
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rhodium(I) complexes derived from tris(isopropyl)-azaphosphatrane—controlling the metal–ligand interplay†
Wei-Chieh Chang‡
 ab,
Fritz Deufel‡§
c,
Thomas Weyhermüllera,
Christophe Farèsc and
Christophe Werlé*ab
ab,
Fritz Deufel‡§
c,
Thomas Weyhermüllera,
Christophe Farèsc and
Christophe Werlé*ab
aMax Planck Institute for Chemical Energy Conversion, Stiftstr. 34–36, 45470 Mülheim an der Ruhr, Germany. E-mail: christophe.werle@cec.mpg.de
bRuhr University Bochum, Universitätsstr. 150, 44801 Bochum, Germany
cMax-Planck-Institut für Kohlenforschung, Kaiser-Wilhelm-Platz 1, D-45470 Mülheim an der Ruhr, Germany
First published on 22nd November 2021
Abstract
Proazaphosphatranes are intriguing ligand architectures comprising a bicyclic cage of flexible nature. They can undergo structural deformations due to transannulation while displaying modular electronic and steric properties. Herein, we report the synthesis and coordination chemistry of rhodium(I) complexes bearing a tris(isopropyl)-azaphosphatrane (TiPrAP) ligand. The molecular structure of the primary complex (1) revealed the insertion of the metal center into a P–N bond of the ligand. The addition of a Lewis acid, i.e., lithium chloride, promoted the dynamic behavior of the complex in the solution, which was studied by state-of-the-art NMR spectroscopy. Substituting the cyclooctadiene ligand at the metal center with triphenylphosphine or 2-pyridyldiphenylphosphine unveiled the adaptive nature of the TiPrAP backbone capable of switching its axial nitrogen from interacting with the phosphorus atom to coordinate the rhodium center. This led the entire ligand edifice to change its binding to rhodium from a bidentate to tridentate coordination. Altogether, our study shows that introducing a TiPrAP ligand allows for unique molecular control of the immediate environment of the metal center, opening perspectives in controlled bond activation and catalysis.
Introduction
In catalysis, finding the right metal–ligand combination to regulate the properties of the resulting system and thus meet the requirements of a specific application is a continuous quest.1,2 For many years, the focus has been on the transition metal; the ligands regarded as spectators were supposed to remain unchanged throughout the lifetime of the catalyst. Research to enhance catalyst activity and selectivity has identified ligands as essential design elements.3 Not only do they exert a steric and electronic influence on the complex, but they are also capable of acting in cooperation with their metal.4,5 As an intrinsic part of the local environment of the metal center, ligands have a significant influence on bond activation processes and catalysis.6In this context, the proazaphosphatrane unit composed of a conformationally flexible bicyclic cage, more commonly referred to as Verkade's superbase, represents an intriguing ligand design.7 This architecture is susceptible to structural deformation caused by transannulation, i.e., an intramolecular interaction between the axial nitrogen (Nax) and phosphorus upon binding to an electrophile which results in the formation of an azaphosphatrane (Fig. 1).
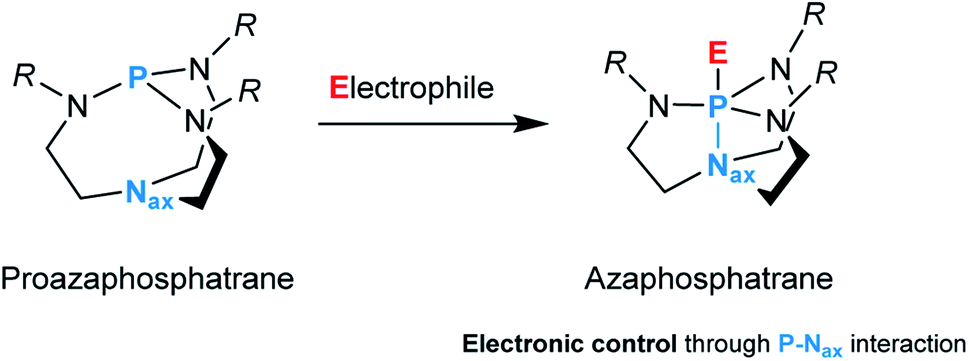 | ||
| Fig. 1 A reaction of a proazaphosphatrane with an electrophile. | ||
Consequently, proazaphosphatranes have attracted considerable interest as stoichiometric or catalytic Lewis bases for various chemical transformations, among which: cyanosilylation of aldehydes and ketones8 or imines,9 selective monoalkylation of active-methylene compounds,10 dehydrohalogenation reactions,11 transesterifications, acylations and deacylations,12 the synthesis of β-hydroxy nitriles,13 or Henry reaction,14 to name a few.
Although the reactivity of proazaphosphatranes has been studied considerably, their coordination chemistry remains largely in its infancy. In this context, Yang and coworkers experimentally determined both Tolman electronic parameters (TEPs) and cone angles for a series of nickel tricarbonyl proazaphosphatrane complexes.15 They found that proazaphosphatranes present variable cone angles depending on the substituents on the equatorial nitrogen (Neq). Additionally, when compared to substituted tertiary phosphine, a higher donor strength was observed, which can be attributed primarily to the presence of amino substituents. In another study, the authors showed that the degree of transannular interaction increases when proazaphosphatrane binds to metal centers with a higher degree of electron deficiency.16 Complementarily, Martinez and coworkers showed by employing DFT calculations that the electron-donating ability of tris(methyl)-azaphosphatrane is better than usual phosphines and similar to N-heterocyclic carbene ligands (NHCs).17
In terms of supporting ligands, proazaphosphatranes and other relevant derivatives have been reported to support their respective metal effectively, as illustrated in cross-coupling reactions.18 Specifically, in their contribution, Johnson and coworkers demonstrated the catalytic relevance of palladium proazaphosphatrane complexes as putative intermediates in C–N cross-couplings.19 The proazaphosphatrane moiety was found to respond to changes in the metal's oxidation state and coordination sphere. The presence of variable transannulation in these complexes unveiled that proazaphosphatranes may accommodate conformational modifications to stabilize catalytic intermediates.18a,20 In a subsequent report, Johnson, Donald, and coworkers studied the factors that influence transannulation.21. They identified that the identity of the electrophile, the substitution at the Neq, and the oxidation state of phosphorus dictate the extent of transannulation. Additionally, they found that ethylene linkers are essential when targeting a strong interaction between the donor (Nax) and acceptor (P) within the molecule.
As part of our research program aimed at developing direct molecular control over bond activation processes and catalysis,22 we speculated that proazaphosphatrane ligands would be attractive molecular handles. The application of these ligands may provide an opportunity for deliberate regulation of the metal's immediate environment by varying the extent of transannulation throughout the catalytic process.
Results and discussion
Treatment of a dichloromethane solution of Rh(COD)2SbF6 (COD = 1,5-cyclooctadiene) with stoichiometric amounts of TiPrAP at room temperature led to the formation of an unprecedented complex (1) whose molecular identity could be accessed by NMR spectroscopy and X-ray diffraction analysis. The solid-state structure of 1 revealed a rhodium(I) center in a square planar geometry, bearing the TiPrAP skeleton and a COD moiety both coordinated in a bidentate manner to the metal center (Fig. 2). The unusual architecture of 1 may result from a tandem reaction under the premise of a phosphorus-directed C–H bond activation,23 followed by a hydride transfer reaction and structural rearrangement (Scheme 1).24 Similar breakdowns of the proazaphosphatrane core, resulting in the formation of a transannulated species lacking a third amido donor at the phosphorus atom, remain rare in the literature25 and have not been observed for a similar system involving [Rh(CO)Cl]2 as the precursor.17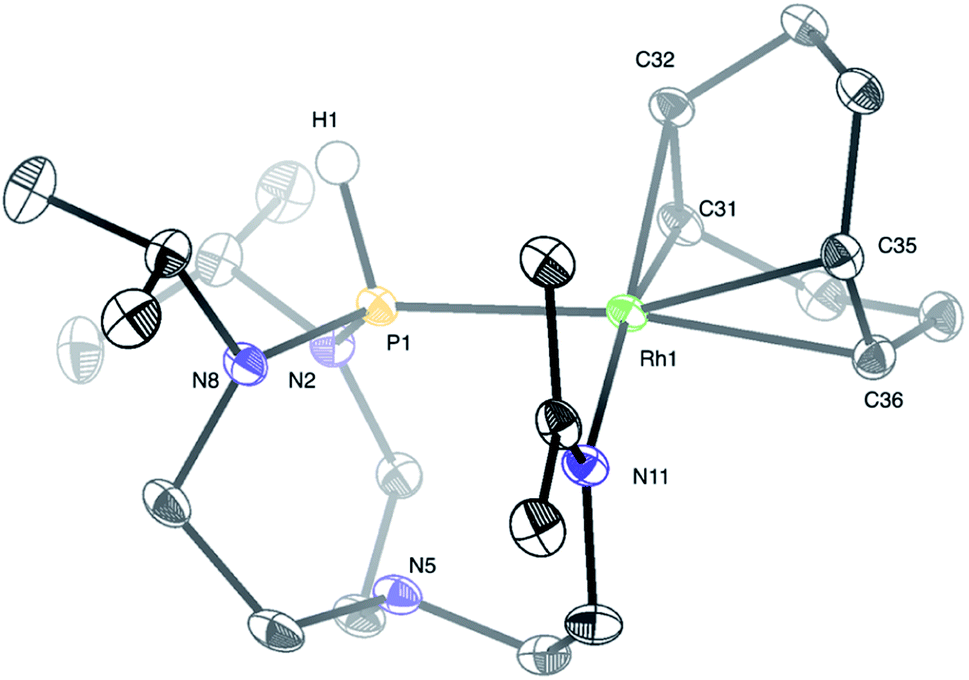 | ||
| Fig. 2 Molecular structure of complex 1 in the solid-state. The SbF6 anion and protons were removed for clarity, except for the proton on phosphorus. Selected interatomic distances (Å) and angles (°): Rh1–P1 2.3042(4), Rh1–N11 2.1091(12), P1–N5 2.608(1), Rh1–C31 2.1403(14), Rh1–C32 2.1441(13), Rh1–C35 2.2302(14), Rh1–C36 2.2577(14), N11–Rh1–P1 90.30(3), N8–P1–Rh1 122.14(5), N2–P1–Rh1 115.87(4), N2–P1–N8 112.26(6), Rh1–P1–N5 84.68(3), H1–P1–N5 168.4(8). | ||
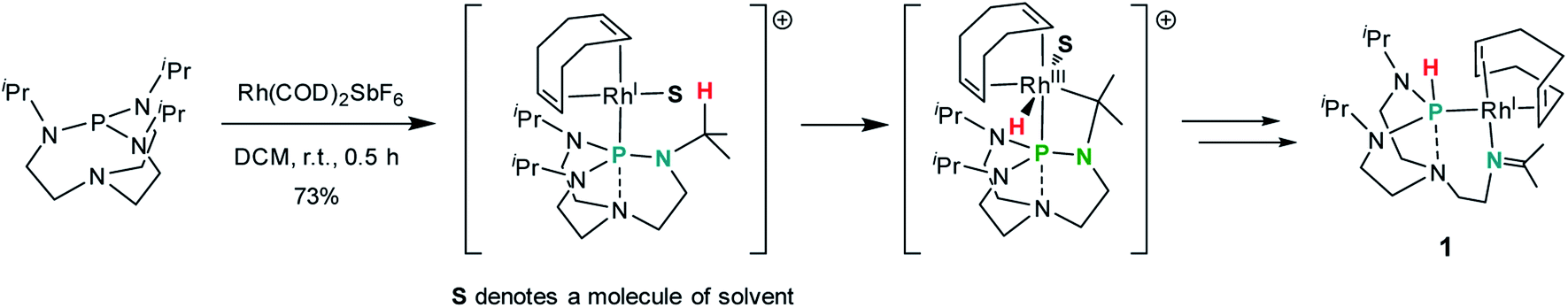 | ||
| Scheme 1 Efforts to rationalize the formation of the complex 1. The anion SbF6 is omitted for clarity. | ||
Structural characterization of complex 1 by means of X-ray diffraction analysis shows a Rh1–P1 bond (2.3042(4) Å) falling into a single bond range, only slightly shorter (ca. 0.030–0.207 Å) than other representative Rh–phosphido bonds reported in the literature.26 The P1 atom nearly lies in the plane of Rh1–N2–N8. The sum of three angles around the phosphorus atom (Rh1–P1–N2, Rh1–P1–N8, and N2–P1–N8) equals 350.26°, expressing a pseudo-trigonal bipyramidal geometry around the phosphorus atom, which deviates from the more common tetrahedral geometries of phosphines. The interatomic distance between P1 and N5 (2.608(1) Å) is longer than for similar azaphosphatranes,7f,27 but shorter than in the TiPrAP starting material,27a suggesting an intermediate transannular interaction between P1 and N5 atoms. Another interesting structural feature is given by the short N11–C18 bond length (1.2837(19) Å) and the trigonal planar nature of C18, advocating for an imine functional group. Finally, a trans-effect of P1 is evidenced by an increased length of the Rh1–C35 and Rh1–C36 bonds, compared to the shorter bond lengths found for the Rh1–C31 and Rh1–C32 bonds located trans to N11.
1H NMR spectroscopy further asserts the asymmetric character of complex 1 and is in good agreement with the solid-state molecular structure. Notably, the 1H NMR spectrum displays a characteristic doublet at 6.36 ppm with a large coupling constant (1JP,H = 331 Hz), resolving in a singlet upon 31P decoupling in the 1H{31P} NMR experiment. This large coupling constant shows a direct attachment of a proton to the phosphorus nuclei and can be confirmed by infrared spectroscopy, where the characteristic band at 2209 cm−1 corresponds to the stretching frequency of the P–H bond (see Fig. S13 in the ESI†). The doublet at 178.4 ppm in the 13C{1H} NMR spectrum confirms the formation of an imine group. Additionally, the corresponding 31P{1H} NMR shows a doublet signal with a coupling constant 1JRh,P = 201.5 Hz, which is in the range of typical Rh–P couplings of square planar rhodium complexes.28 Importantly, 1H–15N HMBC spectrum unveiled strong correlations of the proton located at the phosphorus atom with the Neq and the Nax (both over a two-bond distance), suggesting the presence of a bond between the P and the Nax (Fig. 3). Under these premises, i.e., presence of a Hax and simultaneous interaction with the Nax, the phosphorus atom exhibits a trigonal bipyramidal geometry.
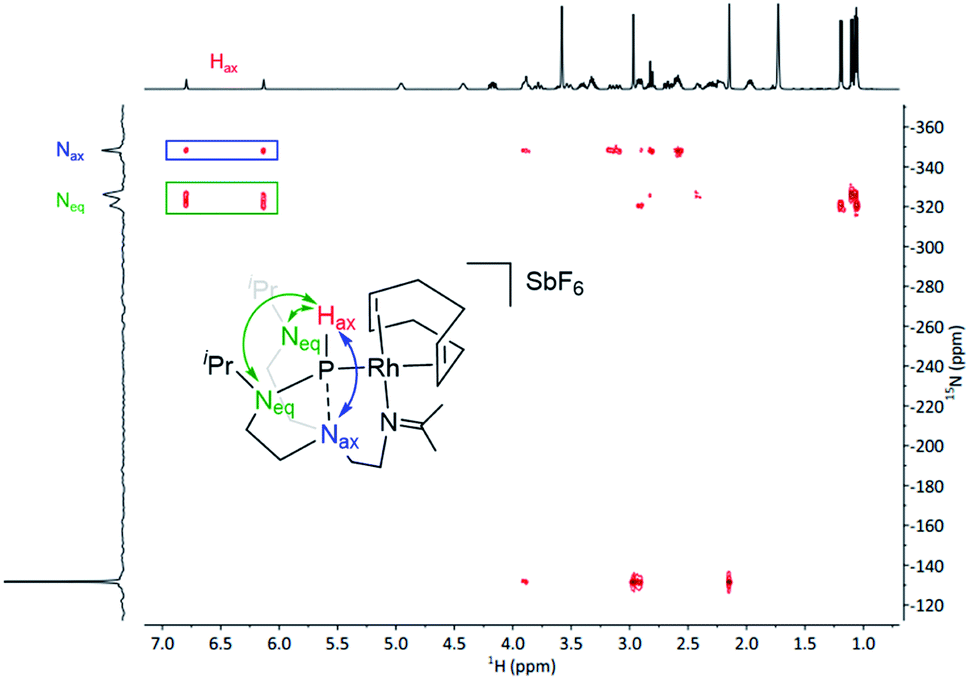 | ||
| Fig. 3 1H–15N HMBC spectrum of complex 1. The double-headed arrow indicates the long-range 2-bond correlations. | ||
A natural bond orbital (NBO) perturbation theory-based approach was chosen to characterize the P–Nax interaction to gain further insights into this interaction. The LP(Nax) → σ*(P–H) stabilization is 10.38 kcal mol−1 (Fig. 4, panel A), which falls into the same range but is slightly stronger than the corresponding interaction in chloro- and iodo-azaphosphatranes (7.10 and 5.16 kcal mol−1, respectively).21 The bonding character is also verified by the atoms-in-molecules (AIM) method, revealing a bond critical point BCP (3,−1) located between the P and the Nax atoms (Fig. 4, panel B).
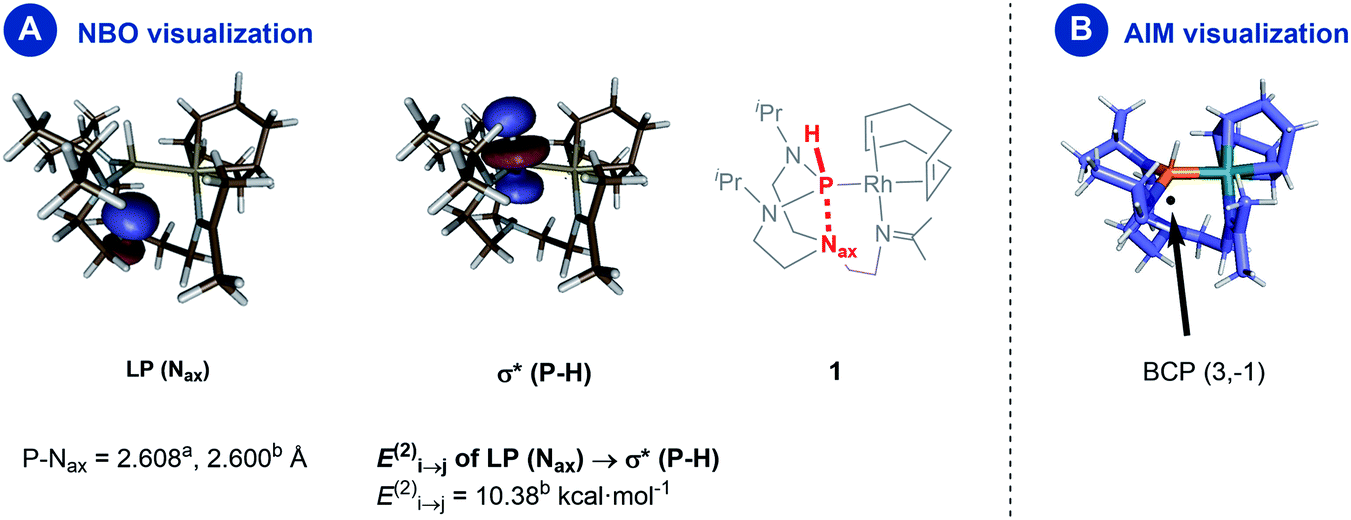 | ||
| Fig. 4 Panel (A) donor–acceptor interaction estimated by second-order perturbation theory; panel (B) the visualization of the bond critical point from the AIM analysis (further BCPs are omitted for clarity). aExperimental value from the molecular structure as determined by X-ray diffraction analysis, btheoretical value from DFT calculations. | ||
On the EXSY time scale (visible in the 2D NOESY spectra, tmix = 1 s, 298 K; see Fig. S4 in the ESI†), exchange peaks between the different protons depicted in identical colors were observed (Fig. 5, panel A), indicating a structural interconversion as illustrated in Fig. 5, panel B.
 | ||
| Fig. 5 Panel (A) schematic representation of complex 1 with nuclei undergoing mutual exchange depicted in the same color; panel (B) shows the dynamic intramolecular interconversion occurring within complex 1. | ||
Variable temperature 1D EXSY experiments provided access to the rate constant, activation entropy and enthalpy for the intramolecular interconversion (see Section 5 in the ESI†). The obtained values correspond to a concentration-independent equilibrium of approximately k293.2 K = 0.113 ± 0.018 s−1 with an activation enthalpy of ΔH‡ = 18.5 ± 2.0 kcal mol−1 and an activation entropy ΔS‡ = −0.8 ± 6.4 cal mol−1 K−1 according to Eyring's theory.29 Given that the interconversion, shown in Fig. 5, panel B, requires ligand dissociation and recoordination of either the imine, phosphine, or cyclooctadiene ligand, we questioned whether the rate of ligand dissociation could be increased by the addition of a Lewis acid capable of interacting with one of the electron-donating atoms present in the complex and thus facilitate the process. The addition of lithium chloride (14 equiv.) resulted in a considerable line broadening of the peaks in the 1H NMR spectrum (Fig. 6, panel A). This is illustrated by the complete coalescence of the methyl peaks of the isopropyl groups at 313 K (Fig. 6, panel B).
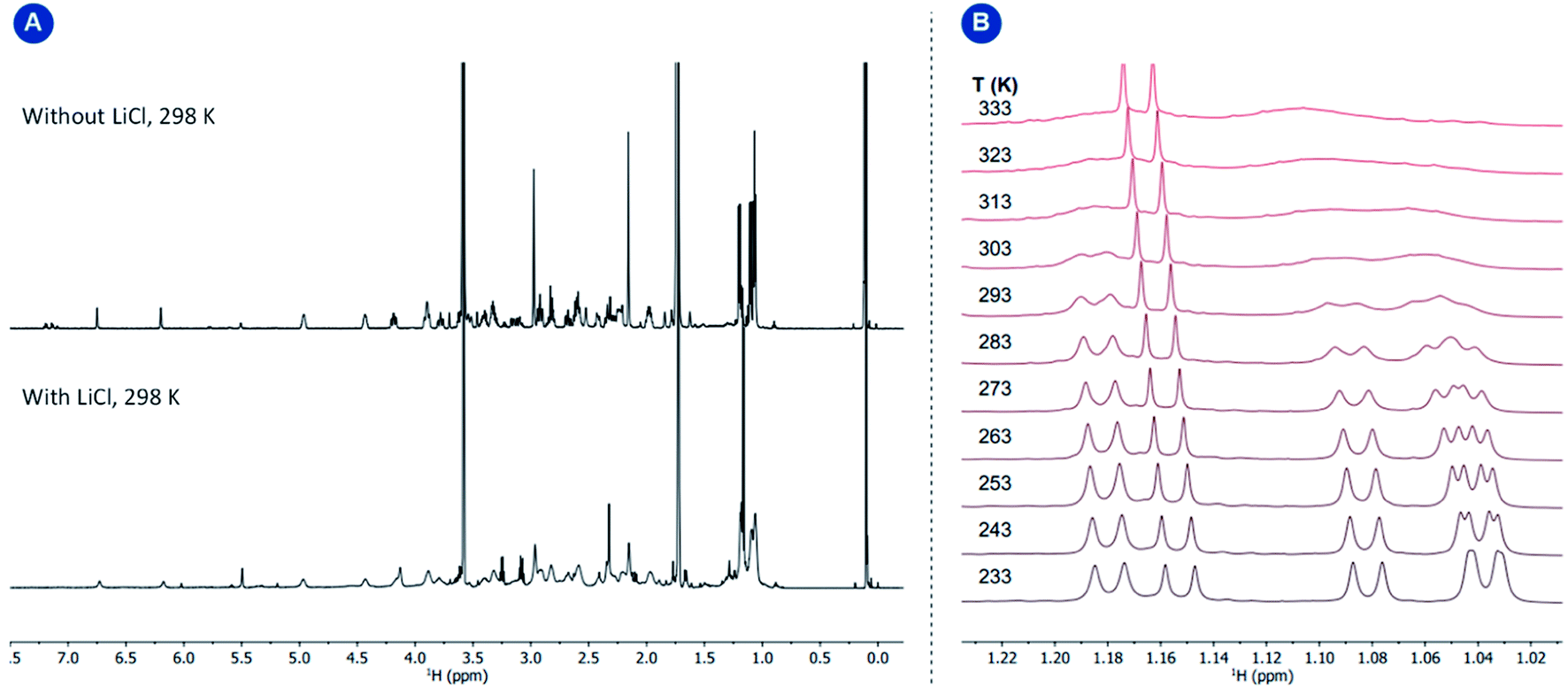 | ||
| Fig. 6 Panel (A) 1H NMR spectra of complex 1 with and without LiCl; panel (B) variable temperature 1H NMR study of complex 1 in the presence of LiCl (14 equiv.). The temperature-independent sharp signal corresponds to the minor amounts of the protonated TiPrAP. | ||
Based on variable temperature 1H NMR experiments, in the presence of lithium chloride (14 equiv.), an activation enthalpy ΔH‡ = 10.3 ± 1.0 kcal mol−1 and entropy of ΔS‡ = −19.5 ± 3.6 cal mol−1 K−1 could be determined. The resulting lower activation enthalpy value illustrates that LiCl supports the dynamic interconversion process. Besides, the decreased and negative activation entropy suggests an associative mechanism, where the interconversion process might involve lithium coordination to the complex on one of the chelating groups (e.g., tertiary nitrogen, phosphorus center). Apart from that, a hypothetic associative mechanism through the formation of a square-pyramidal geometry with chloride coordination to the rhodium center cannot be ruled out at this stage. Fig. 7 summarizes the free activation energy for the intramolecular interconversion of complex 1 with and without lithium chloride as a function of temperature.
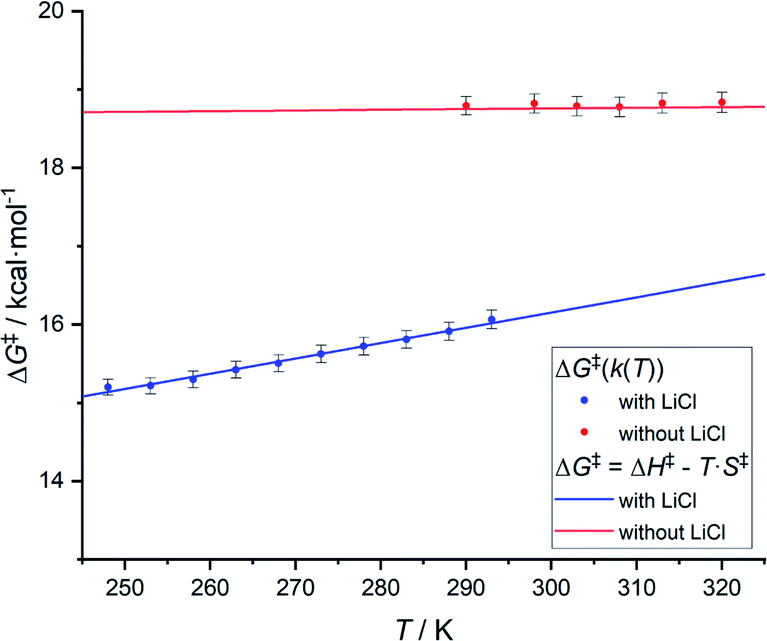 | ||
| Fig. 7 Graph showing the free energy of activation for the intramolecular interconversion of complex 1 with and without lithium chloride as a function of temperature. | ||
This result is important for homogeneous catalysis, where the generation of inorganic salts as a byproduct is well-documented.30 Their presence in the reaction medium may radically change the performance of a catalyst by accelerating ligand coordination and dissociation and affecting the reaction kinetics of a given transformation.31
In attempts to further control the environment around the rhodium center, we hypothesized that replacing the bidentate COD ligand in complex 1 with a monodentate ligand might trigger the reorganization of the TiPrAP ligand framework. This was done by selecting triphenylphosphine as a candidate, carrying a soft phosphorus donor atom in high affinity with the soft rhodium center.
Reacting complex 1 with a stoichiometric amount of triphenylphosphine in dichloromethane at 70 °C led to complex 2 (Scheme 2).
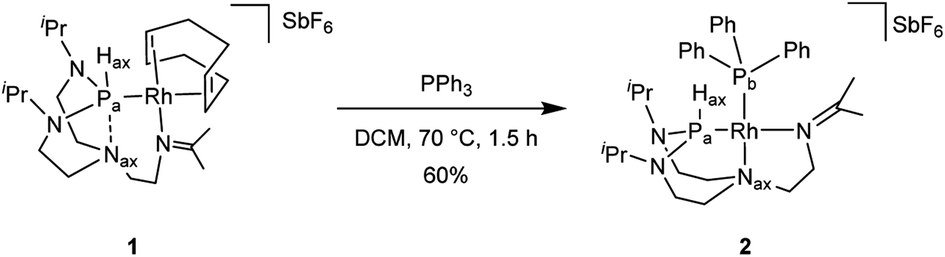 | ||
| Scheme 2 Preparation of complex 2. | ||
Coordination of the triphenylphosphine ligand is evidenced by 31P{1H} NMR spectroscopy and the presence of two doublets of doublets. The coupling constants 1JPa,Rh = 163 Hz and 1JPb,Rh = 202 Hz are falling in the range of a typical square planar complex.28 The magnitude of the coupling constant 1JPa,Pb = 60 Hz reveals a cis conformation. The 13C{1H} NMR spectrum hints at the coordination of the imine group to the rhodium center with a coupling constant 2JC,Rh = 1.6 Hz, which is similar to the value observed for complex 1 (2JC,Rh = 1.9 Hz). The square planar geometry of the rhodium(I) center is satisfied by the occupation of its fourth coordination site by the Nax, shifting from the phosphorus atom to the rhodium center. This is reflected by the absence of correlation peaks between the Hax and Nax atoms in the 1H–15N HMBC spectrum of complex 2 (see Fig. S26 in the ESI†). Moreover, the shift of the Nax leads to a significant geometric change at the Pa atom, evolving from a trigonal bipyramid (complex 1) to a tetrahedral geometry (complex 2). This results in the higher s-character in Pa–Hax bond as shown by an increased coupling constant with the Hax (from 1JPa,H = 331 Hz in complex 1 to 413 Hz in complex 2). Besides, the protons close to the Nax show strong correlations with the Pb, further corroborating its trans-position to the triphenylphosphine as confirmed by the 1H–31P HMBC experiment (see Fig. S25 in the ESI†). Finally, a direct correlation of these protons (i.e., Hax and the protons close to the Nax) with the rhodium center at −8430 ppm in a 1H–103Rh-HMBC was observed (see Fig. S27 in the ESI†).
The 1H NMR spectrum suggests that complex 2 has a more symmetric molecular structure when compared to the structure of 1. It was initially hypothesized that a fast-intramolecular exchange averages signals on the NMR time scale. However, the variable temperature experiments indicate only a slight change in the 1H NMR spectra and a negligible line broadening for the signals in the 13C{1H} NMR spectra are observed (see Fig. S20 in the ESI†).
Diffusion experiments were conducted to inquire whether complex 2 exists in an oligomeric state in solution (see Section 4 in the ESI†). The diffusion constant (D = 6.4 ± 0.2 × 10−10 m2 s−1) matched the predicted volume of a monomeric structural model of 2 (Vhydrodyn,exp = 746 ± 84 Å3, VVdW,calc,monomer = 555 Å3, VVdW,calc,dimer = 1105 Å3). The slightly larger experimental volume can be explained by additional flexibility due to the rotatability of the PPh3 group, stronger ion pairing of SbF6,32 or stronger solvent association. In addition, the computational studies combined with 2D NOESY were carried out to get further insight into the detailed structure of the complex (see Section 6 in the ESI†). The Conformer–Rotamer Ensemble Sampling Tool (CREST)33 with the xtb1 semi-empirical tight-binding method34 was implemented to produce possible conformers and rotamers, which were then used as the inputs for the program DISCON35 under the Janocchio36 interface. Careful evaluation of the interproton distances within complex 2 was obtained employing a set of 2D z-filtered NOESY spectra with increasing mixing time (see Section 6 in the ESI†). The calculated and experimental inter-proton distances are summarized and compared in Table 1. The obtained values are mostly within the experimental error.37
| i | j | Ri,j,calc/Å | ri,j,exp/Å | |Δr|/Å |
|---|---|---|---|---|
| 15 | 18#,18′# | 4.97 | 5.17 | 0.20 |
| 13# | 18#,18′# | 3.85 | 4.04 | 0.19 |
| 2# | 18#,18′# | 3.60 | 3.27 | 0.33 |
| 14 | 18#,18′# | 3.29 | 3.50 | 0.21 |
| 11 | 19#,19′# | 4.92 | 4.28 | 0.64 |
| 11 | 18#,18′# | 3.54 | 3.24 | 0.30 |
| 14 | 19#,19′# | 4.18 | 3.81 | 0.37 |
| 15 | 19#,19′# | 5.73 | 5.13 | 0.60 |
| 2# | 13# | 2.70 | 2.46 | 0.24 |
| 11 | 14 | 5.14 | 4.86 | 0.28 |
| 12 | 8 | 2.63 | 2.67 | 0.04 |
| 8 | 11 | 4.65 | 4.42 | 0.23 |
Considering the monomeric molecule suggested by the DOSY experiments, together with the comparison of the interatomic distances of the computed rotamers with the ones of complex 2, enables us to propose the structure of complex 2 as shown in Fig. 8.
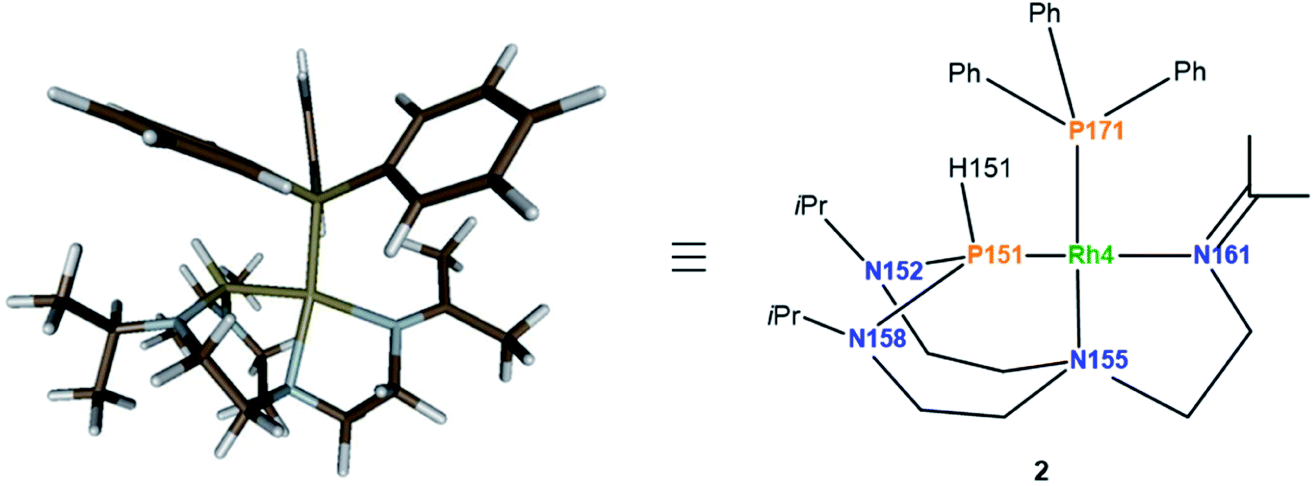 | ||
| Fig. 8 Computed structure of complex 2 at the D3BJ-PBE/def2-SVP level of theory (left) and the associated drawing with labels (right). SbF6 anion is left out for clarity. Selected interatomic distances (Å) and angles (°): Rh4–P151 2.207, Rh4–P171 2.218, Rh4–N155 2.247, Rh4–N161 2.109, N158–P151–Rh4 106.14, N152–P151–Rh4 107.76, N158–P151–N152 109.91. | ||
In turn, single crystals of 2 could be obtained by slow evaporation of a diluted solution of the complex solubilized in a dichloromethane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :diethyl ether (1:4) mixture of solvents. Only one complex of the four independent molecules present in the asymmetric unit is shown for clarity (Fig. 9). The rhodium(I) center displays a square planar geometry with a P–N–N tridentate and triphenylphosphine coordination. X-ray diffraction analysis conclusively shows the shift of the Nax atom to the rhodium center, with the soft monodentate triphenylphosphine ligand promoting a change of the TiPrAP coordination mode from bidentate to tridentate. The shifted coordination of N155 atom to rhodium is evidenced by the formation of the Rh4–N155 bond (2.191(6) Å). This confirms the observations made in 1H–31P HMBC, with the protons close to N155 having strong correlations with the phosphorus of triphenylphosphine (see Fig. S25 in the ESI†). Besides, the angles around the P151 atom, ranging from 107.4(2)° to 109.6(4)°, suggest a tetrahedral structure, which is in good agreement with the larger P–H coupling constant of 2 compared to complex 1. Other structural parameters provided by X-ray diffraction analysis of 2 are compared to the computed data and summarized in Table 2. Altogether, it transpires that the combination of NMR experiments and DFT calculations provides an accurate molecular description of the complex.
:diethyl ether (1:4) mixture of solvents. Only one complex of the four independent molecules present in the asymmetric unit is shown for clarity (Fig. 9). The rhodium(I) center displays a square planar geometry with a P–N–N tridentate and triphenylphosphine coordination. X-ray diffraction analysis conclusively shows the shift of the Nax atom to the rhodium center, with the soft monodentate triphenylphosphine ligand promoting a change of the TiPrAP coordination mode from bidentate to tridentate. The shifted coordination of N155 atom to rhodium is evidenced by the formation of the Rh4–N155 bond (2.191(6) Å). This confirms the observations made in 1H–31P HMBC, with the protons close to N155 having strong correlations with the phosphorus of triphenylphosphine (see Fig. S25 in the ESI†). Besides, the angles around the P151 atom, ranging from 107.4(2)° to 109.6(4)°, suggest a tetrahedral structure, which is in good agreement with the larger P–H coupling constant of 2 compared to complex 1. Other structural parameters provided by X-ray diffraction analysis of 2 are compared to the computed data and summarized in Table 2. Altogether, it transpires that the combination of NMR experiments and DFT calculations provides an accurate molecular description of the complex.
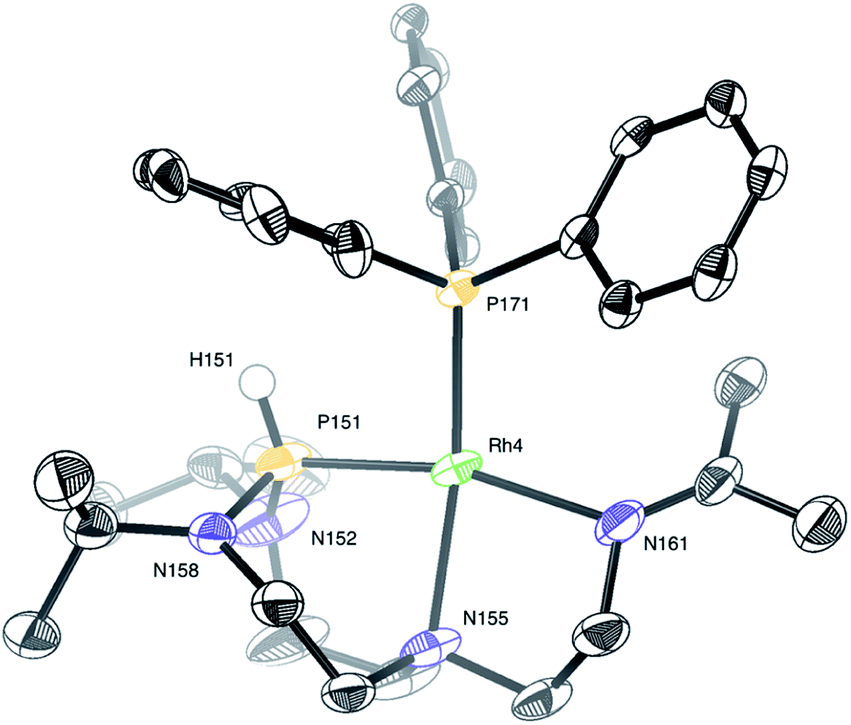 | ||
| Fig. 9 Molecular structure of complex 2 in the solid state. The SbF6 anion and protons were removed for clarity, except for the proton on phosphorus. Selected interatomic distances (Å) and angles (°): Rh4–P151 2.162(2), Rh4–P171 2.2158(18), Rh4–N155 2.191(6), Rh4–N161 2.137(6), N158–P151–Rh4 107.4(2), N152–P151–Rh4 109.1(3), N158–P151–N152 109.6(4). | ||
| Selected interatomic distances | Rcalc (Å) | rexp (Å) | |Δr| (Å) | Selected angles | ∠calc (°) | ∠exp (°) | |Δ∠| (°) |
|---|---|---|---|---|---|---|---|
| Rh4–P151 | 2.207 | 2.162(2) | 0.045 | N158–P151–Rh4 | 106.14 | 107.4(2) | 1.3 |
| Rh4–P171 | 2.218 | 2.2158(18) | 0.002 | N152–P151–Rh4 | 107.76 | 109.1(3) | 1.3 |
| Rh4–N155 | 2.247 | 2.191(6) | 0.056 | N158–P151–N152 | 109.91 | 109.6(4) | 0.3 |
| Rh4–N161 | 2.109 | 2.137(6) | 0.028 |
Combining hard and soft donor atoms into a ligand environment can be used as a proxy to control a given complex's thermodynamic and kinetic stability. Thus, we considered 2-pyridyldiphenylphosphine as another ancillary ligand and investigated the effect of an additional hard pyridyl donor on the coordination chemistry and thermodynamic properties of the resulting complex. The reaction of complex 1 with 2-pyridyldiphenylphosphine in tetrahydrofuran afforded complex 3 (Scheme 3). The 31P{1H} NMR spectrum shows two doublet of doublets with the constants (1JPa,Pb = 60 Hz, 1JPa,Rh = 161 Hz, 1JPb,Rh = 204 Hz). The value of the 1JPa,Pb coupling constant reveals a cis conformation to each other. The 1JPa,Hax coupling increases from 331 Hz (complex 1) to 426 Hz (complex 3), indicating a higher s-character of the P–H bond. The 13C{1H} NMR spectrum reveals that the imine group coordinated to the rhodium center has a coupling constant (2JC,Rh = 1.6 Hz) similar to the one of complex 2 (2JC,Rh = 1.6 Hz). The correlation between the Hax and the Nax atoms present in complex 1 is absent in the 1H–15N HMBC spectrum (see Fig. S39 in the ESI†). The diffusion constant of 3 (D = 6.33 ± 0.08 × 10−10 m2 s−1) is comparable to 2 (D = 6.4 ± 0.2 × 10−10 m2 s−1). Besides, the 1H NMR spectrum of complex 3 shows considerable similarities with complex 2. Based on these observations, it can be reasonably assumed that the two complexes feature an analogous molecular structure.
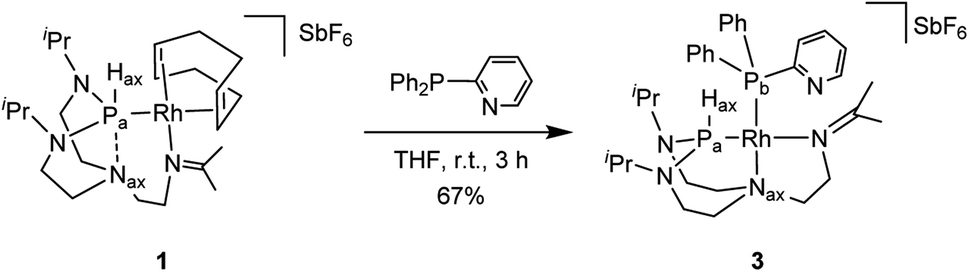 | ||
| Scheme 3 Preparation of complex 3. | ||
We then hypothesized whether, in contrast to complex 2, the pyridyl donor in complex 3 might displace the imine at the rhodium center resulting in its bidentate chelation to the metal center.38 This, in turn, may cause the TiPrAP ligand backbone to further rearrange its coordination mode from tridentate to bidentate and generate a complex similar to 3′. Therefore, computational calculations at the ωB97X-D3BJ/def2-TZVPP level of theory were carried out (see Section 7 in the ESI†).39 Theory suggests that the pyridine coordinated complex 3′ is higher in energy by only ΔG = 6.2 kcal mol−1 when compared to complex 3 (Scheme 4). It might thus constitute a relevant intermediate at elevated temperatures. Attempts to structurally characterize 3′ have, however, been unsuccessful at this stage. In terms of perspectives, introducing a spatial distance between the pyridine and phosphorus atom might reduce the ring strain and enhance the structural flexibility of the resulting complex. This might shift the equilibrium towards the formation of a complex comparable to 3′ by enhancing its intrinsic stability.
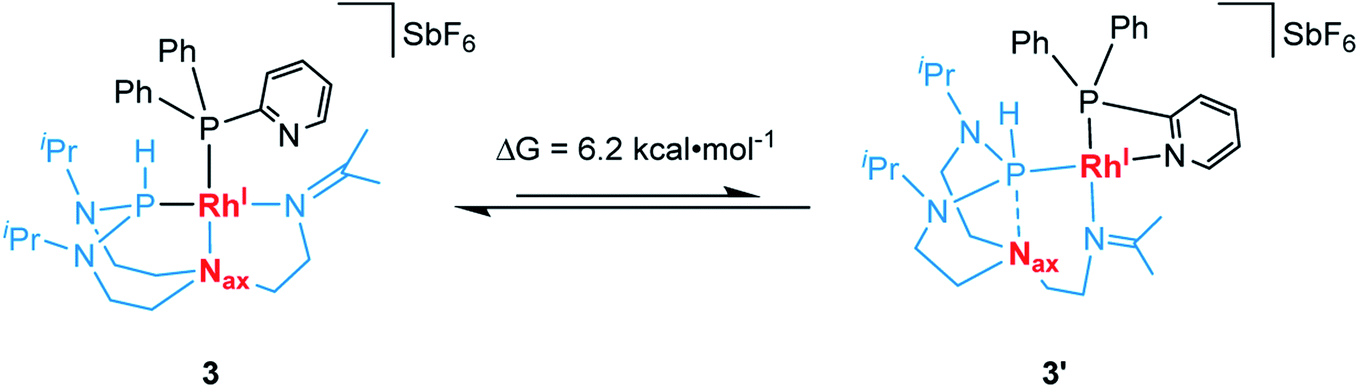 | ||
| Scheme 4 Suggested dynamic equilibrium between complex 3 and 3′ and the associated variation in free energy. | ||
The propensity of the TiPrAP ligand backbone to purposely adapt itself and accommodate different geometries and coordination modes around the metal center offers strategies for controlling the coordination environment at the metal center and subsequent activity of the entire molecular edifice.
Conclusion
In summary, the synthesis and coordination chemistry of rhodium(I) complexes with a tris(isopropyl)-azaphosphatrane (TiPrAP) ligand architecture are reported. The molecular structure of complex 1 indicated the insertion of the metal center into one of the P–N bonds of the ligand and proton transfer from a methine group to the phosphorus atom. A transannular interaction was confirmed by NMR spectroscopy, single-crystal X-ray diffraction study, and NBO analysis. The addition of lithium chloride resulted in the altered dynamic behavior of complex 1 in solution, which was studied by NMR spectroscopy. Substituting the cyclooctadiene ligand at the metal center with triphenylphosphine or 2-pyridyldiphenylphosphine unveiled the adaptive nature of the TiPrAP backbone. This adaptability resides in the axial nitrogen, which can shift from an interaction with the phosphorus atom to the coordination of the rhodium center. The ligand architecture is therefore capable of binding the rhodium center both in a bidentate or tridentate manner. This reversible and apparent hemilability demonstrates that the TiPrAP ligand framework can accommodate electronic or steric changes around the rhodium center by opening or closing a coordination site at the metal center. Altogether, our study shows that precise molecular control of the metal center's immediate environment can develop. Application of this peculiarity in controlled bond activation reactions is currently underway.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the Max Planck Society for financial support and open access funding. We thank Prof. Dr Walter Leitner for insightful conversations and generous support. We are also grateful to Dr Markus Leutzsch for valuable NMR support. Finally, we thank Dr Giovanni Bistoni for guidance on implementing initial calculations and conformational sampling.References
- For selected examples showing the importance of the ligand environment to meet the requirements of specific applications, please refer to ref. 2 a–g.
- (a) R. Noyori and T. Ohkuma, Angew. Chem., Int. Ed., 2001, 40, 40–73 CrossRef CAS; (b) R. Noyori, M. Koizumi, D. Ishii and T. Ohkuma, Pure Appl. Chem., 2001, 73, 227–232 CAS; (c) C. P. Casey, G. A. Bikzhanova, Q. Cui and I. A. Guzei, J. Am. Chem. Soc., 2005, 127, 14062–14071 CrossRef CAS PubMed; (d) T. Ikariya and I. D. Gridnev, Top. Catal., 2010, 53, 894–901 CrossRef CAS; (e) D. Morales-Morales and C. G. M. Jensen, The Chemistry of Pincer Compounds, in The chemistry of pincer compounds, Elsevier Science, Oxford, 2011, pp. 87–105 Search PubMed; (f) A. E. Allen and D. W. Macmillan, Chem. Sci., 2012, 2012, 633–658 RSC; (g) S. Sahu, L. R. Widger, M. G. Quesne, S. P. de Visser, H. Matsumura, P. Moenne-Loccoz, M. A. Siegler and D. P. Goldberg, J. Am. Chem. Soc., 2013, 135, 10590–10593 CrossRef CAS PubMed.
- B. Chatterjee, W. C. Chang, S. Jena and C. Werlé, ACS Catal., 2020, 10, 14024–14055 CrossRef CAS.
- For selected examples on cooperativity, refer to ref. 5a–m.
- (a) A. Vigalok and D. Milstein, Acc. Chem. Res., 2001, 34, 798–807 CrossRef CAS PubMed; (b) D. Milstein, Pure Appl. Chem., 2003, 75, 445–460 CAS; (c) H. Grutzmacher, Angew. Chem., Int. Ed., 2008, 47, 1814–1818 CrossRef PubMed; (d) D. Milstein, Top. Catal., 2010, 53, 915–923 CrossRef CAS; (e) C. Gunanathan and D. Milstein, Acc. Chem. Res., 2011, 44, 588–602 CrossRef CAS PubMed; (f) V. Lyaskovskyy and B. de Bruin, ACS Catal., 2012, 2, 270–279 CrossRef CAS; (g) D. Gelman and S. Musa, ACS Catal., 2012, 2, 2456–2466 CrossRef CAS; (h) K. Hindson, Eur. J. Inorg. Chem., 2012, 2012, 340 CrossRef CAS; (i) O. R. Luca and R. H. Crabtree, Chem. Soc. Rev., 2013, 42, 1440–1459 RSC; (j) C. Gunanathan and D. Milstein, Chem. Rev., 2014, 114, 12024–12087 CrossRef CAS PubMed; (k) H. F. Li, B. Zheng and K. W. Huang, Coord. Chem. Rev., 2015, 293, 116–138 CrossRef; (l) J. R. Khusnutdinova and D. Milstein, Angew. Chem., Int. Ed., 2015, 54, 12236–12273 CrossRef CAS PubMed; (m) S. Elangovan, C. Topf, S. Fischer, H. Jiao, A. Spannenberg, W. Baumann, R. Ludwig, K. Junge and M. Beller, J. Am. Chem. Soc., 2016, 138, 8809–8814 CrossRef CAS PubMed.
- B. Chatterjee, W. C. Chang and C. Werlé, ChemCatChem, 2021, 13, 1659–1682 CrossRef CAS.
- (a) H. Schmidt, C. Lensink, S. K. Xi and J. G. Verkade, Z. Anorg. Allg. Chem., 1989, 578, 75–80 CrossRef CAS; (b) C. Lensink, S. K. Xi, L. M. Daniels and J. G. Verkade, J. Am. Chem. Soc., 1989, 111, 3478–3479 CrossRef CAS; (c) M. A. H. Laramay and J. G. Verkade, J. Am. Chem. Soc., 1990, 112, 9421–9422 CrossRef CAS; (d) J. G. Verkade, Coord. Chem. Rev., 1994, 137, 233–295 CrossRef CAS; (e) P. B. Kisanga, J. G. Verkade and R. Schwesinger, J. Org. Chem., 2000, 65, 5431–5432 CrossRef CAS PubMed; (f) C. Lensink, S. K. Xi, L. M. Daniels and J. G. Verkade, J. Am. Chem. Soc., 2002, 111, 3478–3479 CrossRef; (g) M. A. H. Laramay and J. G. Verkade, J. Am. Chem. Soc., 2002, 112, 9421–9422 CrossRef.
- (a) Z. Wang, B. Fetterly and J. G. Verkade, J. Organomet. Chem., 2002, 646, 161–166 CrossRef CAS; (b) B. M. Fetterly and J. G. Verkade, Tetrahedron Lett., 2005, 46, 8061–8066 CrossRef CAS.
- J. Yang, B. Chatelet, F. Ziarelli, V. Dufaud, D. Herault and A. Martinez, Eur. J. Org. Chem., 2018, 2018, 6328–6332 CrossRef CAS.
- S. Arumugam, D. McLeod and J. G. Verkade, J. Org. Chem., 1998, 63, 3677–3679 CrossRef CAS.
- (a) S. Arumugam and J. G. Verkade, J. Org. Chem., 1997, 62, 4827–4828 CrossRef CAS; (b) X. Liu and J. G. Verkade, J. Org. Chem., 1999, 64, 4840–4843 CrossRef CAS PubMed.
- (a) B. A. D'Sa and J. G. Verkade, J. Org. Chem., 1996, 61, 2963–2966 CrossRef PubMed; (b) P. Ilankumaran and J. G. Verkade, J. Org. Chem., 1999, 64, 3086–3089 CrossRef CAS PubMed.
- P. Kisanga, D. McLeod, B. D'Sa and J. Verkade, J. Org. Chem., 1999, 64, 3090–3094 CrossRef CAS PubMed.
- P. B. Kisanga and J. G. Verkade, J. Org. Chem., 1999, 64, 4298–4303 CrossRef CAS.
- Z. Thammavongsy, I. M. Kha, J. W. Ziller and J. Y. Yang, Dalton Trans., 2016, 45, 9853–9859 RSC.
- Z. Thammavongsy, D. W. Cunningham, N. Sutthirat, R. J. Eisenhart, J. W. Ziller and J. Y. Yang, Dalton Trans., 2018, 47, 14101–14110 RSC.
- B. Chatelet, P. Nava, H. Clavier and A. Martinez, Eur. J. Inorg. Chem., 2017, 2017, 4311–4316 CrossRef CAS.
- (a) J. V. Kingston and J. G. Verkade, J. Org. Chem., 2007, 72, 2816–2822 CrossRef CAS PubMed; (b) C. V. Reddy, J. V. Kingston and J. G. Verkade, J. Org. Chem., 2008, 73, 3047–3062 CrossRef CAS PubMed; (c) S. M. Raders, J. V. Kingston and J. G. Verkade, J. Org. Chem., 2010, 75, 1744–1747 CrossRef CAS PubMed; (d) Y. Zhou and J. G. Verkade, Adv. Synth. Catal., 2010, 352, 616–620 CrossRef CAS; (e) S. H. Kim, M. Kim, J. G. Verkade and Y. Kim, Eur. J. Org. Chem., 2015, 2015, 1954–1960 CrossRef CAS.
- A. D. Matthews, G. M. Gravalis, N. D. Schley and M. W. Johnson, Organometallics, 2018, 37, 3073–3078 CrossRef CAS.
- T. C. Johnstone, A. I. Briceno-Strocchia and D. W. Stephan, Inorg. Chem., 2018, 57, 15299–15304 CrossRef CAS PubMed.
- A. D. Matthews, S. Prasad, N. D. Schley, K. J. Donald and M. W. Johnson, Inorg. Chem., 2019, 58, 15983–15992 CrossRef CAS PubMed.
- (a) H. H. Cramer, B. Chatterjee, T. Weyhermüller, C. Werlé and W. Leitner, Angew. Chem., Int. Ed., 2020, 59, 15674–15681 CrossRef CAS PubMed; (b) B. Chatterjee, S. Jena, V. Chugh, T. Weyhermüller and C. Werlé, ACS Catal., 2021, 7176–7185 CrossRef CAS; (c) H. H. Cramer, S. Ye, F. Neese, C. Werlé and W. Leitner, JACS Au, 2021 DOI:10.1021/jacsau.1c00350.
- (a) S. Oi, S. Watanabe, S. Fukita and Y. Inoue, Tetrahedron Lett., 2003, 44, 8665–8668 CrossRef CAS; (b) R. B. Bedford, S. J. Coles, M. B. Hursthouse and M. E. Limmert, Angew. Chem., Int. Ed., 2003, 42, 112–114 CrossRef CAS PubMed; (c) R. B. Bedford, M. Betham, A. J. Caffyn, J. P. Charmant, L. C. Lewis-Alleyne, P. D. Long, D. Polo-Ceron and S. Prashar, Chem. Commun., 2008, 990–992 RSC; (d) C. U. Grunanger and B. Breit, Angew. Chem., Int. Ed., 2008, 47, 7346–7349 CrossRef PubMed; (e) J. F. Yang, R. H. Wang, Y. X. Wang, W. W. Yao, Q. S. Liu and M. Ye, Angew. Chem., Int. Ed., 2016, 55, 14116–14120 CrossRef CAS PubMed; (f) X. Qiu, M. Wang, Y. Zhao and Z. Shi, Angew. Chem., Int. Ed., 2017, 56, 7233–7237 CrossRef CAS PubMed; (g) X. Luo, J. Yuan, C. D. Yue, Z. Y. Zhang, J. Chen, G. A. Yu and C. M. Che, Org. Lett., 2018, 20, 1810–1814 CrossRef CAS PubMed; (h) J. Wen, D. Wang, J. Qian, D. Wang, C. Zhu, Y. Zhao and Z. Shi, Angew. Chem., Int. Ed., 2019, 58, 2078–2082 CrossRef CAS PubMed; (i) D. Wang, B. Dong, Y. Wang, J. Qian, J. Zhu, Y. Zhao and Z. Shi, Nat. Commun., 2019, 10, 3539 CrossRef PubMed.
- A. Tanushi and A. T. Radosevich, J. Am. Chem. Soc., 2018, 140, 8114–8118 CrossRef CAS PubMed.
- (a) S. Urgaonkar and J. G. Verkade, J. Org. Chem., 2004, 69, 9135–9142 CrossRef CAS PubMed; (b) S. Mummadi, D. Kenefake, R. Diaz, D. K. Unruh and C. Krempner, Inorg. Chem., 2017, 56, 10748–10759 CrossRef CAS PubMed.
- (a) A. M. Geer, A. L. Serrano, B. de Bruin, M. A. Ciriano and C. Tejel, Angew. Chem., Int. Ed., 2015, 54, 472–475 CAS; (b) U. Fischbach, M. Trincado and H. Grutzmacher, Dalton Trans., 2017, 46, 3443–3448 RSC.
- (a) A. E. Wróblewski, J. Pinkas and J. G. Verkade, Main Group Chem., 1995, 1, 69–79 CrossRef; (b) P. B. Kisanga and J. G. Verkade, Tetrahedron, 2001, 57, 467–475 CrossRef.
- (a) P. S. Pregosin and R. W. Kunz, 31P and 13C NMR of transition metal phosphine complexes, Springer-Verlag, Berlin, Heidelberg, 1979 CrossRef; (b) P. S. Pregosin, NMR in Organometallic Chemistry, Wiley-VCH-Verl., Weinheim, 2013 Search PubMed.
- J. H. Espenson, Chemical kinetics and reaction mechanisms, McGraw-Hill, New York, 1981, p. 117 Search PubMed.
- B. Cornils, W. A. Herrmann, M. Beller and R. Paciello, Applied homogeneous catalysis with organometallic compounds a comprehensive handbook in four volumes, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2018 Search PubMed.
- (a) G. Marcotullio and W. De Jong, Green Chem., 2010, 12, 1739–1746 RSC; (b) C. Sambiagio, D. Schonbauer, R. Blieck, T. Dao-Huy, G. Pototschnig, P. Schaaf, T. Wiesinger, M. F. Zia, J. Wencel-Delord, T. Besset, B. U. W. Maes and M. Schnurch, Chem. Soc. Rev., 2018, 47, 6603–6743 RSC; (c) R. Szpera, D. F. J. Moseley, L. B. Smith, A. J. Sterling and V. Gouverneur, Angew. Chem., Int. Ed., 2019, 58, 14824–14848 CrossRef CAS PubMed; (d) B. Niu, K. Yang, B. Lawrence and H. Ge, ChemSusChem, 2019, 12, 2955–2969 CrossRef CAS PubMed; (e) M. A. Mellmer, C. Sanpitakseree, B. Demir, K. Ma, W. A. Elliott, P. Bai, R. L. Johnson, T. W. Walker, B. H. Shanks, R. M. Rioux, M. Neurock and J. A. Dumesic, Nat. Commun., 2019, 10, 1132 CrossRef PubMed; (f) M. Kapoor, P. Chand-Thakuri and M. C. Young, J. Am. Chem. Soc., 2019, 141, 7980–7989 CrossRef CAS PubMed; (g) Y. Q. Chen, S. Singh, Y. Wu, Z. Wang, W. Hao, P. Verma, J. X. Qiao, R. B. Sunoj and J. Q. Yu, J. Am. Chem. Soc., 2020, 142, 9966–9974 CrossRef CAS PubMed; (h) B. Li, B. Lawrence, G. Li and H. Ge, Angew. Chem., Int. Ed., 2020, 59, 3078–3082 CrossRef CAS PubMed.
- P. G. Anil Kumar, P. S. Pregosin, T. M. Schmid and G. Consiglio, Magn. Reson. Chem., 2004, 42, 795–800 CrossRef CAS PubMed.
- P. Pracht, F. Bohle and S. Grimme, Phys. Chem. Chem. Phys., 2020, 22, 7169–7192 RSC.
- C. Bannwarth, E. Caldeweyher, S. Ehlert, A. Hansen, P. Pracht, J. Seibert, S. Spicher and S. Grimme, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2020, 11, e1493 Search PubMed.
- O. Atasoylu, G. Furst, C. Risatti and A. B. Smith 3rd, Org. Lett., 2010, 12, 1788–1791 CrossRef CAS PubMed.
- D. A. Evans, M. J. Bodkin, S. R. Baker and G. J. Sharman, Magn. Reson. Chem., 2007, 45, 595–600 CrossRef CAS PubMed.
- See |Δr| values in the right column of Table 1.
- (a) K. Wajda-Hermanowicz, Z. Ciunik and A. Kochel, Inorg. Chem., 2006, 45, 3369–3377 CrossRef CAS PubMed; (b) M. Angoy, M. V. Jimenez, F. J. Modrego, L. A. Oro, V. Passarelli and J. J. Perez-Torrente, Organometallics, 2018, 37, 2778–2794 CrossRef CAS.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 2, 73–78 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, copies of NMR spectra, diffusion measurement, kinetic studies, quantitative NOESY, computational details, and crystallographic data (PDF). CCDC 2100808 and 2100809. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra07126b |
| ‡ W.-C. C. and F. D. contributed equally to this work. |
| § Current address: Organisch-Chemisches Institut, Corrensstraße 36, D-48149 Münster, Germany. |
| This journal is © The Royal Society of Chemistry 2021 |