum after the azo-polymer was washed with methanol (Fig. S8†). The calculated content was 8 mol%.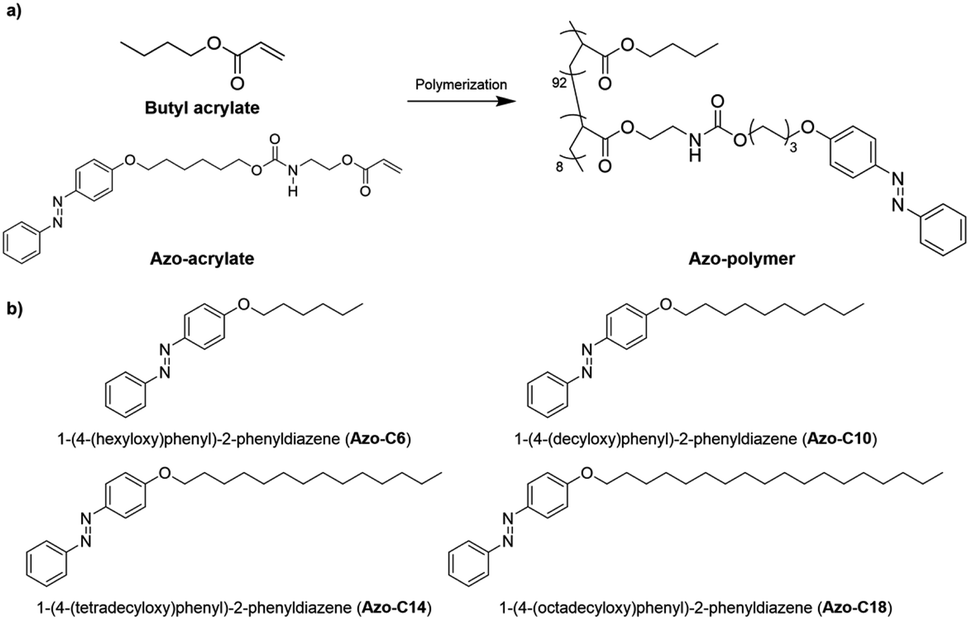 | ||
| Fig. 1 Chemical structures of the (a) azo-polymer and (b) azo-compounds. | ||
Probe tack tests were applied to evaluate the adhesion switching characteristics of the switchable PSAs. Because the intermolecular interaction and arrangement are dependent on the chain length, the influence of the compound hydrocarbon chain length on the switching characteristics was evaluated by measuring the tack value variation according to UV intensity. A UV irradiation time was 30 s. After applying the probe tack test to each UV-irradiated specimen, all specimens were irradiated with visible light to “switch off” the adhesive force; then, the adhesive force was measured again to evaluate whether reversible switching was possible (Fig. S9†). In Fig. 2a, the results of the “switched off” adhesive force were omitted to highlight the trend with respect to UV intensity.
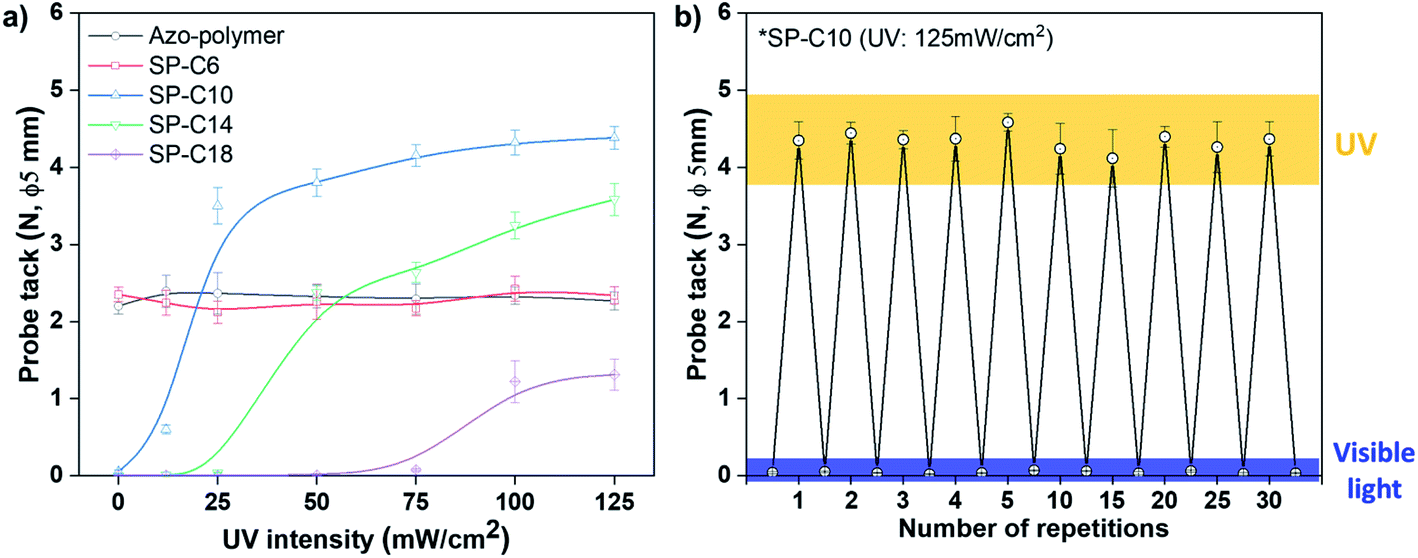 | ||
| Fig. 2 (a) Switchable PSAs probe tack force as a function of UV intensity, and (b) 30-cycle adhesion switching test results for SP-C10 (UV irradiation time: 30 s). | ||
The azo-polymer, which had a 2.2 N tack value before UV irradiation, exhibited no change in adhesion, even when the UV intensity was increased. Specifically, there was no change in the adhesion or switching characteristics even when Azo-C6 was added (SP-C6). The adhesive forces of the azo-polymer and SP-6 were maintained even when under visible light irradiation (Fig. S9†). Alternatively, when an azo-compound had more than 10 hydrocarbon chains, an initial adhesive force was not detected before UV irradiation. In the case of SP-10, weak adhesion was detected when 12 mW cm−2 of UV radiation was applied; the strength of the adhesion began to rapidly increase at 25 mW cm−2. The tack force gradually increased at UV intensities above 25 mW cm−2, but tended to begin saturating at 100 mW cm−2. The tack force increased to 4.4 N under the UV radiation condition of 125 mW cm−2. Additionally, longer hydrocarbon chains of the azo-compound necessitated stronger UV intensities for adhesion. The adhesive forces of SP-C14 and SP-C18 sharply increased at 50 and 100 mW cm−2, respectively (Fig. 2a). The tack value also decreased as the length of the hydrocarbon chain increased. The adhesive forces of SP-C10, SP-C14, and SP-C18 were found to sharply decline in response to visible light irradiation (Fig. S9†). Thus, adhesion switching was possible when the azo-compound had more than 10 hydrocarbon chains. Furthermore, as the hydrocarbon chain length increased, the UV intensity required for adhesion switching increased, and the adhesive force decreased. Reversible adhesion switching was found to be possible for SP-10, SP-14, and SP-18. Note that the 30-cycle adhesion switching repetition test for SP-C10, which had the highest tack force, confirmed no decrease in the adhesive force (Fig. 2b).
As mentioned, adhesion was enabled by wetting the adhesive on the substrate. Although the wetting of an ideal solid with a low-viscosity liquid can be simply explained by surface energy, complex factors must be considered to determine the wetting of a viscoelastic material. In particular, modulus is main factor in determining wetting of viscoelastic material.40 The modulus limits its wetting, and materials with a high modulus have poor wettability.
After adhesion was initially established by wetting, the response of a PSA to various external forces that try to separate it is expressed as an adhesive force. The equation for the probe tack force F is as follows:
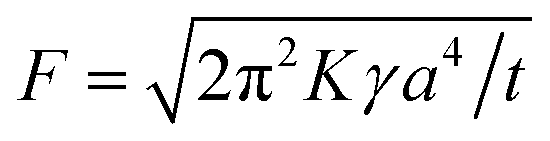 | (2) |
Photoisomerization of the azobenzene moiety
Because any change in adhesive force in response to UV or visible light irradiation was expected to be related to the photoisomerization occurring between the trans and cis forms of the azobenzene moiety, the isomerization was measured at different UV intensities by using a UV/Vis spectrometer. The photoisomerization of the azobenzene moiety was monitored by focusing on the π → π* and n → π* absorption bands. Stable trans-azobenzene tended to have a π → π* absorption band near 360 nm, whereas the cis form had a lower-energy absorption band (n → π*) near 450 nm.42Fig. 3 shows the UV/Vis spectra of the switchable PSAs according to UV intensity. The UV irradiation time was 30 s as in the probe tack test. The photoisomerization process was quantified based on the absorbance near 360 nm, which is the primary absorption band of trans-azobenzene. However, as shown in Fig. 3, the 360 nm absorbance of the switchable PSAs was out of measurement range. To obtain a trans-azobenzene absorption band that is within the measurable range, it is necessary to measure much thinner switchable PSAs. However, according to the Beer–Lambert law, because light intensity is more strongly attenuated through thicker materials, the photoisomerization process, particularly with respect to UV intensity, may be carried out differently for relatively thinner switchable PSAs. Thus, we obtained the UV/Vis spectra of PSA specimens with the same thickness as that previously applied, and monitored the photoisomerization trend by focusing on the n → π* absorption band near 450 nm.
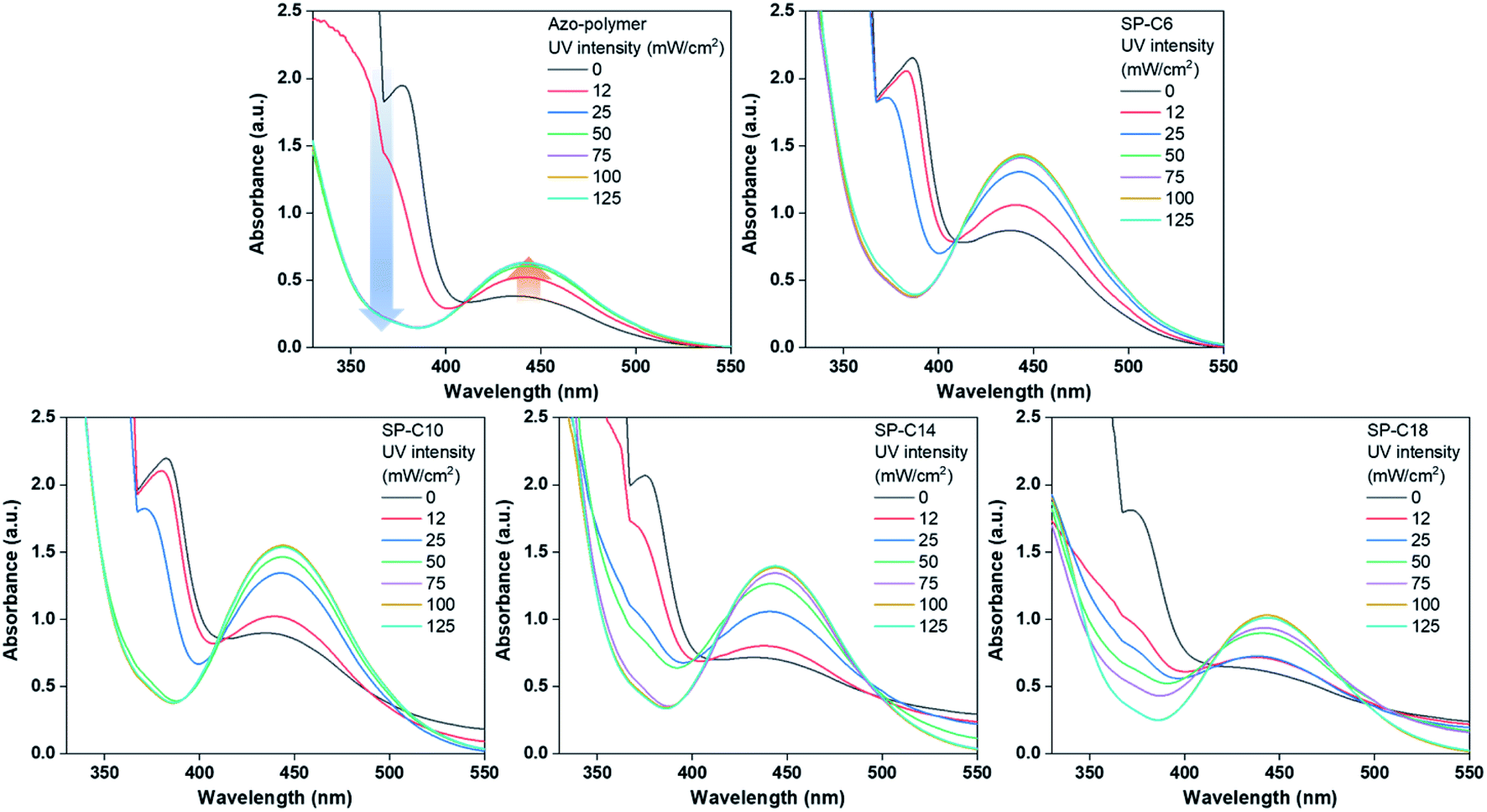 | ||
| Fig. 3 UV/Vis spectra of the switchable PSAs according to UV intensity (UV irradiation time: 30 s). | ||
In the case of the azo-polymer, isomerization of the azobenzene pendants in the polymer chain was observed. Regarding the isomerization results for the trans to cis form, absorption near 450 nm and 360 nm increased and decreased, respectively. The changes in both absorption bands revealed saturation at 25 mW cm−2. This means that most azobenzene groups in the azo-polymer were isomerized at 25 mW cm−2. The degree of SP-C6 isomerization increased gradually at 12 and 25 mW cm2; it was saturated at 50 mW cm−2. The UV intensity associated with isomerization saturation in the switchable PSAs increased with increasing length of the azo-compound hydrocarbon chain. In the case of SP-C18, the degree of photoisomerization gradually increased up until a UV intensity of 100 mW cm2; additionally, under 125 mW cm−2 UV radiation conditions, the absorbance near 450 nm was lower than that in the cases of the other switchable PSAs. It means that more energy was required for photoisomerization of the azobenzene as the aliphatic chain length increased. Although photoisomerization of the azobenzene group was observed in all specimens, the adhesion switching was possible only in SP-C10, SP-C14, and SP-C18. It could be defined through evaluation of chemical and mechanical characteristics.
UV light-induced changes in Tg, modulus, and surface energy of the switchable PSAs
As mentioned in the Introduction, a PSA must have a low Tg to have adhesive properties at the service temperature. Thus, the Tg values for the pre- and post-UV-irradiated switchable PSAs were measured by using DSC (Fig. 4). UV irradiation shifted the initial Tg of the azo-polymer at −10.2 °C to −16.3 °C. This is because the interaction between the trans-azobenzene pendant groups was weakened as they were isomerized into the cis form. The size and flexibility of polymer side groups are factors that determine Tg.43 When the azobenzene pendant groups existed in the trans state, there were interactions between the pendants groups due to their planar structure and the π–π interaction. The interactions limit the mobility of the side groups. Alternatively, when they were isomerized to cis-form, the interactions were reduced due to its bent structure; the Tg could be shifted to a lower temperature. For SP-C6, the initial-state Tg was −16.0 °C, which was lower than the initial Tg of the azo-polymer. Azo-C6 was expected to disperse between the polymer chains and act as a plasticizer. The azo-polymer and SP-C6, which did not have adhesion switchability, had the initial-state Tg; this was not clearly detectable in the DSC curves for SP-C10 and SP-C14, although there was slight slope transition in the DSC curves of SP-C10 and SP-C14. However, under UV irradiation, SP-C10 and SP-C14 was measured to have clear Tg transition of −15.6 °C and −15.5 °C, respectively. The manifestation of Tg by UV radiation constitutes one of the reasons that the adhesive forces of SP-C10 and SP-C14 were “switched on”. Under DSC measurement, the transition in slope of heat flow curve is detected at specific temperature range by a segmental movement of polymer molecule. It is defined as Tg. Thus, the phenomenon that clearly distinguished Tg became non-detectable by external stimuli signified that the molecular mobility of the switchable PSA became constrained. The SP-C18 had a non-sticky surface in the initial state. However, it had relatively clear Tg than those of SP-C10 and SP-C14 even before UV radiation in the DSC curve. The initial-state Tg of SP-C18 (−14.6 °C), which was between those of the azo-polymer (−10.2 °C) and SP-C6 (−16.0 °C), and a small magnitude of gradient change in the DSC curve were results of the partial incompatibility between SP-C18 and azo-polymer.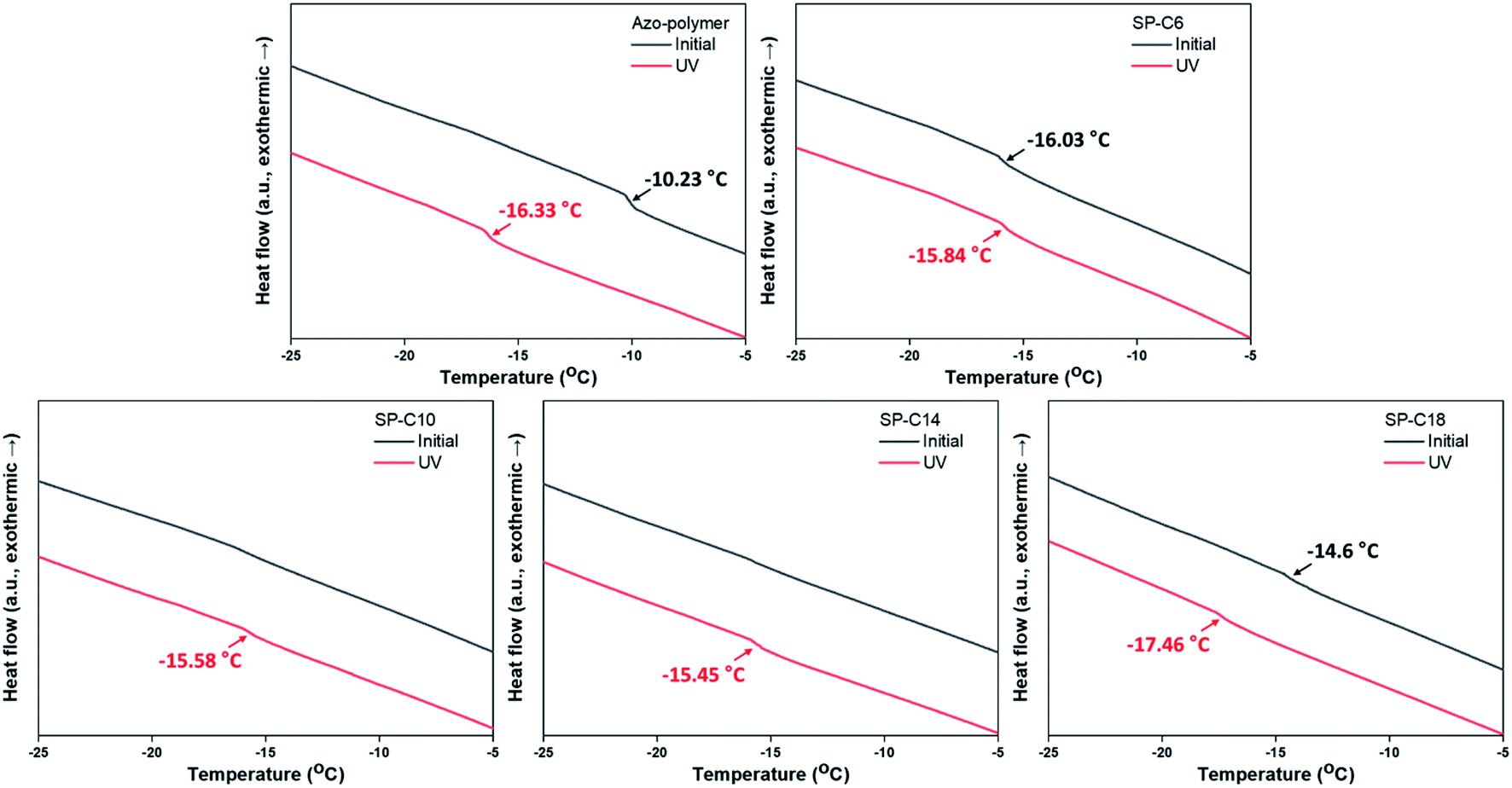 | ||
| Fig. 4 Pre- and post-UV exposure DSC curves for the switchable PSAs. | ||
Fig. 5 shows the lap shear test results for the switchable PSAs. The azo-polymer and SP-C6 had low moduli within the 10–20 kPa range, even before UV exposure. The lower shear modulus of SP-C6 compared to that of the azo-polymer is attributable to the plasticizing effect of Azo-C6, as indicated by the DSC results. The pre-UV-exposure shear moduli of SP-C10, SP-C14, and SP-C18 were 79, 112, and 106 kPa, respectively, which were much higher than those of the azo-polymer and SP-C6. As the hydrocarbon chain length of the azo-compound increased, the modulus tended to increase; however, the modulus of SP-18 is thought to have been lower than that of SP-14 because of the lower compatibility between Azo-C18 and the azo-polymer. In the cases of the specimens that exhibited adhesion switching characteristics, i.e., SP-C10, SP-C14, and SP-C18, the shear modulus decreased as the UV intensity increased. In particular, there was an intensity range in which the modulus rapidly decreased (0–25 mW cm−2 for SP-C10, 25–50 mW cm−2 for SP-C14, and 25–75 mW cm−2 for SP-C18); this range coincided with the range in which the tack forces and absorbance of the cis-azobenzene moiety rapidly increased. SP-C10 and SP-C14 had shear moduli below 10 kPa after UV irradiation at 125 mW cm−2. Alternatively, SP-C18, which had a narrow adhesion modulation range, had a relatively high shear modulus (25 kPa) even after UV irradiation at 125 mW cm−2.
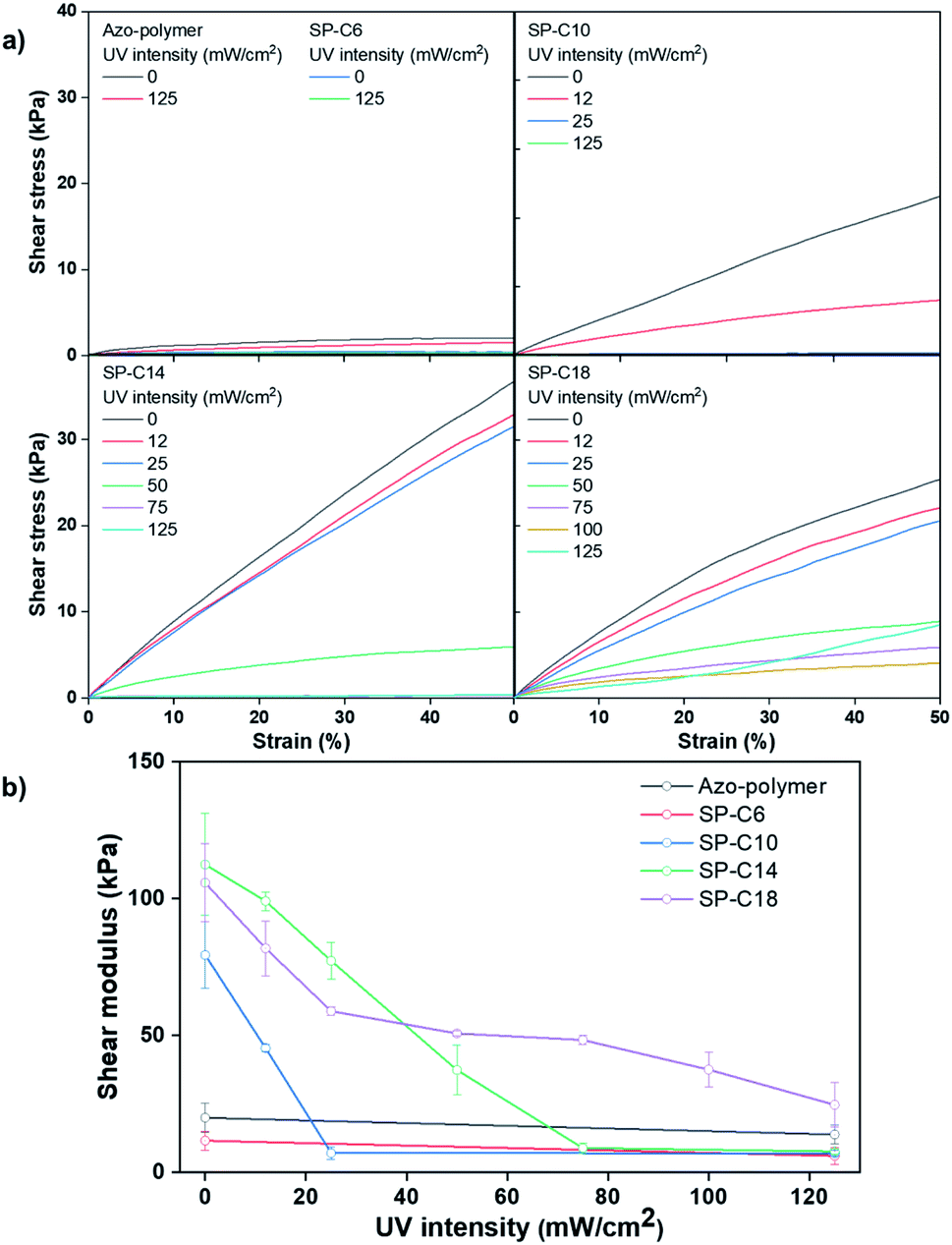 | ||
| Fig. 5 (a) Stress–strain curves and (b) shear modulus results for the switchable PSAs according to UV intensity. | ||
The chemical characteristics of the switchable PSAs were evaluated in terms of their water contact angles. The contact angles of the azo-polymer and SP-C6 were 93–94°; they were maintained even under the conditions of UV and visible light exposure (Fig. 6). The initial contact angles of SP-C10, SP-C14, and SP-C18 increased to 101.4°, 105.5°, and 105.9°, respectively. In general, an increase in the water contact angle means a decrease in surface energy. The contact angles of SP-C10 and SP-C14 decreased to 93° after UV irradiation (125 mW cm−2); this value was found to be similar to those for the azo-polymer and SP-6. The results of measuring the contact angles of SP-C10, SP-C14, and SP-C18 according to the UV intensity revealed that the contact angles for each switchable PSA decreased within a specific intensity range in a way that was similar to the shear modulus results (0–25 mW cm−2 for SP-C10, 25–50 mW cm−2 for SP-C14, and 50–100 mW cm−2 for SP-C18).
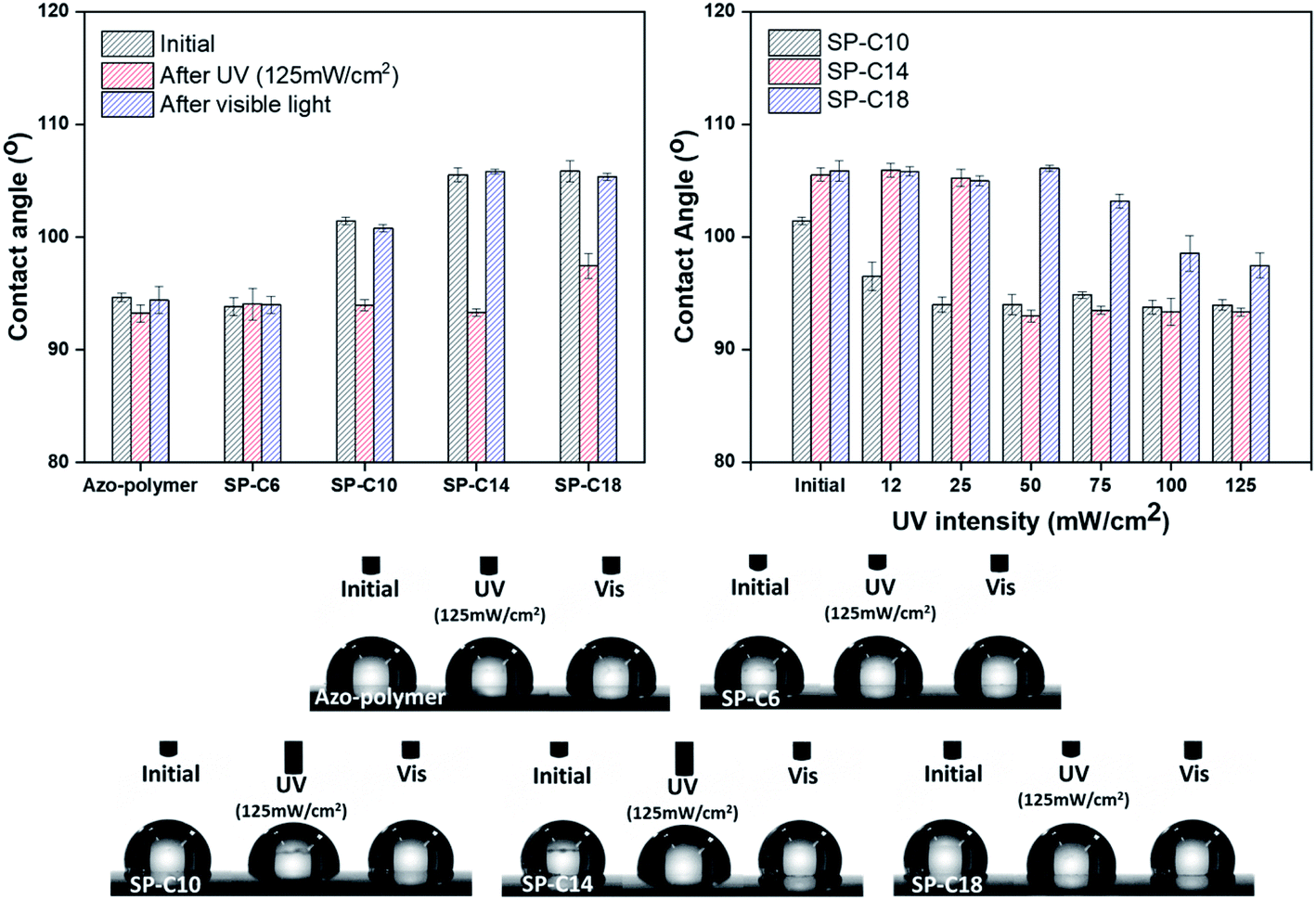 | ||
| Fig. 6 Water contact angle results for the switchable PSAs. | ||
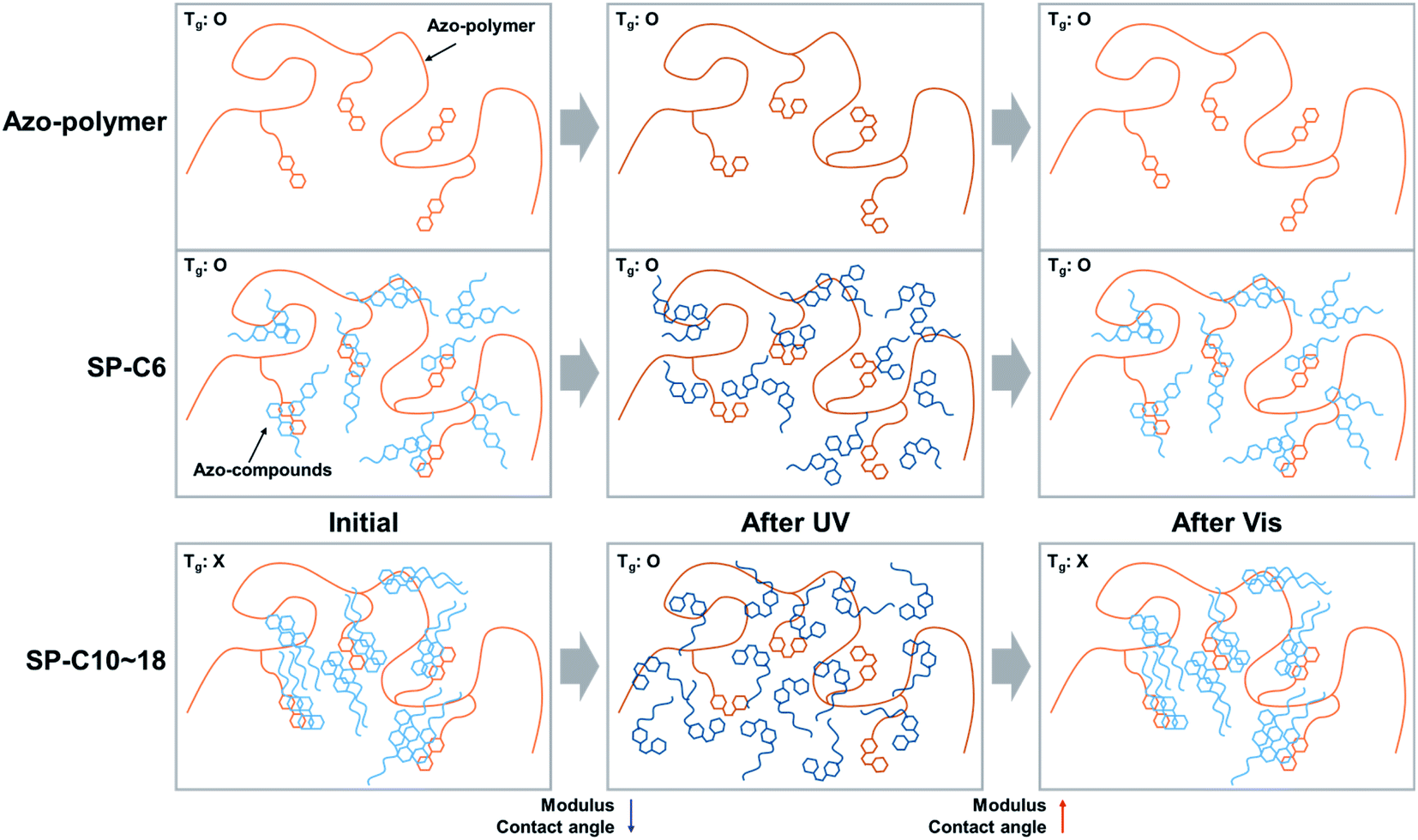
In the cases of the PSAs that were found to be capable of adhesion switching (i.e., SP-C10, SP-C14, and SP-C18), the changes in Tg, shear modulus, and contact angle were confirmed to have been induced as a result of UV exposure. The deactivation of the adhesive force in the initial and visible light-irradiated states of the switchable PSAs was attributable to the non-detectable Tg, an increase in the shear modulus, and a decrease in surface energy. A decrease in surface energy can decrease the adhesive force as the both cases of wetting and eqn (2). However, the visible light-irradiated switchable PSA shear modulus values were 4–6 times larger than that of the azo-polymer; this higher modulus resulted in less wetting, which is considered to be the main cause of adhesion deactivation. The non-detectable Tg in the DSC curves also verified that the molecular mobility of the PSA was restricted and insufficient to form wetting on the substrate. Alternatively, when UV irradiation was applied to the switchable PSAs, the photoisomerization of the azobenzene moiety from the trans to cis form clear Tg transition, as well as caused the modulus to decrease, and surface energy to increase. These complex changes led to the activation/deactivation of the adhesive forces of the switchable PSAs.
Changes in the crystalline structures of the switchable PSAs
Changes in the crystalline structure are believed to be the primary reason for the changes in the Tg, modulus, and surface energy that occurred during photoisomerization of the azobenzene moiety in the switchable PSAs. As shown in Fig. 7, the azo-polymer and SP-C6 were transparent in initial state and maintained transparency under the condition of UV exposure. However, the “switched off” SP-10, SP-14, and SP-18 were opaque before UV irradiation. The SP-C10 became transparent under the conditions of low intensity (12–25 mW cm−2) of UV radiation. SP-C14 became transparent at UV intensities above 50 mW cm−2, whereas SP-C18 appeared to be partially transparent at 100 mW cm−2, and was still not completely transparent at 125 mW cm−2. The UV intensity ranges in which the film transparency changed were the same as those in which the adhesion, trans-to-cis photoisomerization, shear modulus, and contact angle of each PSA were rapidly changed.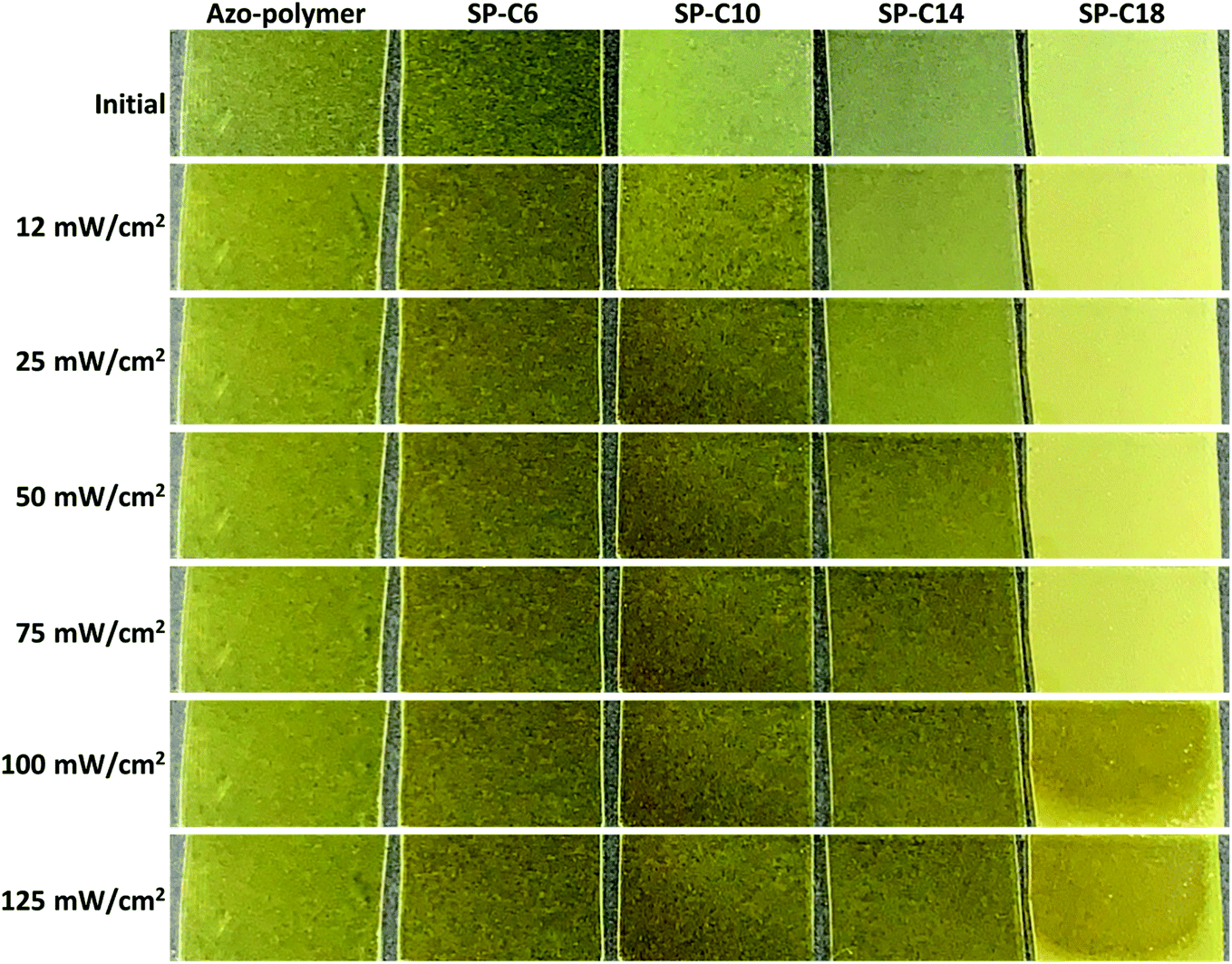 | ||
| Fig. 7 Transition in transparency of the switchable PSAs according to UV intensity. | ||
The opaqueness of the amorphous azo-polymer-containing switchable PSAs was attributable to the crystalline structure that was formed by each azo-compound. The crystallinity of each azo-compound was confirmed by the corresponding DSC curve results (Fig. 8a). Azo-C6 and Azo-C10 had two phase-transition peaks on each DSC curve. This means that these two compounds had a mesophase between the two phase-transition peaks, such as a liquid crystal phase. However, as the length of the hydrocarbon substituent increased (Azo-C14 and Azo-C18), the phase-transition temperature increased, and the mesophase disappeared. The melting enthalpies of Azo-C6, Azo-C10, Azo-C14, and Azo-C18 obtained via integration of the phase-transition peaks were 20.1 kJ mol−1, 38.3 kJ mol−1, 51.2 kJ mol−1, and 62.5 kJ mol−1, respectively (Fig. 8c). The intermolecular interactions of the azo-compounds were found to be the π–π interaction of the azobenzene moiety, and the dispersion interaction of the hydrocarbon chains. Thus, as the length of the hydrocarbon chain increased, the intermolecular interaction and crystalline structure became stronger. This led to an increase in the melting temperature and enthalpy. SP-C6 did not exhibit adhesion switching characteristics because Azo-C6, which had a relatively weak intermolecular interaction, did not form a crystalline structure in the azo-polymer. Alternatively, Azo-C10, C14, and C18 formed a crystalline structure, and adhesion switching was realized by a transition in the crystalline structure by UV and visible light radiation (Scheme 1). The crystalline structures formed by the interaction between Azo-C10, C14, and C18 molecules are stronger than the molecular arrangement of amorphous azo-polymer and SP-C6. The crystalline structures require more stress for deformation and induce an increase in shear modulus. The surface energy is also reduced due to the intermolecular stacking of the crystalline structure formed by dispersion interaction of hydrocarbon chain and the π–π interaction of azobenzene.44
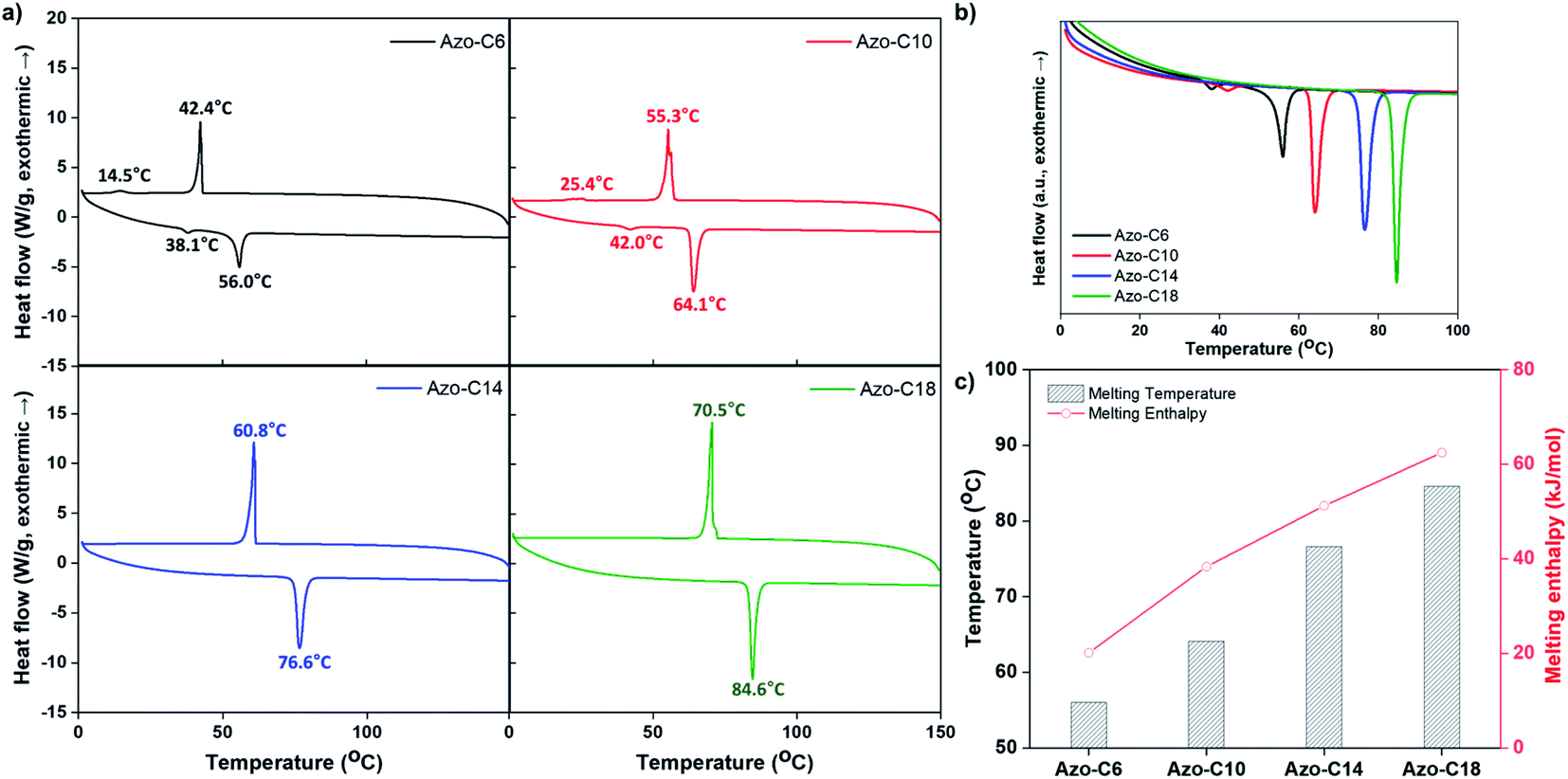 | ||
| Fig. 8 (a) DSC curves; (b and c) comparison of the azo-compound melting temperatures and melting enthalpies. | ||
 | ||
| Scheme 1 Schematic illustration of crystalline structure formation of azo-compounds and transition in crystalline structure by light irradiation. | ||
Consequently, the reason that the UV intensity required for switchable PSAs to activate their adhesive forces increased as the azo-compound aliphatic chain length increased is believed to be related to the energy required to induce phase switching in the crystalline structures of the azo-compounds. The isomerization of the azobenzene moiety was affected by the phase and viscosity of the surrounding substances. Isomerization is known to become difficult when the viscosity surrounding the azobenzene moiety increases.45 Furthermore, the photoisomerization from trans-to cis-azobenzene may not be possible if it exists in the solid phase.46 Thus, photoisomerization may be related to molecular mobility and free volume. This is why the Azo-C10-containing SP-C10, which had a low melting enthalpy, and a mesophase, was capable of adhesion switching at the lowest UV intensity.
Conclusions
Azo-compounds with substituents with different hydrocarbon chain lengths were synthesized on the azobenzene group. The switchable PSAs were prepared by mixing the compounds with an azo-polymer. When the length of the aliphatic chain exceeded 10, adhesion switching was possible. Moreover, longer chain lengths were found to necessitate stronger UV intensity for adhesion switching. The reason for this is related to the phase-transition temperature and melting enthalpy of the compound. Changes in the crystalline structure of the azo-compound in the polymer matrix were induced as a result of UV exposure, and the adhesive forces of the switchable PSA were activated in response to the detection of Tg, a decrease in the shear modulus, and an increase in surface energy. In the case of SP-C10, adhesion switching was possible at a relatively low UV intensity of 25 mW cm−2, even under the condition of a short UV irradiation time of 30 s. In addition, there was no loss of adhesive force even after 30 continuous cycles adhesion switching. It is expected that our novel switchable PSA, which is reusable and capable of rapid switching even at low UV intensities, can be applied in the transfer printing process in electrical and electronic industries. The adhesive force of the PSAs was activated by trans to cis isomerization of azobenzene moiety. Thus, the activated adhesive properties cannot be maintained permanently because of the metastable nature of the cis-azobenzene. Although it is expected to be applicable to processes requiring short-term adhesive force since it was confirmed that the adhesive properties were maintained during the probe tack test (within a few minutes), it would be worth studying the adhesion maintaining time to expand applications and gain a better understanding of the material.Author contributions
Tae-Hyung Lee: conceptualization, data curation, investigation, methodology, writing – original draft. Gi-Yeon Han: data curation, formal Analysis. Mo-Beom Yi: formal analysis, investigation. Jae-Ho Shin: investigation, methodology. Hyun-Joong Kim: conceptualization, supervision.Conflicts of interest
There are no conflicts to declare.References
- C. Creton and M. Ciccotti, Rep. Prog. Phys., 2016, 79, 46601 CrossRef PubMed.
- C. Creton, MRS Bull., 2003, 28, 434–439 CrossRef CAS.
- E. P. Chang, J. Adhes., 1991, 34, 189–200 CrossRef CAS.
- A. B. Croll, N. Hosseini and M. D. Bartlett, Adv. Mater. Technol., 2019, 4, 1900193 CrossRef.
- M. Kamperman and A. Synytska, J. Mater. Chem., 2012, 22, 19390–19401 RSC.
- G. de Crevoisier, P. Fabre, J. M. Corpart and L. Leibler, Science, 1999, 285, 1246–1249 CrossRef CAS PubMed.
- K. Cho, J. H. Cho, S. Yoon, C. E. Park, J. C. Lee, S. H. Han, K. B. Lee and J. Koo, Macromolecules, 2003, 36, 2009–2014 CrossRef CAS.
- T. Ohzono, M. O. Saed and E. M. Terentjev, Adv. Mater., 2019, 31, 1902642 CrossRef PubMed.
- T. Ohzono, Y. Norikane, M. O. Saed and E. M. Terentjev, ACS Appl. Mater. Interfaces, 2020, 12, 31992–31997 CrossRef CAS PubMed.
- Y. Zheng, A. Hashidzume, Y. Takashima, H. Yamaguchi and A. Harada, Nat. Commun., 2012, 3, 831 CrossRef PubMed.
- Y. Zheng, A. Hashidzume and A. Harada, Macromol. Rapid Commun., 2013, 34, 1062–1066 CrossRef CAS PubMed.
- H. Yamaguchi, Y. Kobayashi, R. Kobayashi, Y. Takashima, A. Hashidzume and A. Harada, Nat. Commun., 2012, 3, 603 CrossRef PubMed.
- Y. Gao, K. Wu and Z. Suo, Adv. Mater., 2019, 31, 1806948 CrossRef PubMed.
- T. Nakamura, Y. Takashima, A. Hashidzume, H. Yamaguchi and A. Harada, Nat. Commun., 2014, 5, 4622 CrossRef PubMed.
- R. LaSpina, M. R. Tomlinson, L. Ruiz -Pérez, A. Chiche, S. Langridge and M. Geoghegan, Angew. Chem., 2007, 119, 6580–6583 CrossRef.
- M. A. Meitl, Z. T. Zhu, V. Kumar, K. J. Lee, X. Feng, Y. Y. Huang, I. Adesida, R. G. Nuzzo and J. A. Rogers, Nat. Mater., 2006, 5, 33–38 CrossRef CAS.
- J. M. Boyne, E. J. Millan and I. Webster, Int. J. Adhes. Adhes., 2001, 21, 49–53 CrossRef CAS.
- K. Ebe, H. Seno and K. Horigome, J. Appl. Polym. Sci., 2003, 90, 436–441 CrossRef CAS.
- S. W. Lee, T. H. Lee, J. W. Park, C. H. Park, H. J. Kim, S. M. Kim, S. H. Lee, J. Y. Song and J. H. Lee, Int. J. Adhes. Adhes., 2015, 57, 9–12 CrossRef CAS.
- J. D. Eisenhaure, T. Xie, S. Varghese and S. Kim, ACS Appl. Mater. Interfaces, 2013, 5, 7714–7717 CrossRef CAS PubMed.
- K. Sim, S. Chen, Y. Li, M. Kammoun, Y. Peng, M. Xu, Y. Gao, J. Song, Y. Zhang, H. Ardebili and C. Yu, Sci. Rep., 2015, 5, 16133 CrossRef CAS PubMed.
- P. Testa, B. Chappuis, S. Kistler, R. W. Style, L. J. Heyderman and E. R. Dufresne, Soft Matter, 2020, 16, 5806–5811 RSC.
- J. H. Kim, B. C. Kim, D. W. Lim and B. C. Shin, J. Mech. Sci. Technol., 2019, 33, 5321–5325 CrossRef.
- G. Kaur, P. Johnston and K. Saito, Polym. Chem., 2014, 5, 2171–2186 RSC.
- Z. Liu, J. Cheng and J. Zhang, Macromol. Chem. Phys., 2020, 222, 2000298 CrossRef.
- L. Shen, J. Cheng and J. Zhang, Eur. Polym. J., 2020, 137, 109927 CrossRef CAS.
- H. Akiyama, Y. Okuyama, T. Fukata and H. Kihara, J. Adhes., 2018, 94, 799–813 CrossRef CAS.
- A. Goulet-Hanssens, F. Eisenreich and S. Hecht, Adv. Mater., 2020, 32, 1905966 CrossRef CAS PubMed.
- H. Akiyama, S. Kanazawa, Y. Okuyama, M. Yoshida, H. Kihara, H. Nagai, Y. Norikane and R. Azumi, ACS Appl. Mater. Interfaces, 2014, 6, 7933–7941 CrossRef CAS PubMed.
- H. Akiyama, T. Fukata, A. Yamashita, M. Yoshida and H. Kihara, J. Adhes., 2017, 93, 823–830 CrossRef CAS.
- S. Ito, A. Yamashita, H. Akiyama, H. Kihara and M. Yoshida, Macromolecules, 2018, 51, 3243–3253 CrossRef CAS.
- S. Ito, H. Akiyama, R. Sekizawa, M. Mori, M. Yoshida and H. Kihara, ACS Appl. Mater. Interfaces, 2018, 10, 32649–32658 CrossRef CAS PubMed.
- Y. Zhou, M. Chen, Q. Ban, Z. Zhang, S. Shuang, K. Koynov, H. J. Butt, J. Kong and S. Wu, ACS Macro Lett., 2019, 8, 968–972 CrossRef CAS.
- L. Kortekaas, J. Simke, D. W. Kurka and B. J. Ravoo, ACS Appl. Mater. Interfaces, 2020, 12, 32054–32060 CrossRef CAS PubMed.
- C. Barrett, A. Natansohn and P. Rochon, Chem. Mater., 1995, 7, 899–903 CrossRef CAS.
- D. Gegiou, K. A. Muszkat and E. Fischer, J. Am. Chem. Soc., 1968, 90, 3907–3918 CrossRef CAS.
- H. Rau and S. Y. Quan, J. Photochem. Photobiol. A, 1988, 42, 321–327 CrossRef CAS.
- E. Titov, G. Granucci, J. P. Gotze, M. Persico and P. Saalfrank, J. Phys. Chem. Lett., 2016, 7, 3591–3596 CrossRef CAS PubMed.
- H. Zhou, C. Xue, P. Weis, Y. Suzuki, S. Huang, K. Koynov, G. K. Auernhammer, R. Berger, H. J. Butt and S. Wu, Nat. Chem., 2017, 9, 145–151 CrossRef CAS PubMed.
- S. Donatas, Handbook of Pressure Sensitive Adhesive Technology, Satas & Associates, Rhode Island, 1999 Search PubMed.
- K. Kendall, J. Phys. D: Appl. Phys., 1971, 4, 1186 CrossRef.
- V. Ladanyi, P. Dvorak, J. Al Anshori, L. Vetrakova, J. Wirz and D. Heger, Photochem. Photobiol. Sci., 2017, 16, 1757–1761 CrossRef CAS PubMed.
- S. A. Jenekhe and M. F. Roberts, Macromolecules, 1993, 26, 4981–4983 CrossRef CAS.
- H. S. Lim, J. T. Han, D. Kwak, M. Jin and K. Cho, J. Am. Chem. Soc., 2006, 128, 14458–14459 CrossRef CAS PubMed.
- D. Gegiou, K. A. Muszkat and E. Fischer, J. Am. Chem. Soc., 1968, 90, 12–18 CrossRef CAS.
- M. Tsuda and K. Kuratani, Bull. Chem. Soc. Jpn., 1964, 37, 1284–1288 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra06596c |
| This journal is © The Royal Society of Chemistry 2021 |