
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceColor-tunable arylaminoanthraquinone dyes through hydrogen-bond-assisted charge transfer interaction†
Takashi Takeda *ab,
Yotaro Kasaharab and
Tomoyuki Akutagawaab
*ab,
Yotaro Kasaharab and
Tomoyuki Akutagawaab
aInstitute of Multidisciplinary Research for Advanced Materials, Tohoku University, Katahira 2-1-1, Aoba-ku, Sendai, 980-8577, Japan. E-mail: takashi@tohoku.ac.jp
bDepartment of Applied Chemistry, Graduate School of Engineering, Tohoku University, Sendai, Miyagi 980-8579, Japan
First published on 9th July 2021
Abstract
We prepared a series of arylaminoanthraquinone derivatives, including those with electron-accepting sulfone units and/or with electron-donating dialkylamino units. A color-tunable anthraquinone library that reached into the NIR region could be prepared through the precise control of frontier orbitals. Fine color-tuning was achieved through proper selection and positioning of the substituents. Effective intramolecular hydrogen-bond-assisted charge transfer interaction between electron-donating aniline/p-phenylenediamine and electron-accepting anthraquinone substructures induced a significant bathochromic shift of anthraquinone. The number and position of the substituents and the molecular conformation also significantly contributed to determining photophysical properties.
Introduction
Recently, π-conjugated molecules with a small HOMO–LUMO gap that absorb long-wavelength light, including near-infrared (NIR) light have attracted attention1–6 due to their potential for use in optoelectronic devices,5,7–11 optical devices,1,6,12 and medical applications.13–15 There are several approaches to develop such molecules, including the expansion of π-conjugation and the introduction of an open-shell biradical unit in π-conjugation. A variety of long-wavelength absorbing dyes have been developed with these strategies, including porphyrin/phthalocyanine derivatives,1 including their conjugated array,16–19 polymethine/cyanine derivatives,1,20–25 boron-dipyrromethene (BODIPY) derivatives,26,27 polyacenes,28–31 zethrenes derivatives,32–35 and indenofluorene derivatives.36–47 However, these compounds often suffer from difficult synthesis and low solubility in common organic solvents.The introduction of intramolecular charge transfer (CT) interaction between an electron-donating group (D) and an electron-accepting group (A) in a π conjugated unit is also widely used to develop long-wavelength absorbing dyes.1–4,48–50 While a variety of long-wavelength absorbing dyes have been developed, quinone and its π-extended derivatives have been especially used for this purpose because of their strong electron-accepting properties.1 Especially, anthraquinone has been used due to its easy functionalization.51 Several commercially available dyes have an anthraquinone skeleton, such as the disperse blues. Some disperse blues contain electron-donating aniline/p-phenylenediamine unit(s) fused with an electron-accepting anthraquinone core. Since the degree of intramolecular CT interaction can be modulated through the electron-donating/accepting properties of D and A, this skeleton could serve as a color-tunable long-wavelength absorbing dye. However, little is known about long-wavelength absorbing dyes based on a aminoanthraquinone scaffold.
We previously studied the photophysical and electrochemical properties of arylsulfonamide-substituted anthraquinones 3, 9, 14, and 20.52,53 Thanks to effective intramolecular N–H⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C hydrogen bonds at the peri-position of anthraquinone through fixation of the conformation by bulky terminal substituents, the electron-accepting properties of the anthraquinone units significantly increased, which induced a significant bathochromic shift in UV-Vis spectra thanks to hydrogen-bond-assisted CT interaction (Fig. 1a).54 We also revealed that N-methylation of these derivatives induced a drastic change in conformation to completely suppress this intramolecular CT interaction.55
C hydrogen bonds at the peri-position of anthraquinone through fixation of the conformation by bulky terminal substituents, the electron-accepting properties of the anthraquinone units significantly increased, which induced a significant bathochromic shift in UV-Vis spectra thanks to hydrogen-bond-assisted CT interaction (Fig. 1a).54 We also revealed that N-methylation of these derivatives induced a drastic change in conformation to completely suppress this intramolecular CT interaction.55
 | ||
| Fig. 1 (a) Hydrogen-bond-assisted CT interaction in aminoanthraquinone derivatives with bulky aryl substituents.52 (b) Chemical structure of aminoanthraquinones described in this manuscript. | ||
In this work, we systematically prepared arylaminoanthraquinones and investigated their photophysical and electrochemical properties (Fig. 1b). Thanks to hydrogen-bond-assisted CT interaction, these compounds showed significant bathochromic absorption compared to anthraquinone. Their photophysical properties could be systematically modulated by the introduction of electron-donating dialkylamino groups at peripheral phenyl groups or the insertion of electron-accepting sulfone groups between an amino unit and phenyl group. As a result, color-tunable aminoanthraquinone dyes that reached into the NIR region could be prepared.
Results and discussion
Preparation of aminoanthraquinones with electron-donating/accepting units
We prepared a series of aminoanthraquinones with different electron-donating/accepting units. Fig. 1 shows the molecular structures of aminoanthraquinones described in this manuscript. Since our previous study revealed that a bulky aryl substituent at the peripheral position of aminoanthraquinones worked effectively to form N–H⋯OC hydrogen bonds, we adopted this strategy to maximize hydrogen-bond-assisted CT interaction. We selected a dialkylamino unit on terminal phenyl groups as an electron-donating unit and a sulfone unit between aminoanthraquinone and terminal phenyl group as an electron-accepting unit. The positional and numerical effects of substituents on the photophysical and electrochemical properties should also be considered, since phenylenediamine and its derivatives have been reported to be strong electron donors. Based on this strategy, we synthesized 23 kinds of aminoanthraquinones for this purpose, including those we previously reported52,55 and those in the literature (1, 7, 13, 19).56–59 A substituent was introduced at the 1,4-, 1,5-, 1,8- or 1,4,5,8-positions in each compound. Terminal methyl, butyl, tert-butyl and hexyl groups were introduced to improve their solubility.
Fig. 2 summarizes the synthesis of 1–23 with/without electron-donating/accepting units. Most of the compounds were synthesized through a Buchwald–Hartwig cross-coupling reaction60–63 or Ullmann coupling reaction64 between the corresponding chloroanthraquinone and arylamine/arylsulfonamide, respectively. The reaction of alkylphenylamine and alkylphenylsulfonamide without terminal electron-donating alkylamino groups proceeded smoothly to give the corresponding arylamino derivatives (1, 13, 19)58,59 and arylsulfonamide derivatives (3, 9, 14, 20) in good yields. In the case of 1,5-disubstituted (13, 14) and 1,4,5,8-tetrasubstituted derivatives (19, 20), a terminal tert-butyl group was necessary to increase the solubility to accomplish the reaction. In the reactions of 1,4- and 1,8-dichloroanthraquinone with stronger electron-donating N,N-dimethyl-1,4-phenylenediamine, the reaction did not proceed completely and gave both the desired disubstituted derivatives (2, 8) and monosubstituted derivatives with unreacted chlorine atom (5, 11). If we consider that a significant amount of unreacted monochloro derivative was recovered only in the reaction with phenylenediamine derivatives, this could be accounted for by suppression of the reactivity of monochloro intermediate with Pd catalyst due to the decreased electron-withdrawing properties of arylanthraquinones by introduction of an electron-donating dimethylaminophenyl unit. In the reaction of 1,5-dichloroanthraquinone, only monosubstituted derivative (16) was obtained, due to the relatively limited solubility of 1,5-disubstituted derivatives (see above), in addition to limited reactivity. In the reaction with N,N-dimethylaminophenylsulfonamide,65 only the corresponding 1,4- and 1,8-substituted derivatives 4, 10 were obtained in moderate yields, whereas the 1,5- and 1,4,5,8-substituted derivatives were not obtained under the same reaction conditions. If we consider the decreased electron-donating nature of the terminal NH2 group suitable for the Buchwald–Hartwig cross-coupling reaction due to the neighboring electron-accepting sulfone unit, this limited reactivity was due to the limited solubility of these derivatives, rather than the reactivity. To obtain 1,5- and 1,4,5,8-substituted derivatives, we planned to introduce long alkyl chains (butyl groups for 15 and hexyl groups for 21) to a terminal amino group, instead of a methyl group. To achieve this, we changed the synthetic strategy and planned to react the corresponding aminoanthraquinone with 4-(dialkylamino)benzene sulfonyl chloride. 4-(Dibutylamino)benzenesulfonyl chloride and (dihexylamino)benzenesulfonyl chloride were newly prepared from dibutylamine and dihexylamine, respectively, by the reaction with (TMSO)2SO2 followed by PCl5. The reaction of the corresponding commercially available aminoanthraquinone with 4-(dialkylamino)benzene sulfonyl chloride in pyridine proceeded smoothly to give the desired dialkylaminophenylsulfonamide-substituted anthraquinones 15 and 21. When we tried the reaction between (dibutylamino)benzenesulfonyl chloride and 1,4,5,8-tetraaminoanthraquinone, prepared in-house by hydrolysis of 20, we unexpectedly obtained partly reacted 1,4-disubstituted-5,8-diaminoanthraquinone 22, although we could not clarify the mechanism or selectivity. The structure of 22 was rigorously characterized by single-crystal X-ray structural analysis (Fig. S1 in ESI†). N-Methylation of arylsulfonamide derivatives 3, 9, 14, 20 gave the corresponding N-methylated derivatives (6, 12, 18, 23). All newly synthesized compounds were characterized by 1H and 13C NMR, IR and HR-MS spectroscopy and elemental analyses.
 | ||
| Fig. 2 Synthesis of 1–23. | ||
Photophysical properties
We measured UV-Vis/NIR spectra of 1–23 to investigate the effect of the electron-donating/accepting substituent and the positional/numerical effects of the substituents. Fig. 3 and 4 show the UV-Vis spectra in CH2Cl2 classified according to the substitution position and the substituent, respectively, and Table 1 summarizes the photophysical and electrochemical properties of 1–23, in addition to their theoretical HOMO and LUMO energies and the lowest-energy absorption peak. As seen in the UV-Vis spectra and Fig. 5, we could prepare a color-tunable anthraquinone library through precise control of the frontier orbital. Fine color-tuning was achieved through proper selection and positioning of the substituents. As shown in Fig. 3, the photophysical properties of aminoanthraquinone could be greatly modulated by selecting the substituents. In the case of 1,8-disubstituted anthraquinones, that with a tolylamino group (1) showed bathochromic absorption at λmax 559 nm, which is red-shifted by 233 nm compared to that for anthraquinone. This could be accounted for by effective intramolecular CT interaction between the electron-donating aniline/p-phenylenediamine substructure and the electron-accepting anthraquinone substructure (see below). Introduction of an electron-accepting sulfone unit between amino and aryl units significantly modulated the photophysical properties. Tolylsulfonamide-substituted anthraquinone 3 showed an absorption peak at 434 nm, which is blue-shifted by 125 nm compared to that of 1, due to a larger decrease in the HOMO energy (1: −5.19 eV; 3: −6.41 eV) than that in LUMO energy (1: −2.59 eV; 3: −3.15 eV) (Table 1). Substitution of a terminal alkyl group with an electron-donating dialkylamino unit also affected the optical properties, which was suitable for fine tuning. Substitution of the terminal methyl groups of 1 and 3 with dimethylamino groups (2 and 4, respectively) induced a slight red shift of the absorption peak (λmax: 17 and 10 nm, respectively) and a moderate-to-large red shift of the absorption end (λend: 95 and 37 nm, respectively). This red shift is accounted for by a increase in the HOMO energy (1: −5.19 eV; 2: −4.65 eV/3: −6.41 eV; 4: −5.75 eV, Table 1). A similar tendency of the substituent effect was observed for other positional isomers. Halogen substituents also significantly contributed to determine the photophysical properties. For example, monosubstituted derivatives 5, 11, and 16 with dialkylphenylamino groups showed a hypochromic shift of ca. 40 nm compared to 2 with the same aniline substructure. While arylsulfonamide derivatives (3, 9, 14, 20) showed unique fluorescence even with a non-fluorescent anthraquinone core,52 other derivatives did not exhibit fluorescence. | ||
| Fig. 3 UV-Vis spectra of 1–23 in CH2Cl2 classified according to the substitution position. The color was determined by the introduced substituent. The dotted line indicates monochloro-monosubstituted derivatives or diamino-disubstituted derivative. | ||
 | ||
| Fig. 4 UV-Vis spectra of 1–22 in CH2Cl2 classified according to the substituent. The color was determined by the position of the introduced substituent. The dotted line indicates monochloro-monosubstituted derivatives or diamino-disubstituted derivative. | ||
| Compd | λmaxa | εb | λendc | Epad | Epcd | HOMOe | LUMOe | Abs. calc.f |
|---|---|---|---|---|---|---|---|---|
| a nm. Measured in CH2Cl2.b M−1 cm−1.c nm. Shortest wavelength with ε < 500.d V vs. Ag/AgCl. Measured in CH2Cl2 containing Bu4NBF4.e eV. Calculated at the B3LYP/6-31G(d) level.f Theoretical HOMO–LUMO transition energy in nm. Calculated at the B3LYP/cc-pVDZ level.g Shoulder.h Irreversible.i Reversible.j Broad.k Not observed.l Forbidden transition (oscillator strength < 0.05). | ||||||||
| 1 | 559 | 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 800 |
654 | 1.61h | −0.92,i −1.27i | −5.19 | −2.59 | 577 |
| 2 | 576 | 10700 |
755 | 0.83h | −0.98,i −1.37i | −4.65 | −2.33 | 651 |
| 3 | 434 | 10200 |
498 | NAk | −0.52,i −0.92i | −6.41 | −3.15 | 463 |
| 4 | 442 | 7200 | 535 | 1.43h | −0.55,i −0.94i | −5.75 | −2.91 | 512 |
| 5 | 534 | 5000 | 707 | 0.73i | −0.87,i −1.25i | −4.93 | −2.64 | 651 |
| 6 | 331 | 2800 | 403 | NAk | −0.87,i −1.36i | −6.30 | −2.72 | 472l |
| 7 | 645 | 15000 |
718 | 1.01, 1.46i | −0.95,i −1.28i | −4.82 | −2.54 | 609 |
| 8 | 652 | 13900 |
795 | 0.77h,j | −1.00,i −1.30i | −4.36 | −2.25 | 659 |
| 9 | 468 | 6800 | 547 | NAk | −0.60i | −6.09 | −3.18 | 495 |
| 10 | 494 | 6600 | 592 | 1.42h | −0.52,i −0.65i | −5.53 | −2.91 | 557 |
| 11 | 529 | 5400 | 693 | 0.77h | −0.87,i −1.28i | −5.02 | −2.62 | 615 |
| 12 | 337 | 3100g | 410 | NAk | −0.82,i −1.29i | −6.33 | −2.74 | 481l |
| 13 | 539 | 15200 |
646 | 1.40h | −0.89,i −1.26i | −5.25 | −2.59 | 555 |
| 14 | 432 | 9800 | 494 | NAk | −0.53i | −6.44 | −3.15 | 449 |
| 15 | 432 | 7900 | 547 | 1.46i | −0.61i | −5.67 | −2.95 | 534l |
| 16 | 530 | 7700 | 721 | 0.77i | −0.87,i −1.30i | −4.96 | −2.64 | 634 |
| 17 | 418 | 3900g | 498 | 1.24h | −0.64,i −0.91i | −5.74 | −2.98 | 526l |
| 18 | 333 | 3400 | 400 | NAk | −0.83,i −1.36i | −2.82 | −6.28 | 495l |
| 19 | 758 | 27900 |
855 | 0.60,i 1.02h | −0.97,i −1.22i | −4.40 | −2.44 | 726 |
| 20 | 539 | 15100 |
624 | NAk | −0.21,i −0.54i | −5.96 | −3.37 | 563 |
| 21 | 568 | 13700 |
686 | 1.46h | −0.35,i −0.66i | −5.44 | −2.98 | 593 |
| 22 | 626 | 32700 |
677 | 0.97,i 1.38h | −0.89i,j | −5.05 | −2.61 | 573 |
| 23 | 355 | 2800g | 431 | NAk | −0.92,i −1.51i | −6.20 | −2.62 | 480l |
 | ||
| Fig. 5 Photographic demonstration of fine color-tuning with aminoanthraquinone-based dyes with 1–23 (arranged by compound number from the left). | ||
In addition to the kinds of substituents, the position and number of the substituents also significantly affected the photophysical properties of these derivatives, as summarized in Fig. 4. The absorption peak/end were significantly different for positional isomers with the same substituents. For example, for alkylphenylamino derivatives (1, 7, 13, 19), the absorption peak differs by ca. 220 nm (13: 539 nm, 19: 758 nm). This large difference in photophysical properties could be understood in terms of the different electron-donating natures of the aniline/p-phenylenediamine substructure. HOMO energies of 7 (−4.82 eV) and 19 (−4.40 eV) with stronger electron-donating p-phenylenediamine substructure are higher than those of 1 (−5.19 eV) and 13 (−5.25 eV) with weaker electron-donating aniline substructure. The 1,4-disubstituted and 1,4,5,8-tetrasubstituted derivatives with a stronger electron-donating p-phenylenediamine substructure contributed to stronger intramolecular CT interaction.
Tetrasubstituted derivatives have longer absorption among the derivatives with same substituents. For example, the absorption of tetrasubstituted derivative 19 with alkylphenylamino groups showed a large red shift of ca. 110 nm compared to that of the corresponding 1,4-disubstituted 7 with the same p-phenylenediamine electron-donating groups. DFT calculation indicates that the orbital degeneracy of electron-donating substructure units contributed to increase the HOMO energy (19: −4.40 eV, 7: −4.82 eV, Fig. S6†). As a result, significant red shift occurred in tetrasubstituted derivatives.
In addition, N-methylation induced a drastic hypochromic shift due to the change in the conformation of a terminal arylamino/arylsulfonamide group.55 For example, the absorption peak at the longest wavelength for N-methylated tetrasubstituted derivative 23 showed a blue shift of 190 nm compared to that of the corresponding N–H derivative 20. N-Methylation at the congested peri-position of the anthraquinone unit induced a drastic change in conformation from a coplanar arrangement to an orthogonal arrangement between the anthraquinone scaffold and arylsulfonamide substituents. This drastic structural change significantly affected its LUMO energy and HOMO structure to suppress the CT interaction between the anthraquinone π unit and sulfonamide substituents. We could prepare a color-tunable anthraquinone dye library (λmax: 331–758 nm, λend: 400–855 nm) that extended into the NIR region.
Electrochemical properties and DFT calculations
To understand the huge differences in absorption among 1–23, we performed cyclic voltammetry and DFT calculations. Fig. 6 and S2 (ESI†) summarize the results according to the substitution position and the substituent, respectively. The compounds show reversible reduction peaks and irreversible (some reversible) oxidation peaks at −0.52 to −1.00 V (Epc1), −0.54 to −1.51 V (Epc2) and 0.60–1.61 V (Epa1) vs. Ag/AgCl, respectively, which is accounted for by reduction of the electron-accepting anthraquinone core and oxidation of the electron-donating terminal arylamino/arylsulfonamide group. In the reduction process, compounds with arylsulfonamide substituents (3, 4, 9, 10, 14, 15, 20, 21) showed a significant anodic shift (Epc1: −0.21 to −0.61 V) compared to other compounds (−0.82 to −1.00 V), which is accounted for by an effective intramolecular N–H⋯OC hydrogen bonding effect between electron-donating arylsulfonamide groups and the electron-accepting anthraquinone core.52 On the other hand, the reduction potential of other compounds was almost equal to that of anthraquinone, which indicates that an intramolecular N–H⋯OC hydrogen bonding effect did not work effectively for arylaminoanthraquinones without an electron-accepting sulfone unit. In the oxidation process, its potentials were mainly governed by the electron-donating unit. For example, compounds with stronger electron-donating dialkylaminophenylamino units showed a significant cathodic shift of the oxidation peak. 1,4-Disubstituted and 1,8-tetrasubstituted derivatives with stronger p-phenylenediamine donor units are stronger electron donors than 1,5- and 1,8-disubstituted derivatives with weaker aniline donor units.
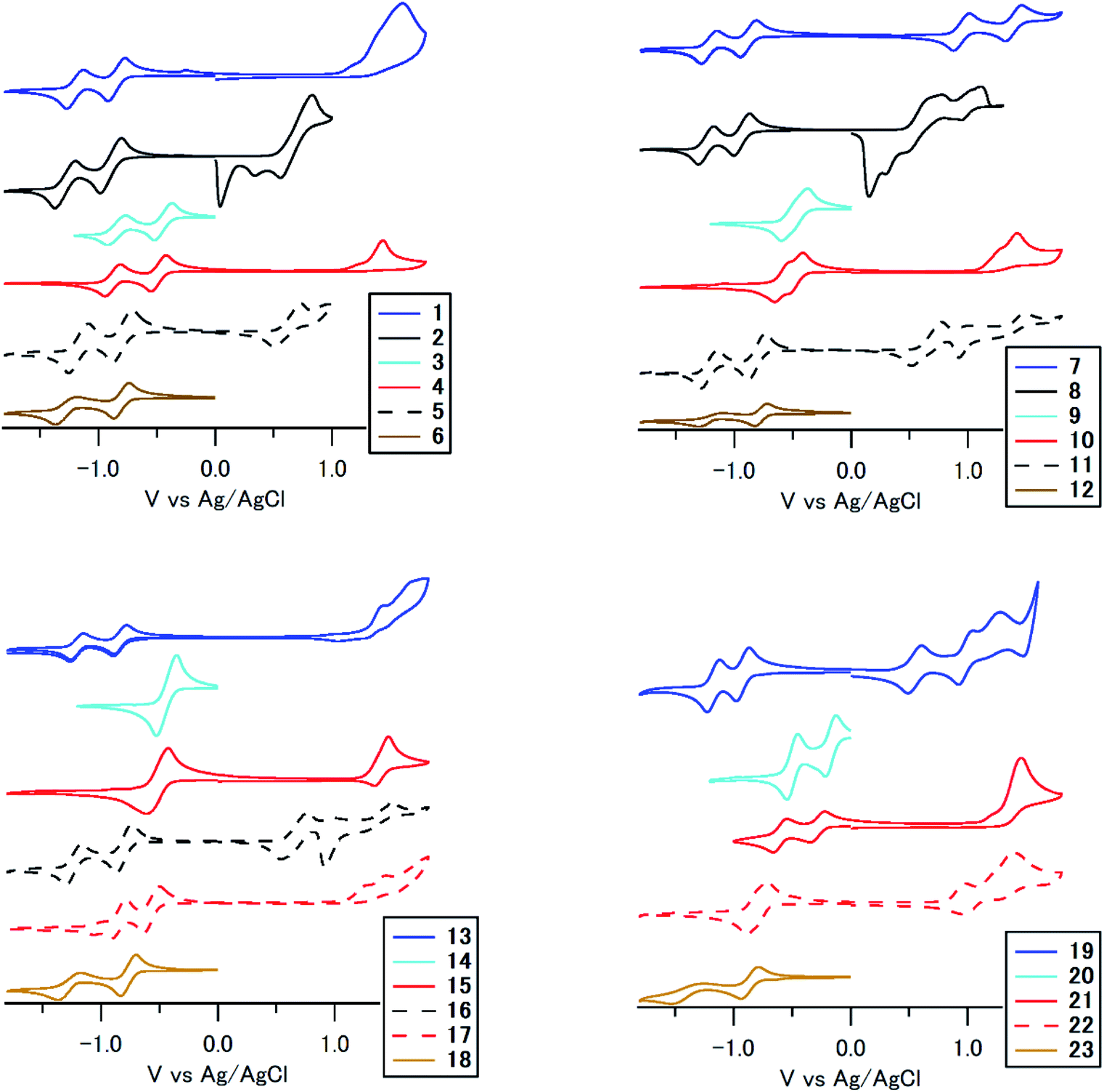 | ||
| Fig. 6 Cyclic voltammograms of 1–23 measured in CH2Cl2 classified according to the substitution position. The color was determined by the introduced substituent. The dotted line indicates monochloro-monosubstituted derivatives or diamino-disubstituted derivative. | ||
The theoretically calculated HOMO–LUMO energy and simulated absorption by DFT calculations well reproduced the trend in the experimental results. Fig. 7 and S3–S6 (ESI†) show the HOMO and LUMO of 1,4-disubstituted derivatives 7–10, 12 and other compounds 1–6, 11, 13–23 for their optimized structures. N–H units faced a carbonyl unit to form intramolecular hydrogen bonding interaction in all non-N-methylated derivatives. HOMO was located on an electron-donating p-phenylenediamine/aniline substructure whereas LUMO was located on an electron-accepting central p-quinone substructure. Since HOMO/LUMO were both co-located on the same π-conjugated plane, transition-allowed intramolecular CT occurred effectively to induce a significant bathochromic shift depending on the electron-donating unit. Since similar intramolecular hydrogen bonding interaction was observed in all N–H compounds, intramolecular N–H⋯OC hydrogen bonding contributed to their frontier orbitals. LUMO was located on N–H nitrogen atoms in all N–H compounds, in addition to an anthraquinone unit, which decreased its energy through intramolecular N–H⋯OC hydrogen bonding, as observed in their cyclic voltammetry measurement, which resulted in a significant bathochromic shift in the UV-Vis spectra (hydrogen-bond-assisted CT interaction). The degree of modulation of LUMO energy through intramolecular hydrogen bonding interaction differs between the compounds depending on the substituents. The LUMO energies of sulfonamide derivatives were more reduced through N–H⋯OC hydrogen bonding, as demonstrated by comparison with the corresponding N–Me derivatives without intramolecular hydrogen bonding interaction. On the other hand, the LUMO energies of other derivatives with arylamino groups was less influenced through intramolecular N–H⋯OC hydrogen bonding, which could be accounted for by the different orbital contributions of frontier orbitals between compounds with/without an electron-accepting sulfone unit. N-Methylation of sulfonamide derivatives also affected the LUMO energy and absorption spectra, since it induced a drastic structural change from a coplanar arrangement to an orthogonal relationship between the anthraquinone scaffold and arylsulfonamide substituents, which suppresses intramolecular N–H⋯OC hydrogen bonding and forbits effective HOMO–LUMO transition.
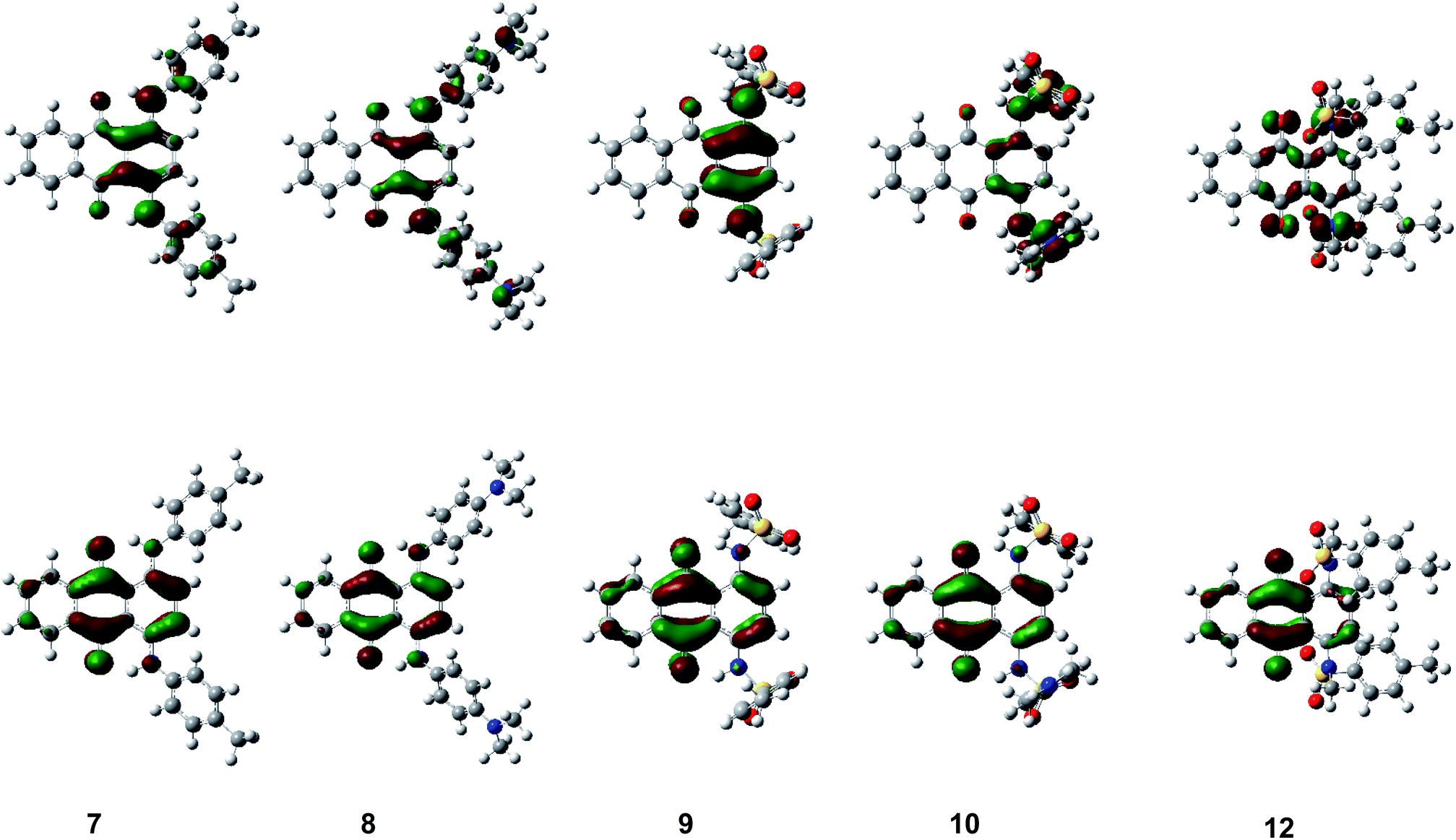 | ||
| Fig. 7 HOMO (top) and LUMO (bottom) of 1,4-disubstituted anthraquinones 7–10 and 12. | ||
Conclusion
We prepared a series of arylaminoanthraquinone derivatives including those with electron-accepting sulfone units and electron-donating dialkylamino units. As demonstrated in UV-Vis spectra, we could prepare a color-tunable anthraquinone library that reached into the NIR region through the precise control of frontier orbitals. Fine color-tuning was achieved through proper selection and positioning of the substituents. Effective intramolecular hydrogen-bond-assisted CT interaction between electron-donating aniline/p-phenylenediamine and electron-accepting anthraquinone substructures induced a significant bathochromic shift for the anthraquinone. The introduction of an electron-accepting sulfone unit between amino and aryl units significantly modulated the photophysical properties. Substitution of a terminal alkyl group with an electron-donating dialkylamino unit also affected the optical properties, which is suitable for fine color-tuning. The number and position of the substituent also significantly contributed to determine the photophysical properties. Especially, tetrasubstituted derivatives showed a larger red shift compared to other derivatives due to the orbital degeneracy of stronger electron-donating p-phenylenediamine substructures. The change in conformation induced by N-methylation at the congested peri-position of the anthraquinone unit suppressed the interaction between the anthraquinone π unit and sulfonamide substituents to induce a significant hypochromic shift.Experimental section
General methods
Commercially available reagents and solvents were used without further purification. NMR spectra were measured in CDCl3 on a Bruker Avance III 400 or a Bruker Avance III 500 NMR spectrometer. Chemical shifts (δ) in ppm were referenced to an internal standard of tetramethylsilane (1H) or residual nondeuterated solvent (13C: 77.0 ppm for CDCl3). Mass spectra were obtained in EI or FAB mode with a JMS-700 mass spectrometer at the NMR and MS Laboratory, Graduate School of Agriculture, Tohoku University. Infrared (IR, 400–4000 cm−1) spectra were measured on a KBr pellet with a Thermo Scientific NICOLET 6700 FT-IR spectrometer with a resolution of 4 cm−1. UV-Vis spectra were measured in CH2Cl2 (for spectrochemical analysis). Cyclic voltammetry was performed in dry CH2Cl2 containing 0.1 M Bu4NBF4 with a scan rate of 100 mV s−1. A glassy carbon electrode, Pt electrode and Ag/AgCl electrode were used as the working, counter, and reference electrodes, respectively.Single-crystal X-ray structure analysis
Crystallographic data were collected using a diffractometer equipped with a rotating anode fitted with a multilayer confocal optic using Cu-Kα (λ = 1.54187 Å) radiation. Structure refinements were carried out using the full-matrix least-squares method on F2. Calculations were performed using the Crystal Structure and SHELEX software packages.66 Parameters were refined using anisotropic temperature factors except for the hydrogen atom.Crystal data of 22
Single crystal of 22 suitable for single-crystal structure analysis was obtained by recrystallization from MeCN. Black plate. C42H48N6O6S2, M = 797.00, P1 bar (#2), a = 10.2153(8) Å, b = 14.5679(11) Å, c = 16.0551(12) Å, a = 107.972(8)°, b = 104.609(7)°, g = 106.675(8)°, V = 2019.1(3) Å3, Z = 2, Dc = 1.311 g cm−3. Independent reflection 7246 (all), T = 173 K, m = 16.46 cm−1, R = 6.57%. CCDC 2063387.†Computational methods
DFT calculations were performed with the Gaussian 09 67 or Gaussian 16 68 program packages. The geometries of the molecules were optimized using the B3LYP/6-31G* basis set. Terminal long alkyl groups were replaced by methyl groups in 13, 15, 19 and 21. Stationary points were assessed by a vibration frequency analysis. TD-DFT calculations were performed for the optimized structure with the B3LYP/cc-pvdz basis set.Preparation of 1
A mixture of 1,8-dichloroanthraquinone (847 mg, 3.06 mmol), toluidine (974 mg, 9.09 mmol) and CsCO3 (3.90 g, 20.2 mmol) in dry toluene (45 mL) was degassed by bubbling with N2. Pd2(dba)3 (109 mg, 119 μmol) and (±)-BINAP (327 mg, 525 μmol) were added at rt. The mixture was stirred at 110 °C for 38 h. The reaction was cooled to rt, and the solvent was removed under reduced pressure. The obtained product was dissolved in CH2Cl2 and H2O. The aqueous layer was separated and extracted with CH2Cl2. The combined organic layer was dried over MgSO4. After evaporation of the solvent under reduced pressure, the obtained crude product was purified by silica gel column chromatography (toluene) to give 1 (1.26 g, 98%) as a dark purple solid. The spectroscopic data were identical to the reported values.58Preparation of 2 and 5
A mixture of 1,8-dichloroanthraquinone (850 mg, 3.07 mmol), N,N-dimethyl-1,4-phenylenediamine (1.15 mL, 9.20 mmol) and CsCO3 (4.14 g, 21.5 mmol) in dry toluene (40 mL) was degassed by bubbling with N2. Pd2(dba)3 (140 mg, 153 μmol) and (±)-BINAP (382 mg, 613 μmol) were added at rt. The mixture was stirred at 110 °C for 36 h. The reaction was cooled to rt, and the mixture was diluted with CHCl3 and H2O. The aqueous layer was separated and extracted with CHCl3. The combined organic layer was washed with brine and dried over MgSO4. After evaporation of the solvent under reduced pressure, the obtained crude product was purified by silica gel column chromatography (CHCl3), followed by GPC to give 2 (908 mg, 62%) and 5 (242 mg, 21%) as a dark blue solid and a dark purple solid, respectively.Preparation of 3 52
A mixture of 1,8-dichloroanthraquinone (5.01 g, 18.1 mmol), p-toluenesulfonamide (9.20 g, 53.7 mmol), copper(II) acetate monohydrate (250 mg, 1.25 mmol) and potassium acetate (4.00 g, 40.8 mmol) in nitrobenzene (50 mL) was stirred at 200 °C under air for 14.5 h. After cooling to rt, the resulting mixture was diluted with water and CHCl3. The aqueous layer was separated and extracted with CHCl3. The combined organic layers were washed with brine and dried over MgSO4. After evaporation of the solvent under reduced pressure, the obtained crude product was recrystallized from benzene to give compound 3 (6.16 g, 62%) as a yellow solid.Mp: 274–275 °C. 1H NMR (400 MHz): δ 11.8 (s, 2H), 8.02 (dd, J = 8.5, 1.2 Hz, 2H), 7.92 (dd, J = 7.5, 1.2 Hz, 2H), 7.86 (d, J = 8.5 Hz, 4H), 7.65 (br t, J = 8.1 Hz, 2H), 7.30 (d, J = 8.1 Hz, 4H), 2.38 (s, 6H). 13C NMR (100 MHz): δ 190.6, 181.5, 144.6, 141.5, 136.2, 135.9, 133.6, 130.0, 127.3, 123.9, 122.3, 118.0, 21.6 IR: 3129, 1668, 1619, 1596, 1575, 1483, 1459, 1386, 1343, 1299, 1263, 1236, 1160, 1090, 993, 886, 863, 844, 812, 743, 718, 664, 563, 544, 513, 464 cm−1. HRMS (FAB) calcd for C28H23N2O6S2: 547.0998 [(M + H)+]; found: 547.0992. Anal. calcd for C28H22N2O6S2: C, 61.52 H, 4.06; N, 5.12. Found: C, 61.49 H, 4.21; N, 5.07.
Preparation of 4
A mixture of 1,8-dichloroanthraquinone (1.11 g, 4.00 mmol), 4-(dimethylamino)benzenesulfonamide (2.00 g, 9.99 mmol),65 copper(II) acetate monohydrate (96.1 mg, 481 μmol) and potassium acetate (995 mg, 10.1 mmol) in nitrobenzene (40 mL) was stirred at 200 °C under air for 16 h. After cooling to rt, the resulting mixture was diluted with water and CHCl3. The aqueous layer was separated and extracted with CHCl3. The combined organic layers were washed with brine and dried over MgSO4. After evaporation of the solvent under reduced pressure, the obtained crude product was suspended in toluene/EtOH (1/1, ∼100 mL) and the insoluble material was collected by filtration. The obtained solid was repeatedly recrystallized from toluene/EtOH to give compound 4 (1.03 g, 43%) as a dark orange solid.Mp: 278–280 °C. 1H NMR (400 MHz): δ 11.72 (s, 2H), 8.03 (dd, J = 8.5, 1.2 Hz, 2H), 7.88 (dd, J = 7.6, 1.2 Hz, 2H), 7.78 (d, J = 9.2 Hz, 4H), 7.63 (t, J = 8.0 Hz, 2H), 6.62 (d, J = 9.2 Hz, 4H), 3.01 (s, 12H). 13C NMR (100 MHz): δ 190.4, 181.9, 153.2, 141.9, 135.6, 133.6, 129.2, 123.9, 123.8, 121.8, 117.9, 110.9, 40.0. IR: 3112, 2904, 2822, 1672, 1618, 1599, 1552, 1522, 1476, 1455, 1380, 1344, 1298, 1264, 1222, 1148, 1089, 989, 944, 873, 852, 815, 70, 749, 715, 701, 674, 661, 643, 557, 544, 516, 460 cm−1. HRMS (FAB) calcd for C30H28N4O6S2Na: 627.1348 [(M + Na)+]; found: 627.1350. Anal. calcd for C30H28N4O6S2·0.5(toluene): C, 61.83 H, 4.96; N, 8.61. Found: C, 61.90 H, 4.95; N8.60.
Preparation of 6 55
To a suspension of NaH (60% in oil, 333 mg, 8.33 mmol) in dry DMF (20 mL) was added 3 (303 mg, 554 μmol) under N2. The mixture was stirred at rt for 45 min. To the resulting solution was added MeI (700 μL, 11.0 mmol) at this temperature. The mixture was stirred at rt for 12 h. The mixture was diluted with an excess amount of water. The resulting precipitate was collected by filtration, washed with water, and then dried under vacuum. Recrystallization from EtOH gave 6 (171 mg, 54%) as an orange crystalline solid.Mp: 236–237 °C. 1H NMR (400 MHz): δ 8.24 (dd, J = 7.8, 1.3 Hz, 2H), 7.70 (d, J = 7.8 Hz, 4H), 7.63 (t, J = 7.6 Hz, 2H), 7.47 (dd, J = 7.8, 1.3 Hz, 2H), 7.31 (d, J = 7.8 Hz, 4H), 3.24 (s, 6H), 2.43 (s, 6H). 13C NMR (100 MHz): δ 182.6, 182.3, 143.5, 140.3, 136.0, 134.6, 133.6, 133.0, 129.6, 127.9, 126.9, 38.8, 21.6. IR: 3072, 2948, 2919, 1680, 1585, 1495, 1461, 1436, 1400, 1345, 1318, 1249, 1216, 1188, 1150, 1089, 1020, 1011, 957, 871, 850, 802, 752, 730, 688, 675, 651, 581, 563, 545, 517 cm−1. HRMS (FAB) calcd for C30H27N2O6S2: 575.1311 [(M + H)+]; found: 575.1309. Anal. calcd for C30H26N2O6S2: C, 62.70 H, 4.56; N, 4.87. Found: C, 62.55 H, 4.73; N, 4.78.
Preparation of 8 and 11
A mixture of 1,4-dichloroanthraquinone (854 mg, 3.08 mmol), N,N-dimethyl-1,4-phenylenediamine (1.25 mL, 10.0 mmol) and CsCO3 (4.34 g, 22.5 mmol) in dry toluene (40 mL) was degassed by bubbling with N2. Pd2(dba)3 (140 mg, 153 μmol) and (±)-BINAP (402 mg, 646 μmol) were added at rt. The mixture was stirred at 110 °C for 47 h. The reaction was cooled to rt, and the mixture was diluted with CHCl3 and H2O. The aqueous layer was separated and extracted with CHCl3. The combined organic layer was washed with brine and dried over MgSO4. After evaporation of the solvent under reduced pressure, the obtained crude product was purified by silica gel column chromatography (CHCl3), followed by recrystallization (EtOH for 8, EtOH/CHCl3 for 11) to give 8 (234 mg, 16%) and 11 (742 mg, 64%) as a dark green solid and a dark purple solid, respectively.Preparation of 9 52
A mixture of 1,4-dichloroanthraquinone (5.00 g, 18.1 mmol), p-toluenesulfonamide (9.20 g, 53.7 mmol), copper(II) acetate monohydrate (250 mg, 1.25 mmol), and potassium acetate (4.00 g, 40.8 mmol) in nitrobenzene (50 mL) was stirred at 200 °C under air for 10 h. After cooling to rt, the resulting mixture was diluted with water and CHCl3. The aqueous layer was separated and extracted with CHCl3. The combined organic layers were washed with brine and dried over MgSO4. The obtained crude solid was washed with a small amount of CHCl3 and then recrystallized from CHCl3 to give compound 9 (2.04 g) as a red solid. Combined filtrate was subjected to silica gel column chromatography (CHCl3) to give an additional amount of compound 9 (5.18 g). Total yield: 7.22 g (73%).Mp: 245–246 °C. 1H NMR (400 MHz): δ 12.3 (s, 2H), 8.22 (dd, J = 9.2, 5.9 Hz, 2H), 7.97 (s, 2H), 7.80 (dd, J = 9.2, 5.9 Hz, 2H), 7.76 (d, J = 8.2 Hz, 4H), 7.24 (d, J = 8.2 Hz, 4H), 2.36 (s, 6H). 13C NMR (100 MHz): δ 186.2, 144.5, 137.4, 136.3, 134.8, 132.9, 129.9, 127.3, 127.2, 126.3, 117.1, 21.5. IR: 3019, 1639, 1590, 1475, 1379, 1352, 1287, 1254, 1186, 1164, 1090, 1072, 971, 920, 896, 859, 839, 818, 726, 714, 656, 579, 558, 544 cm−1. HRMS (FAB) calcd for C28H22N2O6S2Na: 569.0817 [(M + Na)+]; found: 569.0817. Anal. calcd for C28H22N2O6S2: C, 61.52 H, 4.06; N, 5.12. Found: C, 61.60 H, 4.31; N,5.05.
Preparation of 10
Mp: 287–288 °C. 1H NMR (400 MHz): δ 12.25 (s, 2H), 8.24 (dd, J = 5.9, 3.3 Hz, 2H), 7.95 (s, 2H), 7.79 (dd, J = 5.9, 3.3 Hz, 2H), 7.69 (d, J = 9.1 Hz, 4H), 6.58 (d, J = 9.1 Hz, 4H), 2.99 (s, 12H). 13C NMR (125 MHz): δ 186.2, 153.1, 137.7, 134.5, 133.1, 129.2, 127.2, 126.2, 123.9, 116.6, 110.9, 40.0. IR: 3448, 3098, 2896, 1639, 1597, 1552, 1521, 1473, 1449, 1375, 1349, 1326, 1286, 1253, 1206, 1153, 1091, 1069, 998, 970, 919, 892, 836, 824, 816, 794, 773, 728, 708, 642, 558, 544 cm−1. HRMS (FAB) calcd for C30H28N4O6S2Na: 627.1348 [(M + Na)+]; found: 627.1347. Anal. calcd for C30H28N4O6S2·0.33 toluene: C, 61.12 H, 4.86; N, 8.82. Found: C, 61.22 H, 5.06; N, 8.86.
Preparation of 12 55
A mixture of 9 (200 mg, 366 μmol), K2CO3 (506 mg, 3.66 mmol) and MeI (455 μL, 7.32 mmol) in DMF (10 mL) was stirred at 50 °C for 13.5 h in a sealed glass pressure vessel. After cooling to rt, the mixture was diluted with H2O. The resulting precipitate was collected by filtration, washed with H2O, and then dried under vacuum. Recrystallization from CHCl3/EtOH gave 12 (195 mg, 93%) as a yellow solid.Mp: 228–229 °C (decomp.). 1H NMR (400 MHz, 50 °C): δ 7.92 (br s, 2H), 7.68 (dd, J = 5.7, 3.2 Hz, 2H), 7.57 (t, J = 8.2 Hz, 4H), 7.42 (s, 2H), 7.23 (d, J = 8.2 Hz, 4H), 3.35 (s, 6H), 2.39 (s, 6H). 13C NMR (125 MHz, 50 °C): δ 182.5, 143.5, 140.3, 136.5, 136.4, 134.2, 134.1, 133.6, 129.6, 127.7, 126.6, 38.8, 21.5. IR: 1670, 1590, 1463, 1348, 1322, 1305, 1254, 1201, 1156, 1087, 931, 910, 877, 809, 795, 727, 713, 679, 636, 596, 549, 516 cm−1. HRMS (FAB) calcd for C30H27N2O6S2: 575.1311 [(M + H)+]; found: 575.1310. Anal. calcd for C30H26N2O6S2: C, 62.70 H, 4.56; N, 4.87. Found: C, 62.88 H, 4.57; N, 4.82.
Preparation of 13
A mixture of 1,5-dichloroanthraquinone (845 mg, 3.05 mmol), 4-tert-butylaniline (1.45 mL, 9.13 mmol) and CsCO3 (3.91 g, 20.3 mmol) in dry toluene (40 mL) was degassed by bubbling with N2. Pd2(dba)3 (117 mg, 127 μmol) and (±)-BINAP (333 mg, 535 μmol) were added at rt. The mixture was stirred at 110 °C for 38 h. The reaction was cooled to rt, and the resulting solid was collected by filtration and washed with toluene to give 13 (1.29 g, 84%) as a dark purple crystalline solid. The spectroscopic data were identical to the reported values.58Preparation of 14 52
A mixture of 1,5-dichloroanthraquinone (4.71 g, 17.0 mmol), 4-tert-butylbenzenesulfonamide (10.0 g, 46.9 mmol), copper(II) acetate monohydrate (218 mg, 1.09 mmol), and potassium acetate (3.49 g, 35.6 mmol) in nitrobenzene (50 mL) was stirred at 200 °C under air for 11.5 h. After cooling to rt, the resulting mixture was diluted with water and CHCl3. The aqueous layer was separated and extracted with CHCl3. The combined organic layers were washed with brine and dried over MgSO4. After evaporation of the solvent under reduced pressure, the obtained solid was subjected to silica gel column chromatography (CHCl3) followed by recrystallization from toluene to give compound 14 as a yellow solid (6.82 g, 63%).Mp: >300 °C. 1H NMR (400 MHz): δ 12.1 (s, 2H), 7.99 (dd, J = 8.5, 1.1 Hz, 2H), 7.96 (dd, J = 7.7, 1.1 Hz, 2H), 7.87–7.83 (m, 4H), 7.68 (t, J = 8.2 Hz), 7.49–7.44 (m, 4H), 1.26 (s, 18H). 13C NMR (100 MHz): δ 185.8, 157.4, 141.3, 136.2, 135.8, 134.5, 127.1, 126.3, 123.4, 122.5, 116.9, 35.2, 30.9. IR: 3080, 2965, 1637, 1593, 1475, 1385, 1348, 1264, 1199, 1163, 1113, 1086, 1055, 889, 835, 770, 753, 711, 654, 624, 570, 548 cm−1. HRMS (FAB) calcd for C34H35N2O6S2: 631.1937 [(M + H)+]; found: 631.1937. Anal. calcd for C34H34N2O6S2: C, 64.74 H, 5.43; N, 4.44. Found: C, 64.90 H, 5.49; N, 4.37.
Preparation of 4-(dibutylamino)benzenesulfonyl chloride
A mixture of N,N-dibutylaniline (8.47 g, 41.2 mmol) and bis(trimethylsilyl) sulfate (10.0 g, 41.2 mmol) was stirred at 170 °C under a nitrogen atmosphere for 4 h. After cooling to rt, the resulting solid was washed with ether and H2O, and then dried under vacuum to give 4-(dibutylamino)benzenesulfonic acid, which was used for the next reaction directly without further purification. To the obtained 4-(dibutylamino)benzenesulfonic acid in dry CH2Cl2 (110 mL) was added PCl5 (8.59 g, 41.2 mmol) at 0 °C. The mixture was warmed gradually to rt and stirred for 24 h under a nitrogen atmosphere. The solvent was removed under reduced pressure and the product was dissolved in ether. The solution was washed with H2O and dried over MgSO4. After evaporation of the solvent under reduced pressure, the crude product was subjected to silica gel column chromatography (hexane/ether = 100/20) to give 4-(dibutylamino)benzenesulfonyl chloride (8.19 g, 65% in two steps) as a yellow oil.1H NMR (400 MHz): δ 7.79 (d, J = 9.4 Hz, 2H), 6.62 (d, J = 9.4 Hz, 2H), 3.36 (t, J = 7.8 Hz, 4H), 1.65–1.55 (m, 4H), 1.38 (sext, J = 7.5 Hz, 4H) 0.98 (t, J = 7.3 Hz, 6H). 13C NMR (100 MHz): δ 152.6, 129.6, 128.4, 110.3, 50.9, 29.0, 20.2, 13.9. IR: 2959, 2933, 2873, 1591, 1516, 1467, 1409, 1368, 1314, 1292, 1165, 1086, 992, 926, 814, 655, 559, 542, 501 cm−1. HRMS (EI) calcd for C14H22ClNO2S: 303.1060 [(M + H)+]; found: 303.1062.
Preparation of 15
A mixture of 1,5-diaminoanthraquinone (991 mg, 4.16 mmol) and 4-(dibutylamino)benzenesulfonyl chloride (2.65 g, 8.74 mmol) in dry pyridine (50 mL) was stirred at 70 °C for 45 h. After cooling to rt, the solvent was removed under reduced pressure. The resulting mixture was dissolved in CHCl3 and the solution was washed with brine and dried over MgSO4. After evaporation of the solvent under reduced pressure, the obtained crude product was recrystallized from toluene/EtOH to give 15 (2.12 g, 66%) as a red-brown solid.Mp: 256–257 °C. 1H NMR (400 MHz): δ 11.97 (s, 2H), 8.01 (dd, J = 8.5, 1.1 Hz, 2H), 7.93 (dd, J = 7.7, 1.1 Hz, 2H), 7.69 (d, J = 9.2 Hz, 4H), 7.66 (sext, J = 8.0 Hz, 2H), 6.52 (d, J = 9.2 Hz, 4H), 3.24 (t, J = 7.8 Hz, 8H), 1.55–1.46 (m, 8H), 1.31 (sext, J = 7.6 Hz, 8H), 0.92 (t, J = 7.3 Hz, 12H). 13C NMR (100 MHz): δ 185.8, 151.4, 141.8, 135.6, 134.6, 129.3, 123.1, 121.8, 116.6, 110.4, 50.7, 29.0, 20.2, 13.9. IR: 3455, 3094, 2957, 2930, 2871, 1637, 1593, 1509, 1474, 1379, 1348, 1261, 1228, 1207, 1151, 1092, 1054, 995, 929, 917, 881, 825, 813, 774, 734, 715, 707, 654, 608, 566, 550, 514, 443, 418 cm−1. HRMS (FAB) calcd for C42H52N4O6S2Na: 795.3226 [(M + Na)+]; found: 795.3228. Anal. calcd for C42H52N4O6S2·toluene: C, 65.26 H, 6.78; N, 7.25. Found: C, 65.42 H, 6.82; N, 7.16.
Preparation of 16
A mixture of 1,5-dichloroanthraquinone (850 mg, 3.07 mmol), N,N-dimethyl-1,4-phenylenediamine (1.25 mL, 10.0 mmol) and CsCO3 (4.14 g, 21.5 mmol) in dry toluene (40 mL) was degassed by bubbling with N2. Pd2(dba)3 (176 mg, 192 μmol) and (±)-BINAP (470 mg, 755 μmol) were added at rt. The mixture was stirred at 110 °C for 42 h. The reaction was cooled to rt, and the resulting mixture was diluted with water and CHCl3. The aqueous layer was separated and extracted with CHCl3. The combined organic layers were washed with brine and dried over MgSO4. After evaporation of the solvent under reduced pressure, the resulting purple solid was subjected to silica gel column chromatography (CHCl3) to give 16 (694 mg, 61%) as a dark purple crystalline solid.Mp: 213–214 °C. 1H NMR (400 MHz): δ 11.04 (s, 1H), 8.34 (dd, J = 7.7, 1.4 Hz, 1H), 7.74 (dd, J = 8.0, 1.4 Hz, 1H), 7.66 (t, J = 7.9 Hz, 1H), 7.60 (dd, J = 7.3, 1.1 Hz, 1H), 7.44 (dd, J = 8.5, 7.5 Hz, 1H), 7.17 (d, J = 8.8 Hz, 2H), 6.78 (d, J = 8.8 Hz, 2H), 2.99 (s, 6H). 13C NMR (100 MHz): δ 183.3, 182.5, 150.8, 148.9, 137.6, 136.5, 135.6, 135.2, 134.5, 133.4, 129.4, 127.8, 126.7, 126.3, 119.4, 117.2, 113.2, 112.5, 40.7. IR: 1665, 1627, 1591, 1577, 1566, 1522, 1500, 1308, 1258, 1224, 1193, 1158, 808, 760, 709 cm−1. HRMS (EI) calcd for C22H17N2O2Cl: 376.0979 (M+); found: 376.0976. Anal. calcd for C22H17N2O2Cl: C, 70.12 H, 4.55; N, 7.43. Found: C, 70.02 H, 4.58; N, 7.34.
Preparation of 17
A mixture of 1,5-dichloroanthraquinone (1.17 g, 4.22 mmol), 4-(dimethylamino)benzenesulfonamide (2.00 g, 10.0 mmol), copper(II) acetate monohydrate (125 mg, 626 μmol), and potassium acetate (900 mg, 9.15 mmol) in nitrobenzene (80 mL) was stirred at 200 °C under air for 21.5 h. After cooling to rt, the resulting mixture was diluted with water and CHCl3. The aqueous layer was separated and extracted with CHCl3. The combined organic layers were washed with brine and dried over MgSO4. After evaporation of the solvent under reduced pressure, the obtained brown solid was recrystallized from toluene/EtOH to remove insoluble material. After evaporation of the solvent of the filtrate under reduced pressure, the obtained solid was suspended in CHCl3 and filtered. The filtrate was subjected to silica gel column chromatography (CHCl3) to give compound 17 as an orange solid (399 mg, 21%).Mp: 245–247 °C. 1H NMR (400 MHz): δ 11.85 (s, 1H), 8.30 (dd, J = 7.8, 1.4 Hz, 1H), 7.99 (dd, J = 8.5, 1.1 Hz, 1H), 7.91 (dd, J = 7.7, 1.1 Hz, 1H), 7.79 (dd, J = 8.0, 1.4 Hz, 1H), 7.76 (d, J = 9.2 Hz, 2H), 7.69 (t, J = 7.8 Hz, 1H), 7.66 (d, J = 8.1 Hz, 1H), 6.59 (d, J = 9.2 Hz, 2H), 2.99 (s, 6H). 13C NMR (100 MHz): δ 185.4, 181.2, 153.1, 141.4, 137.8, 136.3, 135.8, 135.3, 134.9, 133.8, 129.2, 129.1, 127.0, 123.9, 122.7, 122.1, 116.6, 110.8, 40.0. IR: 3097, 1675, 1636, 1598, 1573, 1553, 1523, 1477, 1468, 1447, 1380, 1340, 1322, 1285, 1268, 1208, 1197, 1149, 1093, 1071, 1036, 997, 935, 909, 846, 839, 812, 759, 708, 644, 557, 544, 431 cm−1. HRMS (FAB) calcd for C22H17N2O4SClNa: 463.0495 [(M + Na)+]; found: 463.0491. Anal. calcd for C22H17N2O4SCl: C, 59.93 H, 3.89; N, 6.35. Found: C, 59.79 H, 3.82; N, 6.23.
Preparation of 18 55
To a suspension of NaH (60% in oil, 380 mg, 9.51 mmol) in dry DMF (30 mL) was added 14 (400 mg, 634 μmol) under N2. The mixture was stirred at rt for 45 min. To the resulting solution was added MeI (790 μL, 12.7 mmol) at this temperature. The mixture was stirred at rt for 16.5 h. The mixture was diluted with an excess amount of water. The resulting precipitate was collected by filtration, washed with water, and then dried under vacuum. Recrystallization from CHCl3/EtOH gave 18 (177 mg, 42%) as a yellow solid.Mp: 268–270 °C (decomp.). 1H NMR (400 MHz, 50 °C): δ 8.08 (br d, J = 7.8 Hz, 2H), 7.65(t, J = 7.8 Hz, 2H), 7.63 (d, J = 8.5 Hz, 4H), 7.50 (d, J = 7.8 Hz, 2H), 7.44 (d, J = 8.5 Hz, 4H), 3.35 (s, 6H), 1.33 (s, 18H). 13C NMR (125 MHz, 50 °C): δ 181.8, 156.4, 140.0, 137.0, 136.8, 136.4, 133.9, 127.9, 127.5, 125.8, 36.7, 35.2, 31.1. IR: 3076, 2957, 1675, 1596, 1580, 1457, 1438, 1400, 1349, 1310, 1267, 1190, 1158, 1112, 1085, 1018, 877, 825, 807, 773, 756, 710, 626, 581, 552 cm−1. HRMS (FAB) calcd for C36H39N2O6S2: 659.2250 [(M + H)+]; found: 659.2250. Anal. calcd for C36H38N2O6S2: C, 65.60 H, 5.81; N, 4.25. Found: C, 62.60 H, 5.86; N, 4.22.
Preparation of 19
A mixture of 1,4,5,8-tetrachloroanthraquinone (1.04 g, 3.01 mmol), 4-tert-butylaniline (2.90 mL, 18.1 mmol) and CsCO3 (7.80 g, 40.8 mmol) in dry toluene (50 mL) was degassed by bubbling with N2. Pd2(dba)3 (110 mg, 120 μmol) and (±)-BINAP (330 mg, 530 μmol) were added at rt. The mixture was stirred at 110 °C for 49 h. The reaction was cooled to rt, and the resulting crystalline solid was collected by filtration, washed with toluene and H2O, and dried under vacuum to give 19 (2.06 g, 86%) as a dark green crystalline solid. The spectroscopic data were identical to the reported values.59Preparation of 20 52
A mixture of 1,4,5,8-tetrachloroanthraquinone (3.00 g, 8.67 mmol), 4-tert-butylbenzenesulfonamide (10.0 g, 46.9 mmol), copper(II) acetate monohydrate (277 mg, 1.39 mmol), and potassium acetate (3.57 g, 36.4 mmol) in nitrobenzene (50 mL) was stirred at 200 °C under air for 18.5 h. After cooling to rt, the resulting mixture was diluted with water and CHCl3. The aqueous layer was separated and extracted with CHCl3. The combined organic layers were washed with brine and dried over MgSO4. The obtained crude solid was subjected to silica gel column chromatography (CHCl3) followed by recrystallization from toluene to give compound 20 as a dark purple solid (5.11 g, 56%).Mp: decomposed above 270 °C. 1H NMR (400 MHz): δ 11.8 (s, 4H), 7.97 (s, 4H), 7.75 (d, J = 8.7 Hz, 8H), 7.50 (d, J = 8.7 Hz, 8H), 1.28 (s, 36H). 13C NMR (100 MHz): δ 188.6, 157.8, 137.4, 136.0, 127.1, 126.6, 126.5, 116.9, 35.3, 30.9. IR: 2964, 1595, 1479, 1377, 1221, 1198, 1167, 1112, 1084, 981, 862, 837, 791, 755, 647, 622, 571, 546 cm−1. HRMS (FAB) calcd for C54H60N4O10S4Na: 1075.3090 [(M + Na)+]; found: 1075.3085. Anal. calcd for C54H60N4O10S4: C, 61.57 H, 5.74; N, 5.32. Found: C, 61.81 H, 5.88; N, 5.24.
Preparation of 4-(dihexylamino)benzenesulfonyl chloride
A mixture of N,N-dihexylaniline (10.8 g, 41.3 mmol) and bis(trimethylsilyl) sulfate (10.0 g, 41.2 mmol) was stirred at 170 °C under a nitrogen atmosphere for 3 h. After cooling to rt, the resulting solid was washed with ether and the dried under vacuum to give 4-(dihexylamino)benzenesulfonic acid, which was used for the next reaction directly without further purification. To the obtained 4-(dihexylamino)benzenesulfonic acid in dry CH2Cl2 (110 mL) was added PCl5 (8.73 g, 41.9 mmol) at 0 °C. The mixture was warmed gradually to rt and stirred for 41 h under a nitrogen atmosphere. The solvent was removed under reduced pressure and the product was dissolved in ether. The solution was washed with H2O and dried over MgSO4. After evaporation of the solvent under reduced pressure, the crude product was subjected to silica gel column chromatography (hexane/ether = 100/20) to give 4-(dihexylamino)benzenesulfonyl chloride (9.40 g, 63% in two steps) as a yellow oil.1H NMR (400 MHz): δ 7.79 (d, J = 9.3 Hz, 2H), 6.61 (d, J = 9.3 Hz, 2H), 3.34 (t, J = 7.8 Hz, 4H), 1.67–1.57 (m, 4H), 1.38–1.29 (m, 4H), 0.91 (t, J = 6.8 Hz, 6H). 13C NMR (125 MHz): δ 152.6, 129.6, 128.6, 110.4, 51.2, 31.6, 26.9, 26.7, 22.6, 14.0. IR: 2957, 2931, 2859, 1591, 1516, 1469, 1409, 1369, 1164, 1087, 814, 656, 561 cm−1. HRMS (EI) calcd for C18H30NO2SCl: 359.1686 (M+); found: 359.1685.
Preparation of 21
To a solution of 1,4,5,8-tetraaminoanthraquinone (Disperse Blue 1, Aldrich, 1.45 g, 5.39 mmol) in dry pyridine (100 mL) was added 4-(dihexylamino)benzenesulfonyl chloride (9.40 g, 24.2 mmol). The mixture was stirred at rt for 77 h under a nitrogen atmosphere. After removal of the solvent under reduced pressure, the resulting crude product was purified by silica gel column chromatography (PhH/AcOEt = 100/0 → 100/4), and recrystallized from toluene/EtOH to give 21 (1.47 g, 18%) as a dark purple solid.Mp: 116–118 °C. 1H NMR (400 MHz): δ 11.87 (s, 4H), 7.95 (s, 4H), 7.67 (d, J = 9.1 Hz, 4H), 6.54 (d, J = 9.1 Hz, 4H), 1,60–1.46 (m, 16H), 1.37–1.25 (m, 16H) 0.89 (t, J = 6.7 Hz, 24 H). 13C NMR (125 MHz): δ 188.6, 151.5, 137.5, 129.3, 126.1, 122.6, 116.4, 110.6, 51.0, 31.6, 26.9, 26.6, 22.6, 14.0. IR: 3119, 2954, 2929, 2857, 1594, 1554, 1514, 1479, 1405, 1367, 1255, 1217, 1156, 1092, 997, 978, 858, 816, 789, 659, 558 cm−1. HRMS (FAB) calcd for C86H128N8O10S4Na: 1583.8534 [(M + Na)+]; found: 1583.8536. Anal. Calcd for C86H128N8O10S4·toluene: C, 66.12 H, 8.26; N, 7.17. Found: C, 66.12 H, 8.40; N, 7.09.
Preparation of 22
A mixture of 20 (4.50 g, 4.27 mmol) and conc. H2SO4 (100 mL) was stirred at 100 °C. After cooling to rt, the mixture was poured into ice water (ca. 600 mL). The mixture was basified with NaOH and the resulting precipitate was collected by filtration, washed with H2O and dried under vacuum to give a blue solid (5.36 g), which was used for the next reaction without further purification. A mixture of the product, 4-(dihexylamino)benzenesulfonyl chloride (5.50 g, 18.1 mmol) in pyridine was stirred at 70 °C for 40 h. After cooling to rt, the solvent was removed under reduced pressure. The product was dissolved in CHCl3. The solution was washed with brine and dried over MgSO4. After evaporation of the solvent under reduced pressure, the resulting blue solid was purified by silica gel column chromatography (PhH/AcOEt = 9/1), followed by recrystallization from toluene/EtOH to give pure 22 (494 mg, 14% in two steps) as a dark purple solid.Mp: 230–231 °C. 1H NMR (400 MHz): δ 13.11 (s, 2H), 7.695 (s, 2H), 7.695 (d, J = 9.2 Hz, 4H), 7.20–6.85 (br s, 4H), 6.63 (s, 2H), 6.55 (d, J = 9.1 Hz, 4H), 3.25 (t, J = 7.6 Hz, 8H), 1.59–1.47 (m, 8H), 1.33 (hext, J = 7.4 Hz, 8H), 0.93 (t, J = 7.4 Hz, 12H). 13C NMR (125 MHz): δ 185.0, 151.2, 146.1, 136.3, 129.2, 128.4, 123.8, 123.1, 119.0, 110.5, 108.8, 50.7, 29.1, 20.2, 13.9. IR: 3440, 3290, 2957, 2931, 2870, 1650, 1601, 1571, 1539, 1515, 1472, 1451, 1408, 1381, 1349, 1318, 1298, 1251, 1204, 1179, 1146, 1092, 995, 983, 926, 900, 873, 847, 821, 776, 720, 656, 561, 516, 507, 490, 441 cm−1. HRMS (FAB) calcd for C42H54N6O6S2Na: 825.3444 [(M + Na)+]; found: 825.3450. Anal. calcd for C42H54N6O6S2: C, 62.82 H, 6.78; N, 10.47. Found: C, 62.83 H, 6.50; N, 10.43.
Preparation of 23 55
To a suspension of NaH (60% in oil, 304 mg, 7.60 mmol) in dry DMF (30 mL) was added 20 (400 mg, 380 μmol) under N2. The mixture was stirred at rt for 45 min. To the resulting solution was added MeI (710 μL, 11.4 mmol) at this temperature. The mixture was stirred at rt for 17 h. The mixture was diluted with an excess amount of water. The resulting precipitate was collected by filtration, washed with water, and then dried under vacuum. Recrystallization from CHCl3/EtOH gave 23 (264 mg, 63%) as a yellow solid.Mp: 253–255 °C (decomp.). 1H NMR (400 MHz): δ 7.76 (d, J = 8.6 Hz, 8H), 7.56 (d, J = 8.6 Hz, 8H), 7.25 (s, 4H), 3.20 (s, 12H), 1.37 (s, 36H). 13C NMR (125 MHz): δ 181.8, 156.8, 139.2, 136.0, 135.0, 134.5, 127.9, 126.0, 39.2, 35.2, 31.2. IR: 2964, 2871, 1702, 1595, 1477, 1397, 1349, 1333, 1307, 1269, 1217, 1160, 1111, 1085, 1042, 936, 888, 865, 842, 812, 793, 762, 671, 630, 601, 580, 547 cm−1. HRMS (FAB) calcd for C58H69N4O10S4: 1109.3897 [(M + H)+]; found: 1109.3899. Anal. calcd for C58H68N4O10S4: C, 62.79 H, 6.18; N, 5.05. Found: C, 62.57 H, 6.13; N, 5.00.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partly supported by JSPS KAKENHI Grant numbers JP17K05769, JP20H04655 and JP20K05442, the Murata Science Foundation, 2020 Mitsui Chemical Award in Synthetic Organic Chemistry, Japan, Tobe Maki Scholarship Foundation, Iketani Science and Technology Foundation, Yamaguchi Educational and Scholarship Foundation, and the research program “Dynamic Alliance for Open Innovation Bridging Human, Environment and Materials” from MEXT, Japan.Notes and references
- J. Fabian, H. Nakazumi and M. Matsuoka, Chem. Rev., 1992, 92, 1197–1226 CrossRef CAS.
- J. Fabian and R. Zahradník, Angew. Chem., Int. Ed. Engl., 1989, 28, 677–694 CrossRef.
- G. Quan and Z. Y. Wang, Chem.–Asian J., 2010, 5, 1006–1029 CrossRef.
- L. Li, X. Dong, J. Li and J. Wei, Dyes Pigm., 2020, 183, 108756 CrossRef CAS.
- Z. Y. Wang, Near-Infrared Organic Materials and Emerging Applications, CRC Press, Boca Raton, FL, 2013 Search PubMed.
- M. Matsuoka, Infrared Absorbing Dyes, Plenum Press, New York, 1990 Search PubMed.
- J. Roncali, P. Leriche and P. Blanchard, Adv. Mater., 2014, 26, 3821–3838 CrossRef CAS PubMed.
- Y. Yang, R. T. Farley, T. T. Steckler, S.-H. Eom, J. R. Reynolds, K. S. Schanze and J. Xue, J. Appl. Phys., 2009, 106, 044509 CrossRef.
- O. Fenwick, J. K. Sprafke, J. Binas, D. V. Kondratuk, F. D. Stasio, H. L. Anderson and F. Cacialli, Nano Lett., 2011, 11, 2451–2456 CrossRef CAS PubMed.
- C. Wang, X.-L. Li, Y. Gao, L. Wang, S. Zhang, L. Zhao, P. Lu, B. Yang, S.-J. Su and Y. Ma, Adv. Opt. Mater., 2017, 5, 1700441 CrossRef.
- A. Zampetti, A. Minotto, B. M. Squeo, V. G. Gregoriou, S. Allard, U. Scherf, C. L. Chochos and F. Cacialli, Sci. Rep., 2017, 7, 1611 CrossRef PubMed.
- M. Emmelius, G. Pawlowski and H. W. Vollmann, Angew. Chem., Int. Ed. Engl., 1989, 28, 1445–1471 CrossRef.
- S. Luo, E. Zhang, Y. Su, T. Cheng and C. Shi, Biomaterials, 2011, 32, 7127–7138 CrossRef CAS.
- P. C. A. Swamy, G. Sivaraman, R. N. Priyanka, S. O. Raja, K. Ponnuvel, J. Shanmugpriya and A. Gulyani, Coord. Chem. Rev., 2020, 411, 213233 CrossRef.
- A. Yuan, J. Wu, X. Tang, L. Zhao, F. Xu and Y. Hu, J. Pharm. Sci., 2013, 102, 6–28 CrossRef CAS PubMed.
- A. Tsuda and A. Osuka, Science, 2001, 293, 79–82 CrossRef CAS PubMed.
- A. Tsuda, H. Furuta and A. Osuka, Angew. Chem., Int. Ed., 2000, 39, 2549–2552 CrossRef CAS PubMed.
- A. Tsuda, H. Furuta and A. Osuka, J. Am. Chem. Soc., 2001, 123, 10304–10321 CrossRef CAS.
- Y. Nakamura, S. Y. Jang, T. Tanaka, N. Aratani, J. M. Lim, K. S. Kim, D. Kim and A. Osuka, Chem.–Eur. J., 2008, 14, 8279–8289 CrossRef CAS.
- J. L. Bricks, A. D. Kachkovskii, Y. LSlominskii, A. O. Gerasov and S. V. Popov, Dyes Pigm., 2015, 121, 238–255 CrossRef CAS.
- M. Panigrahi, S. Dash, S. Patel and B. K. Mishra, Tetrahedron, 2012, 68, 781–805 CrossRef CAS.
- A. V. Kulinich and A. A. Ishchenko, Russ. Chem. Rev., 2009, 78, 141–164 CrossRef CAS.
- K. Asai, A. Fukazawa and S. Yamaguchi, Angew. Chem., Int. Ed., 2017, 56, 6848–6852 CrossRef CAS.
- S. Yamazawa, M. Nakashima, Y. Suda, R. Nishiyabu and Y. Kubo, J. Org. Chem., 2016, 81, 1310–1315 CrossRef CAS.
- Y. Ishigaki, T. Harimoto, K. Sugimoto, L. Wu, W. Zeng, D. Ye and T. Suzuki, Chem.–Asian J., 2020, 15, 1147–1155 CrossRef CAS PubMed.
- Y. Ni and J. Wu, Org. Biomol. Chem., 2014, 12, 3774–3791 RSC.
- S. Shimizu, T. Iino, A. Saeki, S. Seki and N. Kobayashi, Chem.–Eur. J., 2015, 21, 2893–2904 CrossRef CAS PubMed.
- M. M. Payne, S. R. Parkin and J. E. Anthony, J. Am. Chem. Soc., 2005, 127, 8028–8029 CrossRef CAS PubMed.
- D. Chun, Y. Cheng and F. Wudl, Angew. Chem., Int. Ed., 2008, 47, 8380–8385 CrossRef CAS PubMed.
- I. Kaur, N. N. Stein, R. P. Kopreski and G. P. Miller, J. Am. Chem. Soc., 2009, 131, 3424–3425 CrossRef CAS PubMed.
- I. Kaur, M. Jazdzyk, N. N. Stein, P. Prusevich and G. P. Miller, J. Am. Chem. Soc., 2010, 132, 1261–1263 CrossRef CAS PubMed.
- Z. Sun, Z. Zeng and J. Wu, Acc. Chem. Res., 2014, 47, 2582–2591 CrossRef CAS PubMed.
- P. Yadav, S. Das, H. Phan, T. S. Herng, J. Ding and J. Wu, Org. Lett., 2016, 18, 2886–2889 CrossRef CAS PubMed.
- R. Huang, H. Phan, T. S. Herng, P. Hu, W. Zeng, S.-q. Dong, S. Das, Y. Shen, J. Ding, D. Casanova and J. Wu, J. Am. Chem. Soc., 2016, 138, 10323–10330 CrossRef CAS PubMed.
- W. Zeng, Z. Sun, T. S. Herng, T. P. Gonçalves, T. Y. Gopalakrishna, K.-W. Huang, J. Ding and J. Wu, Angew. Chem., Int. Ed., 2016, 55, 8615–8619 CrossRef CAS PubMed.
- C. K. Frederickson, B. D. Rose and M. M. Haley, Acc. Chem. Res., 2017, 50, 977–987 CrossRef CAS.
- A. Shimizu, S. Nobusue, H. Miyoshi and Y. Tobe, Pure Appl. Chem., 2014, 86, 517–528 CAS.
- A. Shimizu and Y. Tobe, Angew. Chem., Int. Ed., 2011, 50, 6906–6910 CrossRef CAS PubMed.
- A. Shimizu, R. Kishi, M. Nakano, D. Shiomi, K. Sato, T. Takui, I. Hisaki, M. Miyata and Y. Tobe, Angew. Chem., Int. Ed., 2013, 52, 6076–6079 CrossRef CAS PubMed.
- H. Miyoshi, S. Nobusue, A. Shimizu, I. Hisaki, M. Miyata and Y. Tobe, Chem. Sci., 2014, 5, 163–168 RSC.
- H. Miyoshi, M. Miki, S. Hirano, A. Shimizu, R. Kishi, K. Fukuda, D. Shiomi, K. Sato, T. Takui, I. Hisaki, M. Nakano and Y. Tobe, J. Org. Chem., 2017, 82, 1380–1388 CrossRef CAS.
- J. J. Dressler, Z. Zhou, J. L. Marshall, R. Kishi, S. Takamuku, Z. Wei, S. N. Spisak, M. Nakano, M. A. Petrukhina and M. M. Haley, Angew. Chem., Int. Ed., 2017, 56, 15363–15367 CrossRef CAS PubMed.
- G. E. Rudebusch, J. L. Zafra, K. Jorner, K. Fukuda, J. L. Marshall, I. Arrechea-Marcos, G. L. Espejo, R. P. Ortiz, C. J. Gómez-García, L. N. Zakharov, M. Nakano, H. Ottosson, J. Casado and M. M. Haley, Nat. Chem., 2016, 8, 753–759 CrossRef CAS PubMed.
- J. J. Dressler, A. C. Valdivia, R. Kishi, G. E. Rudebusch, A. M. Ventura, B. E. Chastain, C. J. Gómez-García, L. N. Zakharov, M. Nakano, J. Casado and M. M. Haley, Chem, 2020, 6, 1353–1368 CAS.
- M. A. Majewski, P. J. Chmielewski, A. Chien, Y. Hong, T. Lis, M. Witwicki, D. Kim, P. M. Zimmerman and M. Stępień, Chem. Sci., 2019, 10, 3413–3420 RSC.
- H. Sharma, P. K. Sharma and S. Das, Chem. Commun., 2020, 56, 11319–11322 RSC.
- Z.-Y. Wang, Y.-Z. Dai, L. Ding, B.-W. Dong, S.-D. Jiang, J.-Y. Wang and J. Pei, Angew. Chem., Int. Ed., 2021, 60, 4594–4598 CrossRef CAS.
- A. Shimizu, Y. Ishizaki, S. Horiuchi, T. Hirose, K. Matsuda, H. Sato and J. Yoshida, J. Org. Chem., 2021, 86, 770–781 CrossRef CAS.
- T. Takeda, H. Sugihara, Y. Suzuki, J. Kawamata and T. Akutagawa, J. Org. Chem., 2014, 79, 9669–9677 CrossRef CAS PubMed.
- G.-G. Luo, H. Lu, Y.-H. Wang, J. Dong, Y. Zhao and R.-B. Wu, Dyes Pigm., 2016, 134, 498–505 CrossRef CAS.
- P. Gregory, Anthraquinone Chromophore, in Industrial Dyes: Chemistry, Properties, Applications, ed. K. Hunger, Wiley-VCH, Weinheim. 2003 Search PubMed.
- T. Takeda, Y. Suzuki, J. Kawamata, S. Noro, T. Nakamura and T. Akutagawa, Phys. Chem. Chem. Phys., 2017, 19, 23905–23909 RSC.
- T. Takeda, S. Noro, T. Nakamura, Y. Suzuki, J. Kawamata and T. Akutagawa, CrystEngComm, 2018, 20, 17–24 RSC.
- Z. Xie, Q. Huang, T. Yu, L. Wang, Z. Mao, W. Li, Z. Yang, Y. Zhang, S. Liu, J. Xu, Z. Chi and M. P. Aldred, Adv. Funct. Mater., 2017, 27, 1703918 CrossRef.
- T. Takeda and T. Akutagawa, Chem. Commun., 2020, 56, 10564–10567 RSC.
- For previously reported photophysical properties of 1, 7, 13 (terminal alkyl groups were replaced with methyl groups), see: T. Hayashi and T. Tokumitsu, Bull. Chem. Soc. Jpn., 1965, 38, 916–922 CrossRef CAS.
- For previously reported photophysical properties of 19 (terminal alkyl groups were replaced with methyl groups), see: Y. C. Chao, Dyes Pigm., 1994, 26, 191–200 CrossRef CAS.
- For a recent synthesis of 1, 13, see: I. P. Beletskaya, A. G. Bessmertnykh, A. D. Averin, F. Denat and R. Guilard, Eur. J. Org. Chem., 2005, 281–305 CrossRef CAS.
- For a recent synthesis of 19, see: R. Duan, D. Scholmeyer, K. Müllen and C. Li, J. Mater. Chem. C, 2018, 6, 1334–1337 RSC.
- J. P. Wolfe, S. Wagaw, J. F. Marcoux and S. L. Buchwald, Acc. Chem. Res., 1998, 31, 805–818 CrossRef CAS.
- J. F. Hartwig, Angew. Chem., Int. Ed., 1998, 37, 2046–2067 CrossRef CAS.
- J. F. Hartwig, Synlett, 2006, 1283–1294 CrossRef CAS.
- D. S. Surry and S. L. Buchwald, Chem. Sci., 2011, 2, 27–50 RSC.
- S. V. Ley and A. W. Thomas, Angew. Chem., Int. Ed., 2003, 42, 5400–5449 CrossRef CAS PubMed.
- C. Binisti, L. Assogba, E. Touboul, C. Mounier, J. Huet, J.-E. Ombetta, C. Z. Dong, C. Redeuilh, F. Heymans and J.-J. Godfroid, Eur. J. Med. Chem., 2001, 36, 809–828 CrossRef CAS.
- G. M. Sheldrick, SHELX 97, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. A. Petersson, et al., Gaussian 09, Revision C01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone and G. A. Petersson, et al., Gaussian 16, Revision A03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: ORTEP drawing of 22. Cyclic voltammograms of 1–23 classified according to the substituent. HOMO and LUMO of 1–6, 11, 13–23. Copies of the 1H and 13C NMR spectra of new compounds. Cartesian coordinates for the optimized structures of 1–23. CCDC 2063387. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra03985g |
| This journal is © The Royal Society of Chemistry 2021 |