
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mining hydroformylation in complex reaction network via graph theory†
Keisuke Takahashi *a and
Maeda Satoshi*ab
*a and
Maeda Satoshi*ab
aDepartment of Chemistry, Hokkaido University, North 10, West 8, Sapporo 060-8510, Japan. E-mail: keisuke.takahashi@sci.hokudai.ac.jp
bInstitute for Chemical Reaction Design and Discovery (WPI-ICReDD), Hokkaido University, Kita 21 Nishi 10, Kita-ku, Sapporo, Hokkaido 001-0021, Japan. E-mail: smaeda@eis.hokudai.ac.jp
First published on 1st July 2021
Abstract
Data science is introduced to identify the reactant, product, and reaction path in the chemical reaction network. Cobalt catalyzed hydroformylation is investigated where the reaction network is built via first principles calculations. The closeness centrality and high frequency node are found to be the reactant cobalt tetracarbonyl hydride. In addition, betweenness centrality uncovers three reaction paths which have the products of aldehyde, CH2O, and CO2, respectively. The energy profile determines that the reaction path leading to aldehyde is energetically favored; thus, the reaction path for cobalt catalyzed hydroformylation is identified without kinetics. Hence, the proposed approach can act as a first step towards understanding the complex chemical reaction network and towards further kinetic understanding of the chemical reaction.
Introduction
Identifying the reaction path within a chemical reaction is a challenging task as a chemical reaction involves complex molecular interactions. For such situations, the introduction of first principles calculations gives insight towards the atomic level understanding of molecular interactions. In particular, the potential energy surface generated by first principles calculations elicits the details of the molecular interactions on an atomic scale.1,2 This essentially allows for the generation of a chemical reaction network in terms of molecular interactions.3–7 In general, chemical kinetics is coupled with a calculated chemical reaction network in order to determine the reaction pathway.8,9 However, one can consider that hidden trends and patterns for identifying the reaction path within the chemical reaction network should be present, considering that the energy landscape created by first principles calculation follows certain rules. In view of how a chemical reaction network is formed, the network can be treated as a graph data structure.5,10 Additionally, it is reported that graph theory can be used in order to extract knowledge from a chemical network.11 Here, data science, particularly graph theory, is implemented in order to search the reaction paths in a chemical reaction network.Hydroformylation is selected as the prototype reaction where the reaction involves the production of aldehydes from alkenes.12,13 In particular, cobalt catalyzed hydroformylation is investigated for considering homogeneous catalysis as the details of the reaction process are rather complex.8,14,15 The chemical reaction network of cobalt catalyzed hydroformylation is constructed via first principles calculations where the atomic interactions of CO dissociated cobalt tetracarbonyl hydride HCo(CO)3 with ethylene (C2H4), hydrogen H2, and carbon monooxide (CO) are considered. Reaction paths in the chemical reaction network for cobalt catalyzed hydroformylation are sought for via data driven analysis based on graph theory.
Methods
Computational method
The chemical reaction path network is explored using the artificial force induced reaction (AFIR) method combined with first principles calculations.16 AFIR induces chemical transformations by applying force and finds reaction paths based on the force-induced paths. The search is performed using the single-component algorithm of AFIR (SC-AFIR) starting from 200 initial structures produced by generating mutual positions and orientations among HCo(CO)3, CO, C2H4, and H2 randomly. Additionally, the model collision energy parameter γ of the AFIR method is set to 300 kJ mol−1, where γ defines an approximate upper limit of the barrier the artificial force can eliminate. During the search, additional weak force with γ = 0.65 kJ mol−1 is applied to all atom pairs in the system in order for the molecules to not separate too far in this system. All AFIR paths were reoptimized using the path optimization method using the locally updated planes (LUP) method, where the network of LUP paths are discussed below.17 The traffic volume is an index showing the total amount of population influx to and outflux from each local minimum within the simulation time tMAX. Therefore, local minima having large traffic volume values are regarded to be kinetically important. The traffic volume Λi is computed for all local minimum structures, and paths are searched preferentially from those having large values.9 For the traffic volume calculations, the initial population is evenly distributed to local minimum structures having the same bond connectivity to the initial species where the reaction time tMAX was set to 3600 seconds, the reaction temperature set to 300, 400, and 500 K, and the model temperature parameter TR set to 4000 K.9 The search is terminated when the latest N successful paths do not update the structural types of the top M traffic volumes, where N and M were set to ten and three times, respectively, of the total number of atoms in the system, a structural type stands for a group of local minimum structures having the same bond connectivity pattern and a successful path corresponds to a path connecting different structural types. All electronic structure calculations are done by the Gaussian 16 program where the ωB97X-D functional and LanL2DZ basis set are implemented.18 All structural displacements were taken by a development version of the GRRM program (version on April 9th, 2020).19 Note that the generated data is a preliminary study of the chemical reaction created for data science applications and requires further study for a more, detailed understanding of the energetics of the chemical reaction. Details of the SC-AFIR method and the traffic volume index are described in previous work.9,16Data science method
The chemical reaction network for cobalt catalyzed hydroformylation is investigated using data science and graph theory. The created reaction network is transformed into a directed graph where source and target nodes are defined as reactants and products, respectively. The activation energy barrier is represented as node edges and is reflected in edge weight. Gephi is then implemented for graph visualization and analysis.20,21 Force Atlas 2 is used for graph visualization while closeness centrality and betweenness centrality are implemented for graph analysis.20,22,23Results and discussion
Data analysis is performed on the data obtained from cobalt catalyzed hydroformylation reaction calculations. The data set consists of 8558 data points with the following information in the columns: reactant node number, product node number, equilibrium energy of the reactant, equilibrium energy of the product, and activation energy barrier. Node number and activation energy barrier are treated as nodes and edges in the network, respectively. The data is treated as a directed graph where reversing the node results in different activation energy barriers. Note that the data and corresponding structural information are listed in the ESI.† Frequency analysis reveals that node number 54 appears 55 times within the 8558 data points. In particular, node number 54, which represents cobalt tetracarbonyl hydride HCo(CO)4 with H2 and C2H4 molecules, is found to be the node with the highest frequency within the map. Understanding which nodes have high frequency in the reaction network allows one to better understand the initial step taken within the reaction as high frequency indicates that many nodes visit this node. Given its frequency, one can therefore see that the molecules represented in node number 54 experience a high level of traffic within the network and thus can be seen as a key step of hydroformylation.Network visualization is performed in order to represent the calculated hydroformylation reaction as a network. In particular, the Force Atlas 2 algorithm is used in order to visualize the network as shown in Fig. 1.20 Network visualization is informed by the continuous algorithm and is force-directed where nodes repel each other while edges attract their respective nodes, making node placement dependent on the other nodes present within the network. Fig. 1 shows the overall reaction network of hydroformylation created by the AFIR method where the reaction network traces how the AFIR method navigates the reaction. Fig. 1 demonstrates that some nodes form clusters at the center of the network while other groups form a branch-like structure that stems out from the center of the network. Note that node numbers 26, 145, 1856, and 1812 are isolated from the network as a result of SCF convergence failure that occurred on paths to these nodes.
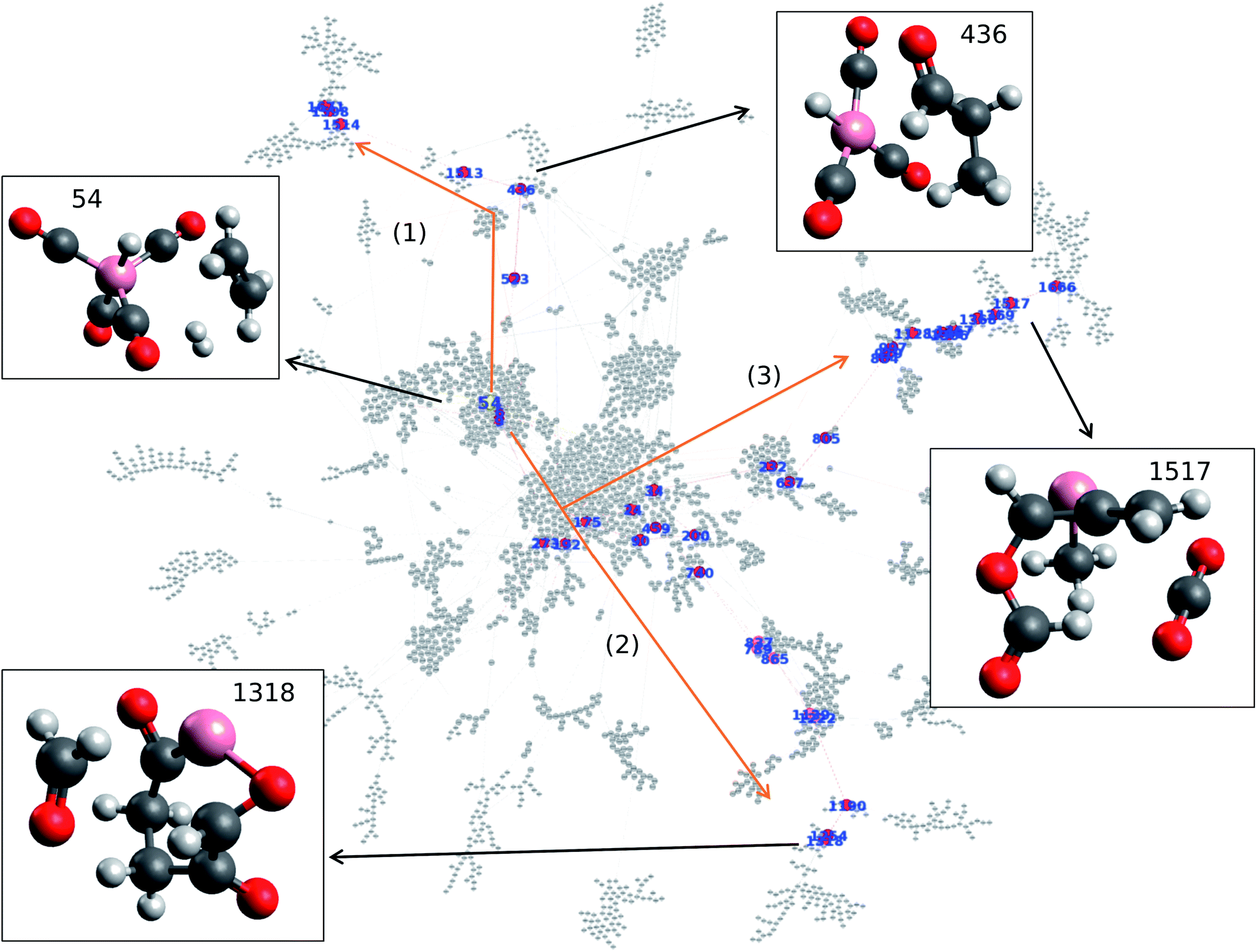 | ||
| Fig. 1 Visualization of the hydroformylation reaction network via Force Atlas 2. Structures of reactant and key products are also represented. Color code: pink: cobalt; gray: carbon; white: hydrogen; red: oxygen. | ||
Here, the question arises concerning how the reactant, product, and reaction paths connecting them will be identified within the reaction network as shown in Fig. 1. In order to find the key nodes within the reaction network, network analysis is performed in terms of graph theory. In particular, harmonic closeness centrality and betweenness centrality are explored where closeness and betweenness represent how close a node is to other nodes and which nodes control the network, respectively.22 In other words, one can consider that harmonic closeness centrality can help indicate energetically stable structures while betweenness centrality can help indicate key intermediate compounds in the reaction. Harmonic closeness demonstrates that node number 54 has the highest score, indicating that node 54 accesses many neighboring nodes and has good agreement with the observation that node 54 has the highest frequency within the network. Note that node 54 is colored in yellow in Fig. 1. HCo(CO)4, the compound represented by node 54, has been previously reported to be an active catalyst for hydroformylation where the dissociation of CO from HCo(CO)4 is considered as the initial step for hydroformylation.24 Hence, analyzing closeness centrality helps determine the reactant within the calculated hydroformylation reaction network.
Similarly, betweenness centrality is investigated where the top 40 betweenness centrality nodes are selected and colored in red as shown in Fig. 1. Please see the ESI† for the top 40 betweenness centrality nodes. It is surprising to find that three paths appear when connecting the top 40 betweenness centrality nodes as shown in Fig. 1. More importantly, each path contains key molecules where paths (1), (2), and (3) result in aldehydes, formaldehyde (CH2O), and carbon dioxide (CO2), respectively. Given that hydroformylation is a reaction that produces aldehydes, this result therefore shows that betweenness centrality can be used to help identify reaction paths for aldehyde formation from node 54 (which contains HCo(CO)4) without kinetics.
Further details of paths (1), (2), and (3) in Fig. 1 are investigated in terms of the energy profiles as shown in Fig. 2. The energy profile of path (1) indicates that aldehydes (node 436) are formed in the node order 54 → 6 → 523 → 436 where an activation barrier of 204.05 kJ mol−1 is required when attempting to move from nodes 523 to 436. In the same fashion, paths (2) and (3) are analyzed via an energy profile. However, these paths encounter multiple high activation energy barriers and endothermic reactions when attempting to arrive at CH2O (node 1254) and CO2 (node 1517) in path (2) and path (3), respectively. Note that node 212 is not in top 40 ranking nodes with high betweenness centrality. Therefore, paths (2) and (3) (illustrated in Fig. 1) can be considered to be unlikely to occur while path (1) is energetically favored. Although the actual reaction path could be more complex with kinetic analysis, data science has provided a near-instant method of providing potential candidates for reactants, products, and reaction paths encountered within a complex reaction network. This approach therefore accelerates the identification of reaction paths within a reaction network without additional kinetics analysis. It must be noted that the frequency of nodes as well as betweenness centrality analysis are able to detect key nodes based on the structure of the network shown in Fig. 1 created by a development version of the GRRM program (version on April 9th, 2020), therefore, different network analysis might be required depending on the structure of network. In other words, the proposed approach can act as the first step towards deeper kinetic analysis into a reaction network and provide insight into where further investigation can occur.
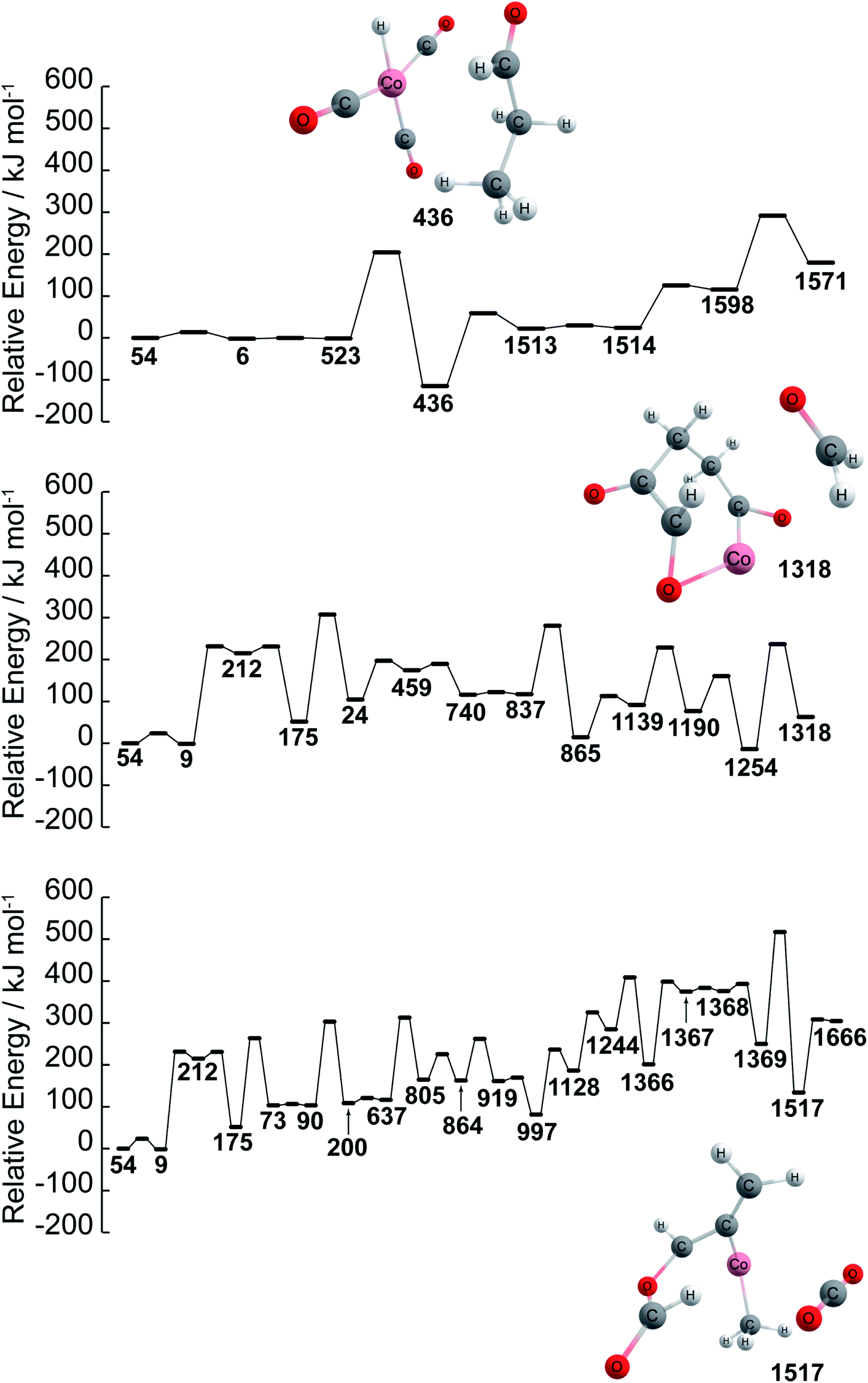 | ||
| Fig. 2 Energy profiles of paths (1), (2), and (3) found by betweenness centrality. Ea is the activation energy barrier. Structures of reactant and key products are also represented. Color code: pink: cobalt; gray: carbon; white: hydrogen; red: oxygen. Note that the energies path is searched and calculated by AFIR with Gaussian 16 and numbers represent the node number in Fig. 1. | ||
The chemical reaction network is then kinetically investigated in order to compare kinetics against the proposed reaction path created using graph theory. The kinetically most feasible path from node 54 to the most stable catalyst–product complex 880 is extracted from the network and depicted in Fig. 3. The path is obtained by combining the shortest path in terms of overall rate constants with local equilibration paths. The local equilibration paths can be seen in the processes from 54 to 1931 and from 527 to 544. The path consists of many steps that include bond reorganization steps and fast steps such as pseudo rotation and conformation change. As can be seen in Fig. 3, the energy profile has three regions: (a) an initial region including node 54 which represents HCo(CO)4 + C2H4 + H2, (b) the intermediate region including node 194 which represents CH3CH2Co(CO)3 + CO + H2, including node 1078 which represents CH3CH2Co(CO)4 + H2, CH3CH2C(O)Co(CO)3 + H2, and (c) the final region which includes node 446 which represents HCo(CO)3 + CH3CH2CHO. This path shows agreement with the well-known Heck–Breslow mechanism, which justifies the use of this network in this study.14 Additionally, aldehyde is found in both the graph network and the kinetic study, where aldehyde is found to be produced at node 436 within the network while aldehyde is found to be produced at node 880 during the kinetic investigation. Thus, these results show that the kinetic study and chemical reaction network are both capable of finding nodes where aldehyde is produced.
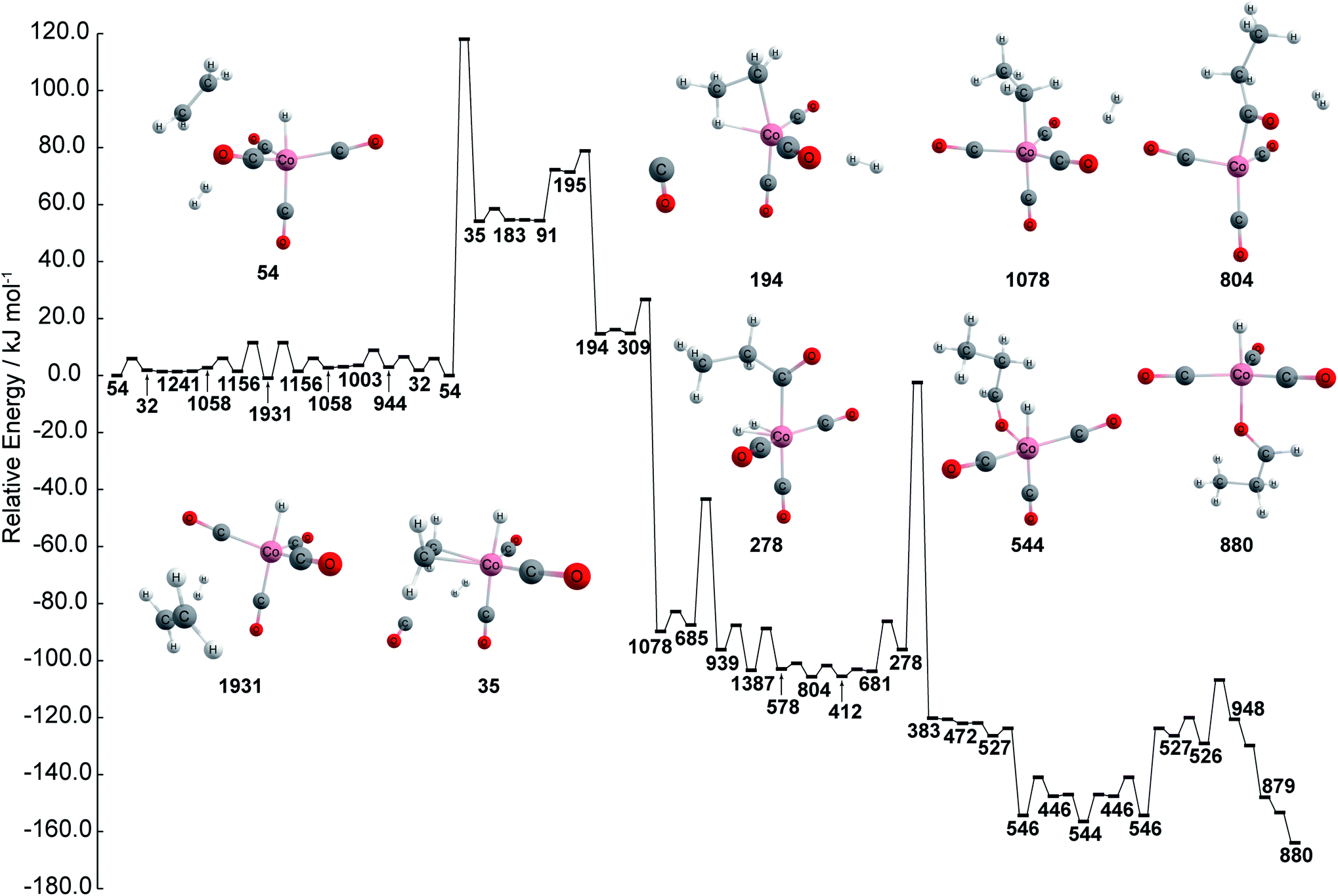 | ||
| Fig. 3 Energy profile of stable path by kinetic analysis. Color code: pink: cobalt; gray: carbon; white: hydrogen; red: oxygen. Note that the energies path is searched and calculated by AFIR with Gaussian 16 and numbers represent the node number in Fig. 1. | ||
Here, one can also understand that the closeness centrality and betweenness centrality of the network shown in Fig. 1 reflects the search procedure used in the SC-AFIR algorithm. First, when Λi is used as an index to rank local minimum structures, the preference in a group that reaches equilibrium in a shorter timescale than tMAX is dependent on the Boltzmann distribution at TR. At the start of the search, only complexes among HCo(CO)3 + CO + C2H4 + H2 are considered and paths are computed from these structures. Once the structures in the intermediate region are found, searches are done preferentially from structures in the intermediate region since the structures in the initial region can transition to the intermediate region within tMAX. Finally, searches are done preferentially from structures in the final region since the structures in the initial and intermediate regions can transition to the final region within tMAX. It should be noted that many other possibilities that originate from the initial region are searched as the other regions are unknown at the start. This can account for why structures having the highest closeness centrality are found within the initial region. Node 54 is quasi-symmetric, having C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and H–H almost on the plane of H, Co, and C in the axial CO, making it a possible transit point among other HCo(CO)4 + C2H4 + H2 complexes in the initial region. Therefore, the closeness centrality is useful for identifying the most important structure within the initial region. As noted, the SC-AFIR searched various possibilities originating from the initial region and created many local areas within the network. By definition, a node that has high betweenness centrality is a node that can be viewed as having more control in the network as many paths lead through it. Given this, structures that have high betweenness centrality should correspond to key intermediates within paths that connect to different areas of the network. Various different chemical transformations are able to be identified by tracing nodes having high betweenness centrality. Identifying possible intermediates, regardless of their kinetic importance, is very important for actual mechanism studies on chemical reactions. Hence, betweenness centrality can be powerful for identifying key intermediates. Chemical reaction network has become possible to create by combining the first principles calculation with data science. However, it has been a challenge to extract knowledge from the network due to the high complexity of the chemical reaction. Here, data science, graph theory in particular, is found to be a powerful approach for quickly extracting knowledge from the reaction network without any kinetics analysis. Thus, combining graph theory and the first principles calculations helps accelerate the determination of the reaction path in a chemical reaction network.
C and H–H almost on the plane of H, Co, and C in the axial CO, making it a possible transit point among other HCo(CO)4 + C2H4 + H2 complexes in the initial region. Therefore, the closeness centrality is useful for identifying the most important structure within the initial region. As noted, the SC-AFIR searched various possibilities originating from the initial region and created many local areas within the network. By definition, a node that has high betweenness centrality is a node that can be viewed as having more control in the network as many paths lead through it. Given this, structures that have high betweenness centrality should correspond to key intermediates within paths that connect to different areas of the network. Various different chemical transformations are able to be identified by tracing nodes having high betweenness centrality. Identifying possible intermediates, regardless of their kinetic importance, is very important for actual mechanism studies on chemical reactions. Hence, betweenness centrality can be powerful for identifying key intermediates. Chemical reaction network has become possible to create by combining the first principles calculation with data science. However, it has been a challenge to extract knowledge from the network due to the high complexity of the chemical reaction. Here, data science, graph theory in particular, is found to be a powerful approach for quickly extracting knowledge from the reaction network without any kinetics analysis. Thus, combining graph theory and the first principles calculations helps accelerate the determination of the reaction path in a chemical reaction network.
Conclusion
In summary, data science is implemented to determine the reaction path in a calculated reaction network. In particular, cobalt catalyzed hydroformylation is selected as a prototype reaction where artificial force induced reaction within the first principles calculations is implemented to create a hydroformylation reaction network. Data science, graph theory, unveils the frequency of nodes as well as closeness centrality determining the reactant which is found to be cobalt tetracarbonyl hydride HCo(CO)4. Furthermore, betweenness centrality reveals the 3 reaction paths which lead to the formation of aldehyde, CH2O, and CO2 where the energy profile indicates that the formation of aldehyde is energetically favored. Thus, it is proposed that data science accelerates identification of the reactant, product, and reaction path from the complex reaction network without kinetics. This approach would act as a first step for understanding a complex reaction network towards further kinetic understanding of a chemical reaction.Author contributions
K. T. performed network and data analysis. S. M. calculated the reaction network.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is funded by Japan Science and Technology Agency (JST) ERATO Grant Number JPMJER1903, JSPS KAKENHI Grant-in-Aid for Young Scientists (B) Grant Number JP17K14803, and JSPS-WPI.Notes and references
- J. P. Doye, M. A. Miller and D. J. Wales, J. Chem. Phys., 1999, 111, 8417 CrossRef CAS.
- D. J. Wales and T. V. Bogdan, Potential energy and free energy landscapes, 2006 Search PubMed.
- S. Maeda, K. Ohno and K. Morokuma, Phys. Chem. Chem. Phys., 2013, 15, 3683 RSC.
- Z. W. Ulissi, A. J. Medford, T. Bligaard and J. K. Nørskov, Nat. Commun., 2017, 8, 14621 CrossRef PubMed.
- G. N. Simm and M. Reiher, J. Chem. Theory Comput., 2017, 13, 6108 CrossRef CAS PubMed.
- C. A. Grambow, A. Jamal, Y.-P. Li, W. H. Green, J. Zador and Y. V. Suleimanov, J. Am. Chem. Soc., 2018, 140, 1035 CrossRef CAS PubMed.
- A. L. Dewyer, A. J. Argüelles and P. M. Zimmerman, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1354 Search PubMed.
- L. E. Rush, P. G. Pringle and J. N. Harvey, Angew. Chem., 2014, 126, 8816 CrossRef.
- Y. Sumiya and S. Maeda, Chem. Lett., 2020, 49, 553 CrossRef CAS.
- R. Van de Vijver and J. Zádor, Comput. Phys. Commun., 2020, 248, 106947 CrossRef CAS.
- L. Takahashi, J. Ohyama, S. Nishimura and K. Takahashi, J. Phys. Chem. Lett., 2021, 12, 558 CrossRef CAS PubMed.
- I. Ojima, C.-Y. Tsai, M. Tzamarioudaki, and D. Bonafoux, Organic Reactions, 2000 Search PubMed.
- R. Franke, D. Selent and A. Börner, Chem. Rev., 2012, 112, 5675 CrossRef CAS PubMed.
- R. F. Heck and D. S. Breslow, J. Am. Chem. Soc., 1961, 83, 4023 CrossRef.
- F. Hebrard and P. Kalck, Chem. Rev., 2009, 109, 4272 CrossRef CAS PubMed.
- S. Maeda, Y. Harabuchi, M. Takagi, K. Saita, K. Suzuki, T. Ichino, Y. Sumiya, K. Sugiyama and Y. Ono, J. Comput. Chem., 2018, 39, 233 CrossRef CAS PubMed.
- C. Choi and R. Elber, J. Chem. Phys., 1991, 94, 751 CrossRef CAS.
- M. Frisch, G. Trucks, H. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, G. Petersson and H. Nakatsuji, et al., Gaussian 16, revision C. 01, 2016 Search PubMed.
- S. Maeda, Y. Harabuchi, Y. Sumiya, M. Takagi, K. Hatanaka, M. Osada, Y. Taketsugu, T. Morokuma, and K. Ohno, GRRM, A Development Version of April 9, 2020 Search PubMed.
- M. Jacomy, T. Venturini, S. Heymann and M. Bastian, PLoS One, 2014, 9, e98679 CrossRef PubMed.
- M. Bastian, S. Heymann and M. Jacomy, ICWSM, 2009, 8, 361 Search PubMed.
- U. Brandes, J. Math. Sociol., 2001, 25, 163 CrossRef.
- K. Okamoto, W. Chen, and X.-Y. Li, in International workshop on frontiers in algorithmics, Springer, 2008, pp. 186–195 Search PubMed.
- I. Wender, H. Sternberg and M. Orchin, J. Am. Chem. Soc., 1953, 75, 3041 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: All structures files and data used in this work. See DOI: 10.1039/d1ra03395f |
| This journal is © The Royal Society of Chemistry 2021 |