
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Coordination complexes of zinc and manganese based on pyridine-2,5-dicarboxylic acid N-oxide: DFT studies and antiproliferative activities consideration†
Hanie Alizadeh a,
Masoud Mirzaei*a,
Amir Sh. Saljooghia,
Vida Jodaianb,
Maryam Bazargana,
Joel T. Maguec,
Rosa M. Gomilad and
Antonio Fronterad
a,
Masoud Mirzaei*a,
Amir Sh. Saljooghia,
Vida Jodaianb,
Maryam Bazargana,
Joel T. Maguec,
Rosa M. Gomilad and
Antonio Fronterad
aDepartment of Chemistry, Faculty of Science, Ferdowsi University of Mashhad, Mashhad 9177948974, Iran. E-mail: mirzaeesh@um.ac.ir
bDepartment of Chemistry, Islamshahr Branch, Islamic Azad University, Islamshahr 3317843154, Iran
cDepartment of Chemistry, Tulane University, New Orleans, LA 70118, USA
dDepartament de Química, Universitat de les Illes Balears, Crta de Valldemossa km 7.5, 07122 Palma de Mallorca (Baleares), SPAIN
First published on 22nd November 2021
Abstract
We report here the design, synthesis, and antiproliferative activity of three coordination complexes [Mn2(pydco)2(bpy)2(H2O)2]·2H2O (1), [Zn(bpy)(Hpydco)2] (2), and [Zn(bpy)Cl(Hpydco)]·2H2O (3) (H2pydco = pyridine-2,5-dicarboxylic acid N-oxide, bpy = 2,2′-bipyridine). Molecular structures of these complexes have been characterized by elemental analysis, Fourier transform infrared spectroscopy, thermogravimetric analysis, and powder and single-crystal X-ray diffraction. According to the structural analysis, 1–3 are discrete complexes containing N- and O-donor ligands (bpy and pydco2−) in which pydco2− can be coordinated to the metal centres via the N-oxide oxygen and one carboxylate oxygen to generate a six-membered chelate ring. Also, these structures benefit from extensive intermolecular interactions such as hydrogen bonds and π-interactions which are the major forces to make them more stable in the solid state. The energetic features of the π-stacking interactions observed in compounds 1–3 have been computed and compared to the H-bonds. The interactions in the solid state have been also studied using the independent gradient model approach (IGM plot). The IGM-δg approach uses a new descriptor (δg) that locally represents the difference between a virtual upper limit of the electron density gradient and the true electron density gradient. This newly developed IGM methodology automatically extracts the signature of interactions between two given fragments. Finally, the antiproliferative properties of these complexes were tested on several cancer cell lines by MTT assay and flow cytometry. Also, to compare the antiproliferative activities of these complexes with common chemotherapy drugs, the antiproliferative property of cisplatin was evaluated as a reference and positive control.
Introduction
Nowadays, metal-based drugs are known as an important class of anti-tumor agents. They can be classified into two groups; (i) those containing the classical noble metals such as Pt, Ru, Au, Ti, and Pd which are the most promising candidates in the treatment of several tumors and (ii) those of low-cost, bio-essential metals such as Cu, Fe, Zn and Mn.1–8 The former group suffers from some clinical problems such as toxicity, solubility and drug half-life while the latter group are better dealt by human physiology and cause less damaging side effects. Despite the variety of physiological roles of the zinc and manganese ions and their complexes, they have been less studied as metal-based anticancer drugs.Apart from the choice of the metal ion, the structure of the organic ligand(s) can have a great influence on the final biological properties by limiting the adverse effects of metal overload, inhibiting selected metalloenzymes and facilitating metal ion redistribution. Up to now, a variety of ligands with various donor atoms have been introduced for bio-essential metals. Among them, O/N-donor ligands are almost the most representative category. Many strategies such as utilizing of one- or mixed-ligand complexes have been reported and due to the variety of accessible arrangements, different probable frameworks have been employed as anticancer agents. For the metal complex-based drugs, two groups of organic ligands are primarily employed: (i) aromatic bidentate N-donors such as 2,2′-bipyridine and 1,10-phenanthroline which are used in molecular biology as DNA intercalation agents, as contrast agents for magnetic resonance imaging, chelation therapies, antiproliferative,9,10 and metalloenzymes. (ii) pyridine polycarboxylates with N/O donor properties which have vast and remarkable applications such as anticancer properties, anti-HIV agents, gas adsorbents, magnetism and luminescence to name just a few.11–16
Recently, the chemistry of pyridine-2,5-dicarboxylic acid (H2pydc) has attracted attention,17–21 due to the position of its carboxylates and aromatic rings which is not only interesting for its diverse coordination complexes but also its potential to create supramolecular structures through intermolecular interactions such as π-stacking and hydrogen bond.12,22,23 It is important to note that N-oxidation of the pyridine ring in the H2pydc can produce an O-donor ligand (pyridine-2,5-dicarboxylic acid N-oxide (H2pydco)) with better electron donor affinity than the nitrogen atom of the pyridine ring in H2pydc because the neutral nitrogen atom can donate one pair of electrons, while the charge-polarized pyridine-N-oxide moiety can donate three pairs of electrons.24 According to the literature survey, this feature can enhance the CO2 separation properties of isoreticular MOFs.11 Several reviews reflect the huge potential of metal-based frameworks as drug delivery and cancer theragnostic platforms.25–28
As part of our study on bio-essential metal complexes as antiproliferative agents, we have synthesized three coordination complexes with H2pydco and bpy under similar condition which are formulated as [Mn2(pydco)2(bpy)2(H2O)2]·2H2O (1), [Zn(bpy)(Hpydco)2] (2), and [Zn(bpy)Cl(Hpydco)]·2H2O (3). The structural description of the three compounds has been done and the interesting π-stacking assemblies have been studied using DFT calculations. In particular, the energetic features of several assemblies have been computed and compared. The interactions in the solid state have been also characterized using the independent gradient model approach (IGM plot), which is a novel methodology to reveal noncovalent interactions in real space. Finally, the antiproliferative potential of 1–3, have investigated against MDA-MD-231, MCF-7, HeLa, HT-29, Neuro-2a cancer cell lines also L-929 as a normal cell, using MTT and TUNEL assay and flow cytometry test.
Experimental
Materials and instruments
All chemicals and solvents were purchased from commercial sources and used without further purification, except for pyridine-2,5-dicarboxylic acid N-oxide (H2pydco) which was synthesized according to a reported method.29 Melting points were determined using a Barnstead Electrothermal 9300 apparatus. The infrared spectra were recorded in the range of 4000 to 400 cm−1 on a Thermo Nicolet/AVATAR 370 Fourier transform spectrophotometer using KBr pellets. Elemental analysis (CHN) was performed using a Thermo Finnigan Flash-1112 EA microanalyzer. Metal content was measured by the Spectro Arcos ICP-OES spectrometer model 76004555 using in the range of 130–770 nm for ICP spectra. Thermal gravimetric analysis (TGA) was carried out under an air atmosphere from ambient temperature up to 950 °C with a heating rate of 10 °C min−1 on a Shimadzu TGA-50 instrument.Synthesis and characterization
X-ray structure determination
Single crystals of 1–3 were mounted on polymer loops with a drop of heavy oil and flash cooled to 150 K in the cold nitrogen stream of an Oxford Cryostream 800 low temperature device attached to a Bruker AXS D8 Quest diffractometer equipped with a PHOTON 3 photon-counting detector. Hemispheres of intensity data were collected under control of the APEX3 (ref. 30) software with the raw data reduced to F2 values with SAINT30 which also performed global least squares refinements of the cell parameters. Application of numerical absorption corrections and merging of equivalent reflections was done with SADABS31 and the structures were solved by dual space methods (SHELXT32). The structures were refined by full-matrix, least-squares methods (SHELXL33) with hydrogen atoms included as riding contributions in idealized positions with isotropic displacement parameters tied to those of the attached atoms. For each structure, a few reflections affected by the beamstop were omitted from the final refinement. Crystal data, data collection, and refinement parameters are given in Table 1.| 1 | 2 | 3 | |
|---|---|---|---|
| Emp. formula | C34H30Mn2N6O14 | C24H16N4O10Zn | C17H16ClN3O7Zn |
| Formula weight | 856.52 g mol−1 | 585.78 g mol−1 | 475.15 g mol−1 |
| Temperature | 150(2) K | 150(2) K | 150(2) K |
| Wavelength | 0.71073 Å | 0.71073 Å | 0.71073 Å |
| Crystal system | Triclinic | Monoclinic | Triclinic |
| Space group | P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/C | P |
| a (Å) | 7.1430(12) | 13.7121(19) | 8.5124(9) |
| b (Å) | 9.3717(15) | 17.737(3) | 9.5030(10) |
| c (Å) | 13.017(2) | 9.5694(13) | 12.0869(13) |
| α (°) | 85.852(2)° | 90 | 75.923(2) |
| β (°) | 75.893(2)° | 93.303(2) | 84.467(2) |
| γ (°) | 89.950(2)° | 90° | 80.995(2) |
| Volume (Å3) | 842.8(2) | 2323.5(6) | 934.96(17) |
| Z | 1 | 4 | 2 |
| Density | 1.688 g cm−3 | 1.675 g cm−3 | 1.688 g cm−3 |
| Abs. coeff. | 0.833 mm−1 | 1.127 mm−1 | 1.504 mm−1 |
| F(000) | 438 | 1192 | 484 |
| Crystal size | 0.117 × 0.121 × 0.250 mm | 0.112 × 0.128 × 0.320 mm | 0.196 × 0.218 × 0.250 mm |
| Theta range | 1.62 to 27.57° | 1.49 to 28.37° | 1.74 to 29.32° |
| Index ranges | −9 ≤ h ≤ 9 | −18 ≤ h ≤ 18 | −11 ≤ h ≤ 11 |
| −12 ≤ k ≤ 12 | −23 ≤ k ≤ 23 | −13 ≤ k ≤ 13 | |
| 16 ≤ l ≤ 16 | −12 ≤ l ≤ 12 | −16 ≤ l ≤ 16 | |
| Reflections collected | 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 261 261 |
21604 |
18159 |
| Independent reflections | 10490 |
5756 | 5008 |
| Rint | 0.037 | 0.0693 | 0.0263 |
| Data/restraints/parameters | 15261/0/254 |
5756/0/352 | 5008/0/262 |
| GOF on F2 | 1.070 | 1.047 | 1.127 |
| Final R indices[I > 2σ(I)] | R1 = 0.0618, wR2 = 0.1660 | R1 = 0.0490, wR2 = 0.1168 | R1 = 0.0255, wR2 = 0.0708 |
| R Indices (all data) | R1 = 0.0913, wR2 = 0.1838 | R1 = 0.0798, wR2 = 0.1298 | R1 = 0.0287, wR2 = 0.0717 |
| Largest diff. peak and hole | 1.139 and −0.743 eÅ−3 | 1.007 and −0.617 eÅ−3 | 0.583 and −0.284 eÅ−3 |
DFT calculations
The interaction energies were evaluated at the RI-PB86-D3/def2-TZVP34–36 level of theory by means of the Turbomole 7.0 program37 and using the crystallographic coordinates. The D3 dispersion36 correction scheme was applied to the interaction energies because it is convenient for the correct evaluation of noncovalent interactions, specially π-stacking interactions. For the Mn(II) metal centre (compound 1) the high spin configuration was used. The QTAIM38 analyses were used to characterize the NCIs using the RI-PB86-D3/def2-TZVP wavefunction.The independent gradient model plot (IGM Plot39), an improved NCIPlot tool, has been used to identify and quantify the noncovalent interactions. The IGM-δg approach is based on a new definition of a non-interacting system in terms of electron density contra-gradience (electron density clash between two sources). A new descriptor, δg, locally accounts for the difference between a virtual upper limit of the electron density gradient (∇ρIGM), representing a non-interacting system, and the true electron density gradient (∇ρ). Therefore, it provides an uncoupling scheme that automatically extracts the signature of interactions between two given fragments (δginter), inside these fragments (δgintra), or even between two atoms (δgpair). The (δginter) description has been used in this work, since we are interested in the intermoelcular interactions. Both the QTAIM and IGM-δginter analyses have been performed using the Multiwfn program40 and represented using the VMD visualization software.41
Biological studies
Analysis of apoptosis for 2 and 3 by cytometry using TUNEL assay. Also, apoptosis was detected using an in situ cell death detection kit (Boehringer Mannheim Corp., Indianapolis, IN) as described by Narla et al. and Zhu et al. (Narla et al., 2000;42 Zhu et al., 1998 (ref. 43)). Cells were incubated with 1–3 in 0.3% DMSO or 1:16-diluted plasma samples from DFX-treated mice for 48 h at 37 °C and were fixed, permeabilized and incubated with the reaction mixture containing TdT- and FITC-conjugated dUTP, and counterstained with propidium iodide. Cells were transferred to slides and viewed with a confocal laser scanning microscope (Bio-Rad MRC 1024) mounted on a Nikon Eclipse E800 series upright microscope as reported previously (Narla et al., 2000;42 Zhu et al., 1998 (ref. 43)).
Results and discussion
Synthesis
Complexes 1–3 were synthesized by the reaction of Zn and Mn salts, H2pydco, and bpy at elevated temperature (see the Experimental section for details). Careful control of reaction time and solvent type are key factors to obtain the complexes reported herein. It is important to note that by changing the accompanied anion from NO3− to Cl−, 3 was obtained. The crystallization time of 1–3 was a few days by slow evaporation of mother liquor. Also, powder X-ray diffraction further demonstrated the bulk purity of 1–3 (Fig. S1–S3† (in the ESI)).† Complexes 1–3 are soluble in water, DMSO and are stable in air.IR spectra
The infrared spectra of 1–3 are consistent with the structures determined by single-crystal X-ray diffraction (See Table 2 and Fig. S4†). The broad and strong bands at 3000–3500 cm−1 can be attributed to the O–H stretching vibrations of lattice and coordinated water molecules and the hydroxyl groups of Hpydco−. They are also indicative of the presence of hydrogen bonds.12,44 In comparison with free H2pydco (1725 and 1413 cm−1), the strong asymmetric and symmetric stretching vibrations of the COO− group in 1–3 are shifted to the lower wavenumbers in the range of 1650–1640 cm−1 and 1400–1390 cm−1, respectively. The separation between νas(COO−) and νs(COO−) has been often used to diagnose the coordination modes for carboxylate ligands. The separation for monodentate carboxylate groups is >200 cm−1, whereas it is >200 cm−1 in bidentate ones. In our complexes this separation shows that the carboxylate group is coordinated monodentately to the metal centre in agreement with the crystal structure. Also, bands in the 1255–1207 cm−1 region were assigned to the N–O stretching vibrations of the pyridine-N-oxide.45,46| Compound | ν(OH) | νas(COO−) | νs(COO−) | ν(N–O) |
|---|---|---|---|---|
| 1 | 3481 | 1640 | 1390 | 1255 |
| 2 | 3130 | 1650 | 1395 | 1210 |
| 3 | 3487 | 1644 | 1400 | 1207 |
| H2pydco | 3138 | 1725 | 1413 | 1229 |
Thermogravimetric study
The thermal stability of 1–3 has been studied by thermogravimetric analysis (TGA) under an air atmosphere. The TG curve of 1 exhibits two weight-loss events (Fig. S5†). The first stage occurs between 25 and 165 °C which corresponds to the removal of three uncoordinated and coordinated water molecules amounting to 6.30 (calcd 6.27%) and the second stage is a double stage sequential process from 170 to 450 °C, due to the combustion of the two pydco2− and two bpy ligands with a weight loss of 79.50 (calcd 78.21%). For 2, the TG curve exhibits a one-step weight loss that is in consistent with its formula (without coordinated or uncoordinated water molecules), occurring between 25 and 540 °C which is attributed to the decomposition of the two Hpydco− and one bpy ligands with a weight loss of 82.76 (calcd 89.57%). Unlike those for 1 and 2, the TG curve for 3 exhibits a three-step weight loss process. The weight losses of 7.58, 7.47, and 71.58%, occurring in the temperature ranges of 25–200, 200–221, and 220–450 °C, respectively, can be attributed to the removal of two uncoordinated water molecules, Cl− as well as Hpydco− and bpy ligands (calcd 7.21, 7.40 and 68.21%).Structure description of 1 and 2
Crystallographic details can be found in the CIF files. The CIF files are available free of charge from the Cambridge Crystallographic Data Centre CCDC47 (1934944, 1979889 and 2009484). Structural analysis indicates 1–3 contain discrete complexes containing N-donor bpy and O-donor pydco2− (or Hpydco−) ligands. Selected bond distances and angles and a list of hydrogen bond geometries are reported in Tables S1 and S2.†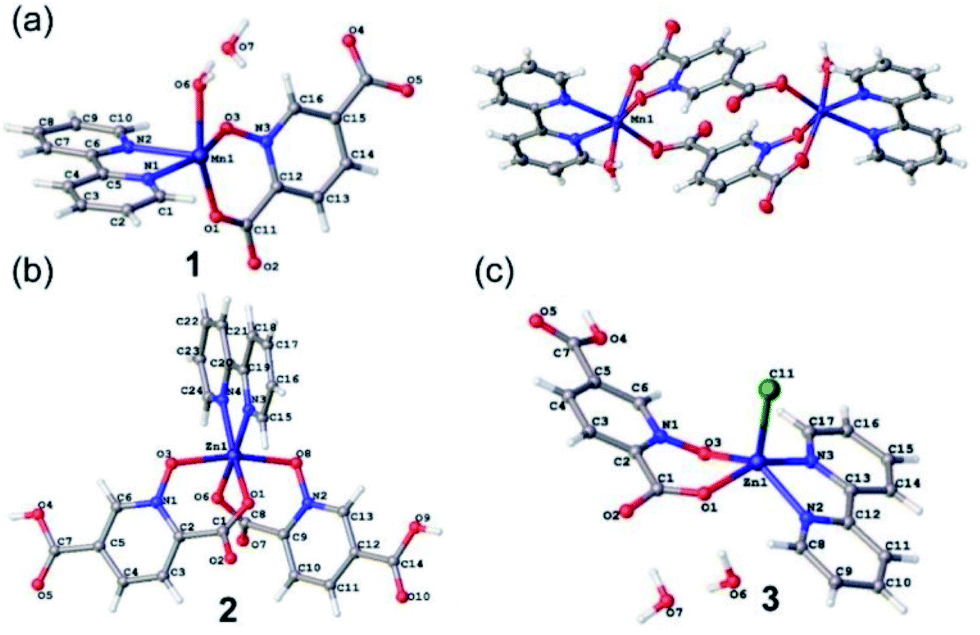 | ||
| Fig. 1 The asymmetric units of 1 (a), 2 (b) and 3 (c) with selected atoms labeled. The centrosymmetric dimer of 1 is also shown (top right). | ||
| Compound | MCF-7 | MDA-MD-231 | HT-29 | HeLa | Neuro-2a | L929 |
|---|---|---|---|---|---|---|
| a The concentration of the complex required to inhibit cell growth by 50%. The experiments were done in triplicate. Data were expressed as the mean of the triplicate. IC50 > 100 μM is considered to be inactive. | ||||||
| 1 | 56.6 ± 7.78 | 28.9 ± 2.47 | 57.9 ± 4.71 | 39.2 ± 2.98 | 79.5 ± 8.44 | >100 |
| 2 | 15.6 ± 3.24 | 5.94 ± 1.98 | 17.3 ± 2.57 | 41.2 ± 5.07 | 81.2 ± 9.28 | >100 |
| 3 | 17.9 ± 2.66 | 9.18 ± 2.25 | 14.6 ± 3.55 | 37.6 ± 4.61 | 87.7 ± 6.93 | >100 |
| H2pydco | 57.9 ± 5.33 | 45.7 ± 4.75 | 58.9 ± 5.01 | 67.1 ± 5.45 | >100 | >100 |
| bpy | 51.3 ± 4.78 | 24.9 ± 1.98 | 67.8 ± 5.54 | 46.5 ± 4.03 | 97.3 ± 8.70 | >100 |
| Cisplatin | 5.94 ± 1.47 | 24.7 ± 4.71 | 19.3 ± 3.46 | 0.45 ± 0.13 | 103 ± 9.8 | 0.7 ± 0.2 |
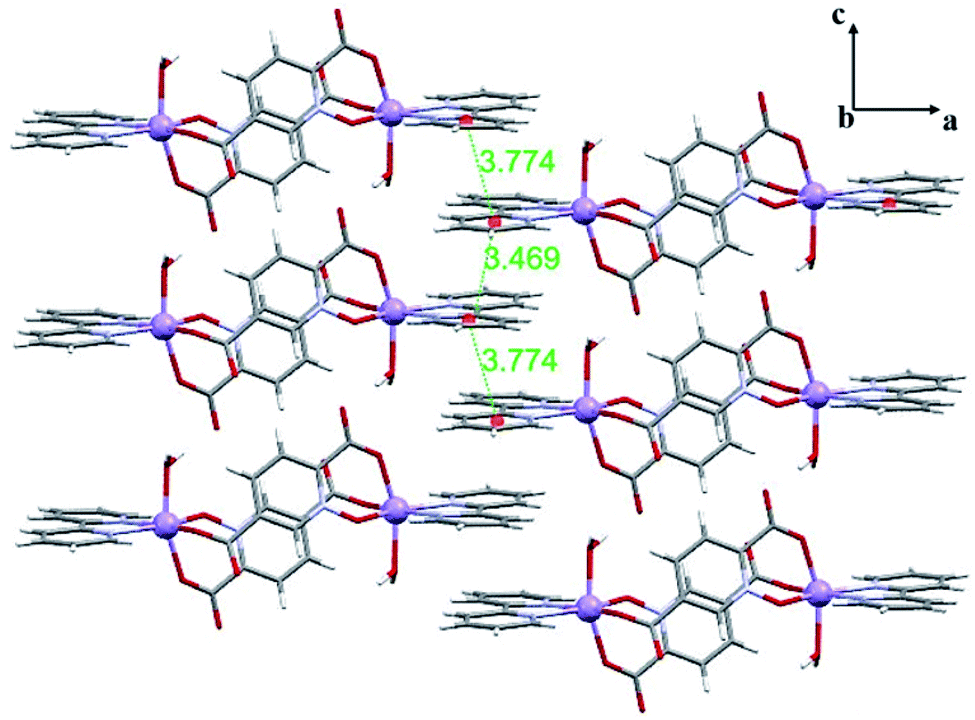 | ||
| Fig. 2 2D representation of 1 in the ac plane formed by π-interactions (distances are shown in Å). | ||
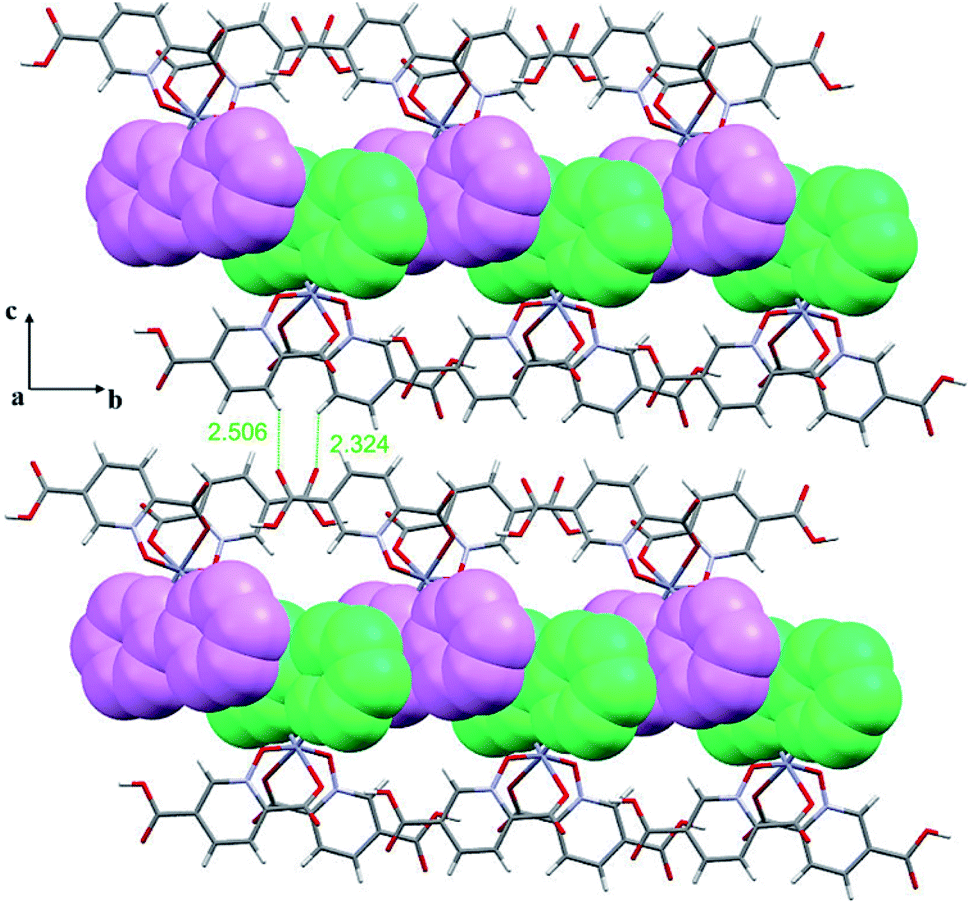 | ||
| Fig. 3 2D representation of 2 in the bc plane formed by π-interactions between adjacent bpy rings where bpy rings from up and down monomers are shown in different colours (purple and green) as well as intermolecular C–H⋯O hydrogen bonding (distances are shown in Å). | ||
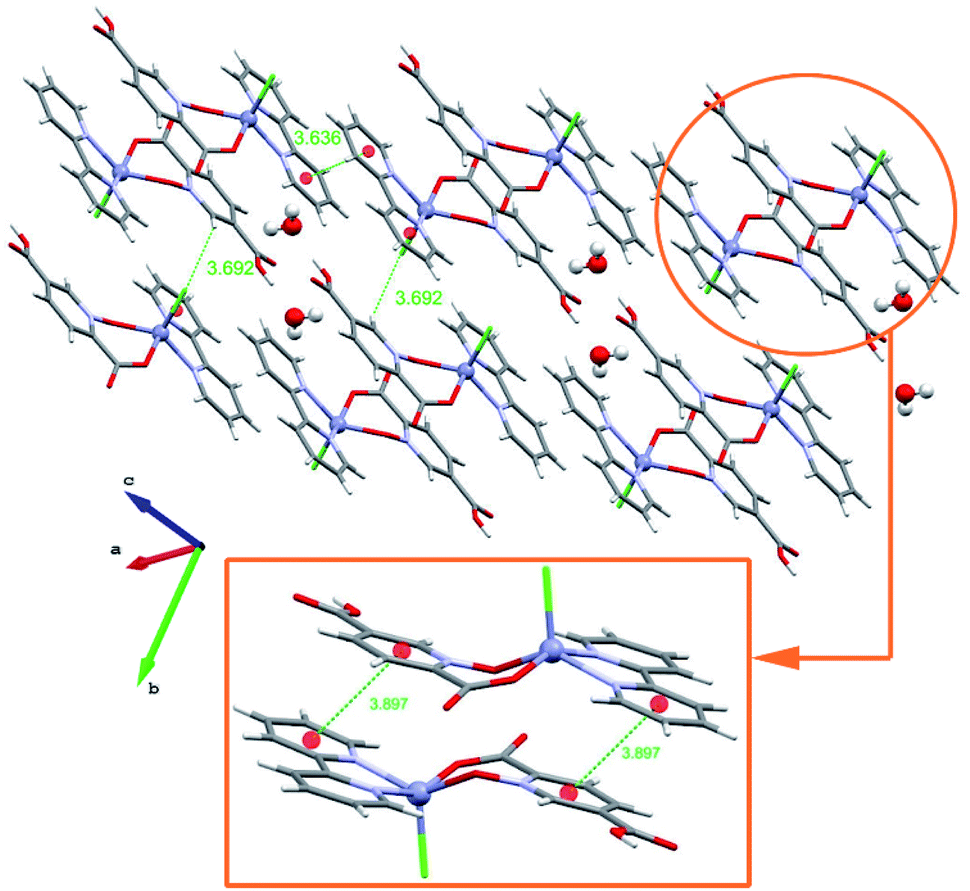 | ||
| Fig. 4 2D representation of 3 that is formed by extensive π-interactions as well as intermolecular O–H⋯O hydrogen bonds (distances are shown in Å). | ||
DFT analysis of noncovalent interactions
In this section we have used the IGM plot method to identify the noncovalent interactions in real space. This representation uses a colour code to identify attractive and repulsive noncovalent contacts that is based on the sign of the middle eigenvalue of the Hessian of ρ(λ2). Blue and green are used to represent strong and weak attractive interactions, respectively and yellow and red to indicate weak and strong repulsive interactions, respectively.The study is focused on the comparison of the relevant structure-guiding π-stacking interactions observed in compounds 1–3, as highlighted before in Fig. 2–4. For compound 1 the QTAIM analysis shows the existence of several bond CPs (small red spheres), bond paths and an extended green IGM isosurface connecting the pypiridine ligands. Moreover, two symmetrically equivalent CH⋯O HBs are also disclosed, each one characterized by a bond CP, bond path and small green isoosurface interconnecting the O and H-atoms. We have evaluated the dissociation energy of each H-bond by using the equation proposed by Espinosa et al. (Edis = ½ × Vr49) that uses the potential energy density (Vr) at the bond CP that characterizes the HB. As a result, the energy of the CH⋯O HBs in 1 is 0.9 kcal mol−1 (1.8 kcal mol−1 the total HB contribution), thus evidencing that the formation of the dimer is dominated by the π-stacking interaction. Such strong interaction energy of 1 (ΔE1 = −32.9 kcal mol−1) is due to the antiparallel orientation of the molecules that maximizes the dipole···dipole interaction. In this type of complexes, where the aromatic rings are coordinated to metal centres, the π-system is very polarized, generating large dipole···dipole attractions, as previously demonstrated.50–54 In compound 2, a similar combination of interactions is observed. That is, the QTAIM/IGM plot analysis shows the existence of a π-stacking interaction characterized by two bond CPs and bond paths connecting two atoms of each aromatic ring and two CH⋯O interactions characterized by the corresponding bond CPs and bond paths. All interactions are also confirmed by the IGM plot isosurfaces. In this case one H-bond is stronger (blue isosurface) with a concomitant dissociation energy of 2.1 kcal mol−1. The other H-bond is weaker (0.7 kcal mol−1) in line with the green colour and small size of the IGM surface. In this case the interaction energy is moderately strong (ΔE2 = −18.0 kcal mol−1) because the overlap is of the π-system is smaller compared to 1.
Finally, in compound 3, the π-stacked dimer represented in Fig. 5c shows an intricate combination of interactions that explains the strong interaction energy (ΔE3 = −34.8 kcal mol−1). That is, in addition to the π-stacking interactions between the π-clouds of the bipyridine and pyridine-2,5-dicarboxylic acid N-oxide ligands, the QTAIM analysis shows the existence of two remarkable π-hole⋯O interactions between the O-atoms of the N-oxide and the carbonyl C-atoms of the coordinated carboxylate groups, characterized by the corresponding bond CPs and bond paths interconnecting the O and C-atoms. Moreover, the IGM plot reveals a blue isosurface between the C and O-atoms confirming the existence and strong nature of these contacts. Furthermore, the QTAIM distribution of bond CPs reveals also the existence of a lp⋯π interaction between the non-coordinated O-atom of the carboxylate group and the π-system of the bipyridine ring.
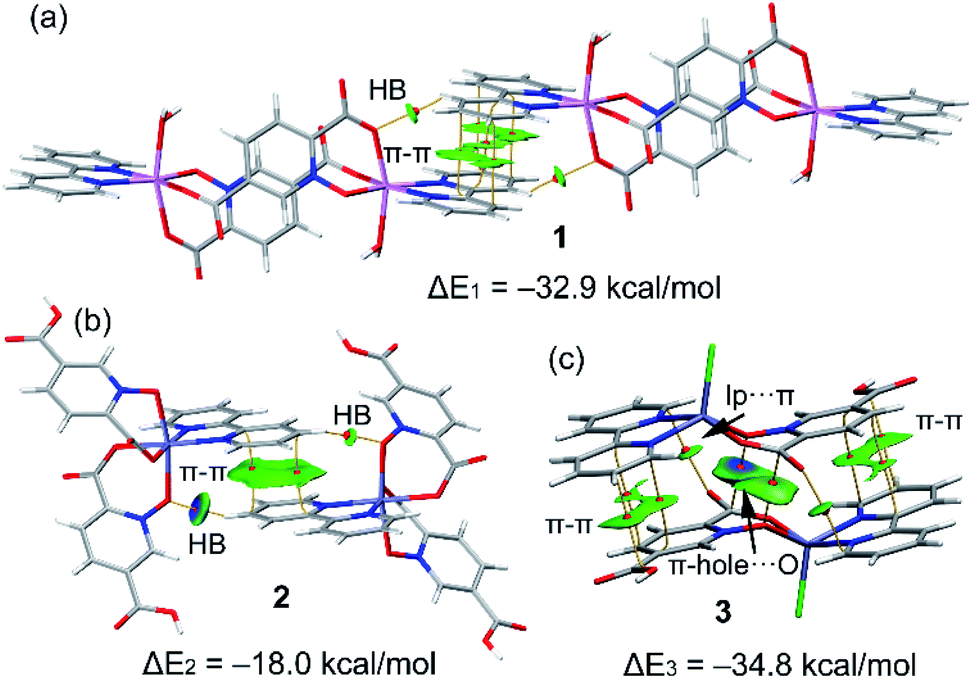 | ||
| Fig. 5 Combined QTIAM (bond CPs in red and bond path as orange lines) and IGM plot (IGM-δginter isosurface 0.085) and colour range −0.03 a. u. ≤ (signλ2)ρ ≤ 0.03 a. u. The dimerization energies are also indicated. Level of theory: PBE0-D3/def2-TZVP. | ||
Biological studies
Clearly from Table 3, all three complexes exhibited remarkable cytotoxic activity against cancer cell lines. In some cases, the IC50 values were lower than those of cisplatin indicating better antiproliferative effects. Among the complexes, 2 and 3 displayed the best results in terms of cytotoxicity. It seems that the presence of the bioactive metal, zinc, H2pydco and bpy ligands play a decisive role in generating cytotoxic effects and consequently antiproliferative activities.Cytotoxic activity was simultaneously measured for mouse fibroblast normal cell line (L929) as control. As shown in Table 4, 2 and 3, displayed cytotoxic activities against L929 significantly higher than against cancer cell lines making them appropriate candidates for anticancer drugs. A notable point is that in the case of cisplatin, the IC50 value against normal cell L929 was so low that it was unable to make a distinction between normal and cancer cells.
| Treatment | Vital cells (%) | Apoptotic cells (%) | Late apoptotic/necrotic cells (%) | Necrotic cells (%) |
|---|---|---|---|---|
| Control | 81.13 | 8.68 | 9.45 | 0.74 |
| Cisplatin | 33.74 | 34.65 | 30.63 | 0.98 |
| 2 | 19.73 | 43.91 | 35.54 | 0.82 |
In this study, the potential of three complexes of Mn and Zn as effective antiproliferative agents have been investigated in vitro. In order to assess this, the in vitro cytotoxicity of these complexes, against MDA-MDA-MD-231, MCF-7, HeLa, HT-29, Neuro-2a and L-929 cell lines were determined by MTT-based assays (Table 3). For 1–3, and cisplatin as a comparative standard, we have measured IC50 values for all cell lines. The measurements were done after 72 h of incubation using concentrations of the several complexes in the range 20 nM and 200 μM. The values determined for these complexes spanned between 5.94 to 100 μM for 2 and 3, while those found for cisplatin as a comparative standard ranged between 0.7 to 100 μM (Table 3).
Based on the results of the in vitro cytotoxicity studies of 2 and 3, those systems having maximum cytotoxicity were selected for apoptosis assay by flow cytometry.
As seen in Tables 4, 5, Fig. 6 and 7, 2 showed a high population of apoptotic cell (79.45%), nearly 1.2-fold higher than cisplatin (65.28%) at the same concentration. Also according to Table 5 and 3 showed a high population of apoptotic cell (63.3%), nearly 1.8-fold higher than cisplatin (35.8%) at the same concentration against MDA-MB-231 cancer cell line.
| Treatment | Vital cells (%) | Apoptotic cells (%) | Late apoptotic/necrotic cells (%) | Necrotic cells (%) |
|---|---|---|---|---|
| Control | 99.00 | 0.30 | 0.10 | 0.60 |
| Cisplatin | 61.00 | 15.80 | 20.00 | 3.20 |
| 3 | 32.00 | 4.30 | 59.00 | 4.70 |
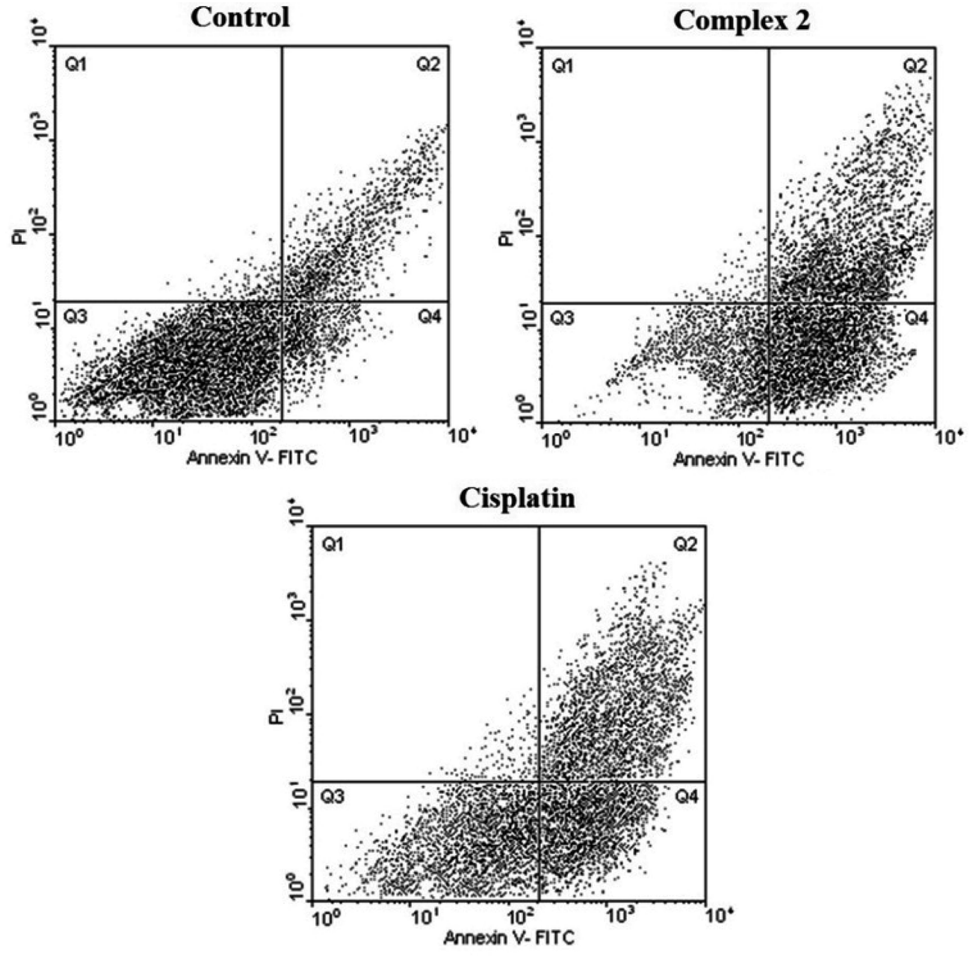 | ||
| Fig. 6 Flow cytometric results after the exposure of MDA-MD-231 cancer cells to 2 and to cisplatin. Four areas in the diagrams represent four different cell states: necrotic cells (Q1), late apoptotic or necrotic cells (Q2), living cells (Q3) and apoptotic cells (Q4). | ||
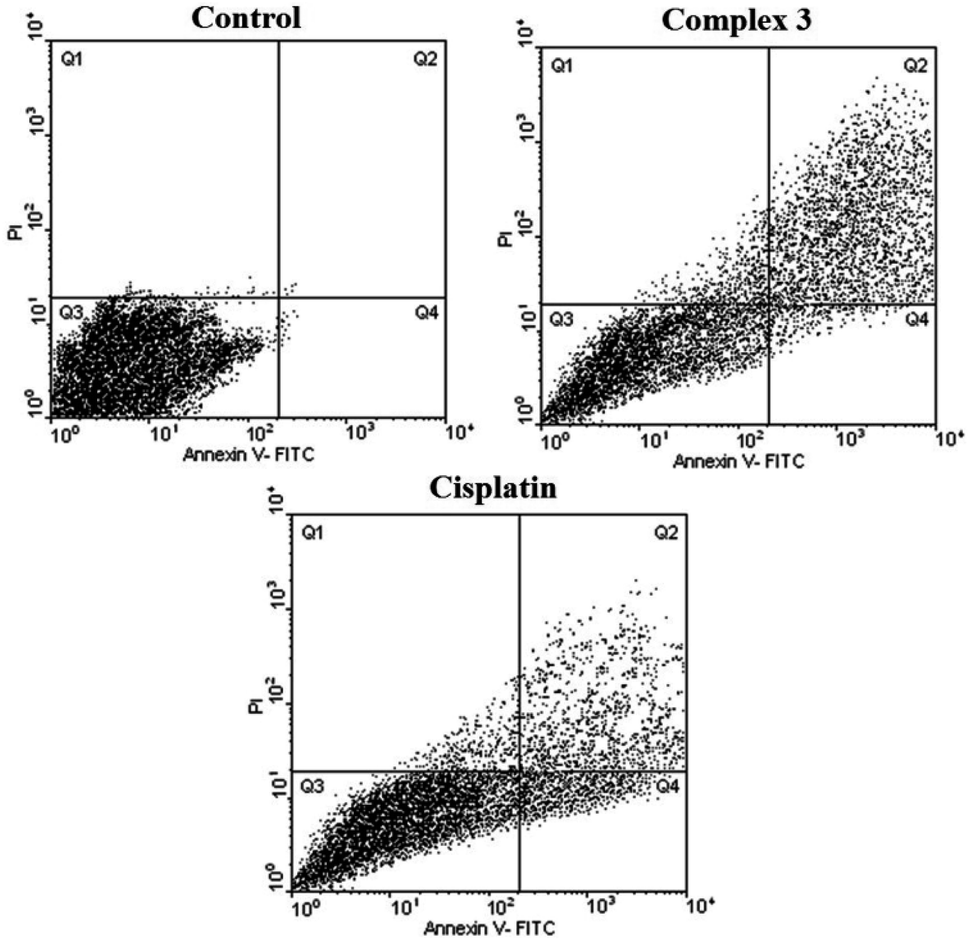 | ||
| Fig. 7 Flow cytometric results after the exposure of MDA-MD-231 cancer cells to 3 and cisplatin. Four areas in the diagrams represent four different cell states: necrotic cells (Q1), late apoptotic or necrotic cells (Q2), living cells (Q3) and apoptotic cells (Q4). | ||
Also, 2- and 3-induced cell death was confirmed to be apoptotic using the TUNEL of exposed 3′-OH termini of DNA with dUTP-FITC. As shown in the confocal laser scanning microscopy images in Fig. 8, complex-treated MDA-MB-231 breast cancer cells, examined for dUTP-FITC incorporation (green fluorescence) and propidium iodide counterstaining (red fluorescence), exhibited many apoptotic yellow nuclei (superimposed green and red fluorescence) at 24 h after treatment.
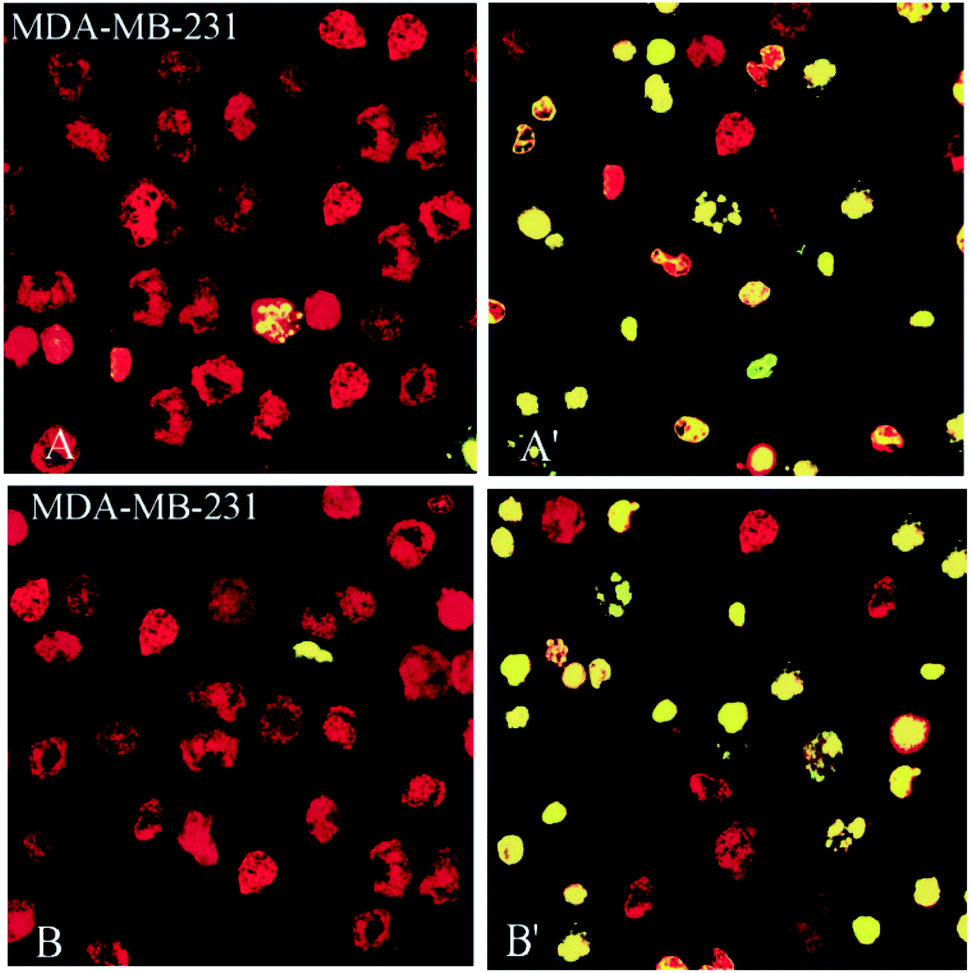 | ||
| Fig. 8 Compounds 2 and 3, induce apoptosis in human breast (MDA-MD-231) cancer cell line. MDA-MB-231 breast cancer (A, A′ for 2 and B, B′ for 3) cells were incubated with 10 mM of 2, 3 for 24 h, fixed, permeabilized and visualized for DNA degradation in a TUNEL assay using dUTP-labeling. Red fluorescence, nuclei stained with propidium iodide. Green or yellow (i.e., superimposed red and green) fluorescence, apoptotic nuclei containing fragmented DNA. When compared with controls, treated with 0.3% DMSO (A and B), several of the cells incubated with 2 and 3 (A′, B′) exhibited apoptotic nuclei. | ||
The results demonstrated that 2 and 3 could induce apoptosis against MDA-MB-231 cancer cell line but the proapoptotic property needs further investigation to better understand the precise mechanism of action of these complexes and basic pre-clinical research is needed before they could be recommended for human administration.
Conclusion
In this work, we report an approach where the combination of N- and O-donor ligands leads to interesting and diverse frameworks incorporating bio-essential metal ions such as manganese and zinc. This study demonstrates that supramolecular frameworks of 1–3 constructed by self-assembling through electrostatic interactions such as strong hydrogen bonds and a variety of π–π stacking interactions. Which have been further studied using DFT calculations including the IGM plot analysis. They revealed the existence of strong π-stacking interactions in the solid state of the three complexes and remarkable π-hole⋯O contacts between the N-oxide and carboxylate groups in compound 3. We have considered the antiproliferative effects of these complexes and compare their biological activity with each other. The obtained results showed that the effect of antiproliferative of 1–3 is due to the presence of metal centres and is somewhat influenced by the auxiliary ligand, and apparently the main ligand did not show any antiproliferative activity. Heteroleptic complexes are preferable to homoleptic complexes due to their size variation, type of interactions, effect on metal oxidation numbers, and many other factors.Author contributions
Hanie Alizadeh: methodology, formal analysis, investigation, data curation, writing-original draft preparation. Masoud Mirzaei: conceptualization, funding acquisition, main idea, supervision, writing-review and editing, project administration, visualization. Amir Sh. Saljooghi: biological studies, funding acquisition, supervision, writing-review and editing. Vida Jodaian: formal analysis, data curation. Maryam Bazargan: formal analysis, software, data curation, writing-original draft preparation. Joel T. Mague: crystallographer. Rosa M. Gomila: DFT studies. Antonio Frontera: DFT studies, funding acquisition, supervision, writing-review-review and editing, project administration, visualization.Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. M. gratefully acknowledges financial support from the Ferdowsi University of Mashhad (Grant No. 3/48157), the Iran Science Elites Federation (ISEF), Zeolite and Porous Materials Committee of Iranian Chemical Society and the Iran National Science Foundation (INSF). M. M. also acknowledges the Cambridge Crystallographic Data Centre (CCDC) for access to the Cambridge Structural Database. This work is supported by Iran Science Elites Federation Grant No. M/98208, M/99397, and M/400230. J. T. M thanks Tulane University for support of the Tulane Crystallography Laboratory. We thank the MICIU/AEI from Spain for financial support (project numbers CTQ2017-85821-R and PID2020-115637GB-I00, FEDER funds).Notes and references
- A. Johnson, L. Iffland, K. Singh, U.-P. Apfel and K. Suntharalingam, Dalton Trans., 2021, 50, 5779–5783 RSC
.
- F. Delavar Mendi, A. Sh. Saljooghi, M. Ramezani, R. Kruszynski, M. Poupon, M. Kucerakova, V. Huch, P. Socha, M. Babaei and M. Alibolandi, J. Biomol. Struct. Dyn., 2020, 38, 1–14 CrossRef PubMed
- A. Eskandari and K. Suntharalingam, Chem. Sci., 2019, 10, 7792–7800 RSC
- C. P. Matos, Y. Addis, P. Nunes, S. Barroso, I. Alho, M. Martins, A. P. Matos, F. Marques, I. Cavaco, J. C. Pessoa and I. Correia, J. Inorg. Biochem., 2019, 198, 110727 CrossRef CAS PubMed
- Y.-P. Zhang, Z.-Y. Ma, C.-Y. Gao, X. Qiao, J.-L. Tian, W. Gu, X. Liu, J.-Y. Xu, J.-Z. Zhao and S.-P. Yan, New J. Chem., 2016, 40, 7513–7521 RSC
- Q.-L. Guan, Z. Liu, W.-J. Wei, Y.-H. Xing, J. Liu, R. Zhang, Y.-N. Hou, X. Wang and F.-Y. Bai, New J. Chem., 2014, 38, 3258–3268 RSC
- Y. Shi, B. B. Toms, N. Dixit, N. Kumari, L. Mishra, J. Goodisman and J. C. Dabrowiak, Chem. Res. Toxicol., 2010, 23, 1417–1426 Search PubMed
- P. F. Liguori, A. Valentini, M. Palma, A. Bellusci, S. Bernardini, M. Ghedini, M. L. Panno, C. Pettinari, F. Marchetti, A. Crispini and D. Pucci, Dalton Trans., 2010, 39, 4205–4212 RSC
- M. Z. Eraj, L. Eriksson, M. Alibolandi, M. Babaei, A. S. Saljooghi and M. Ramezani, New J. Chem., 2020, 44, 20101–20114 RSC
- S. C. Marker, S. N. MacMillan, W. R. Zipfel, Z. Li, P. C. Ford and J. J. Wilson, Inorg. Chem., 2018, 57, 1311–1331 CrossRef CAS PubMed
- Y. Xiong, Y.-Z. Fan, R. Yang, S. Chen, M. Pan, J.-J. Jiang and C.-Y. Su, Chemcomm, 2014, 50, 14631–14634 RSC
- F. A. Mautner, J. H. Albering, R. Vicente, C. Andrepont, J. G. Gautreaux, A. A. Gallo and S. S. Massoud, Polyhedron, 2013, 54, 158–163 CrossRef CAS
- M. Mirzaei, H. Eshtiagh-Hosseini, A. Hassanpoor, T. Szymańska-Buzar, J. Mague, M. Korabik and A. Kochel, Inorg. Chim. Acta, 2012, 391, 232–238 CrossRef CAS
- T. S. Kamatchi, N. Chitrapriya, H. Lee, C. F. Fronczek, F. R. Fronczek and K. Natarajan, Dalton Trans., 2012, 41, 2066–2077 RSC
- J. Balzarini, M. Stevens, E. De Clercq, D. Schols and C. Pannecouque, J. Antimicrob. Chemother., 2005, 55, 135–138 CrossRef CAS PubMed
- S. Lis, Z. Hnatejko, P. Barczynski and M. Elbanowski, J. Alloys Compd., 2002, 344, 70–74 CrossRef CAS
- M. Mirzaei, F. Sadeghi, K. Molcanov, J. K. Zareba, R. M. Gomila and A. Frontera, Cryst. Growth Des., 2020, 20, 1738–1751 CrossRef CAS
- M. Bazargan, M. Mirzaei, M. Aghamohamadi, M. Tahmasebi and A. Frontera, J. Mol. Struct., 2020, 1202, 127243 CrossRef CAS
- Z. Hosseini-Hashemi, M. Mirzaei, A. Jafari, P. Hosseinpour, M. Yousefi, A. Frontera, M. L. Dashtbayaz, M. Shamsipur and M. Ardalani, RSC Adv., 2019, 9, 25382–25404 RSC
- M. Shahbazi, F. Mehrzad, M. Mirzaei, H. Eshtiagh-Hosseini, J. T. Mague, M. Ardalani and M. Shamsipur, Inorg. Chim. Acta, 2017, 458, 84–96 CrossRef CAS
- H. Eshtiagh-Hosseini, M. Mirzaei, M. Biabani, V. Lippolis, M. Chahkandi and C. Bazzicalupi, CrystEngComm, 2013, 15, 6752–6768 RSC
- S.-L. Huang, L. Zhang, Y.-J. Lin and G.-X. Jin, CrystEngComm, 2013, 15, 78–85 RSC
- Y. Wei, H. Hou, L. Li, Y. Fan and Y. Zhu, Cryst. Growth Des., 2005, 5, 1405–1413 CrossRef CAS
- H. S. Moradi, E. Momenzadeh, M. Asar, S. Iranpour, A. R. Bahrami, M. Bazargan, H. Hassanzadeh, M. M. Matin and M. Mirzaei, J. Mol. Struct., 2021, 1249, 131584 CrossRef
- A. Hashemzadeh, G. P. C. Drummen, A. Avan, M. Darroudi, M. Khazaei, R. Khajavian, A. Rangrazi and M. Mirzaei, J. Mater. Chem. B, 2021, 9, 3967–3982 RSC
- M. Falsafi, A. S. Saljooghi, K. Abnous, S. M. Taghdisi, M. Ramezani and M. Alibolandi, Biomater. Sci., 2021, 9, 1503–1529 RSC
- S. Iranpour, A. R. Bahrami, A. S. Saljooghi and M. M. Matin, Coord. Chem. Rev., 2021, 442, 213949 CrossRef CAS
- M. Arefian, M. Mirzaei, H. Eshtiagh-Hosseini and A. Frontera, Dalton Trans., 2017, 46, 6812–6829 RSC
- Z. Hosseini Hashemi, M. Mirzaei, H. Eshtiagh-Hosseini, F. Sadeghi, M. Shamsipur, M. Ardalani and A. J. Blake, J. Coord. Chem., 2018, 71, 4058–4071 CrossRef CAS
- B. A. L. APEX3 and SAINT, Madison, WI., 2000.
- L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2015, 48, 3–10 CrossRef CAS PubMed
- N. Sebbar, M. Ellouz, E. Essassi, Y. Ouzidan and J. Mague, Acta Crystallogr. E, 2015, 71, o999 CAS
- G. M. Sheldrick, Acta Crystallogr. C, 2015, 71, 3–8 CrossRef PubMed
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed
- R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS
- C. Lefebvre, H. Khartabil, J. C. Boisson, J. Contreras-García, J. P. Piquemal and E. Hénon, ChemPhysChem, 2018, 19, 724–735 CrossRef CAS PubMed
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed
- R. K. Narla, Y. Dong, O. J. D'Cruz, C. Navara and F. M. Uckun, Clin. Cancer Res., 2000, 6, 1546–1556 CAS
- D. M. Zhu, R. K. Narla, W. H. Fang, N. C. Chia and F. M. Uckun, Clin. Cancer Res., 1998, 4, 2967–2976 CAS
- Z.-Q. Xia, Q. Wei, S.-P. Chen, X.-M. Feng, G. Xie, C.-F. Qiao, G.-C. Zhang and S.-L. Gao, J. Solid State Chem., 2013, 197, 489–498 CrossRef CAS
- H.-L. Sun, X.-L. Wang, L. Jia, W. Cao, K.-Z. Wang and M. Du, CrystEngComm, 2012, 14, 512–518 RSC
- K. Nakamoto, Handbook of vibrational spectroscopy, 2006 Search PubMed
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr. B, 2016, 72, 171–179 CrossRef CAS PubMed
- M. Bazargan, M. Mirzaei, A. Franconetti and A. Frontera, Dalton Trans., 2019, 48, 5476–5490 RSC
- E. Espinosa, E. Molins and C. Lecomte, Chem. Phys. Lett., 1998, 285, 170–173 CrossRef CAS
- H. Nath, P. Sharma, A. Frontera, M. Barcelo-Oliver, A. K. Verma, J. Das and M. K. Bhattacharyya, J. Mol. Struct., 2021, 1248, 131424 CrossRef
- A. Das, P. Sharma, A. Frontera, M. Barceló-Oliver, A. K. Verma, R. Ahmed, S. Hussain and M. K. Bhattacharyya, New J. Chem., 2021, 45, 13040–13055 RSC
- S. Chetry, P. Sharma, A. Frontera, U. Saha, A. K. Verma, B. Sarma, P. J. Kalita and M. K. Bhattacharyya, New J. Chem., 2021, 45, 3699–3715 RSC
- P. Sharma, P. Sarma, A. Frontera, M. Barceló-Oliver, A. K. Verma, B. Sarma, T. Barthakur and M. K. Bhattacharyya, J. Mol. Struct., 2021, 1229, 129486 CrossRef CAS
- P. Sharma, P. Sarma, A. Frontera, S. Hussain, A. K. Verma and M. K. Bhattacharyya, Inorg. Chim. Acta, 2021, 516, 120082 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1934944, 1979889, 2009484. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra08258b |
| This journal is © The Royal Society of Chemistry 2021 |