
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ruthenium-centred btp glycoclusters as inhibitors for Pseudomonas aeruginosa biofilm formation†
Ciaran O'Reillyab,
Salvador Blasco a,
Bina Parekhb,
Helen Collinsc,
Gordon Cookebc,
Thorfinnur Gunnlaugssona and
Joseph P. Byrne*d
a,
Bina Parekhb,
Helen Collinsc,
Gordon Cookebc,
Thorfinnur Gunnlaugssona and
Joseph P. Byrne*d
aSchool of Chemistry, Trinity Biomedical Sciences Institute, Trinity College Dublin, Ireland
bSchool of Medicine, University College Dublin, Belfield, Dublin 4, Ireland
cDepartment of Applied Science, Tallaght Campus, Technological University Dublin, Ireland
dSchool of Chemistry, National University of Ireland Galway, University Road, Galway, Ireland. E-mail: joseph.byrne@nuigalway.ie
First published on 4th May 2021
Abstract
Carbohydrate-decorated clusters (glycoclusters) centred on a Ru(II) ion were synthesised and tested for their activity against Pseudomonas aeruginosa biofilm formation. These clusters were designed by conjugating a range of carbohydrate motifs (galactose, glucose, mannose and lactose, as well as galactose with a triethylene glycol spacer) to a btp (2,6-bis(1,2,3-triazol-4-yl)pyridine) scaffold. This scaffold, which possesses a C2 symmetry, is an excellent ligand for d-metal ions, and thus the formation of the Ru(II)-centred glycoclusters 7 and 8Gal was achieved from 5 and 6Gal; each possessing four deprotected carbohydrates. Glycocluster 8Gal, which has a flexible spacer between the btp and galactose moieties, showed significant inhibition of P. aeruginosa bacterial biofilm formation. By contrast, glycocluster 7, which lacked the flexible linker, didn't show significant antimicrobial effects and neither does the ligand 6Gal alone. These results are proposed to arise from carbohydrate–lectin interactions with LecA, which are possible for the flexible metal-centred multivalent glycocluster. Metal-centred glycoclusters present a structurally versatile class of antimicrobial agent for P. aeruginosa, of which this is, to the best of our knowledge, the first example.
Introduction
Pseudomonas aeruginosa is a ubiquitous Gram-negative pathogenic bacteria, which is of great medical significance as a result of its multidrug resistance and effects on immune-compromised patients, such as those suffering from cystic fibrosis. It has been identified as one of the ‘ESKAPE’ pathogens, which are the cause of many healthcare-acquired infections, and for which development of new antimicrobial agents is vital.1 Biofilm formation is a key part of pathogenic behaviour for many bacteria, and its inhibition is an important target.2–4 P. aeruginosa expresses two carbohydrate-binding proteins on its surface which have been identified as essential to its pathogenesis, namely lectins LecA and LecB (also known as PA-IL and PA-IIL).5,6 These lectins are pivotal in processes including adhesion to epithelial and endothelial cells, as well as biofilm formation. Biofilms are structured aggregates of bacteria linked together by these lectins and encapsulated in a complex extracellular matrix,2,7 and their formation is implicated in chronic P. aeruginosa infections, which exhibit marked resistance to antibiotics and the capacity to evade host defences.8 The physical barrier of the biofilm may be sufficient to prevent or slow penetration by conventional antimicrobial agents, which are effective on the bacteria in its planktonic form. Novel compounds that prevent biofilm-based infection without directly killing the bacteria could restore sensitivity to established antibiotics.These surface lectins, LecA and LecB, show specific binding for galactosides and fucosides respectively and thus targeting these proteins with carbohydrate-appended therapeutic molecules is a valuable strategy (e.g. for disrupting biofilm formation).9–12 In 2008, Hauber et al. showed that inhalation of galactose and fucose monosaccharides was a safe treatment, and subsequently research has focussed on developing more selective or potent antimicrobial agents.9,13–15 Carbohydrate multivalency has been shown to be very important for providing high-affinity interactions, represented by a range glycopeptide dendrimers, glyco-nanoparticles and glycoclusters, including compounds built upon macrocyclic scaffolds.11,15–22
A recent example of such an antiadhesive glycocluster system, described by Vidal and co-workers, was based on a calix[4]arene scaffold, that was derivatised with galactose and fucose monosaccharide units.19 These carbohydrates selectively bind LecA and LecB and, as a result, this macrocyclic system was shown to inhibit biofilm formation, and was also studied in an in vivo mouse model where it was shown to demonstrate protection against P. aeruginosa lung infection. The topology of carbohydrate presentation from the calix[4]arene scaffold was shown to play an important role, with tetravalent clusters presenting two pairs of epitopes from opposite faces of the calix[4]arene scaffold proving most potent.23,24 In this example, triethylene glycol was used successfully as a flexible spacer, however different spacers, both rigid and flexible, have also been utilised to allow for tuning of avidity of lectin inhibitors.10,14,25,26 In order to present the saccharide units in a three-dimensional way, various different scaffold-types have been utilised to design ligands for LecA, including glycopeptide dendrimer, cyclodextrins and carbohydrate-based scaffolds. To the best of our knowledge, coordination of a metal ion has yet to be exploited in design of inhibitors targeting the P. aeruginosa lectins. Such a strategy would facilitate access to three-dimensional geometries of carbohydrate epitopes defined by the metal coordination geometry, and allow for immediate increase of multivalency of a ligand system, upon coordinating multiple ligands to the same metal centre, providing complexes with potential to inhibit biofilm formation by the bacteria. Cationic charge has also been shown to be a characteristic of many antimicrobial dendrimers, which can disrupt bacterial membranes; this presents another possible added advantage to introducing metal ions into glycocluster structures for targeting bacteria.27–29
Ligands containing the glycosyl-triazolyl-pyridyl motif have come to light in recent years, mostly applied as ligands for catalysis,30–32 and enzyme inhibitors.33,34 The only metal complexes reported with such promising ligands are Pd(II)-containing catalysts for C–C coupling reactions35,36 and Ru(II) complexes.37 We have an interest in triazole ligands, such as those based on the 2,6-bis(1,2,3-triazol-4-yl)pyridine (btp) motif.38 Having exploited such ligands to coordinate transition metal ions including Ru(II), Ni(II), Ir(III) and Pt(II)39 as well as forming luminescent self-assemblies and MOFs with lanthanide ions,40–44 and mechanically interlocked molecules,45 we set out to investigate the use of btp systems as potential antimicrobial agents. In previous studies, we and others have crystallographically demonstrated that Ru(II) forms 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 complexes with btp ligands, which would allow for the incorporation of a minimum of four glycans into a single complex.38,39,46–48 In this article, we describe a series of Ru(II)-centred tetravalent glycoclusters, synthesised from carbohydrate-derived btp ligands, and assess their suitability as inhibitors for P. aeruginosa biofilm formation.
:2 complexes with btp ligands, which would allow for the incorporation of a minimum of four glycans into a single complex.38,39,46–48 In this article, we describe a series of Ru(II)-centred tetravalent glycoclusters, synthesised from carbohydrate-derived btp ligands, and assess their suitability as inhibitors for P. aeruginosa biofilm formation.
Results and discussion
Synthesis of ligands and Ru(II) complexes
The synthesis of acetyl-protected btp compounds, hydrolysed ligands, and target Ru(II) complexes are shown in Scheme 1. The CuAAC reaction is typically used to prepare btp ligands, and as such azide functionality was introduced to monosaccharides. Stereoselective azidation of peracetylated glycosides via treatment with TMS-N3 under Lewis acid catalysed conditions gave access to azide precursors 1,49 while 2Gal was synthesised from a chloro-precursor, as described in the literature.50 Triethylene glycol spaced galactosides have previously been included in calix[4]arene-based inhibitors for LecA, and as such were also included in the family of new compounds designed here.19,23 These carbohydrate azides were then used as substrates for the CuAAC reaction with 2,6-bis(TMS-ethynyl)pyridine, yielding protected btp compounds 3 in good yields (63–82%) and 4Gal in 79% yield. The 1H NMR spectra of triazole compounds each displayed a single set of resonances, corresponding to a single anomer, for instance with doublets observed at 6.40 ppm (J = 9.2 Hz) for β-anomer 3Gal,32 and 4.48 ppm (J = 8.0 Hz) for β-anomer 4Gal. HRMS analysis further supported the formation of the proposed structures (see Experimental and ESI† for full characterisation).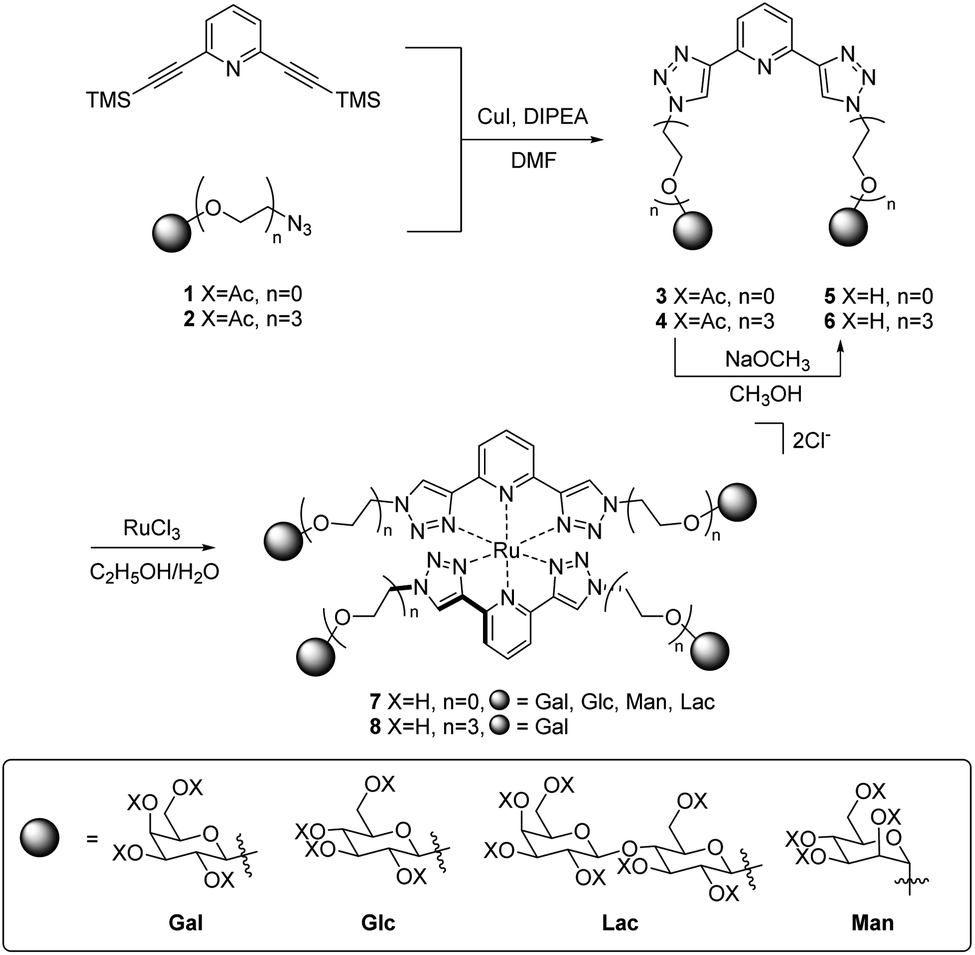 | ||
| Scheme 1 Synthesis of Ru(II)-centred glycoclusters 7 and 8Gal. | ||
In addition to the above characterisation of the acetyl protected ligands, single crystals of 3Gal were grown by slow diffusion of (CH3)2CHOH into a DMSO-d6 solution as colourless hexagonal plates, which were found to be suitable for X-ray diffraction analysis. The ligand crystallised in the trigonal space group P3221. The asymmetric unit contains two distinct residues, each being half a molecule of 3Gal, the other half of each molecule being generated by symmetry. These two complete molecules are shown in Fig. 1(a). The btp motifs of the two crystallographically distinct molecules were also shown to intertwine by non-classical hydrogen bonding interactions between the triazolyl C–H and the pyridyl nitrogen atoms of the other btp motif, with hydrogen bond lengths of C–H⋯N = 3.511(10) and 3.482(9) Å, Fig. 1(b). This dimeric interaction matched our previous observations, which have been exploited to form interlocked supramolecular architectures.40,45
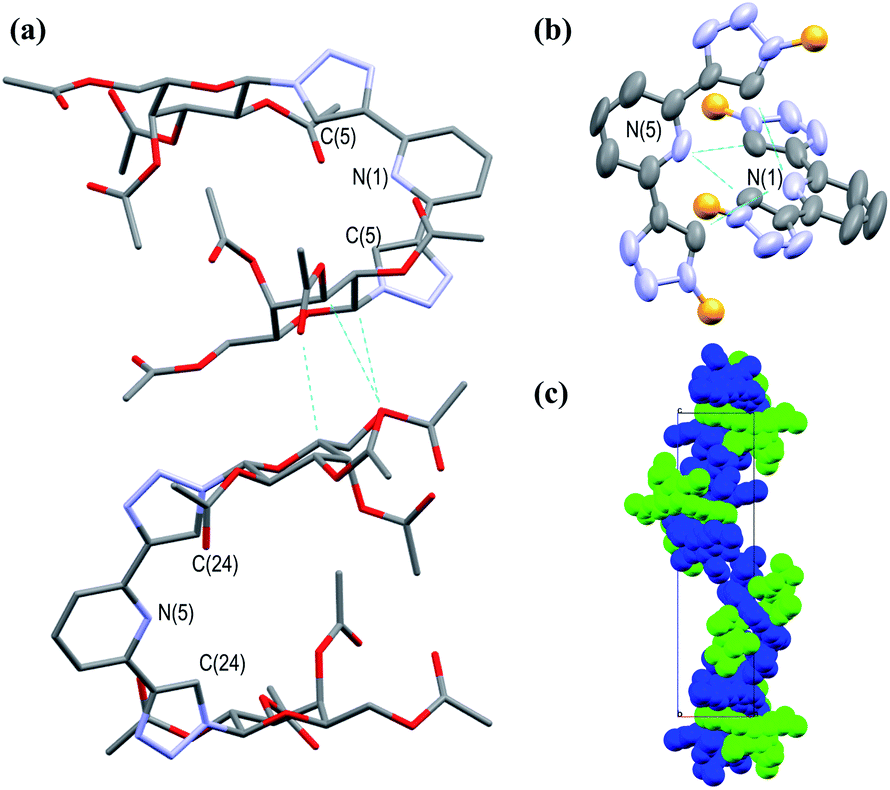 | ||
| Fig. 1 Projections of the X-ray crystal structure of 3Gal,‡ showing various features of the structure (hydrogen atoms omitted for clarity, CCDC deposition number 2004944†): (a) capped stick model showing the two distinct molecules in the structure packing due to weak supramolecular interactions between the adjacent sugar moieties; (b) thermal ellipsoid model showing the non-classical hydrogen bonding interactions between the two btp motifs; the carbohydrate arms are omitted for clarity (represented as orange spheres); (c) a space-filling model, showing the packing of the molecules with a C32 screw-axis. | ||
Crystallographic packing analysis also showed weak supramolecular interactions between the carbohydrate moieties of the adjacent molecules, causing them to stack in the manner seen in Fig. 1(c); all C–H⋯O distances were of the order of 3.5 Å and are shown in a table in ESI.† The triazole rings of btp were found to be slightly twisted out of the plane of the pyridyl rings by ca. 13° in this elegant structure. The unit cell of this crystal structure contained 6 molecules (Z = 6); the two molecules shown in Fig. 1(a) repeat in a left-handed 32 screw axis, Fig. 1(c).
O-Deacetylation under Zemplén conditions gave access to the fully deprotected carbohydrate derivatives 5 and 6Gal in good yields (63–78%), which were soluble in aqueous media. 1H NMR spectra confirmed the removal of acetyl groups (note the absence of proton resonances at ca. 2 ppm in Fig. 2) and HRMS analysis was also consistent with the formation of btp structures decorated with unprotected saccharide moieties.
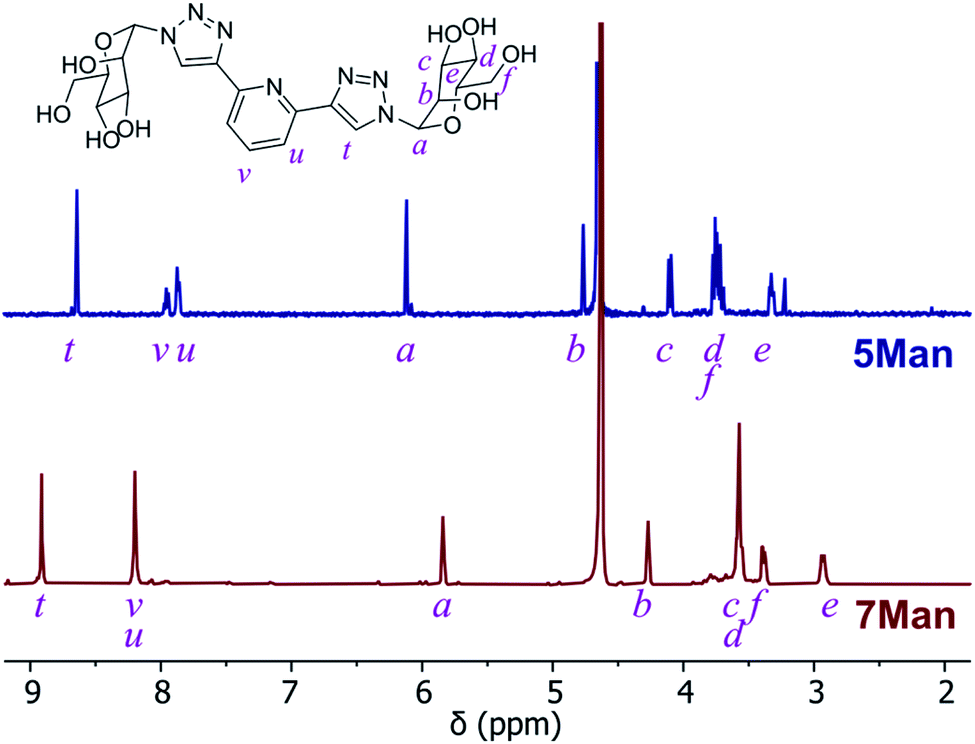 | ||
| Fig. 2 1H NMR spectra (D2O) of btp ligand 5Man (top) and the resulting Ru(II) complex 7Man (bottom). | ||
The corresponding Ru(II) complexes 7 and 8Gal were prepared upon heating 2 equivalents of the ligand (5 or 6Gal) with RuCl3·3H2O in aqueous ethanol solution under microwave irradiation at 120 °C for 40 minutes. Complexes 7 and 8Gal, of the general structure [Ru·(L)2]Cl2, were formed as a single species. Changes observed in the 1H NMR spectrum (400 MHz, D2O) of 7Man, for instance (Fig. 2), including a shift in the resonance arising from the triazolyl CH from 8.35 to 8.91 ppm, and the coalescence of the proton resonances from the pyridyl ring into a multiplet centred at 8.2 ppm, are indicative of coordination of Ru(II) by the btp ligand. HRMS analysis gave signals corresponding to the doubly charged [M−2Cl]2+ ion in all cases. Single crystals suitable for X-ray diffraction were not obtained for these complexes, but based on known structures of related [Ru(btp)2]X2 complexes, it is anticipated that the two tridentate btp ligands coordinate Ru(II) in an approximately octahedral geometry,38,39,47,51 and as such the carbohydrate epitopes from each divalent ligand would be presented orthogonally.
Biofilm inhibition
To the best of our knowledge, only a few ruthenium complexes have reported significant inhibition of biofilm formation by P. aeruginosa,52–55 with these usually being bimetallic complexes. However in general Ru(II)–polypyridyl complexes have shown lower activity towards Gram-negative species (particularly P. aeruginosa) when compared to Gram-positive bacteria (such as MRSA).55–57In order to determine the potential of Ru(II) glycoclusters 7 and 8Gal to inhibit bacterial biofilm formation, samples of P. aeruginosa (PAO1) were incubated with 5 mM of Ru(II) complexes for 24 hours and the biofilm biomass determined by staining with crystal violet. Inhibition was characterised by decrease in absorbance at 590 nm, compared to control experiments in the absence of any glycocluster (see Fig. 3). None of the tested complexes were either bacteriostatic nor bactericidal to PAO1 at these concentrations (see ESI† for MIC and MBC data).
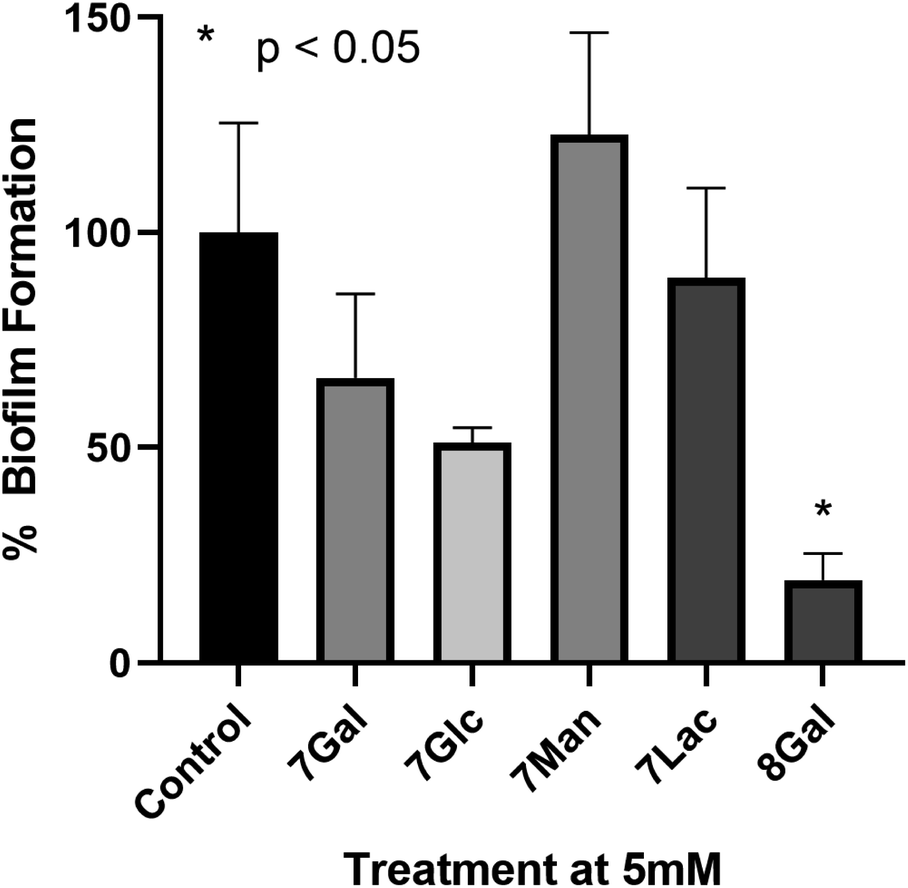 | ||
| Fig. 3 Percentage biofilm formation determined by crystal violet staining of biofilms upon incubation of P. aeruginosa with 5 mM of a range of Ru(II) glycoclusters. Decreased absorbance at 590 nm indicated inhibition of biofilm formation. One way ANOVA on ranks performed, *p < 0.05 versus control. | ||
When complexes 7, where the carbohydrates are directly bonded to the triazole moieties, were incubated with P. aeruginosa, none of the Ru(II) complexes had statistically significant impact on biofilm formation, regardless of the identity of the carbohydrate. This would suggest that the saccharide motifs were not able to interact with the target lectin in this configuration, either due to lack of flexibility, or due to the distances between carbohydrate units not matching the distance between lectin carbohydrate-binding sites. When designing LecA inhibitors, Pieters and co-workers describe a separation of 26 Å between the carbohydrates in neighbouring binding sites of the lectin,14,25,58 and while X-ray crystal structures of Ru(II) complexes 7 were not obtained, the analogous galactose–galactose distance in ligand 3Gal, measured between the anomeric carbons, was significantly shorter (ca. 8 Å). Based on analogy to previously-reported octahedral Ru(II) btp complexes, the distance between anomeric carbons in complexes 7 would be expected to be 8–11 Å. Hence, it is possible that this significant deviation from the ideal multivalent binding geometry for LecA explains the lack of interaction, even with a carbohydrate epitope known to interact with the lectin.
Complex 8Gal, on the other hand, where triethylene glycol chains confer the structure with further flexibility,19,25,59 demonstrated significant inhibition of biofilm formation, with ca. 80% decrease versus the control (p < 0.05). Although highly-multivalent glyconanoparticles have been able to achieve these results at much lower concentrations (μM),22 the activity of 8Gal compares favourably with other similar-valency calix[4]arene and triazine-based glycoclusters, which also showed statistically significant biofilm reduction at 5 mM concentration.13,19 The ligand 6Gal alone does not lead to biofilm inhibition under analogous conditions (see ESI, Fig. S28†), suggesting that the multivalent glycocluster structure is required for this effect to be observed.
The striking difference in behaviour between complexes 7 and 8Gal, all of which are cationic Ru(II)-containing compounds, suggests that antimicrobial properties of this complex do not arise simply from the presence of the cationic metal ion. Nor is the ligand structure alone responsible for the effect on P. aeruginosa biofilm formation. Rather, the combination of the flexible ligand and the mode of presentation of the carbohydrate units in the tetravalent cluster structure is proposed to be the origin of the observed effect. This result confirms the potential of using metal ions as scaffolds for the construction of antimicrobial multivalent glycoclusters from ligands of lower valency, giving rise to biofilm inhibition activity which is not primarily caused by the metal ion. This will form a basis for future developments, which could build on the variety of geometric structures available through coordination chemistry of alternative d- and f-metal ions to explore the relationship between complex topology and biofilm inhibition in detail.
Conclusions
We have successfully synthesised a range of protected carbohydrate-derived tridentate btp ligands and characterised them, including by single crystal X-ray diffraction in the case of 3Gal. These ligands were conveniently deprotected under Zemplén conditions and formed Ru(II) complexes of the form [Ru·(L)2]Cl2, which were tetravalent glycoclusters.Complex 8Gal, containing flexible triethylene glycol spacers between the btp and galactose moieties, showed significant inhibition of P. aeruginosa biofilm formation at 5 mM concentration, while neither complexes 7 nor ligand 6Gal had an impact on biofilm formation under the same conditions. None of the complexes were bacteriostatic or bactericidal. This behaviour is rationalised in terms of the known affinity of soluble lectin LecA for galactose and the advantages of multivalency reported in the literature for inhibitors of this protein that are able to span adjacent carbohydrate-binding sites. As such, it is clear that the topology of presentation of carbohydrate units, templated by coordination chemistry plays an important role in antimicrobial activity of such metal-centred glycoclusters.
These results indicate that coordination chemistry may be used to conveniently direct the topology of multivalent glycoclusters, built up from simpler lower-valency building block ligands. As the Ru(II) centre does not appear to play a significant role in any antimicrobial properties of 7 or 8Gal, it is conceivable that other metal ions could easily be used, either for increased economy, or to exploit complementary properties like magnetism or luminescence (e.g. for use in applications such as imaging of biofilms). We will further develop on this work by investigating the biofilm inhibition properties, and impact on biofilm-related antibiotic resistance, of related systems with differing geometries, metal centres, and spacers in future studies.
Experimental
Materials and methods
All chemicals and reagents were purchased from commercial sources and used without further purification. Electrospray mass spectra (ESI) were acquired using a Micromass time of flight mass spectrometer (TOF), interfaced to a Waters 2690 HPLC or a Waters LCT Premiere XE (with leucine enkephalin used as an internal lock mass). MALDI Q-Tof mass spectra were carried out on a MALDI Q-Tof Premier (Waters Corporation, Micromass MS technologies, Manchester, UK) and high-resolution mass spectrometry was performed using Glu-Fib as an internal reference. NMR spectra were recorded on a 400 MHz Bruker Avance III spectrometer or a 500 MHz Agilent spectrometer. Chemical shifts expressed in parts per million (ppm/δ) are reported relative to internal tetramethylsilane in CDCl3 or CD3OD or relative to HOD in D2O. Coupling constants (J) are expressed in Hz. Infrared spectra were recored on a PerkinElmer Spectrum 100 or 400 FT-IR spectrometer with universal ATR sampling accessory. All microwave reactions were carried out Biotage Microwave Vials in a Biotage Initiator Eight EXP microwave reactor.X-ray diffraction data for 3Gal was measured on a Bruker Apex2 Duo using a high intensity Cu-Kα radiation source (λ = 1.54178 Å). The datasets were collected and processed using Bruker APEX3 suite of programs. All structures were solved by direct methods (SHELXS-2018/3) and refined against all F2 data (SHELXL-2018/3).60 All H-atoms, were positioned geometrically and refined using a riding model.
Biofilm formation assay
Biofilm formation assays were used based on the methodology from previous published work.61 In brief, starting from an overnight liquid culture, a dilution containing approximately 108 CFUs mL−1 was made of Pseudomonas aeruginosa, PAO1 strain (a kind donation from Prof. Seamas Donnelly, School of Medicine, Trinity College Dublin). For each biofilm experiment, 8 wells of a round-bottomed polypropylene 96-well micro plate (Corning® Costar® purchased from Sigma Aldrich, Dublin Cat#CLS3596) were inoculated with 100 μl of this dilution, 8 wells were inoculated with the dilution and treated with 5 mM of the Ru(II) glycoclusters and 8 control wells were filled with sterile medium (bacteria alone). Following 4 hours of adhesion, the supernatant, containing non-adhered cells, was removed from each well and plates rinsed using phosphate buffered saline (PBS) solution. Following this 100 μl of fresh media was added to the control wells and fresh media with 5 mM of the Ru(II) glycoclusters was added to the appropriate wells, the plate was then incubated for a further 24 hours. After 24 hours biofilm formation, the supernatants were again removed, and the wells rinsed with PBS again. Once the wells were washed, 100 μl of a 0.5% crystal violet (CV) solution was added to all wells. After 20 min, the excess CV was removed by washing the plates under running tap water. Finally, bound CV was released by adding 150 μl of 33% acetic acid (Sigma). The absorbance was measured at 590 nm. All steps were carried out at room temperature.General synthesis of protected ligands 3 and 4Gal
The relevant peracetylated carbohydrate was treated with TMS-N3 and SnCl4 according to an established protocol to introduce azide functionality into the anomeric position.49 This carbohydrate azide (2 equiv.), CuI (0.5 equiv.), DIPEA (3 equiv.) and 2,6-bis(trimethylsilylethynyl)pyridine (1 equiv.) were suspended in DMF and heated under microwave irradiation at 100 °C for 20 minutes. The reaction mixture was poured into an aqueous solution of EDTA/NH4OH, extracted three times into CH2Cl2, washed with water and brine, dried over MgSO4, filtered and concentrated under reduced pressure; the product was purified by flash chromatography (CH2Cl2–CH3OH gradient) or trituration with cold CH3OH, yielding 3 or 4Gal as an off-white solid, which was used without further purification (3Gal and 3Glc were previously reported32).![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O qt); FT-IR (ATR, cm−1): 2943, 1743 (s), 1612, 1576, 1435, 1368, 1212 (s), 1045, 915, 810, 732.O); FT-IR (ATR, cm−1): 2884, 1742 (s), 1608, 1575, 1435, 136, 1216 (s), 1174, 1133, 1042 (s), 915, 813, 730.
O qt); FT-IR (ATR, cm−1): 2943, 1743 (s), 1612, 1576, 1435, 1368, 1212 (s), 1045, 915, 810, 732.O); FT-IR (ATR, cm−1): 2884, 1742 (s), 1608, 1575, 1435, 136, 1216 (s), 1174, 1133, 1042 (s), 915, 813, 730.General synthesis of deprotected ligands 5 and 6
To a suspension of the relevant protected ligand 3 or 4 in CH3OH (10 mL) was added a 1 M solution of NaOCH3 in CH3OH (0.1 mL). The reaction mixture was stirred at room temperature. Upon completion, the solution was neutralised by addition of DOWEX® 50X8 H+ resin, filtered and concentrated under reduced pressure, yielding the free sugar ligands as off-white solids (5Glc and 5Gal), or amber oils (5Man, 5Lac and 6Gal).Preparation of Ru(II) complexes of btp ligands
Carbohydrate ligands, 5 or 6 (2 equiv.) were dissolved in 7 mL 70% aqueous ethanol solution and RuCl3·3H2O (1 equiv.) added. The reaction mixture was heated under microwave irradiation to 120 °C for 40 minutes and concentrated under reduced pressure to yield the product as very hygroscopic orange or red solids 7 or 8.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from Science Foundation Ireland: JPB (Starting Investigator Research Grant 18/SIRG/5501), and TG (PI Award, 13/IA/1895). Preliminary work was carried out as part of a UCD School of Medicine Student Summer Research Award (GC, BP, COR, JPB). We thank Dr Samuel Bradberry, Dr Sandra Bright, Karolina Wojtczak, Seán Hennessey and June Lovitt for assistance. JPB thanks Prof. Paul V. Murphy for support and mentoring, and Prof. Eoin Scanlan for helpful advice and discussion.Notes and references
- L. B. Rice, J. Infect. Dis., 2008, 197, 1079–1081 CrossRef PubMed.
- M. N. Hurley, Drugs Future, 2010, 35, 1007–1014 CrossRef.
- M. N. Hurley, M. Cámara and A. R. Smyth, Eur. Respir. J., 2012, 40, 1014–1023 CrossRef CAS PubMed.
- A. Tynan, L. Mawhinney, M. E. Armstrong, C. O'Reilly, S. Kennedy, E. Caraher, K. Jülicher, D. O'Dwyer, L. Maher, K. Schaffer, A. Fabre, E. F. McKone, L. Leng, R. Bucala, J. Bernhagen, G. Cooke and S. C. Donnelly, FASEB J., 2017, 31, 5102–5110 CrossRef CAS PubMed.
- A. Imberty, M. Wimmerová, E. P. Mitchell and N. Gilboa-Garber, Microbes Infect., 2004, 6, 221–228 CrossRef CAS PubMed.
- N. Gilboa-Garber, Methods Enzymol., 1982, 83, 378–385 CAS.
- S. P. Diggle, R. E. Stacey, C. Dodd, M. Cámara, P. Williams and K. Winzer, Environ. Microbiol., 2006, 8, 1095–1104 CrossRef CAS PubMed.
- T. Bjarnsholt, O. Ciofu, S. Molin, M. Givskov and N. Høiby, Nat. Rev. Drug Discovery, 2013, 12, 791–808 CrossRef CAS PubMed.
- H. P. Hauber, M. Schulz, A. Pforte, D. Mack, P. Zabel and U. Schumacher, Int. J. Med. Sci., 2008, 5, 371–376 CrossRef CAS PubMed.
- A. V. Grishin, M. S. Krivozubov, A. S. Karyagina and A. L. Gintsburg, Acta Naturae, 2015, 7, 29–41 CrossRef CAS PubMed.
- R. Visini, X. Jin, M. Bergmann, G. Michaud, F. Pertici, O. Fu, A. Pukin, T. R. Branson, D. M. E. Thies-Weesie, J. Kemmink, E. Gillon, A. Imberty, A. Stocker, T. Darbre, R. J. Pieters and J. L. Reymond, ACS Chem. Biol., 2015, 10, 2455–2462 CrossRef CAS PubMed.
- G. Yu, A. C. Vicini and R. J. Pieters, J. Org. Chem., 2019, 84, 2470–2488 CrossRef CAS PubMed.
- M. Smadhi, S. de Bentzmann, A. Imberty, M. Gingras, R. Abderrahim and P. G. Goekjian, Beilstein J. Org. Chem., 2014, 10, 1981–1990 CrossRef PubMed.
- F. Pertici, N. J. De Mol, J. Kemmink and R. J. Pieters, Chem.–Eur. J., 2013, 19, 16923–16927 CrossRef CAS PubMed.
- E. Kolomiets, M. A. Swiderska, R. U. Kadam, E. M. V. Johansson, K. E. Jaeger, T. Darbre and J. L. Reymond, ChemMedChem, 2009, 4, 562–569 CrossRef CAS PubMed.
- C. Ligeour, O. Vidal, L. Dupin, F. Casoni, E. Gillon, A. Meyer, S. Vidal, G. Vergoten, J. M. Lacroix, E. Souteyrand, A. Imberty, J. J. Vasseur, Y. Chevolot and F. Morvan, Org. Biomol. Chem., 2015, 13, 8433–8444 RSC.
- M. Reynolds, M. Marradi, A. Imberty, S. Penadés and S. Pérez, Chem.–Eur. J., 2012, 18, 4264–4273 CrossRef CAS PubMed.
- A. Bernardi, J. Jiménez-Barbero, A. Casnati, C. De Castro, T. Darbre, F. Fieschi, J. Finne, H. Funken, K.-E. Jaeger, M. Lahmann, T. K. Lindhorst, M. Marradi, P. Messner, A. Molinaro, P. V. Murphy, C. Nativi, S. Oscarson, S. Penadés, F. Peri, R. J. Pieters, O. Renaudet, J.-L. Reymond, B. Richichi, J. Rojo, F. Sansone, C. Schäffer, W. B. Turnbull, T. Velasco-Torrijos, S. Vidal, S. Vincent, T. Wennekes, H. Zuilhof and A. Imberty, Chem. Soc. Rev., 2013, 42, 4709–4727 RSC.
- A. M. Boukerb, A. Rousset, N. Galanos, J.-B. Méar, M. Thépaut, T. Grandjean, E. Gillon, S. Cecioni, C. Abderrahmen, K. Faure, D. Redelberger, E. Kipnis, R. Dessein, S. Havet, B. Darblade, S. E. Matthews, S. de Bentzmann, B. Guéry, B. Cournoyer, A. Imberty and S. Vidal, J. Med. Chem., 2014, 57, 10275–10289 CrossRef CAS PubMed.
- S. Wang, L. Dupin, M. Noël, C. J. Carroux, L. Renaud, T. Géhin, A. Meyer, E. Souteyrand, J. J. Vasseur, G. Vergoten, Y. Chevolot, F. Morvan and S. Vidal, Chem.–Eur. J., 2016, 22, 11785–11794 CrossRef CAS PubMed.
- K. V. Mariño, M. L. Uhrig, J. M. Benito, A. Imberty, O. Renaudet, D. Goyard, B. Thomas and E. Gillon, Front. Chem., 2019, 7, 666 CrossRef PubMed.
- T. R. Flockton, L. Schnorbus, A. Araujo, J. Adams, M. Hammel and L. J. Perez, Pathogens, 2019, 8, 55 CrossRef CAS PubMed.
- S. Cecioni, R. Lalor, B. Blanchard, J. P. Praly, A. Imberty, S. E. Matthews and S. Vidal, Chem.–Eur. J., 2009, 15, 13232–13240 CrossRef CAS PubMed.
- D. Sicard, S. Cecioni, M. Iazykov, Y. Chevolot, S. E. Matthews, J. P. Praly, E. Souteyrand, A. Imberty, S. Vidal and M. Phaner-Goutorbe, Chem. Commun., 2011, 47, 9483–9485 RSC.
- F. Pertici and R. J. Pieters, Chem. Commun., 2012, 48, 4008–4010 RSC.
- C. Ligeour, O. Vidal, L. Dupin, F. Casoni, E. Gillon, A. Meyer, S. Vidal, G. Vergoten, J. M. Lacroix, E. Souteyrand, A. Imberty, J. J. Vasseur, Y. Chevolot and F. Morvan, Org. Biomol. Chem., 2015, 13, 8433–8444 RSC.
- S. Alfei and A. M. Schito, Nanomaterials, 2020, 10, 1–50 Search PubMed.
- G. Michaud, R. Visini, M. Bergmann, G. Salerno, R. Bosco, E. Gillon, B. Richichi, C. Nativi, A. Imberty, A. Stocker, T. Darbre and J. L. Reymond, Chem. Sci., 2016, 7, 166–182 RSC.
- D. H. Simpson, A. Hapeshi, N. J. Rogers, V. Brabec, G. J. Clarkson, D. J. Fox, O. Hrabina, G. L. Kay, A. K. King, J. Malina, A. D. Millard, J. Moat, D. I. Roper, H. Song, N. R. Waterfield and P. Scott, Chem. Sci., 2019, 10, 9708–9720 RSC.
- C. Eidamshaus and H. U. Reissig, Tetrahedron: Asymmetry, 2011, 22, 1644–1652 CrossRef CAS.
- C. Shen, P. F. Zhang and X. Z. Chen, Helv. Chim. Acta, 2010, 93, 2433–2438 CrossRef CAS.
- J. Kraft, D. Schmollinger, J. Maudrich and T. Ziegler, Synth, 2015, 47, 199–208 CAS.
- S. Wang, J. A. Cuesta-Seijo, D. Lafont, M. M. Palcic and S. Vidal, Chem.–Eur. J., 2013, 19, 15346–15357 CrossRef CAS PubMed.
- I. Carvalho, P. Andrade, V. L. Campo, P. M. M. Guedes, R. Sesti-Costa, J. S. Silva, S. Schenkman, S. Dedola, L. Hill, M. Rejzek, S. A. Nepogodiev and R. A. Field, Bioorg. Med. Chem., 2010, 18, 2412–2427 CrossRef CAS PubMed.
- H. Shen, C. Shen, C. Chen, A. Wang and P. Zhang, Catal. Sci. Technol., 2015, 5, 2065–2071 RSC.
- K. J. Kilpin, E. L. Gavey, C. J. McAdam, C. B. Anderson, S. J. Lind, C. C. Keep, K. C. Gordon and J. D. Crowley, Inorg. Chem., 2011, 50, 6334–6346 CrossRef CAS PubMed.
- D. Schmollinger, J. Kraft, C. Ewald and T. Ziegler, Tetrahedron Lett., 2017, 58, 3643–3645 CrossRef CAS.
- J. P. Byrne, J. A. Kitchen and T. Gunnlaugsson, Chem. Soc. Rev., 2014, 43, 5302–5325 RSC.
- J. P. Byrne, J. A. Kitchen, O. Kotova, V. Leigh, A. P. Bell, J. J. Boland, M. Albrecht and T. Gunnlaugsson, Dalton Trans., 2014, 43, 196–209 RSC.
- J. P. Byrne, M. Martínez-Calvo, R. D. Peacock and T. Gunnlaugsson, Chem.–Eur. J., 2016, 22, 486–490 CrossRef CAS PubMed.
- E. P. McCarney, J. P. Byrne, B. Twamley, M. Martínez-Calvo, G. Ryan, M. E. Möbius and T. Gunnlaugsson, Chem. Commun., 2015, 51, 14123–14126 RSC.
- S. J. Bradberry, J. P. Byrne, C. P. McCoy and T. Gunnlaugsson, Chem. Commun., 2015, 51, 16565–16568 RSC.
- J. P. Byrne, J. A. Kitchen, J. E. O'Brien, R. D. Peacock and T. Gunnlaugsson, Inorg. Chem., 2015, 54, 1426–1439 CrossRef CAS PubMed.
- E. P. McCarney, C. S. Hawes, J. A. Kitchen, K. Byrne, W. Schmitt and T. Gunnlaugsson, Inorg. Chem., 2018, 57, 3920–3930 CrossRef CAS PubMed.
- J. P. Byrne, S. Blasco, A. B. Aletti, G. Hessman and T. Gunnlaugsson, Angew. Chem., Int. Ed., 2016, 55, 8938–8943 CrossRef CAS PubMed.
- R. M. Meudtner, M. Ostermeier, R. Goddard, C. Limberg and S. Hecht, Chem.–Eur. J., 2007, 13, 9834–9840 CrossRef CAS PubMed.
- M. Ostermeier, M.-A. Berlin, R. M. Meudtner, S. Demeshko, F. Meyer, C. Limberg and S. Hecht, Chem.–Eur. J., 2010, 16, 10202–10213 CrossRef CAS PubMed.
- B. Zhang, H. Wang, X. Yan, Y. A. Duan, S. Guo and F. X. Luo, Transition Met. Chem., 2020, 45, 99–110 CrossRef CAS.
- H. Paulsen, Z. Györgydeák and M. Friedmann, Chem. Ber., 1974, 107, 1568–1578 CrossRef CAS.
- C. Bouillon, A. Meyer, S. Vidal, A. Jochum, Y. Chevolot, J. P. Cloarec, J. P. Praly, J. J. Vasseur and F. Morvan, J. Org. Chem., 2006, 71, 4700–4702 CrossRef CAS PubMed.
- Y. Li, J. C. Huffman and A. H. Flood, Chem. Commun., 2007, 2692–2694 RSC.
- S. Li, C. Wu, X. Tang, S. Gao, X. Zhao, H. Yan and X. Wang, Sci. China: Chem., 2013, 56, 595–603 CrossRef CAS.
- P. Rogala, G. Czerwonka, S. Michałkiewicz, M. Hodorowicz, B. Barszcz and A. Jabłońska-Wawrzycka, Chem. Biodiversity, 2019, 16, e1900403 CrossRef CAS PubMed.
- G. Czerwonka, D. Gmiter, A. Guzy, P. Rogala, A. Jabłońska-Wawrzycka, A. Borkowski, T. Cłapa, D. Narożna, P. Kowalczyk, M. Syczewski, M. Drabik, M. Dańczuk and W. Kaca, Biofouling, 2019, 35, 59–74 CrossRef CAS PubMed.
- X. Li, K. Heimann, F. Li, J. M. Warner, F. Richard Keene and J. Grant Collins, Dalton Trans., 2016, 45, 4017–4029 RSC.
- F. Li, J. G. Collins and F. R. Keene, Chem. Soc. Rev., 2015, 44, 2529–2542 RSC.
- S. V. Kumar, S. O. Scottwell, E. Waugh, C. J. McAdam, L. R. Hanton, H. J. L. Brooks and J. D. Crowley, Inorg. Chem., 2016, 55, 9767–9777 CrossRef CAS PubMed.
- A. Imberty, M. Wimmerová, E. P. Mitchell and N. Gilboa-Garber, Microbes Infect., 2004, 6, 221–228 CrossRef CAS PubMed.
- P. Braun, B. Nägele, V. Wittmann and M. Drescher, Angew. Chem., Int. Ed., 2011, 50, 8428–8431 CrossRef CAS PubMed.
- G. M. Sheldrick and IUCr, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- E. Peeters, H. J. Nelis and T. Coenye, J. Microbiol. Methods, 2008, 72, 157–165 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: NMR spectra, biological testing data & supplementary crystallographic data. CCDC 2004944. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra05107a |
| ‡ Selected crystallographic and refinement data for crystal of 3Gal: a, b, c (Å): 13.033(15), 13.033(15), 44.95(5); α, β, γ (°): 90, 90, 120; V (Å3): 6613(17); Z: 6; F(000): 2748; Dc (Mg m−3): 1.316; μ (mm−1): 0.91; GOF, R1, wR2, flack: 1.075, 0.062, 0.151, 0.03(9). |
| This journal is © The Royal Society of Chemistry 2021 |