
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew water-soluble isoxazole-linked 1,3,4-oxadiazole derivative with delocalized positive charge†
Urszula Bąchor a,
Ewa Drozd-Szczygieł*a,
Remigiusz Bąchor*b,
Lucjan Jerzykiewiczb,
Robert Wieczorekb and
Marcin Mączyńskia
a,
Ewa Drozd-Szczygieł*a,
Remigiusz Bąchor*b,
Lucjan Jerzykiewiczb,
Robert Wieczorekb and
Marcin Mączyńskia
aDepartment of Organic Chemistry, Faculty of Pharmacy, Wroclaw Medical University, Borowska 211A, 50-556 Wroclaw, Poland. E-mail: ewa.drozd-szczygiel@umed.wroc.pl
bFaculty of Chemistry, University of Wroclaw, F. Joliot-Curie 14, 50-383 Wrocław, Poland. E-mail: remigiusz.bachor@chem.uni.wroc.pl
First published on 3rd September 2021
Abstract
Herein we present a synthesis and characterization of a new and unique low-weight heterocyclic compound 5-amino-2-(5-amino-3-methyl-1,2-oxazol-4-yl)-3-methyl-2,3-dihydro-1,3,4-oxadiazol-2-ylium bromide with the unusual electron charge delocalization owing the local positive charge at the carbon atom of oxadiazole moiety. X-ray single crystal of C7H10N5O2·Br− showed the molecule crystalized in monoclinic, space group P21/c. Both five membered rings are planar and twisted forming the ring motif with the counter ion where H⋯Br interactions are one of the dominant. The presented compound is characterized by high ionization efficiency in ESI-MS mode and undergoes dissociation within oxadiazole moiety under ESI-MS/MS conditions even under low collision energies. The presented compound is an interesting example of heterocyclic stable carbocation which may serve as a new lead structure.
Introduction
The synthesis of heterocycles is possibly one of the oldest and at the same time one of the youngest branches of organic chemistry. Chemistry of heterocycles is an ever-expanding subject of organic chemistry and plays a crucial role in drug design. Among all known pharmaceuticals a predominant number of them are small molecules with heterocyclic moieties therefore there is a strong need to design a new, rapid and cost-efficient synthetic protocols. Isoxazole/oxadiazole-based molecules have been characterized as compounds possessing anti-inflammatory, antibacterial, anti-viral or anti-cancer activities.1–4 Despite significant progress that has been achieved in many fields of medicine including anticancer or anti-inflammatory therapy, the development of new potential drugs represents a major challenge to medicinal chemist researchers. Taking into account, our efforts have continued in the last years studies on isoxazole and oxadiazole derivatives in searching for new biological active derivatives.5–7 Although, there are known molecules with positive charge localized at the nitrogen atom,8–11 in the literature there is less mentioned about compounds possessing local positive charge at the carbon atom, and they are limited to small molecules.12,13 Tailoring the chemical and physical properties of molecules by rational planning to improve overall stability, solubility or to reduce toxicity when designing new compounds with expecting application in medicine, continues to be an important research attempt. It seems obvious that structural modification of neutral organic compounds by obtaining its salts by pairing organic cations with different counterions lead to the preparation of a molecule with unique properties exhibiting by e.g. increased water solubility. An examination of oxadiazolium salts obtained and characterized by Boyd et al.,14 showed the positively charged nitrogen atom but all compounds decomposed to diacylhydrazines in the presence of water. Motivated by the aforementioned biological and pharmacological importance of the heterocyclic compounds, especially in those with directly directed rings, and as a continuation with our previous research on isoxazoles, herein we report the synthesis of new stable and water-soluble 5-amino-2-(5-amino-3-methyl-1,2-oxazol-4-yl)-3-methyl-2,3-dihydro-1,3,4-oxadiazol-2-ylium bromide (ED) with a positive charge localized predominantly on a carbon atom of oxadiazole moiety.Results and discussion
X-Ray analysis
The molecular structure of the investigated compound with numbering scheme is presented in Fig. 1. The crystallographic data for both 1,3,4-oxadiazole ring and isoxazole ring show that the carbon-nitrogen, carbon–oxygen and nitrogen–nitrogen bond lengths are shorter than average respective single bonds (Tables 1–3).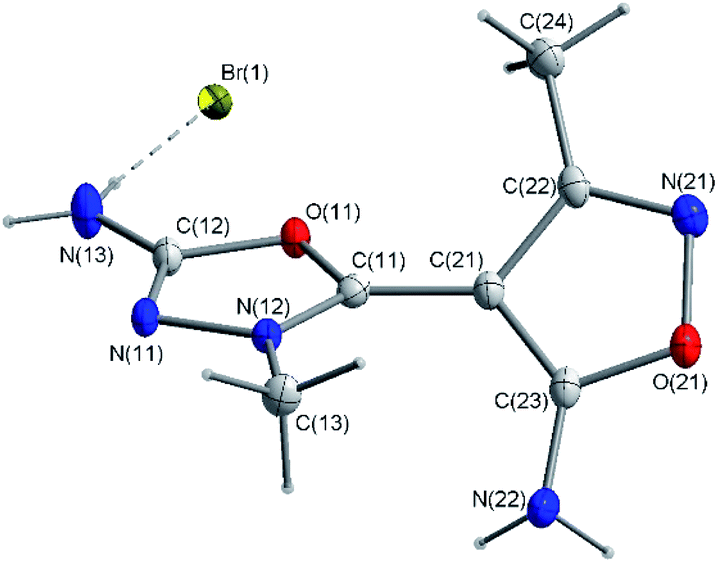 | ||
| Fig. 1 Molecular structure of ED. C-bound hydrogen atoms are omitted for clarity. | ||
| Identification code | 3e | |
| Empirical formula | C7H10BrN5O2 | |
| Formula weight | 276.10 | |
| Temperature | 100(2) K | |
| Wavelength | 1.54184 Å | |
| Crystal system | Monoclinic | |
| Space group | P21/c | |
| Unit cell dimensions | a = 15.2177(2) Å | α = 90° |
| b = 6.76340(10) Å | β = 109.445(2)° | |
| c = 11.0057(2) Å | γ = 90° | |
| Volume | 1068.13(3) Å3 | |
| Z | 4 | |
| Density (calculated) | 1.717 Mg m−3 | |
| Absorption coefficient | 5.209 mm−1 | |
| F(000) | 552 | |
| Crystal size | 0.192 × 0.104 × 0.088 mm3 | |
| Theta range for data collection | 3.080 to 75.438° | |
| Index ranges | −18 ≤ h ≤ 19, −8 ≤ k ≤ 7, −13 ≤ l ≤ 13 | |
| Reflections collected | 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 170 170 |
|
| Independent reflections 2189 | [R(int) = 0.0242] | |
| Completeness to theta = 67.684° | 100.0% | |
| Absorption correction | Analytical | |
| Max. and min. transmission | 0.917 and 0.816 | |
| Refinement method | Full-matrix least-squares on F2 | |
| Data/restraints/parameters | 2189/0/157 | |
| Goodness-of-fit on F2 | 1.119 | |
| Final R indices [I > 2σ(I)] | R1 = 0.0197, wR2 = 0.0541 | |
| Largest diff. peak and hole | 0.314 and −0.458 e Å−3 | |
| N(11)–C(12) | 1.299(2) |
| N(11)–N(12) | 1.3921(18) |
| N(12)–C(11) | 1.303(2) |
| N(12)–C(13) | 1.458(2) |
| N(13)–C(12) | 1.319(2) |
| N(21)–C(22) | 1.3015(18) |
| N(21)–O(21) | 1.4407(16) |
| N(22)–C(23) | 1.3333(19) |
| O(11)–C(11) | 1.3481(19) |
| O(11)–C(12) | 1.3818(18) |
| O(21)–C(23) | 1.3436(17) |
| C(11)–C(21) | 1.431(2) |
| C(21)–C(23) | 1.3848(19) |
| C(21)–C(22) | 1.431(2) |
| C(22)–C(24) | 1.4854(19) |
| C(12)–N(11)–N(12) | 102.59(13) |
| C(11)–N(12)–N(11) | 111.64(14) |
| C(11)–N(12)–C(13) | 129.02(14) |
| N(11)–N(12)–C(13) | 119.30(13) |
| C(22)–N(21)–O(21) | 106.02(11) |
| C(11)–O(11)–C(12) | 104.39(12) |
| C(23)–O(21)–N(21) | 108.69(10) |
| N(12)–C(11)–O(11) | 108.20(13) |
| N(12)–C(11)–C(21) | 130.31(15) |
| O(11)–C(11)–C(21) | 121.48(13) |
| N(11)–C(12)–N(13) | 129.72(15) |
| N(11)–C(12)–O(11) | 113.14(13) |
| N(13)–C(12)–O(11) | 117.13(14) |
| C(23)–C(21)–C(11) | 127.35(14) |
| C(23)–C(21)–C(22) | 104.78(12) |
| C(11)–C(21)–C(22) | 127.76(13) |
| N(21)–C(22)–C(21) | 111.40(12) |
| N(21)–C(22)–C(24) | 119.91(12) |
| C(21)–C(22)–C(24) | 128.68(12) |
| N(22)–C(23)–O(21) | 117.33(12) |
| N(22)–C(23)–C(21) | 133.62(13) |
| O(21)–C(23)–C(21) | 109.05(12) |
| C(12)–N(11)–N(12)–C(11) | −1.93(13) |
| C(12)–N(11)–N(12)–C(13) | 176.10(11) |
| C(22)–N(21)–O(21)–C(23) | 0.02(14) |
| N(11)–N(12)–C(11)–O(11) | 1.64(13) |
| C(13)–N(12)–C(11)–O(11) | −176.15(11) |
| N(11)–N(12)–C(11)–C(21) | −179.43(13) |
| C(13)–N(12)–C(11)–C(21) | 2.8(2) |
| C(12)–O(11)–C(11)–N(12) | −0.65(13) |
| C(12)–O(11)–C(11)–C(21) | −179.69(12) |
| N(12)–N(11)–C(12)–N(13) | −177.98(14) |
| N(12)–N(11)–C(12)–O(11) | 1.50(13) |
| C(11)–O(11)–C(12)–N(11) | −0.61(14) |
| C(11)–O(11)–C(12)–N(13) | 178.94(12) |
| N(12)–C(11)–C(21)–C(23) | 41.3(2) |
| O(11)–C(11)–C(21)–C(23) | −139.88(14) |
| N(12)–C(11)–C(21)–C(22) | −134.27(15) |
| O(11)–C(11)–C(21)–C(22) | 44.5(2) |
| O(21)–N(21)–C(22)–C(21) | 1.44(15) |
| O(21)–N(21)–C(22)–C(24) | −178.33(12) |
| C(23)–C(21)–C(22)–N(21) | −2.33(16) |
| C(11)–C(21)–C(22)–N(21) | 174.04(14) |
| C(23)–C(21)–C(22)–C(24) | 177.41(13) |
| C(11)–C(21)–C(22)–C(24) | −6.2(2) |
| N(21)–O(21)–C(23)–N(22) | 178.65(11) |
| N(21)–O(21)–C(23)–C(21) | −1.50(15) |
| C(11)–C(21)–C(23)–N(22) | 5.7(3) |
| C(22)–C(21)–C(23)–N(22) | −177.92(15) |
| C(11)–C(21)–C(23)–O(21) | −174.13(13) |
| C(22)–C(21)–C(23)–O(21) | 2.26(15) |
The 1.299(2) Å C12–N11 bond is the shortest whilst the 1.458(2) Å C13–N12 bond is the longest among the C–N bonds of the two heterocyclic rings (Table 2). These differences in bond lengths may be explained by the occurrence to the distribution of the positive charge between the atoms of heterocyclic rings and the fixation of the conjugate bond system. As a consequence of conjugation both five-membered rings are planar with maximum deviation of the carbon atoms from the least-squares planes by 0.008(2) Å and 0.014(1) Å for 1,3,4-oxadiazole ring and 0.014(1) Å for isoxazole ring, respectively. Moreover, as expected, the amino and methyl groups lie in the planes of these heterocyclic rings, with the torsion angles in the range of 1.1(1) to 5.7(3)° (Table 3). These observations confirm the presence of the electron delocalization through the chemical bonds. This delocalization of the electron, also support shortening the bond length between the 1,3,4-oxadiazole and isoxazole rings (C(11)–C(21) 1.431(2) Å). In addition, these rings are twisted about 75.5(3)°. The crystal structure of the analyzed compound is stabilized by hydrogen bonds and the presence of other weak intermolecular interactions (Fig. 2). The percentages of main interactions, quantified by Hirshfeld surface analysis is shown in Fig. 3.
 | ||
| Fig. 2 Packing diagram of crystal ED showing the hydrogen bonds and intermolecular interactions. | ||
 | ||
| Fig. 3 The Hirshfeld surface analysis of the compound ED. | ||
The amino group of the cation and the bromide anion are connected to form a ring R24(8) motif (Fig. 4). These ring motifs are arranged along the b-axis of the unit cell. Moreover, cations of the analyzed compound are connected each other via N(22)–H(3)⋯N(21)[−x + 1,y − 1/2,−z + 3/2] hydrogen bonding generates C(8) chains of cations propagating in [010] with adjacent ions related by the screw axis (Fig. 5). This C(8) chains are stabilized by additional the N(22)–H(4)⋯Br(1)[x,−y + 1/2,z + 1/2] interaction.
 | ||
| Fig. 4 Characteristic ring motif formed by the amino group of the cation and the bromide anion. | ||
 | ||
| Fig. 5 Chains of cations formed by hydrogen bonding network. | ||
ESI-MS analysis
The obtained heterocycle was also analyzed by ESI-MS method and the obtained spectrum is presented below (Fig. 6). | ||
| Fig. 6 ESI-MS spectrum of compound ED in positive ion mode. m/z range from 50–1000. | ||
On the presented mass spectrum obtained in the positive ion mode (Fig. 6) high intense signal at m/z 196.0830 corresponding to the M+ ion of model compound is observed. The obtained m/z value is in good agreement with calculated m/z ratio which is equal to 196.0829. The enlarged isotope pattern confirms the ion charge and low amount of carbon atom in the analyzed cation. Other signal have not been identified. The observed even m/z value is in agreement with the nitrogen rule for even-electron ions in ESI-MS mode. Ionization of this compound is the consequence of its chemical structure containing positive charge. To determine the chemical structure of the analyzed compound the ESI-MS/MS analysis under different collision energies was performed (Fig. 7).
 | ||
| Fig. 7 ESI-MS/MS spectra of analyzed compound obtained at different collision energy. Parent ion m/z 196.0830, collision energy (A) 10 eV, (B) 20 eV, (C) 30 eV. | ||
The obtained MS/MS spectra present a small amount of signals corresponding to the parent ion even under higher collision energy (Fig. 7). The most common signals are at m/z 154.0615, m/z 127.0507, m/z 110.0242 and m/z 82.0287. The fragmentation pathways and the possible structure of formed fragment ions are presented in the Fig. 8. The obtained m/z values of fragment ions are in a good agreement with the calculated ones and meet the nitrogen rule for even-electron ions.
 | ||
| Fig. 8 Schematic presentation of fragmentation pathways of parent ion at m/z 196.0830. | ||
NMR analysis
1H NMR spectroscopy shows only the presence of signals characterized the hydrogen atoms in the methyl and amino groups. In the 13C NMR spectrum we identified the signals corresponding to all of the carbon atoms presented in the molecule with typical chemical shifts. However, the chemical shift for the C7 is not reflected in an upfield shift typical for carbocations, we did not observe any hydrogen atom bonded to this carbon. Due to the large difference in chemical shifts characterizes carbon atoms form methyl groups (6 and 14) it can be assumed that a quaternary nitrogen atom is present in the molecule. Additionally, 13C DEPT135 analysis confirmed that there is no attached hydrogens to C7 carbon (Table 4) in oxadiazole ring due to the presence of signals corresponding to the carbon atom from methyl groups: 11.24 ppm and 38.09 ppm for C6 and C14 carbons, respectively. To proof clearly the charge location in the analyzed molecule the computational analysis was performed.
|
|||
|---|---|---|---|
| No | 1H NMR | 13C NMR | 13C DEPT 135 |
| δH | δC | δC | |
| 1 | |||
| 2 | |||
| 3 | 159.38 C | ||
| 4 | 73.90 C | ||
| 5 | 162.37 C | ||
| 6 | 2.22 (s) CH3 | 11.25 CH3 | 11.24 CH3 |
| 7 | 151.80 C+ | ||
| 8 | |||
| 9 | 170.35 C | ||
| 10 | |||
| 11 | |||
| 12 | 8.25 (s) NH2 | ||
| 13 | 8.51 (s) NH2 | ||
| 14 | 3.72 (s) CH3 | 38.09 CH3 | 38.08 CH3 |

Computational studies
Computational methods of theoretical chemistry have been used as useful tool to predict structure and properties of organic and inorganic compounds.15–19 The molecular orbital studies on isolated molecule has been done on the DFT level of theory. Gaussian 16 C.01 (ref. 20) suite of programs using the ωB97X-D21 long-range corrected hybrid density functional with damped atom–atom dispersion corrections was used with triple-ζ 6-311G(2d,2p) basis set. The presented structure was fully optimized with demanding convergence criteria (RMS force = 1 × 10−5, RMS displacement = 4 × 10−5, max force = 2 × 10−5, max displacement = 6 × 10−5) predefined as “opt = tight” in the Gaussian package, in atomic units. The atomic charges have been calculated on base of full NBO (Natural Bond Orbital) analysis, graphic has been done with GaussView 6.0.16 (ref. 22) program.The analyzed compound is thermodynamically stable dual-ring ion (see Fig. 9) with enthalpy H = −696.461666 hartree.
 | ||
| Fig. 9 The structure of ED. | ||
Compound consists of one oxadiazole and one oxazole rings, connected by C–C bond (1.413 Å). Please note that the rings planes are twisted, as expected, and the N6–C2–C15–C4 dihedral angle equals 35.2°.
The OCNNC oxadiazole ring builds interatomic distances 1.362 Å, 1.294 Å, 1.380 Å, 1.310 Å and 1.343 Å where atoms number 14, 1, 3, 6 and 2 are used respectively (see Table 5). The second, smaller in diameter oxazole ring, builds interatomic distances 1.412 Å, 1.312 Å, 1.388 Å, 1.442 Å and 1.291 Å between N, O, C, C, C of the ring atoms as presented in Table 5.
| Atoms | DFT | Exp. |
|---|---|---|
| C1–N11 | 1.329 | 1.319 |
| C1–O14 | 1.362 | 1.382 |
| C1–N3 | 1.294 | 1.299 |
| N3–N6 | 1.380 | 1.392 |
| O14–C2 | 1.343 | 1.348 |
| N6–C7 | 1.452 | 1.458 |
| C2–C15 | 1.413 | 1.431 |
| C15–C5 | 1.442 | 1.431 |
| C5–C18 | 1.488 | 1.486 |
| C5–N17 | 1.291 | 1.301 |
| N17–O16 | 1.412 | 1.441 |
| O16–C4 | 1.312 | 1.343 |
| C4–C15 | 1.388 | 1.385 |
| C4–N22 | 1.343 | 1.333 |
Each ring is substituted with methyl and amino group. Due to different bond type between methyl groups and rings only comparison involving amino groups is accessible here. Interestingly the C1–N11 distance ∼1.33 Å is shorter that C4–N22 ∼ 1.34 Å that suggests increase of the double bond character between C1–N11 in comparison to C4–N22 bond.
The DFT calculated parameters of the structure are in good agreement with presented experimental values.
| Atom | Charge | ||
|---|---|---|---|
| 0 | +1 | Delta | |
| C 1 | 0.72 | 0.74 | 0.02 |
| C 2 | 0.36 | 0.64 | 0.28 |
| N 3 | −0.41 | −0.36 | 0.05 |
| C 4 | 0.53 | 0.64 | 0.12 |
| C 5 | 0.22 | 0.25 | 0.04 |
| N 6 | −0.26 | −0.17 | 0.09 |
| C 7 | −0.35 | −0.36 | −0.01 |
| H 8 | 0.18 | 0.22 | 0.04 |
| H 9 | 0.21 | 0.23 | 0.01 |
| H 10 | 0.20 | 0.24 | 0.04 |
| N 11 | −0.81 | −0.78 | 0.04 |
| H 12 | 0.41 | 0.43 | 0.02 |
| H 13 | 0.40 | 0.42 | 0.02 |
| O 14 | −0.52 | −0.46 | 0.06 |
| C 15 | −0.32 | −0.38 | −0.06 |
| O 16 | −0.36 | −0.32 | 0.04 |
| N 17 | −0.19 | −0.12 | 0.06 |
| C 18 | −0.60 | −0.61 | −0.01 |
| H 19 | 0.21 | 0.23 | 0.01 |
| H 20 | 0.21 | 0.22 | 0.01 |
| H 21 | 0.22 | 0.25 | 0.03 |
| N 22 | −0.82 | −0.78 | 0.04 |
| H 23 | 0.39 | 0.42 | 0.03 |
| H 24 | 0.38 | 0.40 | 0.02 |
The NBO analysis describes the C2 atom as the most electrophilic with 28% positive charge accumulated.
Conclusions
In summary, we synthesized new water soluble and stable heterocyclic molecule owing the local positive charge at the carbon atom of oxadiazole moiety. The structure of this new stable cation was confirmed by using by 1H NMR, 13C NMR, 13C DEPT135, HMQC, X-ray, mass spectrometry and computational studies.Experimental
Materials and methods
Melting point was determined on Büchi apparatus (Laboratoriums-Technik AG, Flawil, Switzerland), heated table Kofler system (Wagner & Munz). Thin layer chromatography (TLC) was carried out on Polygram SIl G/UV 254 nm glass silica gel plates (Macherey-Nagel), using the developing system CHCl3–EtOAc 7:3, and detected with UV Krüss Optronic 254 nm lamp. NMR spectra were recorded on high-field spectrometer Bruker Avance 500 MHz using TMS as the internal standard. Complete 1H NMR, 13C NMR, 13C NMR-DEPT135 and HMQC analysis were performed on samples dissolved in DMSO-d6. All ESI-MS experiments were performed on the LCMS-9030 qTOF Shimadzu (Shimadzu, Kyoto, Japan) device, equipped with a standard ESI source and the Nexera X2 system. Analysis was performed in the positive ion mode between 50–1000 m/z. LCMS-9030 parameters: nebulizing gas–nitrogen, nebulizing gas flow 3.0 L min−1, drying gas flow – 10L min−1, heating gas flow – 10L min−1, interface temperature 300 °C, desolvation line temperature – 400 °C, detector voltage – 2.02 kV, interface voltage 4.0 kV, collision gas–argon, collision energy was optimized between 10 and 30 eV. The injection volume was optimized depending on the intensity of the signals observed on the mass spectrum within the range of 0.1 to 1 μl. All obtained signals had a mass accuracy error in the range of 1 ppm.
Single crystals of analyzed compound suitable for X-ray analysis were grown at room temperature by slow evaporation of methanol solution. Summary of structure determination is given in Table 1. Single crystal data collection was performed on a Rigaku XtaLAB Synergy-S single crystal X-ray diffractometer with CuKα radiation. at 100 (2) K. Cell refinement, data reduction, analysis, and absorption correction were carried out with CRYSALISPro (Rigaku Oxford Diffraction, Wrocław, Poland) software. Cell refinement, data reduction, analysis, and absorption correction were carried out with CRYSALISPro (Rigaku Oxford Diffraction, Wrocław, Poland) software. The structures were solved by direct methods with SHELXS23 and refined with full-matrix least-squares techniques on F2 with SHELXL.24 The C-bonded hydrogen atoms were calculated in idealized geometry riding on their parent atoms.
The elemental analysis was performed on the elemental analyzer Vario EL III CHNS (Elementar, Germany).
Synthetic procedure
A mixture of 5-amino-N,3-dimethyl-isoxazolo-4-carbohydrazide (n = 0.0176 mol, 3 g) and cyanogen bromide (3 g) in ethanol (30 ml) was refluxed for 2 h. The completion of reaction was monitored by TLC. Then the reaction mixture was cooled to the room temperature and the suspension was filtered. The crude product was recrystallized from water to get 5-amino-2-(5-amino-3-methyl-1,2-oxazol-4-yl)-3-methyl-2,3-dihydro-1,3,4-oxadiazol-2-ylium bromide. Water solubility is 45 mg ml−1. Yield 90.9%, mp 242 °C.Elemental analysis
Found: C, 31.06%; H, 3.58%; N, 25,78%.
Author contributions
E. D. performed the chemical synthesis. U. B. and RB made MS and NMR analysis. L. J performed X-ray analysis. R. W. performed computational studies. U. B. wrote the manuscript. M. M. revised the manuscript. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was financially supported by the Wroclaw Medical University (grant numbers: SUB.D090.21.065). The authors would like to thank Andrzej Reszka (Shim-Pol, Poland) for providing the access to Shimadzu LCMS-9030 instrument.Notes and references
- J. Jampilek, Molecules, 2019, 24, 3839 CrossRef CAS PubMed.
- S. Alsalameh, M. Burian, A. G. Mahr, B. G. Woodcock and G. Geisslinger, Aliment. Pharmacol. Ther., 2003, 17, 489–501 CrossRef CAS PubMed.
- R. I. Fox, M. L. Herrmann, C. G. Frangou, G. M. Wahl, R. E. Morris, V. Strand and B. J. Kirschbaum, Clin. Immunol., 1999, 93, 198–208 CrossRef CAS PubMed.
- A. Vaidya, D. Pathak and K. Shah, Chem. Biol. Drug Des., 2021, 97, 572–591 CrossRef CAS PubMed.
- U. Bąchor, S. Ryng, M. Mączyński, J. Artym, M. Kocięba, E. Zaczyńska, I. Kochanowska, E. Tykarska and M. Zimecki, Acta Pol. Pharm., 2019, 76, 251–263 CrossRef.
- M. Zimecki, U. Bąchor and M. Mączyński, Molecules, 2018, 23, 2724 CrossRef PubMed.
- M. Mączyński, J. Artym, M. Kocięba, I. Kochanowska, S. Ryng and M. Zimecki, Pharmacol. Rep., 2016, 68, 894–902 CrossRef PubMed.
- M. Akkurt, G. S. Duruskari, F. A. A. Toze, A. N. Khalilov and A. T. Huseynova, Acta Crystallogr., Sect. E: Crystallogr. Commun., 2018, 74, 1168–1172 CrossRef CAS PubMed.
- A. A. Hassan, N. K. Mohamed, A. A. Aly, H. N. Tawfeek, S. Bräse and M. Nieger, J. Macromol. Sci., 2019, 1176, 346–356 CAS.
- P. Yin, J. Zhang, D. A. Parrish and J. M. Shreeve, J. Mater. Chem. A, 2015, 3, 8606–8612 RSC.
- Y. Jia, Q. Ma, Z.-Q. Zhang, W. Geng, J. Huang, W. Yang, G.-J. Fan and S. Wang, Cryst. Growth Des., 2020, 20, 3406–3412 CrossRef CAS.
- G. A. Olah and P. W. Westerman, J. Am. Chem. Soc., 1973, 95(11), 3706–3709 CrossRef CAS.
- G. A. Olah, L. Heiliger and G. K. S. Prakash, J. Am. Chem. Soc., 1989, 111, 8020–8021 CrossRef CAS.
- G. V. Boyd and S. R. Dando, J. Chem. Soc. C, 1970, 10, 1397–1401 RSC.
- Z. Mielke, Z. Latajka, A. Olbert-Majkut and R. Wieczorek, J. Phys. Chem. A, 2000, 104, 3764–3769 CrossRef CAS.
- R. Wieczorek, Z. Latajka and J. Lundell, J. Phys. Chem. A, 1999, 103, 6234–6239 CrossRef CAS.
- T. K. Olszewski, E. Wojaczyńska, R. Wieczorek and J. Bąkowicz, Tetrahedron: Asymmetry, 2015, 26, 601–607 CrossRef CAS.
- S. Pedro, R. Wieczorek and J. J. Dannenberg, J. Phys. Chem. B, 2007, 111, 2398–2403 Search PubMed.
- M. Rudowska, R. Wieczorek, A. Kluczyk and P. Stefanowicz, J. Am. Soc. Mass Spectrom., 2013, 24, 846–856 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- D. Roy, T. A. Keith, and J. M. Millam, GaussView, Version 6.1, Semichem Inc., Shawnee Mission, KS, 2016 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 13C, 13C DEPT135, ESI-MS, X-ray crystallographic data of the analyzed compound (CIF) see supplementary materials. CCDC 2084686. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra05116d |
| This journal is © The Royal Society of Chemistry 2021 |