
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTowards a selective synthetic route for cobalt amino acid complexes and their application in ring opening polymerization of rac-lactide†
Andrés Castro Ruiz ,
Krishna K. Damodaran and
Sigridur G. Suman*
,
Krishna K. Damodaran and
Sigridur G. Suman*
Science Institute, University of Iceland, Dunhagi 3, 107 Reykjavik, Iceland. E-mail: sgsuman@hi.is
First published on 4th May 2021
Abstract
Catalysts based on cobalt amino acids and 2,2 bipyridine (bipy) present an attractive and cost-effective alternative as ring opening polymerization catalysts, yet this system remains underexplored despite the advantageous coordination properties of amino acids and bipy as ligands combined with the variety of accessible oxidation states and coordination geometries of cobalt. Here, metal complexes of type [Co(aa)2(bipy)] with amino acids (aa: glycine, leucine and threonine) as ligands are reported. The complexes were characterized spectroscopically (IR, UV-vis and 1H, 13C NMR for diamagnetic species), and by MS spectrometry and elemental analysis. The data reveal that the 2,2 bipyridine acts as a neutral bidentate donor coordinating to the metal ion through two nitrogen atoms and the amino acid acts as a bidentate ligand coordinating through the carboxylate and amino group forming a stable five membered ring and a pseudo-octahedral geometry around the Co center. The activity of the complexes for the ring opening polymerization (ROP) of rac-lactide is presented. The complexes are effective initiators for the ROP of rac-lactide (Kobs = 9.05 × 10−4 s−1) at 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 [rac-lactide]:[catalyst] 1 M overall concentration of lactide in toluene at 403 K.
:1 [rac-lactide]:[catalyst] 1 M overall concentration of lactide in toluene at 403 K.
Introduction
Biodegradable and biocompatible polyesters such as poly(lactic acid) (PLA) are a promising class of oxygenated polymers that have received considerable attention due to their essential properties in diverse applications, ranging from packaging, fibers for bone scaffolds and drug carriers in the controlled release of active compounds.1–5 PLA is most efficiently synthesized by ring opening polymerization (ROP) of rac-lactide (LA) using homogeneous metal complexes as initiators or catalyst.6 The most active and selective complexes contain metal centers of Al(III),7 Fe(II)8 In(III),9,10 Zn(II)11–13 and Y(III).14Cobalt based catalysts have been widely used in polymerization reactions and more recently as bio-inspired photocatalysts,15–22 but they are far less investigated in ROP of cyclic esters. Only a few examples are known of cobalt complexes as catalysts in ROP of LA. These systems include 2-guadinobenzimidazole, amino mono and bis(phenolate), as shown in Fig. 1, and related amino-ethoxide chelating ligands.23–26 These catalysts have exhibited good activity and well-controlled ROP of LA affording polymers with high molecular weight in absence of rigorously dry solvents and inert atmospheres, making them an interesting system for industrial application. Nonetheless, they underperform in terms of stereoselective control of PLA microstructure, in comparison with other systems, independently of the nature of donor atoms and substituents on the ligands.27
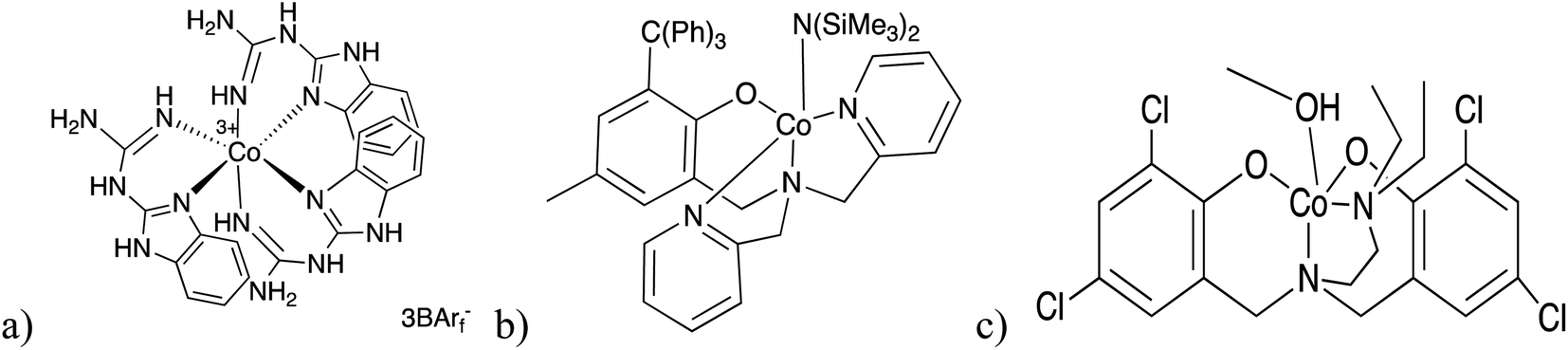 | ||
| Fig. 1 (a) Cobalt chelate complex of 2-guadinobenzimidazole reported by Gladysz and co-workers.26 (b and c) Efficient initiators cobalt complexes containing tripodal amino mono and bis(phenolate) ligand.23,24 | ||
Nature uses metalloenzymes as catalysts to convert small molecules selectively with outstanding efficiency. These biological catalysts often feature earth-abundant metals such as Fe, Zn or Co surrounded by protein scaffolds whose incredible performance is attributable to the dynamic coordination environment provided by the first and outer coordination sphere.28,29 Cobalt is one of the few transition metals identified with significant bio-essential organometallic chemistry, e.g. vitamin B12 and related coenzymes.28,30,31 Taking inspiration from nature, increased interest in the synthesis of coordination compounds constructed from biologically based ligands such as amino acid offers advantages such as intrinsic chirality, structural diversity and diverse coordination capabilities making them an attractive class of bio-ligands for the development of new compounds.32,33 The heterocyclic diamine 2,2′-bipyridine (bipy) and its derivatives provide important functions in coordination and organometallic chemistry due to its strong chelation property and π-accepting effects.34,35 Moreover, several reports of coordination complexes feature bipy in various applications including biological studies and ROP.35,36 Early studies reported high reactivity and stereocontrolled ROP of lactide mediated by a chiral amino acid as an organocatalyst.37 Over the last decade research on metal-amino acid compounds has focused on the formation of metal organic framework (MOFs)38 that exhibit interesting applications in research field such as CO2 capture,39 gas separation, and catalysis.40,41 However, applications of chiral catalysts with single a hexacoordinated metallic center bearing amino acids and bipy as ligands have been limited due to lack of vacant coordination site that is a crucial feature in ring opening polymerization reaction catalysts. Nevertheless this limitation may be circumvented by employing ligands theoretically predicted to be labile in solution.42–45
Capitalizing on precedence of successful cobalt based catalysts in ring opening polymerization of rac-lactide, a series well-defined mononuclear cobalt complexes containing bipy and commercially available amino acids as ligands were synthesized. In that context, this work describes selective synthesis and detailed characterization of chiral amino acidato bis(2,2- bipyridine)cobalt(II)/(III) complexes. For clarification, separations of geometrical isomer mixtures along with synthesis of analogous amino acid complexes using heterocyclic diamines as ligands the reader is referred to previously reported works in this field.46–49 As previously stated by Yasui et al., three diastereoisomers are possible for [Co(aa)2(bipy)] complexes which have achiral amino acidato ligand and possible six diastereoisomers if optically active amino acids are used.46,47 Hartshorn and co-workers48 established that a solvent dependence in the equilibria will exist between any secondary reaction products and selective formation of [Co(aa)2(bipy)]+ when the reaction is performed in aqueous or non-aqueous solvents. Also, results from electrochemical studies of these types of compounds suggest to carry out this reaction under kinetic control in the presence of a strong oxidant such as dioxygen in aqueous solutions if cationic species [Co(aa)2(bipy)]+ is desired. Based on these observations a synthetic route in an efficient yield towards neutral complexes was designed and different approaches were attempted. In the syntheses, optimization of reaction parameters such as temperature, solvent ratio and order of reagent addition was performed in order to control distribution and formation of thermodynamic or kinetic reaction side-products. This work hypothesizes that an electron rich cobalt center favors coordination-insertion mechanism and that the sigma donor atoms of the amino acids may provide an electronic environment around the metallic center increasing the cobalt center Lewis acidity.50 Furthermore, the side chains such as hydroxyl/alkyl in the outer coordination sphere may have synergistic effects on the catalytic performance and activity of these compounds.42,51 With this in mind, to the best of our knowledge, this is the first intentional application of octahedral stereogenic cobalt amino acid complexes employed as a chiral catalyst in ring opening polymerization of rac-lactide.
Results and discussion
Optimization of parameters and synthesis
The synthetic strategies employed are illustrated in the Scheme 1. The major interest in the synthesized cationic species is their use as a spectroscopic benchmark for the subsequent preparation and characterization of neutral Co(II) analogous complexes.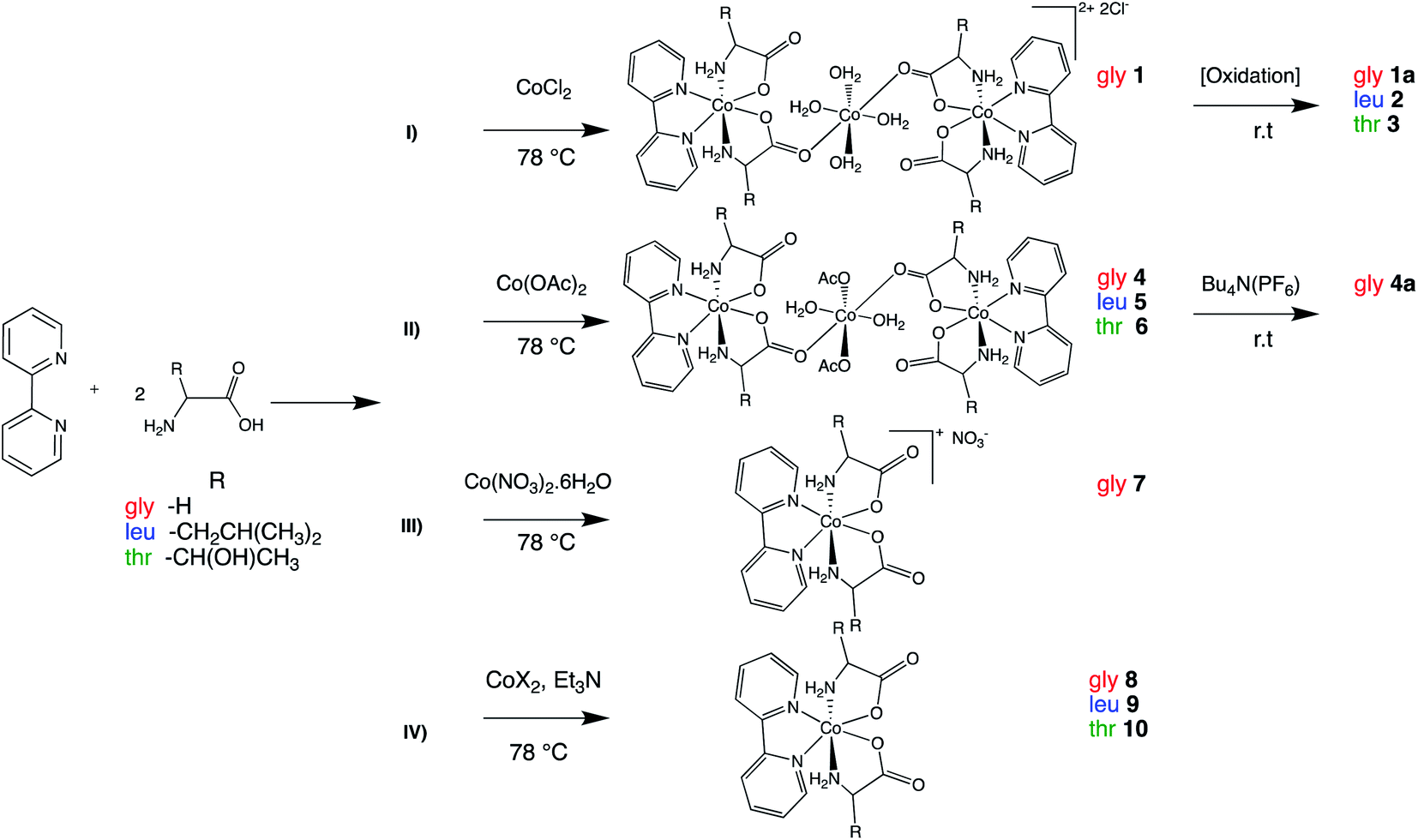 | ||
| Scheme 1 Synthetic routes (I–IV) for [Co(aa)2(bipy)] 1–10. | ||
Cobalt amino acid complexes were synthesized using a one-pot synthesis that consisted of 1:2 ratios of bipy, and amino acid and with a subsequent addition of 1 equivalent of CoX2 (X = Cl−, OAc−, NO3−) in aqueous ethanol. The complexes isolated were trinuclear Co(II) complexes with the formula [Co(H2O)2(X)2{trans(N)-[Co(aa)2(bipy)]}2]Y2 (X = H2O, Y = Cl; 1, X = OAc; 4–6) (bipy: 2,2′ bipyridine; aa: glycine, leucine or threonine), cationic species trans(N)-[Co(aa)2(bipy)]X (X = Cl; 1a–3, X = PF6−; 4a, X = NO3−; 7) and the neutral species 8–10.
Synthesis of 1 at room temperature followed by slow addition of CoCl2 led to formation of a mixture of products, which were cis-[Co(bipy)2Cl2]52 as the major product, and glycinato complex with trans-(N) geometry as a secondary product, as well as uncomplexed amino acid and bipy in minor quantities. The products were confirmed using 1H NMR or HRMS-ESI analysis. This observation is in good agreement with stability constants of Co(II) complexes in aqueous solution with bipy (logβ3 = 17.6) and glycine (logβ3 = 10.8).44,53,54 In view of theoretical and experimental evidence it is reasonable to expect that the equilibrium kinetically favors the formation of cis-[Co(bipy)2Cl2].48 Therefore, it was of interest to evaluate the influence of temperature on the formation of trans-(N) isomer.55 Assuming the reaction rate for the coordination of bipy to Co(II) is faster than the in situ deprotonation and subsequent N,O chelation of glycine; increasing the temperature and employing rapid addition of CoCl2 may reduce the formation of cis-[Co(bipy)2Cl2]. Indeed, this approach was found to readily form two of the desired neutral glycinato complexes with trans-(N) geometry isomer units linked via tetra-aqua cobalt bridge, with two chloride counter anions. Hence, the formula of 1 can be expressed as [Co(H2O)4{trans(N)-[Co(gly)2(bipy)]}2]Cl2 that was confirmed by structural analysis using X-ray crystallography. Similar reaction followed by air oxidation led to isolation of 1a, 2 and 3 (Scheme 1(I)) that were also spectroscopically characterized.
As part of the synthesis optimization process, employing cobalt acetate also resulted in successful synthesis of trimetallic complexes 4–6 (Scheme 1(II)). The compound compositions were confirmed by microanalysis of the pink solids obtained. Aerobic reaction conditions during an ion exchange reaction of 4 with (Bu4N)(PF6) or a complexation reaction using a cobalt salt with non-coordinating counter-anion such as NO3− resulted in cationic species 4a and 7 (Scheme 1(II) and (III)), respectively.
These experiments resulted in highly reproducible route in good yields to form tri-metallic species. The complexes are air-sensitive and minor air exposure is sufficient to complete oxidation of Co(II) to Co(III).
From a practical perspective, an analogous procedure using CoX2 (X = Cl, OAc) and triethylamine was carried out in order to evaluate the reactivity of cobalt with other amino acids and bypass the formation of both tri-metallic and ionic compounds. The neutral complexes (8–10, Scheme 1(IV)) were isolated as pale pink powders in reasonable >60% yield and were characterized by FT-IR and with high resolution mass spectrometry (HRMS-ESI). The results from this approach indicate that temperature control and the base employed are two major factors that influence whether the trans-(N) isomer or any other intermediate species is formed. The neutral complexes are stable as solids under ambient conditions but react slowly with oxygen in the air. In solution 8–10 appear stable for about 24 hours, and after this time an oxidation is observed through a color change of their solutions from pinkish-orange to brownish-red.
Characterization of solid complexes
An ellipsoidal plot of 1 at 50% probability is shown in Fig. 2. The structures of 2 and 9a share same cation; however, the two structures were obtained from different synthetic routes and resulted in crystallization of a chiral molecule 2 and achiral molecule 9a. The ellipsoidal plot of 2 is given in Fig. 3. Complex 9a crystallized in a triclinic crystal system, compared to a monoclinic crystal system for direct crystallization of 2. The structure of 9a has a disorder that appears solvent dependent and was not completely refined. Fig. S1† depicts the connectivity of the molecule with two crystallographically independent molecules in the asymmetric unit.
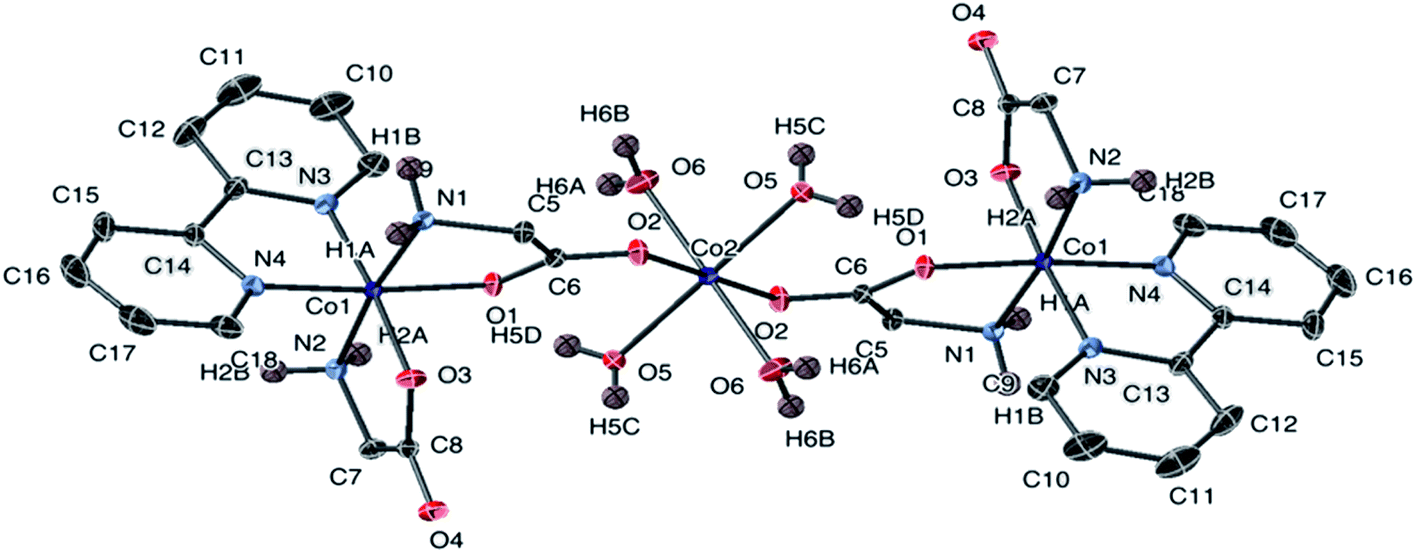 | ||
| Fig. 2 The structure of the cation of 1 with ellipsoids at 50% probability. Non-amine bound hydrogen atoms are omitted. The counter anion is Cl− (not shown). | ||
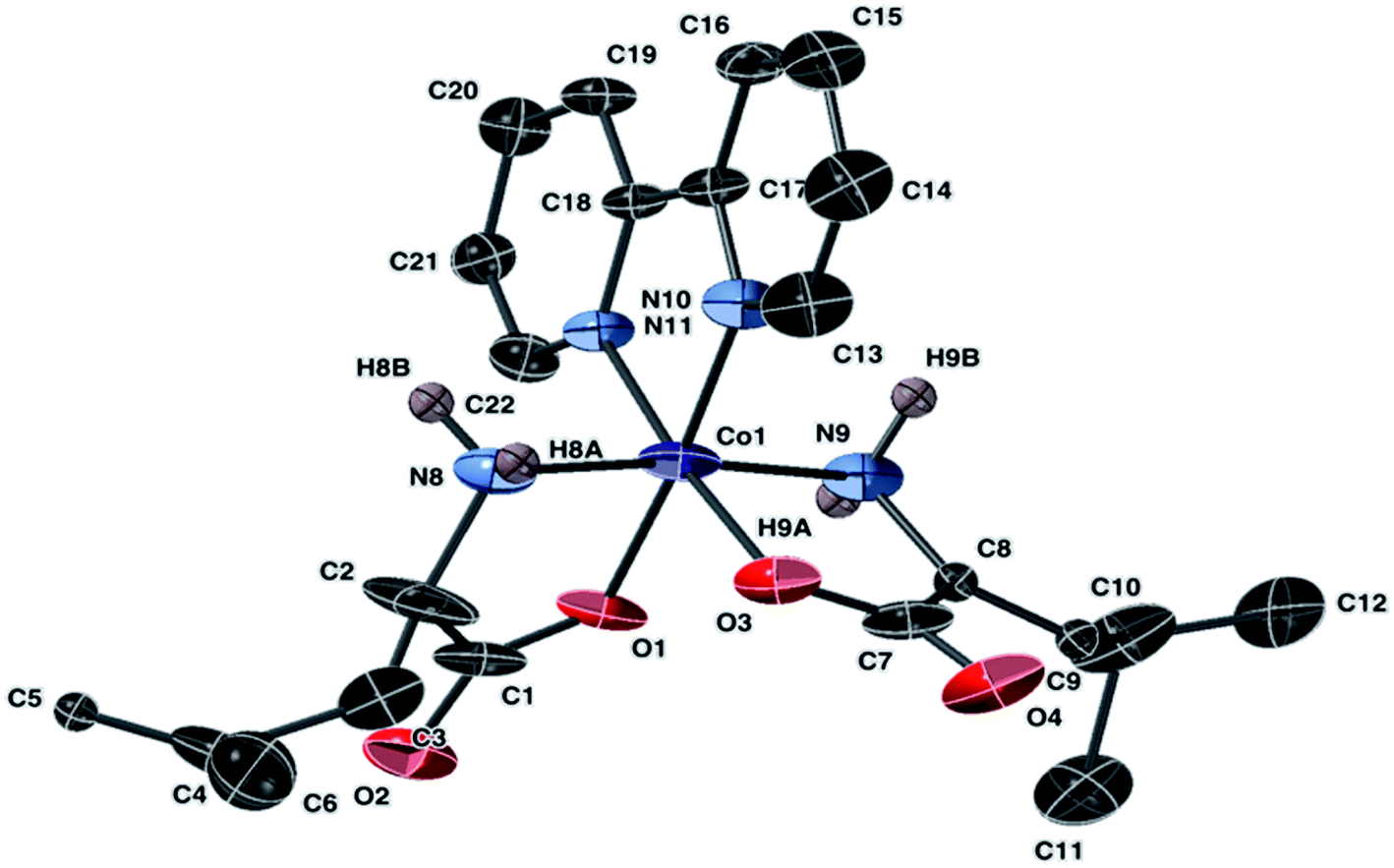 | ||
| Fig. 3 Molecular structure of the cation of 2, [Co(leu)2(bipy)]Cl, with ellipsoids at 50% probability. Non amine bound hydrogen atoms are omitted. The counter anion is Cl− (not shown). | ||
Complexes 1a, 4a, and 7 share a common cation. Fig. 4 and 5 depict ellipsoidal plots of 1a and 7. Figure depicting the ellipsoidal plot of 4a was placed in the ESI Fig. S2.†
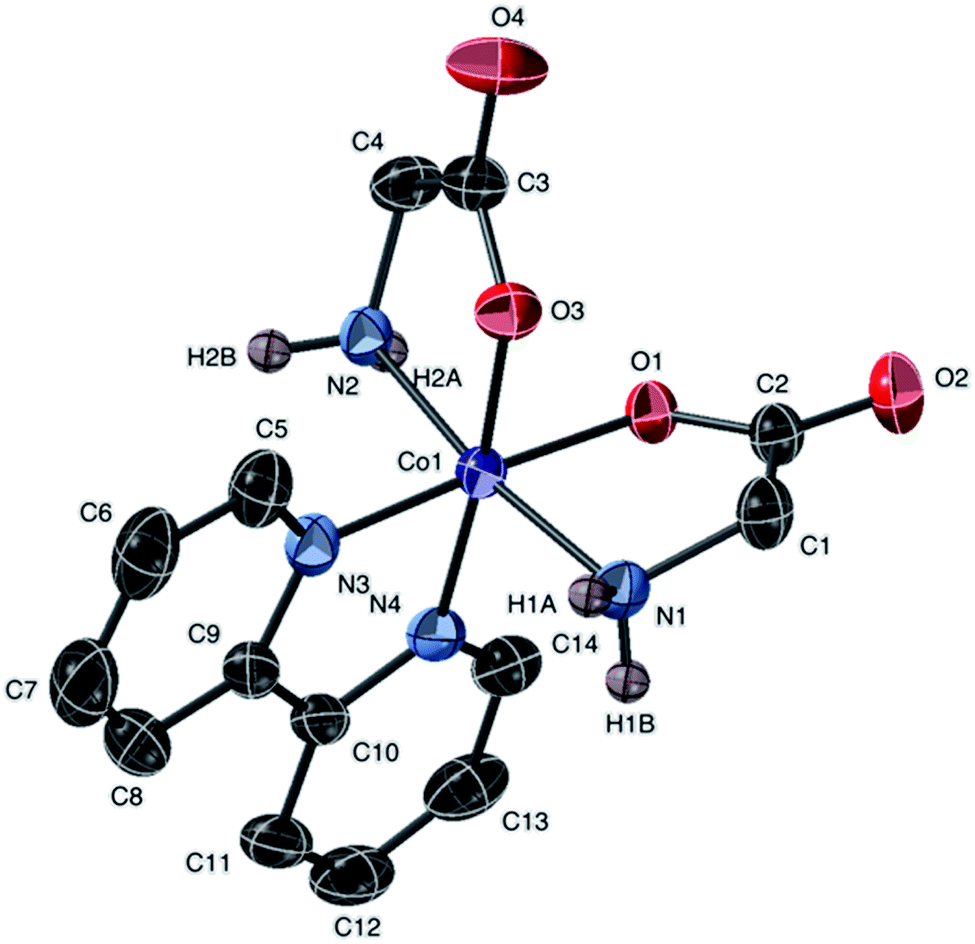 | ||
| Fig. 4 Molecular structure of the cation of 1a, [Co(gly)2(bipy)]Cl, with ellipsoids at 50% probability. Non amine-bound hydrogen atoms are omitted. The counter anion is Cl− (not shown). | ||
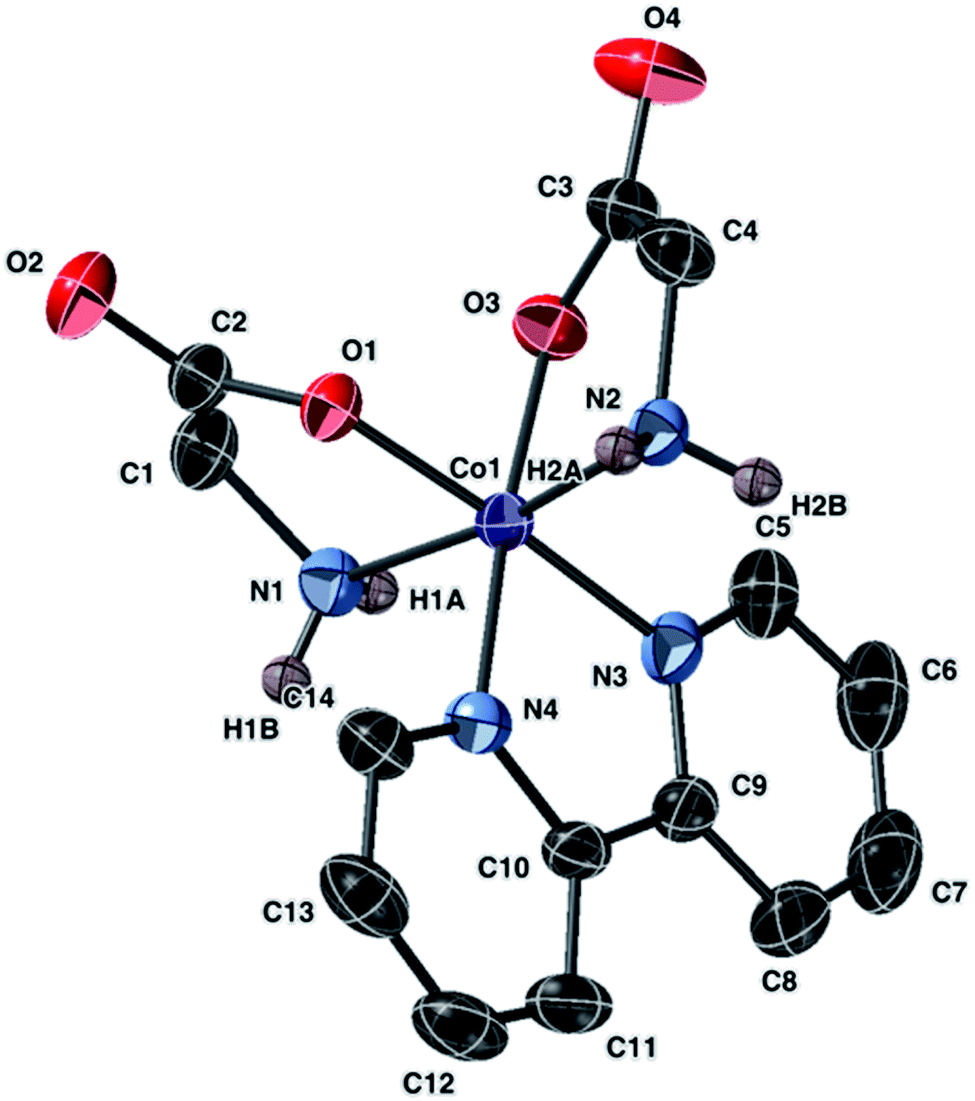 | ||
| Fig. 5 Molecular structure of the anion of 7 with ellipsoids at 50% probability. Non amine-bound hydrogen atoms are omitted. The counter anion is NO3− (not shown). | ||
A side product, Co(bipy)2Cl2 was also crystallized (ESI Fig. S3†) and the structural analysis revealed the formation of Co(bipy)2Cl2, which was crystallized in monoclinic P21/c. The structure of Co(bipy)2Cl2 in the solvated form [Co(bipy)2Cl2]·3H2O has been reported.52 Therefore, details regarding the crystal data and selected bond lengths and angle are given in Tables S3 and S10.†
Complex 1 was crystallized from aqueous acetone. The X-ray analysis of 1 revealed a trinuclear structure. Two [Co(gly)2(bipy)] complexes are bridged by tetraaquacobaltate(II) chloride [Co(H2O)4]Cl2 molecule co-crystallized in the lattice, as shown in Fig. 2. The Co(II) center adopts hexacoordinated {CoO6} in octahedral geometry between two oxygen atoms of the carbonyl of glycine and the other four positions occupied by oxygen atoms belonging to the four water molecules. The structure shows two hexacoordinated Co(II) center on each side adopting slightly distorted octahedral geometry. Each Co(II) center is coordinated by two nitrogen atoms and two oxygen atoms from the α-amino acid and two nitrogen atoms from 2,2-bipyridine respectively, forming a monomeric unit with three five-membered chelate rings. Selected bond lengths and bond angles values obtained for this complex were found to be in a good agreement with previously reported cobalt complexes of the same category.56–58
Complex 2 crystallized in a monoclinic space group C2/c. The Co(III) center has a distorted octahedral geometry with the leucine ligands arranged in a trans-N,N-cis-O,O coordination. The leucine ligands exhibited disorder that was addressed during refinement. Crystals of 9a revealed a Co(III) center and an overall formula identical to 2. The data obtained for 9a appears accurate but contains disordered leucine molecules. The structure was solved but the R-factor was high even after the removal of the disordered dichloromethane molecule. Therefore, the data for 9a is presented in the ESI.† Bond lengths and angles in the molecular unit appear similar for 9a and for 2. Co–N bond lengths in 2 and 9a fall within the range of 1.959 Å to 1.919 Å, and Co–O bond lengths in the range of 1.869 Å to 1.898 Å.
Complexes 1a and 7 exhibit comparable bond lengths and angles around the Co(III) center. They crystallized in different space groups presumably resulting from the different hydrogen-bond-acceptor strengths of counter anions employed. The glycine bite angles (NH2–Co–O) fall within the range of 84.77° to 86.40° for all four glycine ligands in 1a and 7. Coordination geometry in both crystals is trans-N,N–cis-O,O with the oxygen atoms of the glycine trans to the bipyridine nitrogen donor atoms. Crystals of 4a (Fig. S3†) have parameters in good agreement with that found for 1a and 7.
X-ray analysis revealed that 7 with the formula [Co(bipy)(gly)2](NO3) crystallized in an orthorhombic P212121 space group. The Co(III) metal center displayed a distorted octahedral geometry and the two glycinato anions and the bipyridine moieties were coordinated to the metal center in a chelate fashion. The axial positions were occupied by the nitrogen atoms of the glycinato ion, and the equatorial positions were occupied by the oxygen atoms of two glycinato and the nitrogen atoms of the bipyridine. The non-coordinated nitrate anion interacted with the amino groups via non-bonding interaction.
Infrared spectroscopy
Selected FT-IR data of complexes 8–10 are given in the Experimental section and in Fig. S22 –S24.† The three complexes showed broad and strong bands located at high frequencies of ca. 3400–3442 cm−1 that may be assigned to the OH stretches of water co-crystallized in the lattice. Complexes 8–10 revealed expected IR bands that are similar with significant differences related to the bands expressing differences in the amino acid side chain; an alkyl group at 2870–2957 cm−1 for 9 and hydroxyl group at 3390 cm−1 for 10. The bands observed in the range of 3280 cm−1 and 2870 cm−1 are designated to NH2 and CH stretching vibrations of amino acids, respectively. These values are slightly shifted to higher energy compared to free α-amino acids (3200–2500), confirming the N-chelation of the α-amino acids to cobalt. Two intense bands with broad profile and a maximum centered between 1640–1660 cm−1 and at 1451–1330 cm−1 correspond to the asymmetric νasym(COO−) and symmetric νsym(COO−) vibrations modes of carboxylate group, respectively.59,60 Thus, the difference between these vibration modes ca (∼290 cm−1) is indicative of monodentate coordination of the carboxylate. Moreover, the complete deprotonation of carboxylic acid was confirmed by the absence of a band normally observed at 1750 cm−1.61As far as bipy ligand is concerned, the vibrations which were attributed to the aromatic stretching vibrations ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) and ν(CN) were observed for complexes 8–10 at 1472–1500 cm−1, respectively. Medium absorption peak at ∼770 cm−1 was assigned to out-of-plane ν(C–H) modes of the ring, shifted to higher frequencies as compared with the free bipy. The IR spectra are consistent with the bidentate coordination of the bipy ligand and N,O-chelation of the amino acids to the metal center.
C) and ν(CN) were observed for complexes 8–10 at 1472–1500 cm−1, respectively. Medium absorption peak at ∼770 cm−1 was assigned to out-of-plane ν(C–H) modes of the ring, shifted to higher frequencies as compared with the free bipy. The IR spectra are consistent with the bidentate coordination of the bipy ligand and N,O-chelation of the amino acids to the metal center.
Characterization of complexes in solution
Conductivity, magnetic moment, and mass spectrometry
Neutral complexes 8–10 are highly soluble in polar solvents, hot toluene and slightly soluble in CH2Cl2. The molar conductance values of 8–10 at 298 K of 10−3 M in ethanol solution are between 11–15 Ω−1 cm2 mol−1, indicating non-electrolytic nature of the complexes. The magnetic moment of 8–10 was obtained using Evans' NMR method,63 giving an overall effective magnetic moment of 4.24, 4.70 and 4.40 B.M, respectively, which is consistent with three unpaired electrons for 3d7 Co(II) and high-spin electronic configuration.64 These values are slightly higher than the spin-only value reported for a high spin cobalt(II) centre (μSO = 3.87 μB, for S = 3/2), suggesting significant spin orbit coupling. Such orbital interactions are typically observed in high-spin Co(II) centers for N4O2 donor atoms sets,65 owing to the degeneracy of the triplet ground state, 4T1g.66 The complexes were further analyzed by high resolution ESI-MS in positive mode using MeOH as solvent (Fig. S27–S29†). Molecular cation peaks at m/z = 363.05, 475.18 and 451.10 were obtained for 8, 9, and 10, respectively, confirming the complexes composition.Electronic absorption spectra
Electronic spectral data for complexes 8–10 are listed in the Table 1. All spectra are quite similar in the UV region where strong ligand-based absorptions predominate. Fig. 6 shows two bands for complexes 8–10 in the 315–230 nm region. All the bands observed in this region exhibit bathochromic shifts compared to the free ligands. The high intensities bands arise from n–π* and (π–π*, LMCT) charge transfer transitions from donating groups of the ligands to the metallic center. Co(II) bands are Laporte forbidden transitions to doublet states and are assumed to be too weak to be visible. The d–d transition bands are rather weak compared to the pronounced n–π* and π–π* absorption bands, thus quantification of d–d transitions for complexes 8–10 was recorded separately at higher compound concentrations in EtOH (5 × 10−3 M). A low-energy absorption band centered at 480–500 nm is attributed to the transition (4T1g(F) 4T1g(P)) of the high-spin cobalt(II) ions in octahedral geometry.67| Compounds | ΛMa (Ω−1 cm2 mol−1) | λmax (nm) | (ε (L cm−1 mol−1)) | μeff (B.M) |
|---|---|---|---|---|
| a Molar conductance values at 298 K of 10−3 M ethanol solution Ω−1 cm2 mol−1.b Recorded in 10−3 M ethanol solutions. | ||||
| 8 | 12.9 | 299 | (7115) | 4.24 |
| 309 | (6698) | |||
| 501 | (36)b | |||
| 9 | 13.1 | 299 | (7740) | 4.70 |
| 308 | (7104) | |||
| 487 | (47)b | |||
| 10 | 13.9 | 301 | (7717) | 4.40 |
| 309 | (7648) | |||
| 488 | (35)b | |||
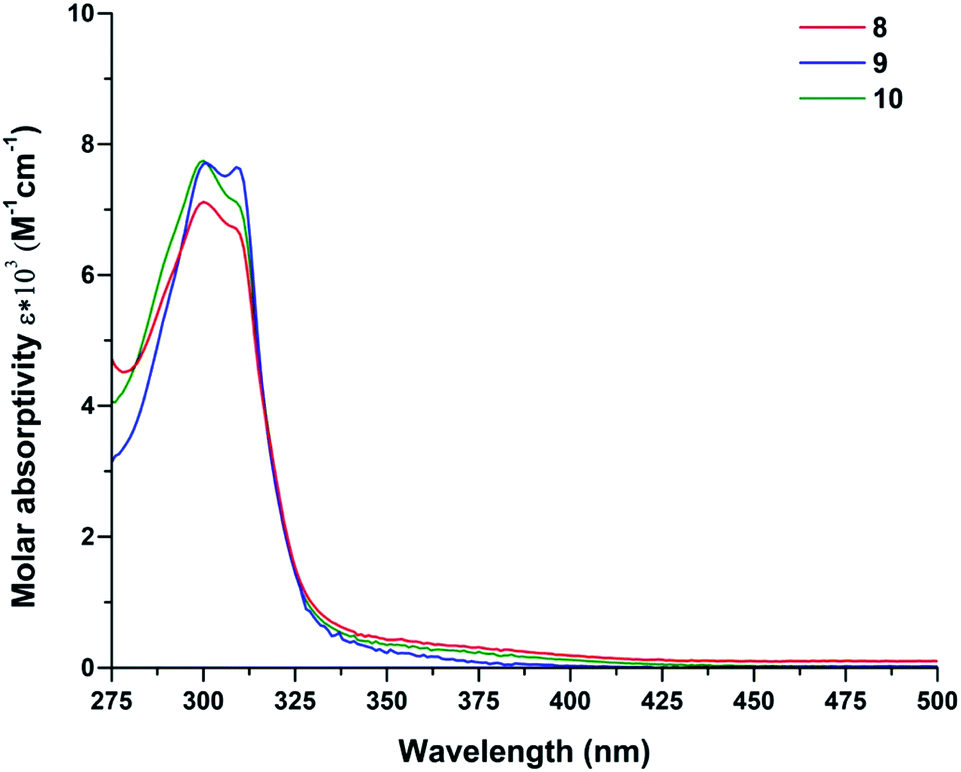 | ||
| Fig. 6 UV-vis absorption spectra of 8–10 in EtOH (5 × 10−5 M). | ||
Ring opening polymerization (ROP)
Complexes 8–10 were evaluated as catalysts in the ROP of rac-lactide (LA). Representative polymerization data are summarized in Table 2. The experiments were conducted using a standard set of conditions which were typically at 403 K in toluene solution ([cat]:[LA] 100:1). When required, benzyl alcohol (BnOH) was added as a co-initiator (1 equivalent, 3.5 μL). Comparison between 8–10 and other cobalt-catalyst system is difficult since different authors report catalyst performance under dissimilar range of conditions.24,26 All three complexes displayed high catalytic activity towards ROP of rac-lactide either in the presence of an exogenous alcohol or without.
| Entry | Catalyst | Time (min) | Mn,theoc (kg mol−1) | Mn,obsd (kg mol−1) | Pie | Đf | Conv.g (%) |
|---|---|---|---|---|---|---|---|
| a Reaction conditions: [LA]:[catalyst] = 100:1, [LA] = 1 M in toluene.b [LA]:[catalyst]:[BnOH] = 100:1:1, 403 K. Unless otherwise stated.c The theoretical molecular weight calculated from conversion {([LA]:[catalyst] × (% conv.) × 144.13)} or {([LA]:[catalyst] × (% conv./no. eq BnOH × 144.13)) + 108.14}.d Determined by GPC analysis calibrated using narrow MW polystyrene standards, in THF and corrected by Mark–Houwink parameter (0.58).e Determined by relative integration of PLA methine tetrads in the 1H{1H} NMR spectrum using values predicted according to Bernoullian statistics.68f Đ = Mw/Mn.g Determined by integration of the methine region of the 1H NMR spectrum at δ 4.98–5.06 ppm (LA) and δ 5.08–5.22 (PLA). |
|||||||
| 1 | 8 | 60 | 13.54 | 12.39 | 0.65 | 1.11 | 94 |
| 2 | 9 | 60 | 10.52 | 15.33 | 0.54 | 1.13 | 73 |
| 3 | 10 | 60 | 13.11 | 19.52 | 0.49 | 1.22 | 91 |
| 4 | 8 | 100 | 14.27 | 15.36 | 0.60 | 1.13 | 99 |
| 5 | 9 | 100 | 13.84 | 34.60 | 0.59 | 1.28 | 96 |
| 6 | 10 | 100 | 13.98 | 21.97 | 0.50 | 1.40 | 97 |
| 7b | 8 | 240 | 12.35 | 0.14 | — | 2.02 | 85 |
| 8b | 9 | 180 | 3.86 | 0.32 | — | 2.04 | 26 |
| 9 | 1a–3 | 240 | — | — | — | — | Traces |
In all cases, polymer kinetics showed a pseudo first-order dependence on rac-lactide concentration on polymerization rates as shown in Fig. 7, as is commonly reported for ROP of lactide proceeding via coordination–insertion mechanism.69,70 The pseudo first order rate coefficients (kobs) were obtained from the slopes of these plots. Comparison of the rate constants revealed that 8 and 10 are slightly faster than 9, which in terms of electronic influence of the ligands substituents implied that complexes containing neutral and electron-withdrawing side chain induced the polymerization more rapidly, reaching nearly complete conversion of the monomer in 1 hour (Table 2, entries 1–3), this observation agrees with previous reports where use of withdrawing groups was proposed to increase the Lewis acidity of the metal and consequently accelerate the polymerization reaction.71,72 However, greater activity of 8 with unsubstituted glycine ligand over 9 and 10 bearing leucine and threonine ligands (R = alkyl and hydroxyl group, respectively) may be tentatively attributed by decreased steric hindrance in the metal coordination sphere, increasing lability of the metal–carboxylate M–OR bond that subsequently increases the rate of lactide insertion.50 It is notable that all catalysts show a short induction period. Typically, it was 20 min, and the relative length was independent of the type of catalyst. It is observed that this time period incorporates the time required for the reaction solution to reach the desired temperature from room temperature and subsequently activate the catalyst.
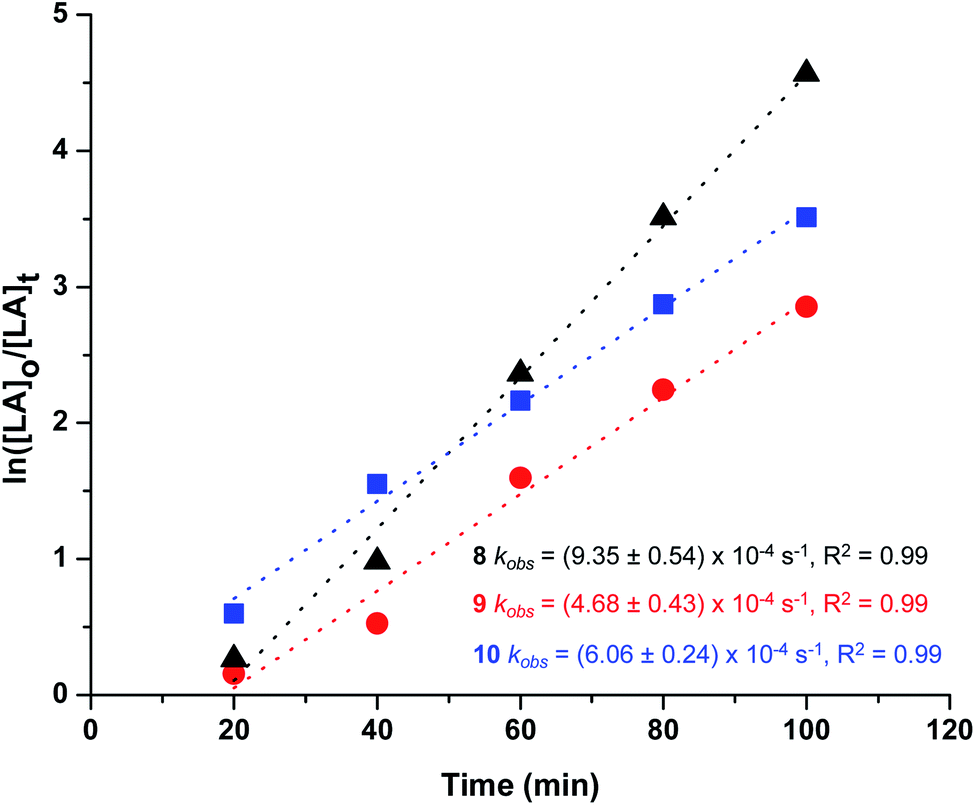 | ||
| Fig. 7 Pseudo first-order kinetic plot of ln([LA]0/[LA]t) vs. time for ROP of rac-lactide using complexes 8–10. | ||
Molecular weights of polymers obtained with 8 (Table 2, entries 1 & 2), as determined experimentally by GPC analysis i.e. (Mn,obs = 12.39 kg mol−1), were found to be in good agreement with theoretical values (Mn,theo = 13.59 kg mol−1) that were calculated assuming the growth of a single polymer chain per active center. This observation is indicative of a well-controlled polymerization. On the contrary, 9 and 10 produced PLA with higher molar mass than expected theoretically. This is most likely due to slow initiation rates relative to propagation rates, usually observed for poor nucleophilicity and sterically hindered initiating group.9,14 Alternatively, experiments were carried out incorporating one equivalent of BnOH which could undergo fast and reversible reaction exchange between free alcohol and ligands, improving the initiation rate efficiency (Table 2, entries 7 & 8). Interestingly, presence of an exogenous alcohol, was found to both increase polymerization times and impart a significant decrement on catalytic activity. The PLA so obtained showed large and bimodal molecular weight distribution (Đ) suggesting that BnOH negatively impacts control of the polymerization. Based on previous literature reports, it is also plausible that two competing mechanisms are active, promoted from two different initiating groups.73–75 Extended reaction times without incorporation of BnOH resulted in observed broadened polydispersities (Đ = 1.13–1.40, Table 2, entries 4–6), suggesting that chain transfer and intermolecular transesterification reactions may predominate at high conversion >60%, which could be ascribable to the presence of distinct amounts of water in commercial grade rac-lactide and the crystal water in the lattice of the complexes accelerating transesterification reaction. Due to the absence of initiator groups or vacant active site in the metal center a mechanistic hypothesis can be proposed to rationalize the formation of PLA as shown in Fig. 8. The initiator is thermally activated where the Co-carboxylate (Co–O) bond is labilized. Dissociation of the Co–O bond is followed by the coordination of the carbonyl oxygen of the monomer to the metal center, activating thereby it towards attack from the metal–carboxylate bond. Following by monomer insertion into the metal–organic bond formed, through the breakdown of the acyl-oxygen or alkyl–oxygen bond in the cyclic compound. Finally, the polymerization process occurs through concerted process with the consecutive insertion of the monomer molecules in the metal–oxygen bond (in this case Co–O) of the propagating species concurrent with acyl oxygen bond cleavage, termination step occurs with hydrolysis of chain end group in presence of excess of methanol.
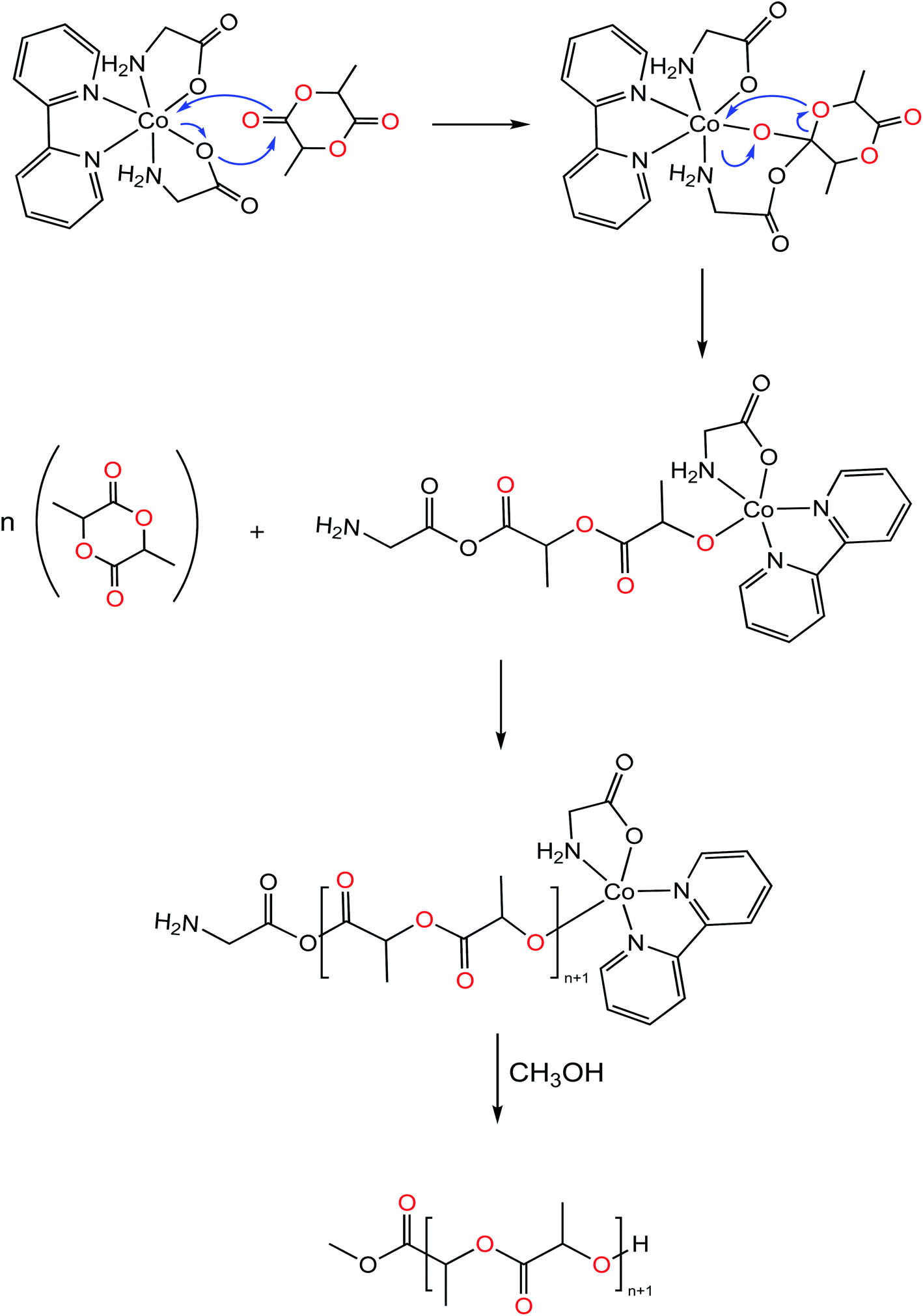 | ||
| Fig. 8 Proposed pathway for the ROP of LA catalyzed by [Co(aa)2(bipy)]. | ||
To gather further information on the polymer microstructure, tacticity of the polymers was assessed by relative integration of methine resonances in the homonuclear decoupled 1H{1H} NMR spectrum (Fig. S36–S38†) for PLA samples, and stereo-sequence (tetrads) probabilities were predicted according to Bernoullian statistical model.68,76 All catalysts induced at least moderate levels of stereocontrolled microstructure, displaying slightly isotactic bias in resulting polymers (Pi = 0.49–0.65). This observation was correlated to the high reaction temperatures used. As an attempt to enhance stereoselectivity, polymerization reactions were conducted at lower temperatures ≤100 °C which, however, did not result in polymer formation. Co(III) species 1a–3 resulted completely inactive for LA ROP presumably due to highly stable electronic environment around the metal center, this observation has been reported for other cationic complexes.77–79
Conclusion
The synthesis of a series of trimetallic, cationic and neutral cobalt complexes with bipy and amino acids as ligands successfully led to isolation and characterization of several Co(II) and Co(III) complexes. Complex 1 was obtained and characterized in the solid state by X-ray diffraction its structure showed a tetraaquacobaltate chloride(II) co-crystallized in the lattice. Trimetallic species were prepared and oxidized in air in the presence of non-coordinative anion such as PF6−. Neutral complexes 8–10 proved to be active as catalysts in the ROP reaction of lactide to form PLA, whereas cationic complexes 1a–3 were inactive. The electronic properties of the complexes can be varied systematically changing the amino acid side chain by a neutral, hydrophilic or hydrophobic and these traits were further assessed in ROP of LA. This system displayed good performance towards lactide polymerization with a moderate isotactic bias. The promising results obtained highlight the potential applicability and future investigation of this catalytic system for mechanistically related polymerizations such as lactone ROP and epoxide/CO2 ring-opening copolymerization (ROCOP). In summary, the cobalt catalysts are composed of biorelevant ligands, inexpensive metal, and prepared in environmentally friendly solvent to produce a biodegradable polyester. Future work needs to target the amino acid side chain optimization so as to enhance stereoregularity of the polymer.Experimental section
Materials and methods
Preparation of compounds
O), 1607(s, CO), 1571(w), 1470(mw), 1444(ms), 1393(ms), 929(mw), 769(ms). UV/vis (EtOH, 5 × 10−5 M), λmax (ε, M−1 cm−1): 307(19100), 316(19860), 491(130).:DCM:MeCN 1:2:1 at 3 °C. Dark red crystals were collected. Yield: (0.22 g, 0.430 mmol, 56%). 1H NMR(D2O): 8.58–8.51 (m, 4H, Ar-H), 8.43 (m, 2H, Ar-H, J = 7.9, 6.3, 2.8 Hz), 7.90 (m, 2H, Ar-H, J = 7.3, 5.9, 2.6, 1.3 Hz), 3.82 (dd, 2H, α-H, J = 10.6, 3.5 Hz), 3.59–3.43 (m, 1H, α-H), 1.91–1.77 (m, 1H), 1.75–1.57 (m, 4H, CH2CH), 1.45 (td, 2H, CH2CH, J = 12.1, 10.5, 3.3 Hz), 0.99–0.81 (m, 12H, CH3) ppm (DMSO-d6): 8.80 (d, 2H, Ar-H, J = 7.9 Hz), 8.37 (td, 4H, Ar-H, J = 14.8, 13.9, 6.8 Hz), 7.89 (dt, 2H, Ar-H, J = 13.7, 6.6 Hz), 6.43 (t, 1H, NH, J = 9.1 Hz), 6.00 (t, 1H, NH, J = 9.3 Hz), 5.54–5.44 (t, 1H, NH), 4.97 (t, 1H, NH, J = 9.4 Hz), 3.29 (m, 1H, α-H, J = 4.8, 4.2 Hz), 3.02 (m, 1H, α-H, J = 12.1, 5.7 Hz), 1.94–1.68 (m, 2H, CH2CH), 1.63–1.42 (m, 2H, CH2CH), 1.30 (m, 2H, CH2CH, J = 14.4, 9.7, 3.8 Hz), 0.87 (dd, 6H, CH3, J = 11.2, 6.5 Hz), 0.78 (dd, 6H, CH3, J = 17.2, 6.5 Hz). 13C NMR (DMSO-d6) 183.27, 182.74 (2 COO−), 157.48, 157.07, 150.87, 150.78, 141.01, 140.95, 126.87, 126.80, 123.37, 123.19, 54.70, 54.57, 42.41, 23.36, 23.30, 23.25, 20.77, 20.71. FT-IR (KBr disc, νmax/cm−1): 3423(m, br), 3219(m), 3061(m), 2957–2869(ms), 1652(s), 1471(mw), 1451(ms), 1369(ms), 928(w), 771(ms). UV/vis (EtOH, 5 × 10−5 M), λmax (ε, M−1 cm−1): 301(7696), 313(8952), 479(109). Elemental analysis for C22H32N4O4CoCl·2H2O (546.94 g mol−1): calculated C, 48.31; H, 6.63; N 10.24%. Found C, 48.57; H, 6.71; N, 10.10%. HRMS (ESI) for C22H32N4O4CoCl 475.1750; Found 475.1750 ([M–Cl]+).Trimetallic species 4–6 were all prepared by a similar procedure. The synthesis of 4 is given as an example.
:EtOH after 2 days. Yield: (0.02 g, 0.043 mmol, 41%). 1H NMR (D2O) 8.59–8.50 (m, 4H, Ar-H), 8.44–8.36 (m, 2H, Ar-H), 7.89 (t, J = 6.7 Hz, 2H, Ar-H), 3.75 (d, 2H, α-H, J = 17.4 Hz), 3.48 (d, 2H, α-H, J = 17.2 Hz) ppm (DMSO-d6): 8.75 (d, 2H, Ar-H, J = 7.9 Hz), 8.48–8.35 (m, 4H, Ar-H), 7.92 (m, 2H, Ar-H, J = 7.3, 5.6, 1.3 Hz), 6.08 (t, 2H, NH, J = 11.3, 8.6, 4.5 Hz), 5.59 (t, 2H, NH, J = 8.1 Hz), 3.33–3.22 (m, 2H, α-H), 3.08 (m, 2H, α-H, J = 16.5, 8.3 Hz). 13C NMR (DMSO-d6): 181.68 (COO), 156.86, 150.80, 141.23, 127.16, 123.47, 45.59. FT-IR (KBr disc, νmax/cm−1): 3431(br), 3304(w), 3060(w), 1663(s), 1369(mw), 1317(mw), 845(s), 774(mw), 556(mw). HRMS (ESI) for C14H16N4O4Co 363.0498; Found 363.0484 ([M–PF6]+).Synthesis of neutral compounds 8–10
Similar procedure of 8 but employing CoCl2. The product is a pink solid. Yield: (1.52 g, 3.200 mmol, 83%), FT-IR (KBr disc, νmax/cm−1): 3417(m, br), 3207(m, br), 3080(mw), 2956–2869(mw), 1647(s), 1564(mw), 1471(w), 1450(m), 1388(s), 928(w), 770(m). HRMS (ESI) for C22H32N4O4Co 475.1750; found 475.1751 [M]+.
Similar procedure of 8. Pink solid. Yield: (1.10 g, 2.430 mmol, 63%), FT-IR (KBr disc, νmax/cm−1): 3396(m, br), 3238(m, br), 3061(w), 2971(mw), 1659(s), 1471 (w), 1421(m), 1373(m), 995(w), 772(m). HRMS (ESI) for C18H24N4O6Co 451.1022; found 451.1019 [M]+.
General procedure of polymerization of rac-lactide
All the polymerizations were conducted under argon. In a typical experiment a Schlenk flask (10 mL) was charged sequentially with rac-lactide (0.357 g, 2.5 mmol), complex (0.025 mmol), and dissolved in toluene (2.5 mL) such that the overall molar ratio was ([LA]:[1] = 100:1). The flask was sealed and placed into a hot oil bath set to 403 K. The reaction was stirred for the desired time and aliquots were withdraw at specific time intervals and quenched with CDCl3 to analyze the conversion by relative integration of the methine proton resonances for the polymer and monomer using 1H NMR spectroscopy. Remaining crude polymer mixture was dissolved in CHCl3 (∼2 mL) and dropwise added into a solution of acidic cold methanol (∼15 mL) to precipitate it. The polymer collected was dried at 30 °C in vacuo.
:DMC:MeCN concentrate solution of the metal complex. The crystals were isolated from the mother liquor, immediately immersed in cryogenic oil and then mounted. The X-ray single crystal data was collected using MoKα (λ = 0.71073 Å) or CuKα (λ = 1.54178 Å) radiation on a Bruker D8Venture (Photon100 CMOS detector) diffractometer equipped with a Cryostream (Oxford Cryosystems) open-flow nitrogen cryostats at the temperature 150(2) K. The unit cell determination, data collection, data reduction, structure solution/refinement and empirical absorption correction (SADABS) were carried out using Apex-III (Bruker AXS: Madison, WI, 2015). The structure was solved by direct method and refined by full-matrix least squares in F2 for all data using SHELXTL80 and Olex2 (ref. 81) software. All non-disordered non-hydrogen atoms were refined anisotropically except for the carbon atoms in 2 which was refined using FVAR instructions and the hydrogen atoms were placed in the calculated positions and refined in riding model. Complex 2 and 7 were refined as an inversion twin we could not model the disordered solvent in 2, 4a and 9a, which was located in the Fourier map, and the contribution of the electron densities from the disordered solvent molecules was excluded from the refinement using PLATON/SQUEEZE.73 This resulted in the removal of the solvent molecules in 2 corresponding to 93 electron per unit cell, which can be assigned as four acetonitrile molecules per unit cell. The disordered solvent molecule in 4a could be assigned as five ethanol molecules per unit cell (124 electron per unit cell) and the 360 electron per unit cell in 9a can be assigned to two dichloromethane molecules. Crystallographic data for the structures have been deposited to Cambridge Crystallographic Data Centre as supplementary publication (CCDC no. 2060291–2060294 and 2062819–2062821†).82Abbreviations
| Bipy | 2,2-Bipyridine |
| BnOH | Benzyl alcohol |
| Đ | Number-average molecular weight distribution |
| DCM | Dichloromethane |
| EtOH | Ethanol |
| gly | Deprotonated glycine |
| glyH | Protonated glycine |
| HRMS | High-resolution mass spectrometry |
| Leu | Deprotonated leucine |
| LeuH | Protonated leucine |
| MeCN | Acetonitrile |
| Mn | Number-average molecular weight |
| OAc | Acetate |
| rac-LA | Lactide dimer(monomer) |
| ROP | Ring opening polymerization |
| thr | Deprotonated threonine |
| thrH | Protonated threonine |
Conflicts of interest
The authors have no conflicts to declare.Acknowledgements
Funding from the Icelandic Centre of Research (Rannis) grant no. 152323 is gratefully acknowledged. We thank Rannís Iceland for infrastructure grant for the single crystal X-ray diffractometer (150998-0031). Dr Sigridur Jonsdottir is thanked for assistance with the collection of mass spectrometry data. Dr Esteban Mejia and MSc. Lea Grefe at the Leibniz Institute for Catalysis, (LIKAT), Germany, are acknowledged for their help with GPC and homonuclear decoupling NMR data collection.References
- M. He, Y. Cheng, Y. Liang, M. Xia, X. Leng, Y. Wang, Z. Wei, W. Zhang and Y. Li, Polym. J., 2020, 52, 567–574 CrossRef CAS.
- M. Fuchs, S. Schmitz, P. M. Schäfer, T. Secker, A. Metz, A. N. Ksiazkiewicz, A. Pich, P. Kögerler, K. Y. Monakhov and S. Herres-Pawlis, Eur. Polym. J., 2020, 122, 109302 CrossRef CAS.
- V. M. Padilla-Gainza, H. Rodríguez-Tobías, G. Morales, E. Saucedo-Salazar, K. Lozano, V. Montaño-Machado and D. Mantovani, J. Appl. Polym. Sci., 2021, 138, 1–13 CrossRef.
- F. Emami, S. J. Mostafavi Yazdi and D. H. Na, J. Pharm. Invest., 2019, 49, 427–442 CrossRef CAS.
- M. N. Abu Hajleh, A. AL-Samydai and E. A. S. Al-Dujaili, J. Cosmet., Dermatol. Sci. Appl., 2020, 19, 2805–2811 CrossRef PubMed.
- M. Shaver and A. Buchard, Eur. Polym. J., 2020, 129, 109600 CrossRef CAS.
- N. Nomura, R. Ishii, Y. Yamamoto and T. Kondo, Chem.–Eur. J., 2007, 13, 4433–4451 CrossRef CAS PubMed.
- P. Marin, M. J. L. Tschan, F. Isnard, C. Robert, P. Haquette, X. Trivelli, L. Chamoreau, V. Guérineau, I. del Rosal, L. Maron, V. Venditto and C. M. Thomas, Angew. Chem., Int. Ed., 2019, 58, 12585–12589 CrossRef CAS PubMed.
- N. Yuntawattana, T. M. McGuire, C. B. Durr, A. Buchard and C. K. Williams, Catal. Sci. Technol., 2020, 10, 7226–7239 RSC.
- D. Myers, A. J. P. White, C. M. Forsyth, M. Bown and C. K. Williams, Angew. Chem., Int. Ed., 2017, 56, 5277–5282 CrossRef CAS PubMed.
- P. M. Schäfer and S. Herres-Pawlis, ChemPlusChem, 2020, 85, 1044–1052 CrossRef.
- Y. Gong and H. Ma, Chem. Commun., 2019, 55, 10112–10115 RSC.
- W. Gruszka, A. Lykkeberg, G. S. Nichol, M. P. Shaver, A. Buchard and J. A. Garden, Chem. Sci., 2020, 11, 11785–11790 RSC.
- C. Bakewell, T.-P.-A. Cao, N. Long, X. F. Le Goff, A. Auffrant and C. K. Williams, J. Am. Chem. Soc., 2012, 134, 20577–20580 CrossRef CAS PubMed.
- M. R. Kember, A. J. P. White and C. K. Williams, Macromolecules, 2010, 43, 2291–2298 CrossRef CAS.
- M. R. Kember, F. Jutz, A. Buchard, A. J. P. White and C. K. Williams, Chem. Sci., 2012, 3, 1245 RSC.
- Y. Hiranoi and K. Nakano, Beilstein J. Org. Chem., 2018, 14, 2779–2788 CrossRef CAS PubMed.
- D. Dolui, S. Das, J. Bharti, S. Kumar, P. Kumar and A. Dutta, Cell Rep. Phys. Sci., 2020, 1, 100007 CrossRef.
- H. Ullah, B. Mousavi, H. A. Younus, Z. A. K. Khattak, S. Chaemchuen, S. Suleman and F. Verpoort, Commun. Chem., 2019, 2, 1–9 CrossRef CAS.
- K. Ambrose, K. N. Robertson and C. M. Kozak, Dalton Trans., 2019, 48, 6248–6260 RSC.
- T. A. Zevaco and A. W. Kleij, in Non-Noble Metal Catalysis, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2018, vol. 19, pp. 529–547 Search PubMed.
- A. C. Deacy, E. Moreby, A. Phanopoulos and C. K. Williams, J. Am. Chem. Soc., 2020, 142, 19150–19160 CrossRef CAS PubMed.
- J. Zhang, B. Wang, L. Wang, J. Sun, Y. Zhang, Z. Cao and Z. Wu, Appl. Organomet. Chem., 2018, 32, 1–12 Search PubMed.
- P. Marin, M. J. L. Tschan, P. Haquette, T. Roisnel, I. del Rosal, L. Maron and C. M. Thomas, Eur. Polym. J., 2019, 120, 109208 CrossRef CAS.
- L. E. Breyfogle, C. K. Williams, V. G. Young, M. A. Hillmyer and W. B. Tolman, Dalton Trans., 2005, 6, 928–936 Search PubMed.
- C. Thomas and J. A. Gladysz, ACS Catal., 2014, 4, 1134–1138 CrossRef CAS.
- M. J. L. Tschan, J. Guo, S. K. Raman, E. Brulé, T. Roisnel, M. N. Rager, R. Legay, G. Durieux, B. Rigaud and C. M. Thomas, Dalton Trans., 2014, 43, 4550–4564 RSC.
- A. K. Renfrew, E. S. O'Neill, T. W. Hambley and E. J. New, Coord. Chem. Rev., 2018, 375, 221–233 CrossRef CAS.
- N. P. Boralugodage, R. J. Arachchige, A. Dutta, G. W. Buchko and W. J. Shaw, Catal. Sci. Technol., 2017, 7, 1108–1121 RSC.
- M. Hapke and G. Hilt, Cobalt Catal. Org. Synth., 2020, 1–23 CAS.
- H. Shimakoshi and Y. Hisaeda, in Advances in Bioorganometallic Chemistry, Elsevier Inc., Fukuoka, Japan, 2019, pp. 379–398 Search PubMed.
- D. J. Darensbourg and O. Karroonnirun, Inorg. Chem., 2010, 49, 2360–2371 CrossRef CAS PubMed.
- S. L. Anderson and K. C. Stylianou, Coord. Chem. Rev., 2017, 349, 102–128 CrossRef CAS.
- X. L. Wang, H. Chao, X. L. Hong, Y. J. Liu and L. N. Ji, Transition Met. Chem., 2005, 30, 305–311 CrossRef CAS.
- T. Kondori, O. Shahraki, N. Akbarzadeh-T and Z. Aramesh-Boroujeni, J. Biomol. Struct. Dyn., 2021, 39, 595–609 CrossRef CAS PubMed.
- J. Börner, U. Flörke, A. Döring, D. Kuckling, M. D. Jones and S. Herres-Pawlis, Sustainability, 2009, 1, 1226–1239 CrossRef.
- A. Sanchez-Sanchez, I. Rivilla, M. Agirre, A. Basterretxea, A. Etxeberria, A. Veloso, H. Sardon, D. Mecerreyes and F. P. Cossío, J. Am. Chem. Soc., 2017, 139, 4805–4814 CrossRef CAS PubMed.
- Y. Yan, E. J. Carrington, R. Pétuya, G. F. S. Whitehead, A. Verma, R. K. Hylton, C. C. Tang, N. G. Berry, G. R. Darling, M. S. Dyer, D. Antypov, A. P. Katsoulidis and M. J. Rosseinsky, J. Am. Chem. Soc., 2020, 142, 14903–14913 CrossRef CAS PubMed.
- Y. Shi, S. Hou, X. Qiu and B. Zhao, Top. Curr. Chem., 2020, 378, 11 CrossRef CAS PubMed.
- S. L. Anderson and K. C. Stylianou, Coord. Chem. Rev., 2017, 349, 102–128 CrossRef CAS.
- I. Burneo, K. C. Stylianou, I. Imaz and D. Maspoch, Chem. Commun., 2014, 50, 13829–13832 RSC.
- W. J. Shaw, Catal. Rev. - Sci. Eng., 2012, 54, 489–550 CrossRef CAS.
- H. Sigel and R. B. Martin, Chem. Rev., 1982, 82, 385–426 CrossRef CAS.
- R. Griesser and H. Sigel, Inorg. Chem., 1971, 10, 2229–2232 CrossRef CAS.
- L. Rulíšek and Z. Havlas, J. Am. Chem. Soc., 2000, 122, 10428–10439 CrossRef.
- B. E. Douglas and T. Yasui, Inorg. Chem., 1971, 10, 97–102 CrossRef CAS.
- T. Yasui, Bull. Chem. Soc. Jpn., 1975, 48, 454–457 CrossRef CAS.
- R. M. Hartshorn and S. G. Telfer, J. Chem. Soc., Dalton Trans., 1999, 3217–3224 RSC.
- A. Wojciechowska, A. Gagor, A. Duczmal, M. Staszak and Z. Ozaroswski, Inorg. Chem., 2013, 52, 4360–4371 CrossRef CAS PubMed.
- L. M. Alcazar-Roman, B. J. O'Keefe, M. A. Hillmyer and W. B. Tolman, Dalton Trans., 2003, 15, 3082–3087 RSC.
- A. Dutta, D. L. Du Bois, J. A. S. Roberts and W. J. Shaw, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 16286–16291 CrossRef CAS PubMed.
- K. Arun Kumar, M. Amuthaselvi and A. Dayalan, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2011, E67(4), m468 CrossRef PubMed.
- A. E. Martell and R. D. Hancock, Metal Complexes in Aqueous Solutions, Springer US, Boston, MA, 1996 Search PubMed.
- J. Na'aliya and H. Aliyu, Bayero J. Pure Appl. Sci., 2011, 3, 52–55 Search PubMed.
- E. Cardozo, C. RR, F. Bellandi, J. Avendaño, C. Araque and J. Vielma, Rev. Politec., 2015, 36, 1–5 Search PubMed.
- B. Ye, X. Chen and T. Zeng, Polyhedron, 1994, 13, 2185–2191 CrossRef CAS.
- U. K. Das, J. Bobak, C. Fowler, S. E. Hann, C. F. Petten, L. N. Dawe, A. Decken, F. M. Kerton and C. M. Kozak, Dalton Trans., 2010, 39, 5462 RSC.
- B. H. Ye, T. X. Zeng, P. Han and L. N. Ji, Transition Met. Chem., 1993, 18, 515–517 CrossRef CAS.
- T. Balakrishnan, P. Revathi, A. Krishnaveni, J. Thirupathy and K. Ramamurthi, J. Mater. Sci.: Mater. Electron., 2018, 29, 16971–16982 CrossRef CAS.
- L. Gobinathan and K. Boopathy, J. Adv. Chem., 2016, 12, 5446–5453 CAS.
- M. M. Alam, S. M. S. Islam, S. M. M. Rahman and M. M. Rahman, J. Sci. Res., 2009, 2, 91–98 CrossRef.
- A. Tatehata, Inorg. Chem., 1982, 21, 2496–2499 CrossRef CAS.
- D. F. Evans, J. Chem. Soc., 1959, 2003–2005 RSC.
- A. C. Rizzi, C. D. Brondino, R. Calvo, R. Baggio, M. T. Garland and R. E. Rapp, Inorg. Chem., 2003, 42, 4409–4416 CrossRef CAS PubMed.
- D. Das, A. Banaspati, N. Das, B. Bora, M. K. Raza and T. K. Goswami, Dalton Trans., 2019, 48, 12933–12942 RSC.
- V. Mishra, F. Lloret and R. Mukherjee, Inorg. Chim. Acta, 2006, 359, 4053–4062 CrossRef CAS.
- F.-L. Yin, R. Xie, J. Hao, Y.-F. Wang and J.-J. Yang, Inorg. Chem. Commun., 2013, 30, 97–104 CrossRef CAS.
- M. Cheng, A. B. Attygalle, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 1999, 121, 11583–11584 CrossRef CAS.
- M. J. Stanford and A. P. Dove, Chem. Soc. Rev., 2010, 39, 486–494 RSC.
- D. Li, B. Gao and Q. Duan, New J. Chem., 2019, 43, 6943–6950 RSC.
- S. L. Hancock, M. F. Mahon and M. D. Jones, Dalton Trans., 2013, 42, 9279–9285 RSC.
- P. M. Schäfer, K. Dankhoff, M. Rothemund, A. N. Ksiazkiewicz, A. Pich, R. Schobert, B. Weber and S. Herres-Pawlis, ChemistryOpen, 2019, 8, 1020–1026 CrossRef PubMed.
- A. J. Gaston, G. Navickaite, G. S. Nichol, M. P. Shaver and J. A. Garden, Eur. Polym. J., 2019, 119, 507–513 CrossRef CAS.
- A. Rae, A. J. Gaston, Z. Greindl and J. A. Garden, Eur. Polym. J., 2020, 138, 109917 CrossRef CAS.
- C. Robert, T. E. Schmid, V. Richard, P. Haquette, S. K. Raman, M. N. Rager, R. M. Gauvin, Y. Morin, X. Trivelli, V. Guérineau, I. Del Rosal, L. Maron and C. M. Thomas, J. Am. Chem. Soc., 2017, 139, 6217–6225 CrossRef CAS PubMed.
- D. C. Aluthge, J. M. Ahn and P. Mehrkhodavandi, Chem. Sci., 2015, 6, 5284–5292 RSC.
- A. B. Biernesser, B. Li and J. A. Byers, J. Am. Chem. Soc., 2013, 135, 16553–16560 CrossRef CAS PubMed.
- J. A. Byers, A. B. Biernesser, K. R. Delle Chiaie, A. Kaur and J. A. Kehl, Adv. Polym. Sci., 2017, 279, 67–118 CrossRef.
- M. Qi, Q. Dong, D. Wang and J. A. Byers, J. Am. Chem. Soc., 2018, 140, 5686–5690 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2015, 71, 3–8 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Tables S1–S3 of crystal data summary, S4–S10 of selected bond lengths and angles for 1, 2, 1a, 7, 9a, 4a and Co(bipy)2Cl2. Fig. S1–S3 of Molecular structures of 9a, 4a and Co(bipy)2Cl2. Fig. S4–S30 of NMR spectroscopy, infrared spectra, and HRMS of compounds, and S30–S39 spectroscopy and analysis of polymers. CCDC codes 2060291–2060294 and 2062819–2062821 contain the supplementary crystallographic data for this paper. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra02909f |
| This journal is © The Royal Society of Chemistry 2021 |