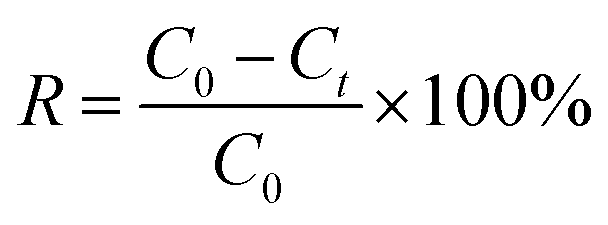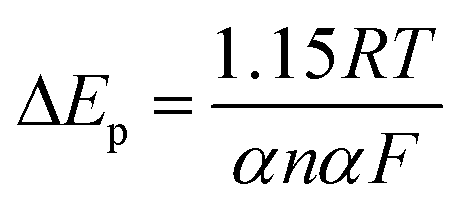DOI:
10.1039/D1RA02505H
(Paper)
RSC Adv., 2021,
11, 23241-23248
Organic acid mediated photoelectrochemical reduction of U(VI) to U(IV) in waste water: electrochemical parameters and spectroscopy†
Received
30th March 2021
, Accepted 3rd June 2021
First published on 1st July 2021
Abstract
The photoelectrochemical reduction of U(VI) is recognized as an economical and effective way to eliminate radioactive pollution. In this study, we construct a α-Fe2O3/TiO2 film electrode-based photoelectrochemical cell to remove U(VI) and recover uranium from aqueous solution. Citric acid and oxalic acid could act as hole scavengers, being favorable for the photocatalytic reduction of U(VI). In the presence of 0.5 mM citric acid and oxalic acid, the uranium removal capacity reached 70% and 58%, respectively, while 24% was achieved for the system in the absence of acid. The XRD, SEM, FT-IR and XPS results revealed that a proportion of U(IV) was also precipitated as surface associated metastudtite. These novel observations have significant implications for the behavior of uranium within engineered and natural environments.
1. Introduction
With the consumption of fossil fuels, uranium is an important nuclear fuel that is widely used in nuclear power plants, nuclear reactors, nuclear weapons and other industries.1 It is reported that the amount of identified uranium on land is only 7.6 million tons, which severely restricts the development of the nuclear industry. However, uranium mining and processing have resulted in the release of considerable amounts of uranium into the natural environment, mostly through migration in ground or surface water systems as the highly soluble uranyl(VI) ion.2–4 It will cause a wide range of environmental pollution and induces a serious threat to human health. Therefore, it is of great significance for environmental protection to explore new technologies and methods for efficient removal and extract of uranium from the uranium wastewater.
There are two main species of uranium in the environment, hexavalent uranium U(VI) has good solubility in aqueous solution, while tetravalent uranium U(IV) has less solubility.5,6 Hence, reduction of the uranyl ion (UO22+) and precipitation as the insoluble U(IV) species have been regarded to be an effective and economic approach to remediate and manage the uranium contamination. Till now, some traditional processing methods such as chemical precipitation,7 chemical reduction,8 ion exchange,9 biological treatment10 and evaporation concentration11 can effectively prevent the pollution of U(VI) in contaminative environments, but these methods generate secondary pollution, incur high production costs, and require harsh processing. Among various proposed methods, photoelectrochemical (PEC) method, a kind of versatile, efficient, cost-effective, and clean technique based on photoinduced electron transfer processes at electrode/interfaces, has received much interest. Kim et al.12 have found that photoelectrochemical (PEC) treatment was more effective in reducing U(VI) than photocatalytic (PC) treatment and electrochemical (EC) treatment. The synergistic effect at −0.4 VSCE was 250% of the PEC U(VI) reduction, because it can enhance the separation of photogenerated electron–hole pairs and prolong the lifetime of charge carriers via applying bias.
TiO2 is very attractive for their fascinating features such as good chemical and thermal stability, plentiful polymorphs, and excellent electronic and optical properties. The conduction band (ECB) of TiO2 is located at −0.29 V versus NHE, which is lower than the redox potential of UO22+/U4+ (0.267 V) and UO22+/UO2 (0.411 V), revealing the photo-excited electrons in TiO2 electrode have enough energy to drive the reduction reaction of U(VI) to U(IV) in theory. Actually, several works reported that photocatalytic reduction of U(VI) species using TiO2 photocatalyst. For instance, Li et al.13 use TiO2 as photocatalyst reduction and removal of U(VI) under UV irradiation, which exhibits a superior reduction performance, and the reduced uranium was easily eluted and recovered by sodium carbonate solution. Li et al.14 have found that uranium extraction from seawater photocatalysis by TiO2 was reached 3960 mg g−1 without saturation. However, considering the inferior transfer ability of photo-generated charges, the insufficient response to visible light, further efforts are needed to construct metal-free photocatalysts with high efficiency. To overcome the intrinsic drawbacks of TiO2, a series of modification methods have been proposed including polymerization of organic compounds,15 elements doping16 and acid treatment.17 At the same time, the high production cost, the instability and toxicity of these advanced materials greatly restricts their practical application in U(VI) remediation. Hematite (α-Fe2O3) has material abundance, chemical stability over a wide pH range, and favourable band gap (2.0–2.2 eV) structure enabling the absorption of a great spectrum of sunlight (27% of the total power). However, the fast recombination of photogenerated electrons and holes weakened its photocatalytic activity. Shejale K. P. et al.18 using core–shell α-Fe2O3–anatase and rutile TiO2 nanostructures as photoanodes demonstrated have enhance 8% and 21% photoconversion efficiency and fill factor of the solar cell compared to that of α-Fe2O3 photoanodes, respectively. Similarly, we use α-Fe2O3/TiO2 as photocatalyst reduction and removal of Cr(VI), which exhibits a superior reduction performance.19
Noteworthily, the combination of oxidation and reduction reactions would markedly improve the separation rate of photogenerated holes and electrons, enhance the quantum efficiency, leading to enhancement of removal rate of U(VI). In order to reduce the compound of hole and electron, the effective method has been proposed though adding hole scavenger in the reaction system. For example, Raivis E. et al.20 investigated the impact of scavenging power on the photochromic properties of TiO2, which shown that solvents such as ethanol have the best hole scavenging properties among the alcohols tested. Denisov N. et al.21 find that hole scavenger, such as: methanol, isopropanol, ethylene glycol, EDTA-Na2, Na2SO3, lead to a 10.0–28.8 times higher H2 production by TiO2 nanotubes than the scavenger-free case. Our previous studies have proven that ethanol as hole scavenger improved the U(VI) reduction efficiency of TiO2-based photoelectrochemical cell.22
Inspired by the prior works, in this work, the α-Fe2O3/TiO2 heterojunction was prepared as photoanode and fluorine-doped tin oxide (FTO) glass cathode for U(VI) photoelectrochemical reduction. Furthermore, the different environmental conditions, such as pH values, concentration of U(VI), action time and hole scavenger, were investigated for U(VI) photoelectrochemical reduction. Addition, the mechanism of U(VI) photoreduction process was investigated via X-ray diffraction (XRD), scanning electron microscopy (SEM), Fourier Transform infrared spectroscopy (FTIR), X-ray photoelectron spectroscopy (XPS). It is expected that this research will pave the way for the further development of efficient photocatalysts for the removal of radionuclide.
2. Material and methods
2.1 Preparation of α-Fe2O3/TiO2 electrode
Ethylene glycol, NH2CONH2, HNO3 were purchased from Chengdu Chemical Reagent Co., Ltd. (Chengdu, China). All chemicals were analytically pure grade. Distilled water was used throughout the whole experiments. The α-Fe2O3/TiO2 film on FTO glass was used as the photoanode, which was prepared according to the solvent-thermal methods in the published literatures.19 Briefly, 1.0 mL TiCl3 solution (15.0–20.0 wt%, Aldrich) was diluted with 5.0 mL ethylene glycol to form a precursor solution. 500 μL precursor solution was drop-cast onto the surface of FTO, and was annealed in air at 150 °C for 40 min followed by 550 °C for 2 h. Next, 1.08 g FeCl3·6H2O (Aladdin, 99.9%) and 0.36 g NH2CONH2 were dissolved in 20 mL distilled water. The precursor solution was transferred into a 30 mL Teflon-lined stainless autoclave and kept at 150 °C for 6 h, followed by natural cooling to room temperature. Finally, the film formed on FTO was annealed at 500 °C for 30 min, a orange/rufous film was formed on the FTO glass substrate.
Before the photoelectrochemical measurements, the morphology and phase structure of α-Fe2O3/TiO2 film were investigated using a Zeiss Supra 55 VP scanning electron microscope (SEM), a Rigaku Spider X-ray diffractometer (XRD, powder sample test mode) and UV-vis diffuse reflectance spectra (DRS) (Fig. S1–S3†). In previous works, it has been proved that α-Fe2O3/TiO2 film with mixed phase of hematite and anatase is advantageous for reducing the recombination of photo-excited electrons and holes and enhancing the visible-light-induced photocatalysis properties.19
2.2 Electrochemical and photoelectrochemical measurement
The electrochemical and photoelectrochemical measurement were performed in a conventional three-electrode system linked with the electrochemical workstation (Princeton Applied Research, USA, model PARSTAT 4000). The as-prepared α-Fe2O3/TiO2 film electrode was used as working electrode, Pt electrode as the counter electrode and saturated calomel electrode (SCE, Shanghai INESA Scientific Instrument Co., Ltd.) as reference electrode. 0.5 mM uranyl nitrate hexahydrate [UO2(NO3)2·6H2O] (Sigma Aldrich) dissolved into 0.1 M NaCl supporting electrolyte was used as U(VI) solution for electrochemical and photoelectrochemical measurements. A 0.1 M NaCl solution without additives was used as a reference. The pH is adjusted with NaOH or HCl (0.1 M). The illumination source was a 300 W xenon lamp with AM 1.5 G filter (Beijing China Education Au-light Co., Ltd.). For efficient charge separation, the photoelectrochemical measurements for the α-Fe2O3/TiO2 films are conducted using back-side illumination.23 For converting the obtained potential (vs. SCE) to reversible hydrogen electrode (RHE) (NHE at pH = 0), the following equation was used.| | |
ERHE = ESCE + 0.059 pH + E0SCE (E0SCE = 0.2458 V vs. NHE at 25 °C)
| (1) |
2.3 Photocatalytic characterization
The photocatalytic experiments were carried out in a three-electrode cell using an 300 W xenon lamp with AM 1.5 G filter as a light source and a three-electrode cell comprising of an α-Fe2O3/TiO2 electrode, FTO (1.5 cm × 2.5 cm), and SCE were used as a working, counter, and reference electrodes, respectively. The uranium solution used in the experiment is obtained by dissolving UO2(NO3)2·6H2O in a 0.01 M HNO3 solution. The concentrations of U(VI) in supernatant were determined using the spectrophotometrically arsenazo III method at a wavelength of 652 nm.24 In brief, the method involved the addition of 1 mL of supernatant into volumetric flask of 25 mL, then adding 5 mL of CH2ClCOOH–CH3COONa buffer and 1 mL of 0.06% arsenazo (III). After 10 min, the absorbance at 651 nm was measured by UV-Vis spectrophotometer (UV-2450, SHIMADZU, China). The linear relationship between absorbance and CU(VI) see in Fig. S4.† The removal percentage (R, %) could be expressed as eqn (2).| |
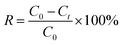 | (2) |
where C0 and Ct are the concentrations of the uranium in the initial and final solutions (mg L−1), respectively. All chemicals are of A.R. grade quality, and the solutions prepared with distilled water.
2.4 XRD, SEM, FTIR, XPS analysis
The structural characterization of uranium was characterized via X-ray diffractometer (XRD, Rigaku D/max TTRIII) with Cu-Kα radiation (λ = 1.5419 Å) at a scanning rate (2θ) of 0.05° s−1. The morphology of the samples was observed using scanning electron microscope (SEM, Zeiss Supra 55 VP). Fourier transform infrared spectroscopy (FTIR, PerkinElmer Nicolet-5700) were collected by spectrophotometer in the wave number range of 400–4000 cm−1 at room temperature. The X-ray photoelectron spectroscopy (XPS, Thermo Escalab 250 XPS, USA) parameters were: Al K-alpha source, spot size of 400 μm, and energy step size of 1.0 eV. The XPS datas were processed using the XPSPEAK software (version 4.1).
3. Results and discussion
3.1 Photoelectrochemical activity of α-Fe2O3/TiO2 in U(VI) solution
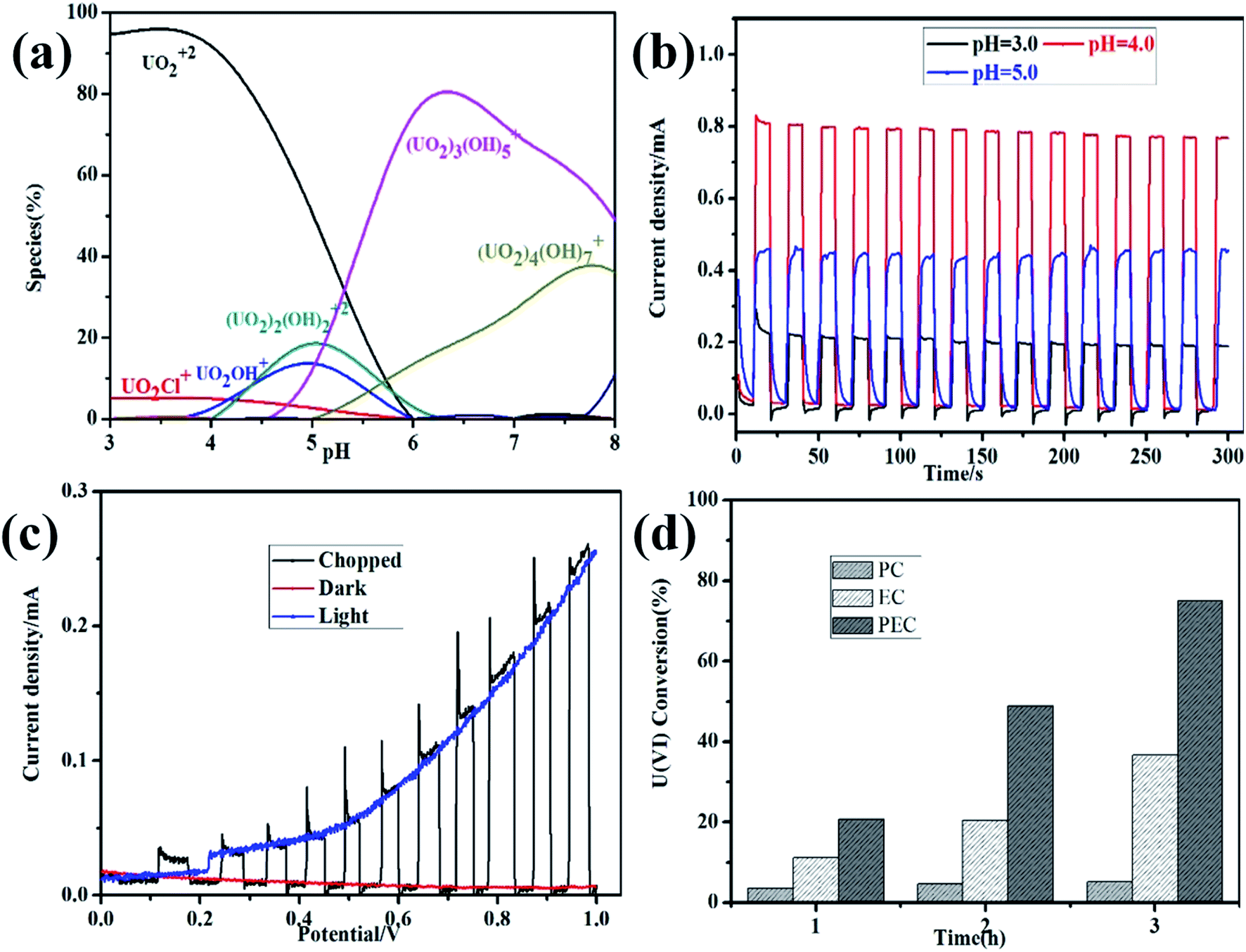
pH of solution is regarded as the most important factor, as it affects not only the speciation of uranium ions in solution, but also the chemical properties and surface characteristics. As shown in Fig. 1a, the distribution of U(VI) species in solution is calculated by Visual MINEQL 3.0 based on Nuclear Energy Agency (NEA) database. The transformation of U(VI) from free UO22+ to series hydroxide compound (e.g. UO2OH+, (UO2)2(OH)22+, (UO2)3(OH)5+ and (UO2)4(OH)7+) with the pH increasing, which might be explanation of the pH-dependent removal. In conclusion, the excellent removal efficiency of U(VI) was primarily ascribed to the electrostatic interaction and surface complexation or precipitation.25 It is worth noting that most of the uranium wastewater is acidic, and subsequent experiments are mainly carried out under acidic conditions. The photoelectrochemical activity of α-Fe2O3/TiO2 film photoanode was investigated by amperometric i–t curves at different pH in Fig. 1b. Notably, the anodic photocurrent intensity far exceeds that of all other samples at pH 4, which reaches 0.8 mA. One possible reason was that the pH induced protonation and deprotonation of surface groups on α-Fe2O3/TiO2.26,27 The H+ will protonize the surface of material and prevent it from combining with the positively charged U(VI) (mainly UO22+). In addition, the protons will compete with U(VI) to consume photogenerated, thus reducing the photoreduction activity. In addition, the cyclic voltammogram of U(VI) was measured at different pH in Fig. S1,† which found that the photocurrent of reduction peak and anodic peak A is maximum at the pH 4.
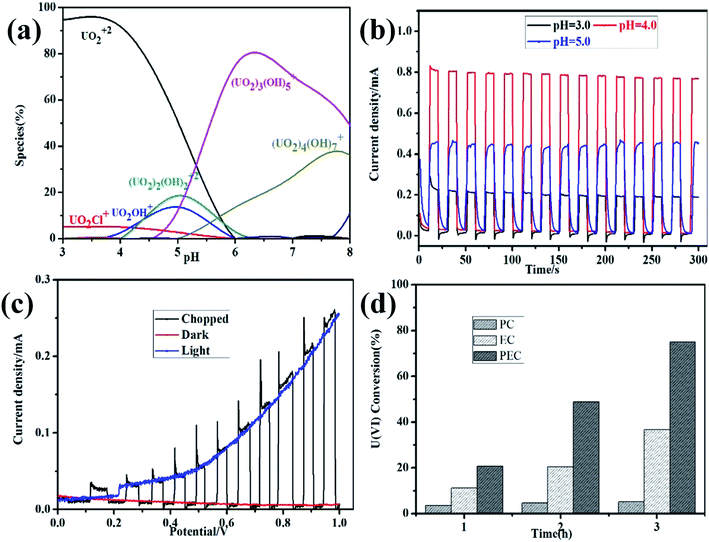 |
| | Fig. 1 pH-dependent speciation of U(VI) in 0.1 M NaCl calculated with MINEQL software (a); amperometric i–t curves at different pH (b); chopped linear sweep voltammetry scans (c); time-profiled changes during the photocatalytic (PC, irradiation only), electrochemical (EC, bias only), and photoelectrochemical (PEC, irradiation + bias) treatment of α-Fe2O3/TiO2 electrode in 0.5 mM U(VI) + 0.1 M NaCl at pH 4.0, Ebias = 1.0 V (d). | |
Fig. 1c shows the chopped linear sweep voltammetry scans for α-Fe2O3/TiO2 electrode in 0.5 mM U(VI) + 0.1 M NaCl (pH 4.0). Under AM 1.5 G irradiation, it is apparent that the α-Fe2O3/TiO2 film has pronounced photocurrent response, revealing a positive photoelectrochemical activity of α-Fe2O3/TiO2 film photoanode for PEC water oxidation. The α-Fe2O3/TiO2 film shows a more pronounced photoresponse, which starts around 0.15 V vs. SCE and gradually increases with applied potential. The photocurrent on the α-Fe2O3/TiO2 film reached 0.25 mA cm−2 at 1.0 V vs. SCE, which is a 25 times enhancement compared to without irradiation at the same potential. This experimental result shows a positive synergistic effect was formed between bias and irradiation for PEC water oxidation, because most of photogenerated holes are expected to transfer for water oxidation. Similar results have been found in other research relating to photocatalysts on FTO-coated glass substrates for water oxidation.28 The standard reduction potential of U(VI) to U(IV) is EΘ = 0.327 V vs. RHE, and O2 is EΘ = 1.229 V vs. RHE.29 This suggests that O2 was an effective electron acceptor, comparing to reduction of U(VI) to U(IV). Thus in our experiments, all the electrolyte was treated by N2 purging for 10 min to remove the dissolved oxygen before measurement. The comparisons of PC, EC, and PEC processes on the reduction of U(VI) were shown in Fig. 1d. Under PC treatment conditions, approximately 5.2% of U(VI) was removed. The EC treatment (Ebias = 1.0 V vs. SCE) for 3 h has removed approximate 36.6% of U(VI), whereas PEC treatment for the same period at 1.0 V vs. SCE has removed the U(VI) about 75%. Hence, the synergistic effect at 1.0 V vs. SCE was 179.4% (ΔUPEC/(ΔUEC + ΔUPC) × 100%). Upon irradiation, a small Ebias can drive effective separation of the photogenerated charges.12,30 Therefore, there were obvious synergistic PEC effects on U(VI) removal.
3.2 Characterization of U(VI) reduction potential
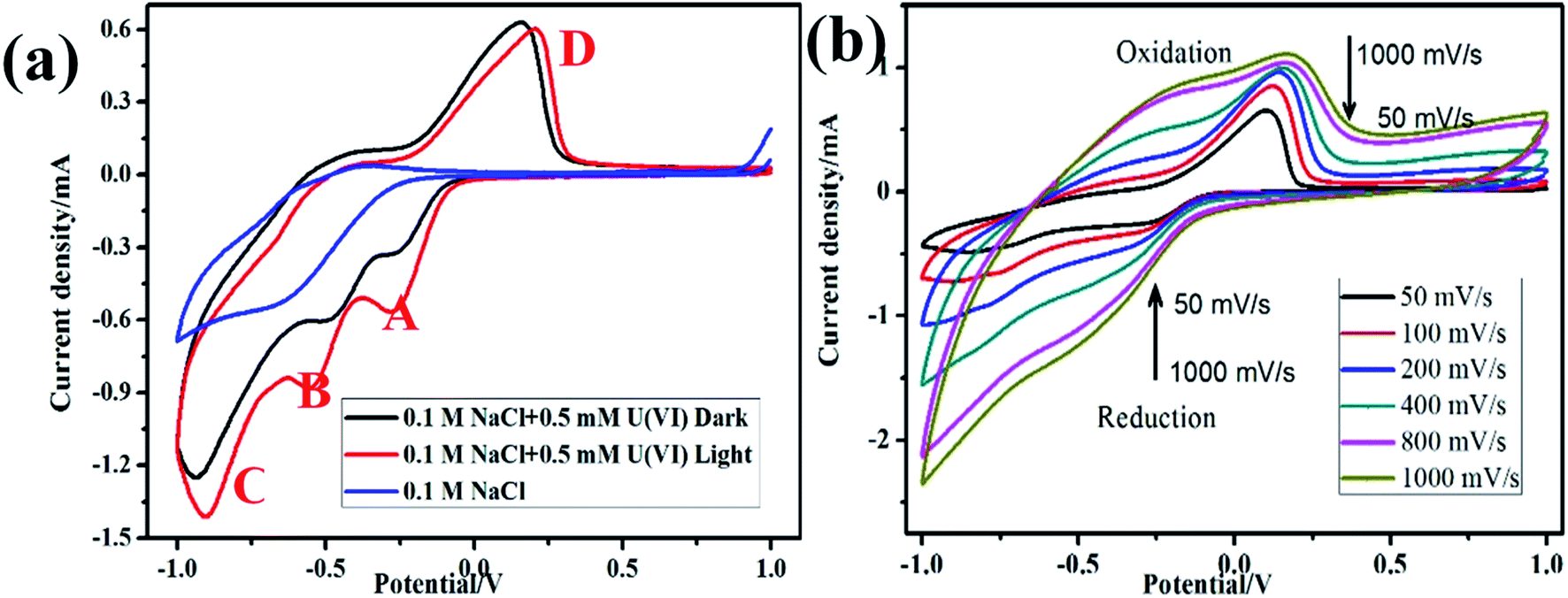
The Redox potential of U(VI) in 0.1 M NaCl solution was determined by cyclic voltammetry, as shown in Fig. 2a In the absence of U(VI), no distinct redox peak was observed on FTO glass electrode in 0.1 M NaCl. However, three significantly large anodic peaks at −0.26 V vs. SCE (peak A), −0.7 V vs. SCE (peak B) and −1.1 V vs. SCE (peak C) were observed. The reduction peak A are presumed to be related to the one-electron reduction of U(VI) to U(V) (e.g., UO22+, reaction (3)). Peak B correspond to the pentavalent uranium (UO2+) further reduction to U(IV) (reaction (4)), and H2 evolution reaction started at peak C (reaction (5)). During the return scan, a distinct oxidation peak at 0.15 V vs. SCE (peak D) was observed, which can be attributed to oxidation of U(IV) to U(VI) (reaction (6)).| | |
UO2+ + 4H+ + e− → U4+ + 2H2O
| (4) |
| | |
U4+ + 2H2O − 2e− → UO22+ + 4H+
| (6) |
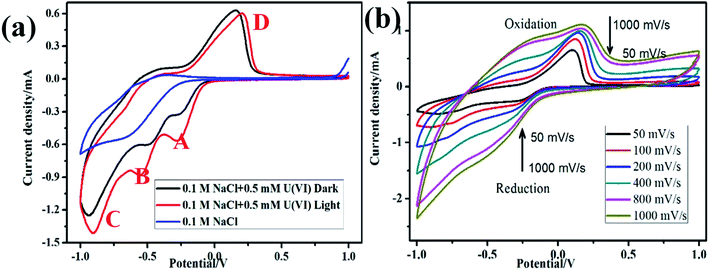 |
| | Fig. 2 Cyclic voltammogram of α-Fe2O3/TiO2 electrode on dark and light in 0.5 mM U(VI) + 0.1 M NaCl at pH 4.0 (a); cyclic voltammogram of U(VI) at different scan rates (b). | |
Fig. 2b presents the cyclic voltammograms of α-Fe2O3/TiO2 electrode in 0.5 mM U(VI) + 0.1 M NaCl solution measured at different scanning rates. A linear relations were identified between the cathodic/anodic peak current values and the square root of scan rates, which indicate that the electro-chemical reduction of U(VI) and the oxidation of U(IV) both were diffusion controlled process.31,32 The peak current ratio ipa/ipc > 1 (ipa and ipc are the anodic peak current and the cathodic peak current, respectively) indicating that the redox behavior of U(VI) is quasi-reversible by eqn (7).
| |
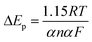 | (7) |
where
n is the number of transferred electrons,
α the electron transfer coefficient. The number of electrons transferred per redox change was fitted using
eqn (7) and the results show that
n equals 1.0, consistent with the one-electron reduction of U(
VI) → U(
V) on the α-Fe
2O
3/TiO
2 surface.
3.3 Organic acid mediated photoelectrochemical reduction of U(VI)
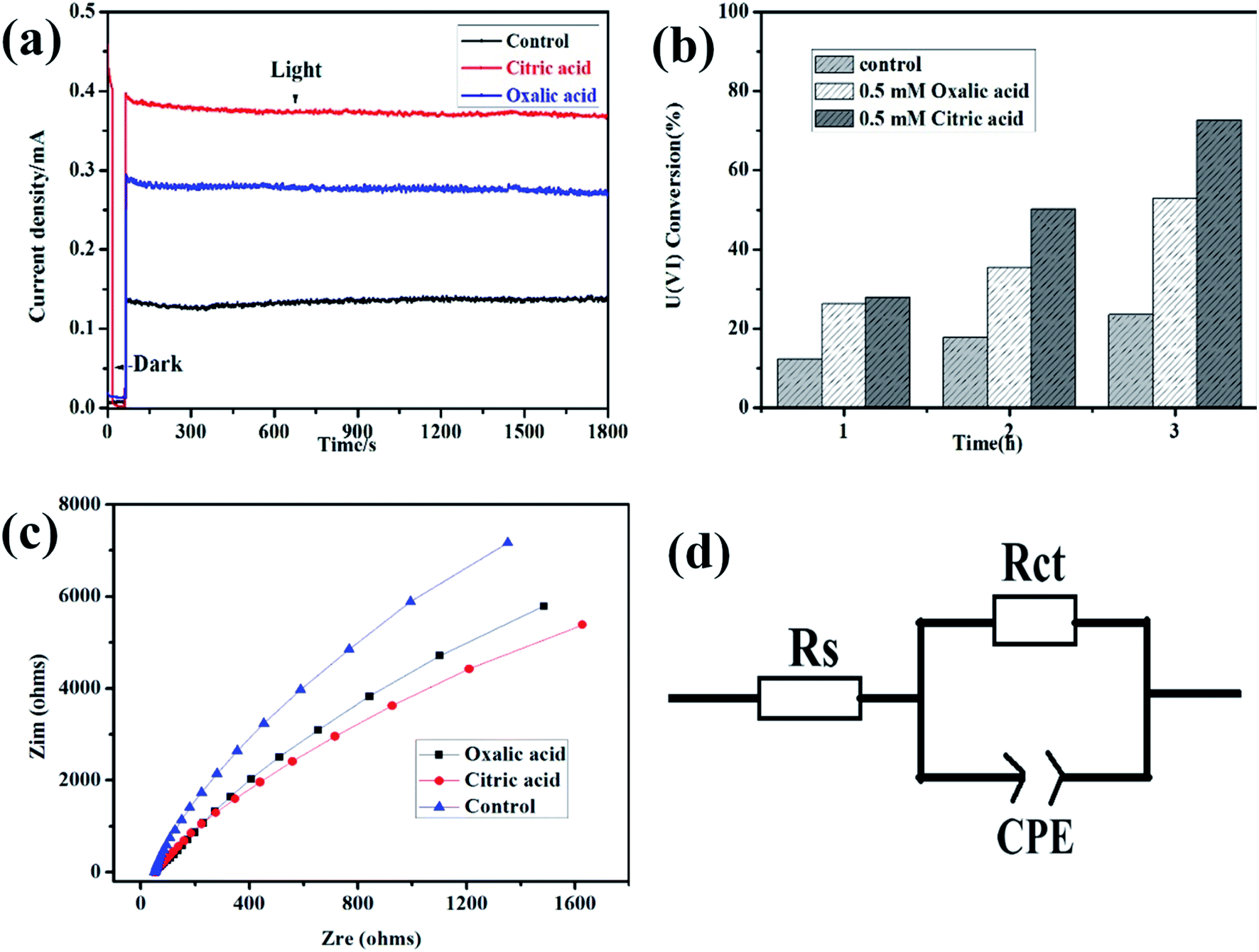
To better understand the effects of hole scavengers to the photoelectrochemical reduction of U(VI), citric acid and oxalic acid were added in the mixed solution. As shown in Fig. 3a, higher photocurrent was observed on α-Fe2O3/TiO2 film photoanode in the water–citric acid and oxalic acid mixed solution compared with that in the water solution, indicating enhanced U(VI) reduction efficiency on counter electrode. Due to adding citric acid and oxalic acid as hole scavenger, which can consume h+/˙OH, and a synergistic effect is expected in photocatalysis. This result was consistent with the above linear sweep voltammetry result. The effect of citric acid and oxalic acid on photoreduction of U(VI) by α-Fe2O3/TiO2 was evaluated, as shown in Fig. 3b. After illuminated for 3 h, the uranium remove capacity in the presence of 0.5 mM citric acid and oxalic acid reached 70% and 58%, respectively, while 24% was achieved for the system in absence acid, which indicates citric acid and oxalic acid greatly improved the photoelectrochemical reduction ability of U(VI). This may be because citric acid and oxalic acid as hole scavenge, which were oxidized by the photogenerated holes, thus reduce the recombination of photogenerated holes and electrons. On the other hand, citric acid and oxalic acid were formed a greater amount of radicals in this process, which enhances U(VI) photoreduction rate. Similarly, it has also been proven preliminary research work that the application of the photocatalytic method showed a high enhance for U(VI) in the presence of other hole scavenger including ethanol, methanol, sodium citrate and formic acid.33,34
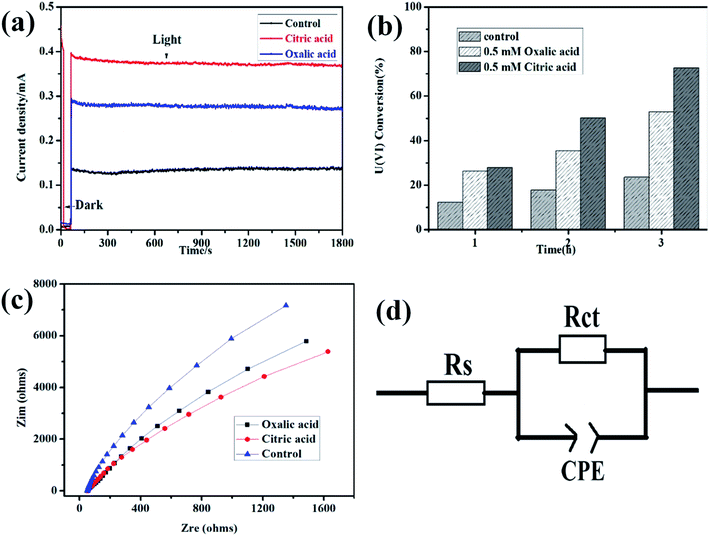 |
| | Fig. 3 Current–time plots (a); effects of hole scavengers to the photoelectrochemical reduction (b); electrochemical impedance spectra (c); the equivalent circuit (d) of α-Fe2O3/TiO2 electrode in 0.5 mM U(VI) + 0.1 M NaCl (pH 4.0). The control condition is absence hole scavengers in 0.5 mM U(VI) + 0.1 M NaCl (pH 4.0). | |
To confirm the possibility, the kinetics of U(VI) reduction at the interface of α-Fe2O3/TiO2 the film electrodes/electrolyte were evaluated by electrochemical impedance spectroscopy (EIS). The measured EIS spectra are shown in the form of Nyquist plots in Fig. 3c. In the presence of citric acid, the semicircle arcs are much smaller than oxalic acid, indicating faster PEC U(VI) reduction kinetics. The arcs are related to charge transfer at the interface of the electrode/electrolyte and the surface of electrode in this typical Nyquist plot. Fig. 3d shows the equivalent circuit for the α-Fe2O3/TiO2 film electrodes. In the equivalent Randle circuit, Rs is the solution resistance, Rsc is the space charge resistance, CPE are the constant phase elements for the electrolyte/electrode interface and electrode surface, respectively. The distribution of U(VI) species in solution is calculated by Visual MINEQL 3.0 based on Nuclear Energy Agency (NEA) database. The fitted values of Rs and Rct for the α-Fe2O3/TiO2 film electrode is calculated by Zsimwin based on the measured impedance data. The fitted values of Rs and Rct for the α-Fe2O3/TiO2 film electrode are 3943 Ω and 65.6 Ω, respectively, which are much lower than those oxalic acid and control for the α-Fe2O3/TiO2 film electrodes. This indicates that the citric acid has higher charge separation efficiency and faster charge transport rate for U(VI) reduction.35
3.4 Mechanisms of photoelectrochemical reduction of U(VI)
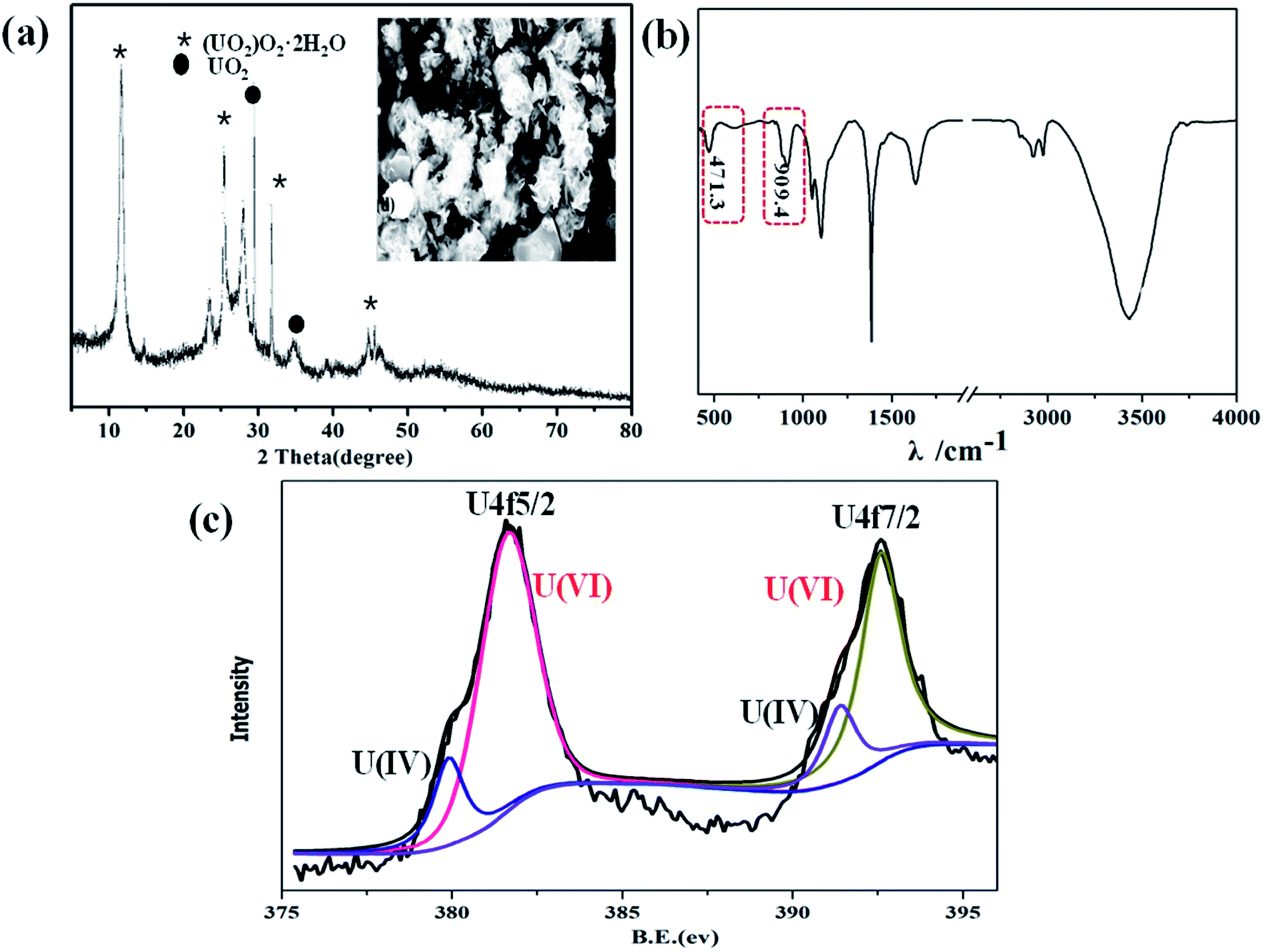
U(VI) was conversed to UO2+, U4+, or UO2 by photocatalytic reduction, which has been proven by previous research work.36,37 To elucidate the interaction mechanisms of U(VI) photoreduction over α-Fe2O3/TiO2, XRD, SEM, FT-IR and XPS were used to explore the possible photocatalytic reduction mechanism. For assessing the reduction product of U(VI), XRD patterns were employed to obtain an inside view into the formed uranium species (Fig. 4a). Under irradiation, obvious diffraction peaks were observed at 12.0°, 28.1°, 32.4°, and 46.8° which identified as metastudtite (UO2)O2·2H2O (PDF-35-0571). The diffraction peaks of UO2 (PDF-46-0952) were observed in the XRD pattern of film. The same results were also observed by previous research, in which photoelectrochemical and electrochemical reduction U(IV) precipitates as UO2, U3O8 and (UO2)6O2(OH)8·6H2O.21,38,39 Furthermore, the micromorphology of the samples after photocatalytic reactions were further researched by SEM, as shown inset of Fig. 4a. Obviously, abundant small nanoparticles were deposited after UV illumination, which indicating that U(VI) are photoelectrochemically reduced to U(IV), and form solid U(IV)-based complexes. FTIR is employed to further investigate the functional groups of U(VI) reduction in Fig. 4b. The FTIR spectra of tetravalent and hexavalent uranium presented a characteristic peak between 400 and 620 cm−1 and 800–1100 cm−1, respectively.40 The results indicate the reduction of U(VI) and U(IV). XPS analysis was further performed to identify the oxidation states of the accumulated uranium (Fig. 4c). The position of the satellite peaks on the high binding-energy side of the U 4f primary peaks is the key criterion to distinguish the oxidation states of uranium from 6+, 5+, and 4+. It is notable that new peaks which appeared at 382.2 eV and 393.0 eV were attributed to U 4f7/2 and U 4f5/2 photoelectrons, correspondingly, indicated the successful photoreduction of U(VI) and U(VI). Among them, the peaks at 381.4 eV and 391.5 eV are the characteristic peaks of U(VI), while the peaks at 382.2 eV and 392.7 eV correspond to the formation of U(IV). The results from XPS suggest that the uranium reaction product is most likely a U(VI) and U(IV) oxidation state. The XPS and XRD results both indicate the photoconversion of metaschoepite precipitates into UO2. Interestingly, similar results were reported in almost all previous studies, where mixed uranium states were finally formed in U(VI) photoreduction.14,41
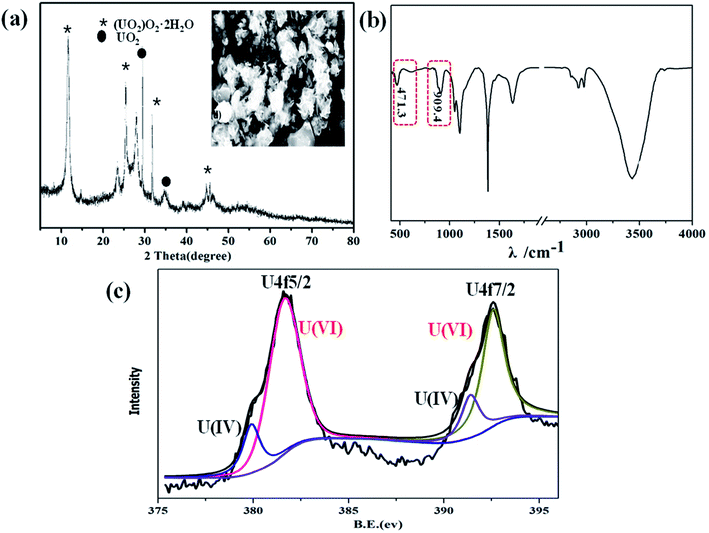 |
| | Fig. 4 XRD ((inset SEM image of the U(VI) reduction)) (a); FT-IR (b); U 4f spectra (c) of U(VI) reduction. | |
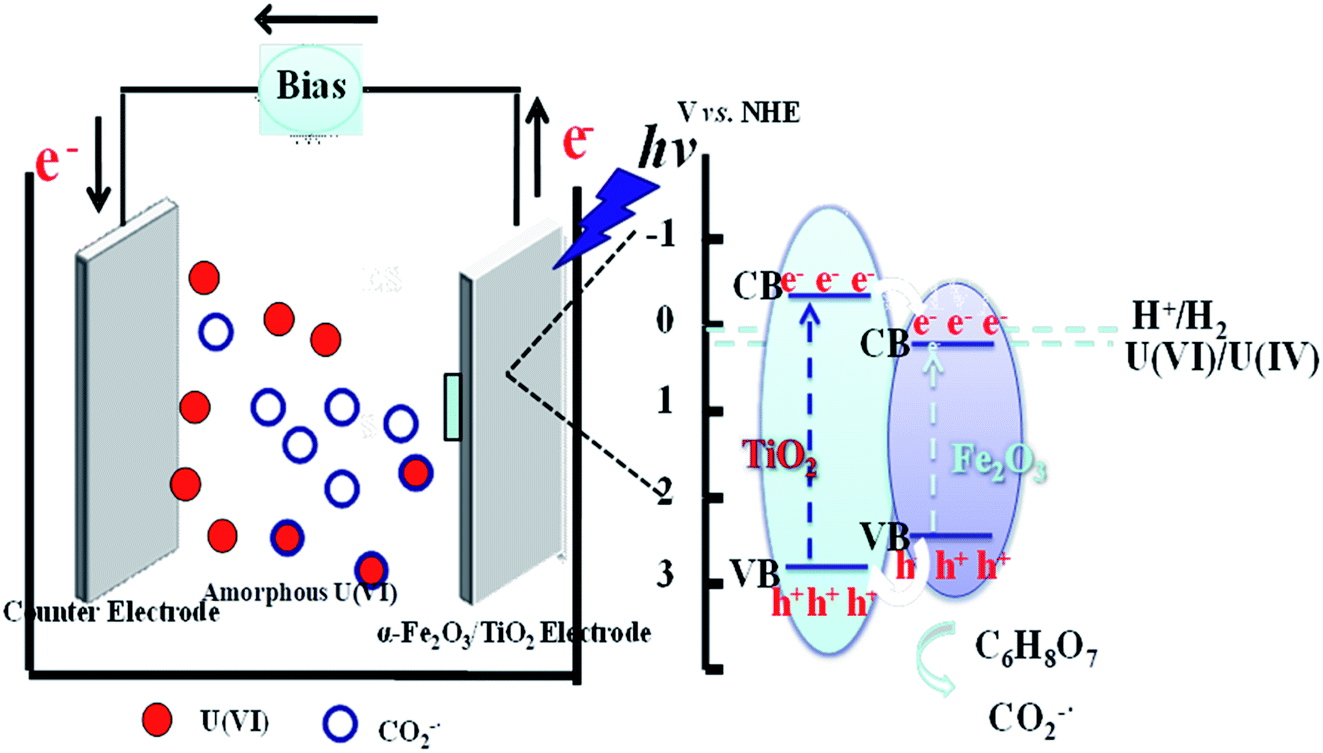
Base on the above results, a plausible mechanism for the photocatalytic reduction of U(VI) was proposed, as shown in Fig. 5. The photoreduction of U(VI) can be described as follows (see reactions (8–12)). Under visible light agitation, α-Fe2O3/TiO2 is excited and the electron–hole pairs are generated (eqn (8)). Then the reduction of U(VI) is achieved by the photogenerated electrons on the conduction band of α-Fe2O3/TiO2 via eqn (9). Subsequently, the holes may oxidize water to give hydroxyl radicals (eqn (10)). Meanwhile, the citric acid can be formed a greater amount of CO2−˙ radicals by photogenerated holes in this process (eqn (11)). The reduction potentials of UO22+/UO2 and UO22+/U4+ are reported to be 0.411 and 0.327 V vs. SHE, respectively.42 High reductibility CO2−˙ [E0(CO2−˙/CO2) = −1.8 V vs. NHE] are advantageous for the reduction of U(VI) to U(IV) (eqn (12)).43
| | |
α-Fe2O3/TiO2 + hv → h+ + e−
| (8) |
| | |
h+ + C6H8O7 + 5OH·→ 6CO2−˙ + 13H+
| (11) |
| | |
U(VI) + CO2−˙ → U(IV) + CO2↑
| (12) |
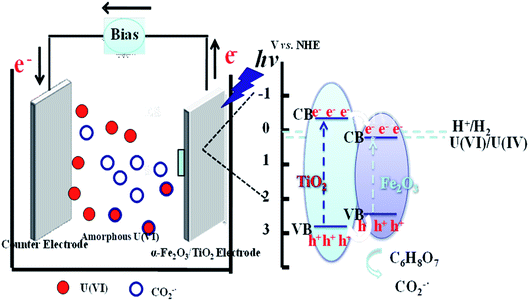 |
| | Fig. 5 Proposed mechanism for photoelectrochemical reduction U(VI) of α-Fe2O3/TiO2 electrode. | |
Therefore, the photoelectrochemical reduction mechanism might be that U(VI) was reduced to form U(IV) by photogenerated electron or CO2−˙, and formed (UO2)O2·2H2O deposits on the FTO cathode.
4. Conclusions
In this study, U(VI) photoreduction on α-Fe2O3/TiO2 was feasible and U(VI) could be partly reduced to U(IV) from wastewater. The batch experiments showed H+ would inhabit U(VI) photoreduction by competing for the photogenerated e−. Citric acid can accelerate U(VI) photoreduction by consuming photogenerated holes. Obviously, compared with traditional photoreduction methods, uranium extraction via photoelectrochemical reduction is more efficient, which has a significantly higher capacity. In similarity, citric acid can promote U(VI) photoreduction under acidic conditions, which has a synergistic effect. In addition, the formation of secondary mineral phase was provided additional reactive sites for U(VI) reduction according to XRD and XPS analysis. These observations are crucial for applying α-Fe2O3/TiO2 composites into the immobilization and preconcentration of radionuclides in environmental cleanup.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the basic Research Programs of Mianyang Normal University (QD2020A05). National Basic Research Program of China (973 Program: 2014CB846003).
References
- K. M. Campbell, J. T. Gallegos and E. R. Landa, Appl. Geochem., 2015, 57, 206–235 CrossRef CAS.
- L. Newsome, K. Morris and J. R. Lloyd, Chem. Geol., 2014, 363, 164–184 CrossRef CAS.
- L. Peng, P. Chen and G. Wang, Chem. Eng. J., 2020, 393, 124819 CrossRef.
- S. Szenknect, A. Mesbah and M. Descostes, J. Hazard. Mater., 2020, 392, 122501 CrossRef CAS PubMed.
- K. H. Williams, J. R. Bargar and J. R. Lloyd, Curr. Opin. Biotechnol., 2013, 24(3), 489–497 CrossRef CAS PubMed.
- Y. Wang, M. Frutschi and E. Suvorova, Nat. Commun., 2013, 4, 2942 CrossRef PubMed.
- S. A. McMaster, R. Ram and S. B Hargava, Miner. Eng., 2015, 81, 58–70 CrossRef CAS.
- J. Xiao, W. Song, R. Hu, L. Chen and X. Tian, ACS Appl. Nano Mater., 2019, 2, 385–394 CrossRef CAS.
- L. Tan, Y. Wang, Q. Liu, J. Wang, X. Jing, L. Liu, J. Liu and D. Song, Chem. Eng. J., 2015, 259, 752–760 CrossRef CAS.
- L. Newsome, K. Morris, S. Shaw, D. Trivedi and J. R. Lloyd, Chem. Geol., 2015, 409, 125–135 CrossRef CAS.
- Z. Niu, X. Wei, S. Qiang, H. Wu, D. Pan, W. Wu and Q. Fan, Chemosphere, 2019, 251, 945–951 Search PubMed.
- Y. K. Kim, S. Lee, J. Ryu and H. Park, Appl. Catal., B, 2015, 163, 584–590 CrossRef CAS.
- Y. Li, A. Lu and C. Wang, Acta Geol. Sin. (Engl. Ed.), 2009, 83(3), 633–639 CrossRef CAS.
- P. Li, J. Wang, Y. Wang and J. Liang, Chem. Eng. J., 2019, 365, 231–241 CrossRef CAS.
- S. R. Yan, T. Gholami, O. Amiri, M. S. Niasari, S. Seifi, M. Amiri, M. Sabet and L. K. Foog, J. Mol. Liq., 2021, 828, 154–163 Search PubMed.
- Z. Zhu, K. Wei, H. Li, X. Li, B. Li, X. Gu, L. Chen, J. Zhou, X. Pan and Y. Wang, J. Phys. D: Appl. Phys., 2021, 54(9), 94–105 CrossRef.
- T. T. H. Tran, H. Kosslick, A. Schulz and Q. L. Nguyen, Adv. Nat. Sci.: Nanosci. Nanotechnol., 2017, 8, 1–9 Search PubMed.
- K. P. Shejale, S. Saxena and S. Shukla, Mol. Syst. Des. Eng., 2020, 5, 797–803 RSC.
- P. Wang, F. Dong, M. Liu, H. He, T. Huo, L. Zhou and W. Zhang, Environ. Sci. Pollut. Res., 2018, 25, 22455–22463 CrossRef CAS PubMed.
- E. Raivis, A. Zukuls, R. Viter and A. Sutka, Photochem. Photobiol. Sci., 2020, 19, 1072–1077 CrossRef PubMed.
- N. Denisov, J. E. Yoo and P. Schmuki, Electrochim. Acta, 2019, 319, 61–71 CrossRef CAS.
- H. He, M. Zong, F. Dong, P. Yang, G. Ke, M. Liu, X. Nie, W. Ren and L. Bian, J. Radioanal. Nucl. Chem., 2017, 313, 59–67 CrossRef CAS.
- H. He, S. P. Berglund, A. J. Rettie, W. D. Chemelewski, P. Xiao, Y. Zhang and C. B. Mullins, J. Mater. Chem. A, 2014, 2, 9371–9379 RSC.
- X. Nie, F. Dong, N. Liu, S. Sun, M. Liu and D. Zhang, Appl. Surf. Sci., 2015, 347, 122–130 CrossRef CAS.
- N. Zhang, L. Y. Yuan, W. L. Guo, S. Z. Luo, Z. F. Chai and W. Q. Shi, ACS Appl. Mater. Interfaces, 2017, 9, 25216–25224 CrossRef CAS PubMed.
- J. Li, Z. Wu, Q. Duan, A. Alsaedi, T. Hayat and C. Chen, J. Cleaner Prod., 2018, 204, 896–905 CrossRef CAS.
- S. Yu, X. Wang, Y. Liu, Z. Chen, W. Yihan, Y. Liu, H. Pang, G. Song, J. Chen and X. Wang, Chem. Eng. J., 2019, 365, 51–59 CrossRef CAS.
- M. Yang, H. He, J. Du, H. Peng and Y. Zhou, J. Phys. Chem. Lett., 2019, 10, 6159–6165 CrossRef CAS PubMed.
- S. R. Yan, T. Gholami, O. Amiri, M. S. Niasari, S. Seifi, M. Amiri, M. Sabet and L. K. Foog, J. Mol. Liq., 2021, 828, 154–163 Search PubMed.
- S. Kim and H. Park, RSC Adv., 2013, 3, 17551–17558 RSC.
- K. Yuan, E. S. Ilton, M. R. Antonio, Z. Li, P. J. Cook and U. Becker, Environ. Sci. Technol., 2015, 49, 6206–6213 CrossRef CAS.
- C. Adán, J. Marugán, E. Sánchez, C. Pablos and R. van Grieken, Electrochim. Acta, 2016, 191, 521–529 CrossRef.
- P. Li, J. Wang, Y. Wang, J. Liang, B. He, D. Pan, Q. Fan and X. Wang, Chem. Eng. J., 2019, 365, 231–241 CrossRef CAS.
- Y. Guo, L. Li, Y. Li, Z. Li, X. Wang and G. Wang, J. Radioanal. Nucl. Chem., 2016, 310, 883–890 CrossRef CAS.
- V. Oskoei, M. H. Dehghani, S. Nazmara, B. Heibati, M. Asif, I. Tyagi, S. Agarwal and V. K. Gupta, J. Mol. Liq., 2016, 213, 374–380 CrossRef CAS.
- C. Ke, C. Chen, X. Ren, A. Alsaedi and T. Hayat, Chem. Eng. J., 2019, 359, 944–954 CrossRef.
- D. L. Zhao, Q. Zhang, H. Xuan, Y. Chen, S. J. Feng, A. Alsaedi, T. Hayat and C. L. Chen, J. Colloid Interface Sci., 2017, 506, 300–307 CrossRef CAS PubMed.
- B. Hu, X. Guo, C. Zheng, G. Song, D. Chen, Y. Zhu, X. Song and Y. Sun, Chem. Eng. J., 2019, 357, 66–74 CrossRef CAS.
- H. Li, F. Zhai, D. Gui, X. Wang, C. Wu, D. Zhang, X. Dai, H. Deng, X. Su, J. Di, Z. Lin, Z. Chai and S. Wang, Appl. Catal., B, 2019, 254, 47–54 CrossRef CAS.
- J. G. Kim, Y. S. Park and Y. K. Ha, Int. J. Nucl. Energy Sci. Technol., 2009, 46, 1188–1192 CAS.
- Z. Dai, Y. Sun, H. Zhang, D. Ding and L. Li, Chemosphere, 2020, 254, 126671 CrossRef CAS PubMed.
- A. J. Bard, R. Parsons and J. Jordan, Standard potentials in aqueous solution, CRC Press, 1985, vol. 6 Search PubMed.
- M. Kouhnavard, S. Ikeda, N. A. Ludin, N. B. A. Khairudin, B. V. Ghaffari, M. A. Matteeridi, M. A. Ibrahim, S. Sepeai and K. Sopian, Renewable Sustainable Energy Rev., 2014, 37, 397–407 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra02505h |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Faqin Dong*b,
Dengliang He
a,
Faqin Dong*b,
Dengliang He