
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDisulfide–yne reaction: controlling the reactivity of a surface by light†
Yuwen Li‡
ab,
Sen Li‡ab,
Xin Du *ab and
Zhongze Gu*ab
*ab and
Zhongze Gu*ab
aState Key Laboratory of Bioelectronics, Southeast University, Nanjing, 210096, China. E-mail: du.xin@seu.edu.cn; gu@seu.edu.cn
bSchool of Biological Science and Medical Engineering, Southeast University, Nanjing, 210096, China
First published on 14th June 2021
Abstract
In this paper we provide new insight into the disulfide–yne photo reaction, which is similar but different from the well-known thiol–yne photoclick reaction. We show that, unlike the stable product generated from thiol–yne chemistry, the vinyl dithioether structure obtained from disulfide–yne reaction exhibits unique reactivity with thiols and disulfides, which can be used for surface photochemistry to fabricate reactive and dynamic surfaces. The possible mechanism for the unique reactivity of vinyl dithioether structure was discussed. We demonstrated that disulfide–yne reactions are highly compatible with thiol–yne chemistry, but offer the flexibility and dynamic nature that is lacking in thiol–yne chemistry, thus could be a good replenishment for the existing thiol–yne toolbox.
Introduction
Being first reported in the 1920s and reinvented in 2009,1 thiol–yne photochemistry, one of the most studied “click” reactions,2 has been widely applied in the last decade.1,3–5 Upon UV irradiation, thiols irreversibly react with alkyne groups to form stable ethyl dithioether structures (1) via a radical addition process. The reaction ends in seconds with high conversion and perfect spatial–temporal controllability.2 Due to these advantages, thiol–yne chemistry is recognized as a reliable and powerful tool for chemists and material scientists, with numerous applications ranging from drug delivery,6–11 hydrogel fabrication12–15 and polymer synthesis and modification,3,10,16–19 to surface functionalization and surface patterning.20–25On the contrary, very little attention has been paid to its sister chemistry, disulfide–yne photochemistry. Disulfides react with alkynes under UV irradiation, forming a unique vinyl dithioether structure (2) that has a very similar structure with the product of thiol–yne chemistry (1). Although first reported in 1960s,26 this chemistry has almost been forgotten during the past half century.27–32
The absence of studies on disulfide–yne photochemistry can be attributed to several reasons. On one hand, as reported by Heiba et al., the disulfide–yne reaction leads to various products when equimolar amounts of disulfides and alkynes are reacted.26 The reaction exhibit better conversion only when disulfides were used with >2 eq. excess. On the other hand, no applications were found for the vinyl dithioether structures forming during the disulfide–yne reaction and the only proposal for the potential application was to synthesize sulfide containing drugs.33 Therefore, compared to the superior thiol–yne chemistry, the disulfide–yne photoreaction has been largely neglected.
In this paper, we first report the unique photo-induced reversible/irreversible exchange reaction on the vinyl dithioether structure. We show that, the double bond in the structure 2 maintains its reactivity and can react with both disulfides and thiols under UV irradiation (Scheme 1). The use of thiol or disulfide as a reactant will lead to a different product with distinct reactivity. We show that disulfide–yne photoreaction could be applied as a novel surface chemistry, to construct smart surfaces with dynamic features. Therefore, disulfide–yne reaction could be considered as a good replenishment for the existed thiol–yne toolbox for the construction of smart surfaces and materials.
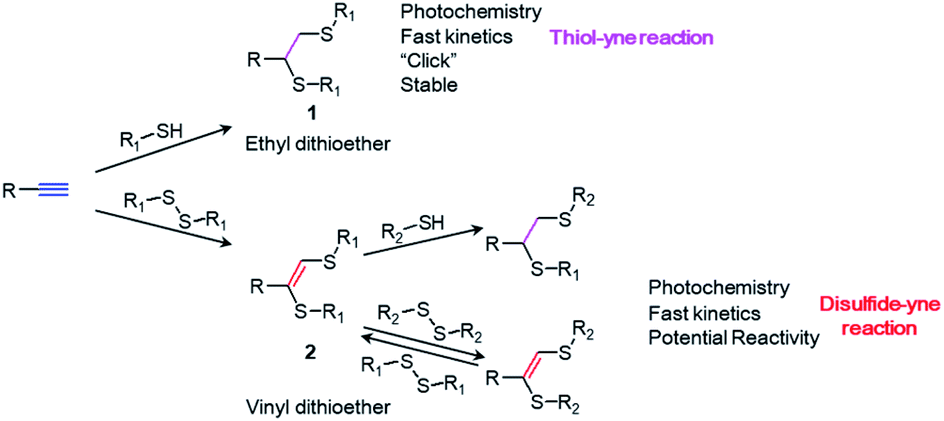 | ||
| Scheme 1 Thiol–yne and disulfide–yne reactions. | ||
Results and discussion
The disulfide–yne photo reaction was first investigated on small molecular level. To perform the reaction, 4-pentynoic acid (PA), dibutyl disulfide (DBD) and a photoinitiator 2,2-bimethoxy-2-phenylacetophenone (DMPA) were added into a 20 mL glass vial containing 4 mL THF, the solution was purged by argon and irradiated under UV (365 nm, 5 mW cm−2) for 1 h. The thiol–yne reaction between 4-pentynoic acid and butanethiol (BT) was also performed as a comparison (Fig. 1a). The products of the reactions were analyzed by mass spectrometry, as shown in Fig. 1b. The reaction between PA and BT exhibited high efficiency, leading to a dithioether product (Fig. 1aii), 4,5-bis(butylthio)pentanoic acid (Fig. 1bi, m/z = 277.12 for negative ion). Disulfide–yne reaction leads to a similar product but with vinyl dithioether structure, 4,5-bis(butylthio)-4-pentenoic acid (Fig. 1aiii), as confirmed by the mass spectrometry (Fig. 1bii, m/z = 275.13 for negative ion) and NMR results (Fig. S1,† from purified product). However, the conversion was rather poor when DBD and PA are in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, as confirmed by the complicated MS results (Fig. 1bii, less than 10% conversion by column chromatography); when the amount of DBD increases, the side reaction significantly decreased, as indicated by the much higher intensity at the peak of 275 (Fig. 1biii and iv) and the higher calculated conversion after column chromatography (40% and 55% for PA with 2 eq. and 4 eq. DBD, respectively). When the amount of DBD reaches 16 eq., clear peak at m/z = 277 is observed, this is probably because in this case, DBD is 16 times excess, thus the little impurity (probably BT) in the chemicals will lead to obvious amount of side product. Reaction between PA and other types of disulfides (bis(carboxyethyl) disulfide, dihydroxyethyl disulfide, phenyl disulfide) were also investigated, the mass spectrometry analysis on the UV-exposed reaction mixture clearly show the formation of vinyl dithiolether structures with different functional groups (Fig. S2†). Disulfide–yne reaction in different solvents was also tested, the reaction was performed using ethanol, toluene and water as a solvent. Mass spectra of the obtained mixtures shown clear peaks of the products and few side products were obtained (Fig. S3†).
:1 ratio, as confirmed by the complicated MS results (Fig. 1bii, less than 10% conversion by column chromatography); when the amount of DBD increases, the side reaction significantly decreased, as indicated by the much higher intensity at the peak of 275 (Fig. 1biii and iv) and the higher calculated conversion after column chromatography (40% and 55% for PA with 2 eq. and 4 eq. DBD, respectively). When the amount of DBD reaches 16 eq., clear peak at m/z = 277 is observed, this is probably because in this case, DBD is 16 times excess, thus the little impurity (probably BT) in the chemicals will lead to obvious amount of side product. Reaction between PA and other types of disulfides (bis(carboxyethyl) disulfide, dihydroxyethyl disulfide, phenyl disulfide) were also investigated, the mass spectrometry analysis on the UV-exposed reaction mixture clearly show the formation of vinyl dithiolether structures with different functional groups (Fig. S2†). Disulfide–yne reaction in different solvents was also tested, the reaction was performed using ethanol, toluene and water as a solvent. Mass spectra of the obtained mixtures shown clear peaks of the products and few side products were obtained (Fig. S3†).
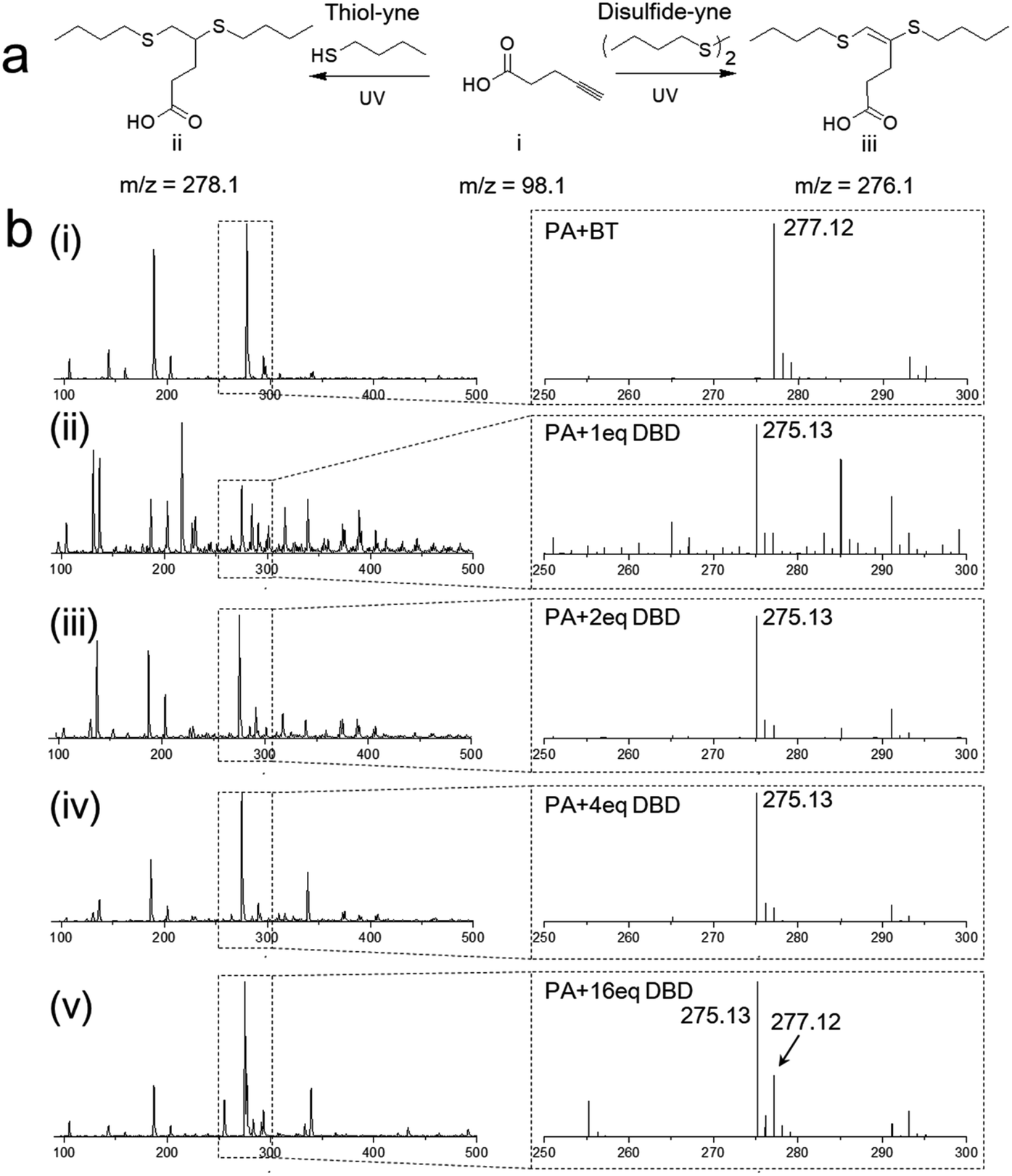 | ||
| Fig. 1 Mass spectroscopy measurement of the disulfide–yne photo reaction. (a) Scheme representing the thiol–yne reaction and disulfide–yne photo reaction. (b) Mass spectrum of the resulted mixture of thiol–yne reaction, and disulfide–yne reaction (with different amount of DBD). PA: 4-pentynoic acid; BT: butanethiol; DBD: dibutyl disulfide. | ||
An interesting question for the disulfide–yne reaction is that whether the C–C double bond on the resulted vinyl dithioether structure keeps reactivity, and if it keeps the reactivity, why does the reaction only lead to 1-step addition product. Indeed, as we check the mass spectrometry results, no peak could be found for the 2-step addition products (Fig. S2†). A similar situation could be found on allyl disulfides, which is known to undergo addition–fragmentation chain transfer (AFCT) reaction, leading to the maintenance of double bond while exhibiting unique dynamic nature (Fig. S4†).34 We assume that vinyl dithioether structure may exhibit similar reactivity as allyl disulfide structure. To confirm our hypothesis, 4,5-bis(butylthio)pent-4-enoic acid was purified from the obtained mixture of PA-DBD reaction and then reacted with 4 eq. of mercaptoethanol or hydroxyethyl disulfide under UV (Fig. 2a). Mass spectrometry analysis was performed on the resulted mixtures. As we expected, further reaction indeed occurred on the vinyl dithioether structures, indicating that the double bond on the molecule still keeps the reactivity. Interestingly, we found that the addition of thiol or disulfide would lead to different results. As shown in Fig. 2b, after UV irradiation in both samples, the peak of 4,5-bis(butylthio)pent-4-enoic acid (m/z = 275 for negative ion) completely disappeared, indicating that further reaction occurred on this molecule. When mercaptoethanol was used in the further reaction, the reaction leads to 5-(butylthio)-4-(2-hydroxyethylthio)pentanoic acid, a saturated product with one butyl sulfide group substituted by hydroxyethyl sulfide group. On the contrary, when hydroxyethyl disulfide was added, the reaction led to 4,5-bis(2-hydroxyethylthio)pent-4-enoic acid (Fig. 2c), with a retained C–C double bond but complete substitution of butyl sulfide group by hydroxyethyl sulfide group. This indicates that vinyl dithioether structure exhibits reactivity and undergo different reactions when reacting with thiols or disulfides.
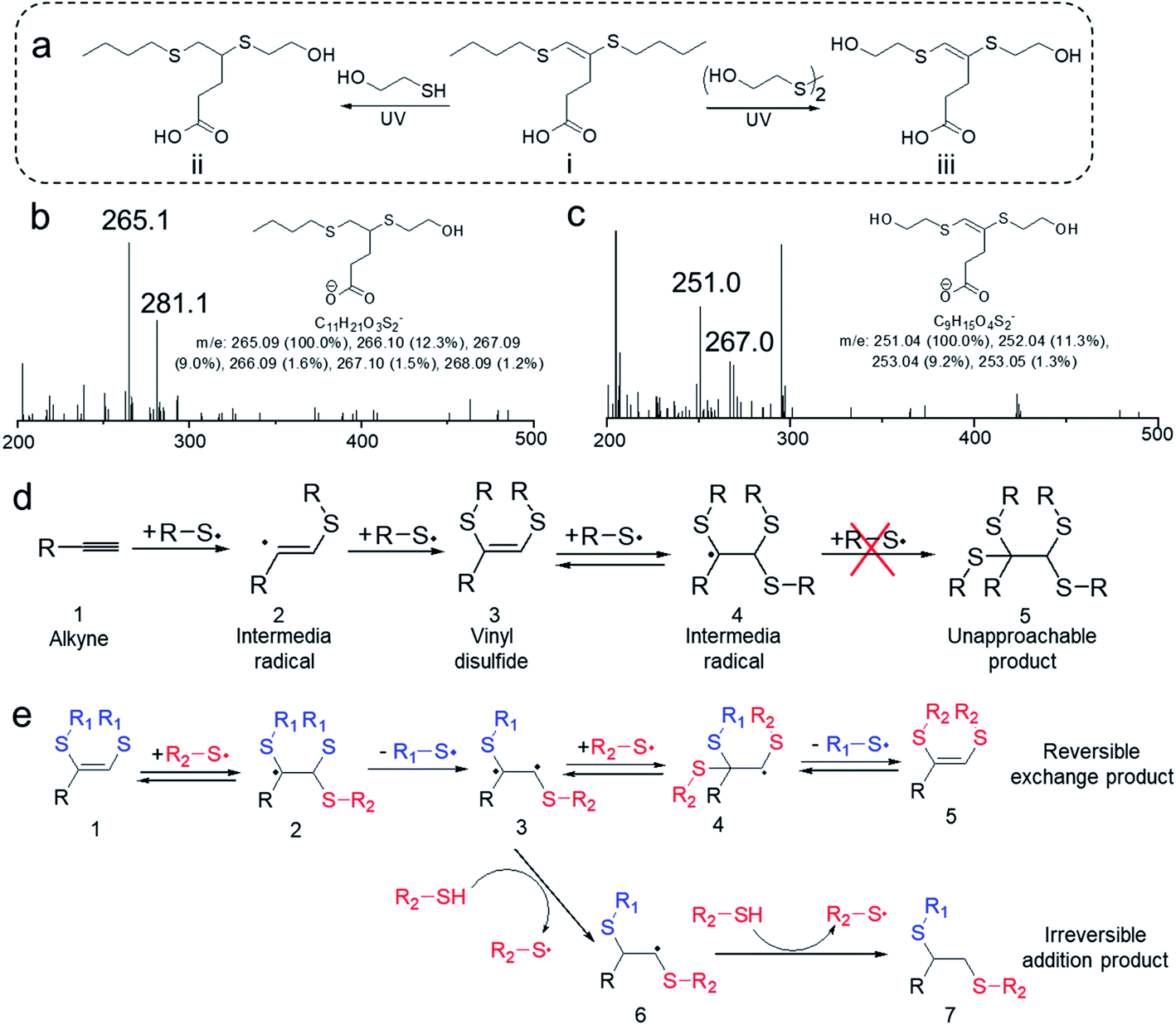 | ||
| Fig. 2 (a) Vinyl dithioether structure keeps further reactivity and can react with thiols or disulfides to results different products. (b) Mass spectrometry result of the reaction mixture ii in (a). The peak at 275 disappeared and peak at 265 appeared, indicating that vinyl dithioethers undergo further reactions with thiols (peak 281 refers to the oxidation product of peak 265). (c) Mass spectrometry result of the reaction mixture iii in (a). The peak at 275 disappeared and peak at 251 appeared, indicating that vinyl dithioethers undergo further reactions with disulfides (peak 267 refers to the oxidation product of peak 251). (d) Proposed mechanism for the disulfide–yne reaction. (e) Proposed mechanism for the reaction of vinyl dithioether with disulfides or thiols, respectively. | ||
The proposed mechanism for the formation of vinyl dithioethers in disulfide–yne reaction is shown in Fig. 2d. When an alkyne group (1) is exposed to a thiyl radical (formed by thiols or disulfides under UV irradiation), the radical will add to the C–C triple bond to form a vinyl dithioether radical (2). In a typical thiol–yne reaction, this radical then absorbs a hydrogen from mercapto group and becomes a vinyl sulfide, which can further react with a thiyl radical to form ethyl disulfide products.17 In disulfide–yne reaction, however, the reaction solution does not contain mercapto groups and the vinyl dithioether radical can only be coupled with another thiyl radical to form vinyl dithioethers (3). This vinyl dithioether can react with a thiyl radical to form an ethyl trisulfide radical (4), however, as in the final product the double bond was retained, in this process the ethyl trisulfide radical probably do not exhibit further reactivity, which is similar as allyl disulfide radical.34 To obtained a stable product, a thiyl radical will then leave from the ethyl trisulfide radical, leading to vinyl dithioether again (3). Therefore, vinyl dithioethers will be obtained by disulfide–yne reaction in spite of the large excess of disulfide molecules.
When the vinyl dithioether (1) is reacted with a thiyl radical from a different disulfide (R2–S˙), as shown in Fig. 2e, an ethyl trisulfide radical (2) without further reactivity would be formed. This ethyl trisulfide radical will then release a thiyl radical to go back to vinyl dithioether. However, in this process, the released thiyl radical can either be the newly added one (R2–S˙) or the former one (R1–S˙), leading to the possible substitution of the sulfide group on vinyl dithioethers. This addition–release process is dynamic and reversible under UV irradiation, thus a new vinyl dithioether (5) with both sulfide chains (R1–S–) substituted by new substituents (R2–S–) would be obtained when the new disulfide is excess.
When thiols were used in the secondary reaction, however, the mechanism of the reaction alters. As shown Fig. 2e, upon UV irradiation, vinyl dithioether (1) reacts with the thiyl radical R2–S˙ to form an ethyl trisulfide radical (2), which can release a thiyl radical to form an intermediate radical (3). The intermediate radical has the opportunity to absorb a hydrogen from the surrounded thiol molecules, to generate a new radical that cannot go back to vinyl dithioether (6). The resulting radicals are still unable to couple with a thiyl radical, thus the only possible reaction for the radical is to absorb another hydrogen, leading to a stable ethyl dithioether (7) with one sulfide group being substituted (when the thiol is in excess). Therefore, the use of thiol or disulfide to react with vinyl dithioether will determine whether the reaction is irreversible (with thiols) or reversible (with disulfides).
To investigate the potential reactivity of vinyl dithioether structure with other groups, 4 eq. butanol, acetic acid and octylamine were allowed to react with vinyl dithioether under UV for 1 h, and the products were analyzed by mass spectrometry. As shown in Fig. S5,† it's clear that no addition or exchange products can be found, indicating that the vinyl dithioether structure is quite stable to these groups. This is beneficial since those groups are commonly existed in bioenvironment, the stability of vinyl dithioether structure to such groups avoid the possible disturb when disulfide–yne reaction is used in bioapplications.
Due to its distinctive property, disulfide–yne reaction may be employed to generate surfaces with unique reactivity. We investigated the performance of this reaction as a surface chemistry. An alkyne surface was generated on a porous HEMA–EDMA surface as reported previously (Fig. 3a).35 to perform the functionalization, 50 μL mL−1 DBD in toluene (with 2.5 mg mL−1 DMPA) was added onto the surface, then the surface was covered with a quartz glass and irradiated under UV for 5 min, resulting in a DBD modified surface (DBD surface). The same process was also applied by using butanethiol (50 μL mL−1 in toluene) as a comparison (BT surface, generated with thiol–yne chemistry). As shown in Fig. 3a, the reaction with both butanethiol (BT surface) and dibutyldisulfide (DBD surface) showed an increase in static WCA from 100° to 130°, confirming the successful attachment of hydrophobic butyl sulfide groups onto the alkyne surface. Disulfides with other functional groups could also be used to modify the alkyne surface, leading to corresponding WCA change after modification. To further characterize the surface, we performed ToF-SIMS measurement on alkyne, BT and DBD surfaces. As shown in Fig. 3b, the alkyne surface exhibits a strong peak at m/z = 97, referring to the anion of the attached 4-pentynioc acetate (Fig. S6†). After functionalization by BT or DBD, the peak at m/z = 97 disappeared and strong peaks at m/z = 89 appeared (butyl sulfide anion), indicating the consumption of alkyne groups and the attachment of butyl sulfide groups on the surface. Clear peak at 277 is observed on BT surface, which comes from the thiol–yne addition product (Fig. S6†), while such peak is not observed on DBD surface. Thus, the attachment of butyl sulfide groups to the DBD surface should follow a different path. The peak at 275, referring to the product of disulfide–yne reaction, is not observed on the DBD surface, this might be because of the low stability of the vinyl dithioether group under the condition of ToF-SIMS experiment.
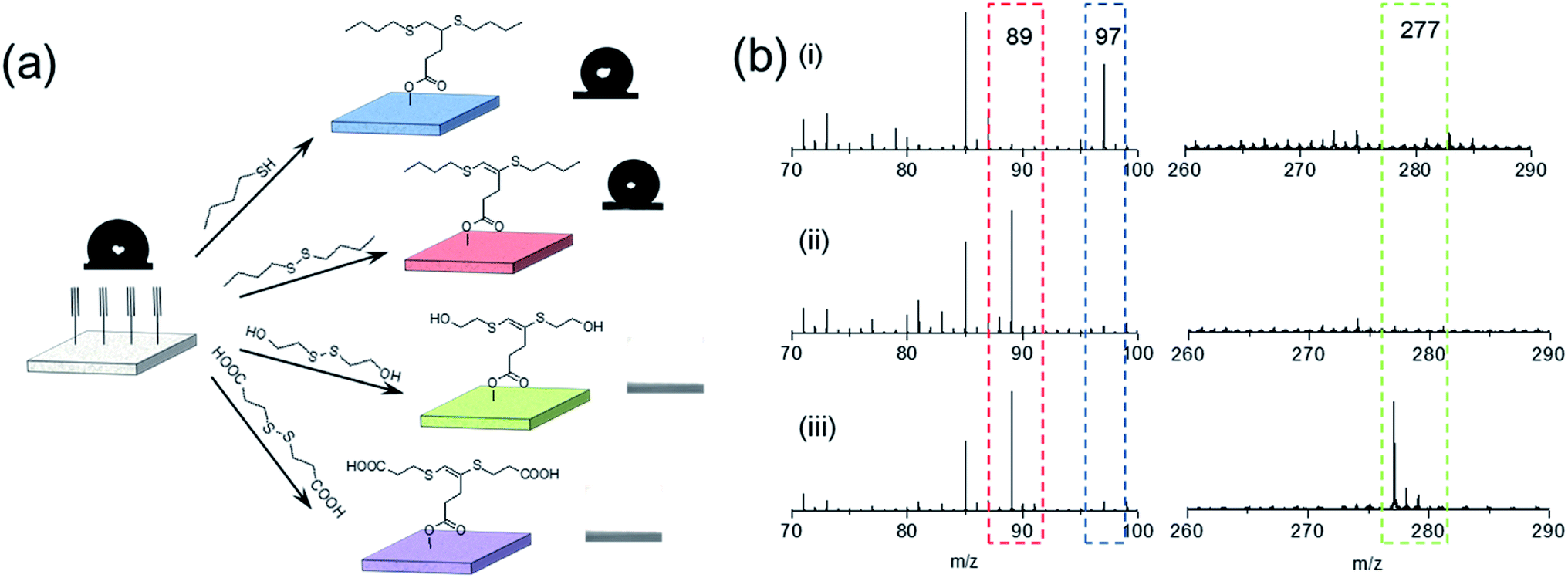 | ||
| Fig. 3 Disulfide–yne reaction on surface. (a) Direct functionalization of an alkyne surface with disulfides leads to different surfaces. (b) ToF-SIMS results of (i) alkyne surface, (ii) alkyne–DBD surface and (iii) alkyne–BT surface. | ||
The kinetics of the disulfide–yne surface photochemistry was investigated by modifying the alkyne surface with DBD, HED or CED, and measuring the WCA change of the surface during modification. As shown in Fig. S7,† the kinetics of the reaction is very fast, in general 180 s is enough for the modification (Fig. S8†). The photo-controlled nature of the reaction also allows the modification to be patterned with a photomask (Fig. S9†).
We then examine the reactivity of the obtained surfaces. As shown in Fig. 4a, thiol–yne reaction on alkyne surface generates a stable BT surface without further reactivity, thus the WCA of the surface is unchangeable with further modification (Fig. 4ai). On the contrary, disulfide–yne reaction generates a reactive DBD surface (Fig. 4aii), which is reactive to thiols and disulfides, as confirmed by the WCA change of the surface after further modification. When thiol is used to functionalize DBD surface, the reactivity of the surface is inhibited due to the elimination of the vinyl dithioether structure (Fig. 4aiii). When disulfide is used, the obtained surface is still reactive due to the remaining vinyl dithioether structure after exchange (Fig. 4aiv). Thus the selection of thiol or disulfide can determine the further reactivity of the surface. The exchange process is also confirmed by a ToF-SIMS test on a patterned surface, where HED modified alkyne surface was secondary modified with DBD under a photomask. The result (Fig. 4b) clear shows the significant decrease of hydroxyethyl sulfide anion and the increase of butyl sulfide anion on the exposed area, describing that an exchange process was occurred between hydroxyethyl sulfide group and butyl sulfide group, which corresponds to the proposed mechanism for the dynamic exchange on vinyl dithioether structure.
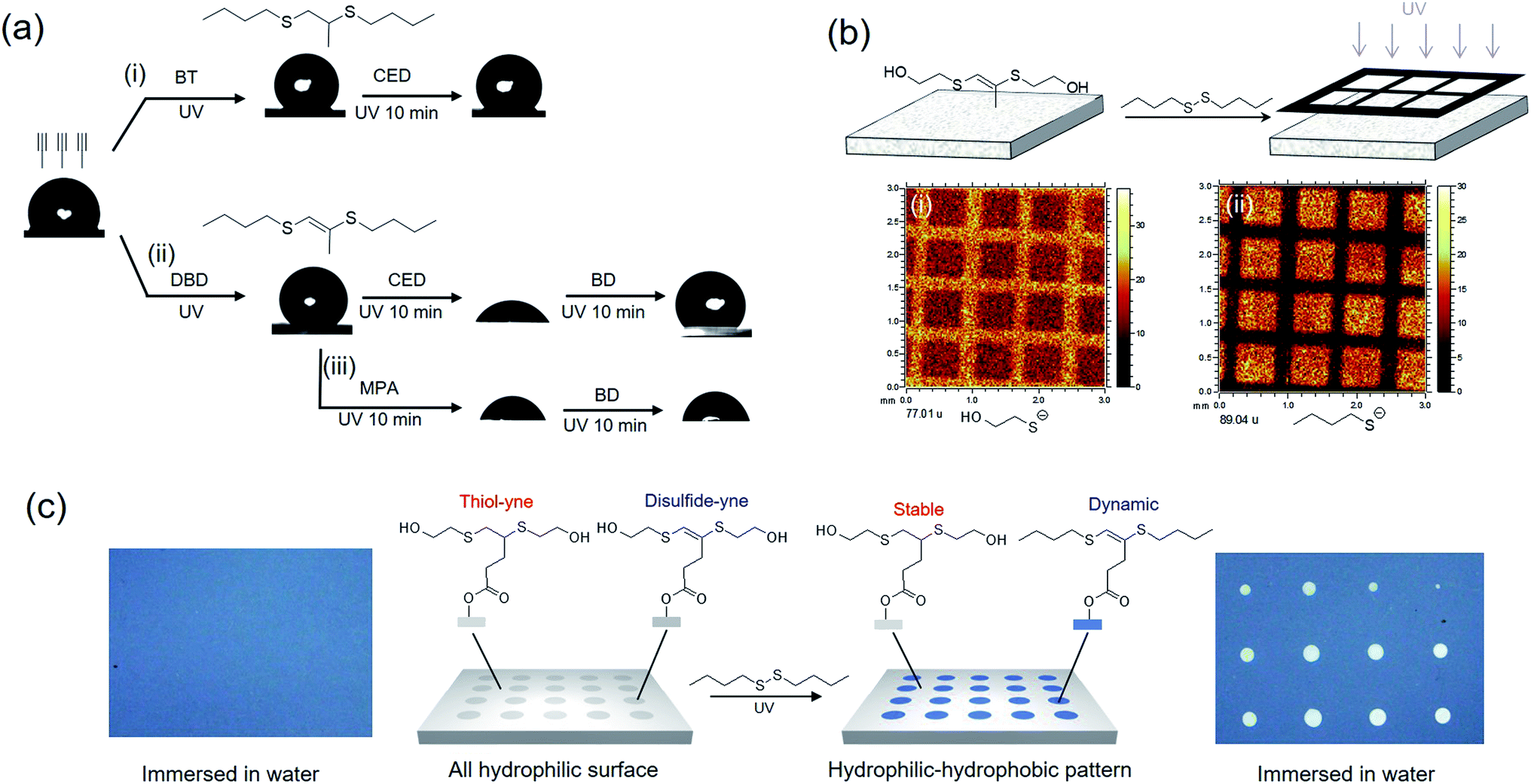 | ||
| Fig. 4 Reactivity of surfaces generated via thiol–yne and disulfide–yne reaction. (a) Reactivity test on the obtained surface using WCA test. BT: butyl thiol; CED: bis(carboxyethyl) disulfide; DBD: dibutyl disulfide; MPA: mercaptopropionic acid. (b) ToF-SIMS test on a patterned vinyl dithioether surface. After UV exposure, the hydroxyethyl sulfide anion on irradiated area decreased, and the butyl sulfide anion on the same area increased, indicating that exchange of sulfide chains were occurred during the secondary modification. (c) Co-functionalization an alkyne surface with thiol–yne chemistry and disulfide–yne reaction, resulting a surface with hidden reactivity at selected area. | ||
Combining the thiol–yne and disulfide–yne reaction, patterned surface with hidden reactivity can be generated. As shown in Fig. 4c, a patterned surface was generated by sequently modifying alkyne surface with mercapto ethanol and hydroxyl ethyl disulfide under a photomask (Fig. S9†). A hydrophilic surface was obtained, when immersing in water, the surface was completely wetted (Fig. 4c, left). After immersing in DBD solution and flood irradiated, butyl sulfide chains were attached on the exposed area, making the area hydrophobic and none wettable in water (Fig. 4c, right). This leads to the reveal of the “hidden pattern”. This makes the disulfide–yne reaction a very good replenishment for the thiol–yne chemistry for the construction of smart surfaces and materials.
Conclusions
In conclusion we proposed a new insight into the disulfide–yne photochemistry, which is similar but also different from the well-known thiol–yne photoclick chemistry. We show the vinyl dithioether structures obtained from disulfide–yne reaction exhibit unique dynamic properties that can further react with thiols and disulfides. The choose of thiols or disulfides for further reaction can selectively eliminate or keep the C–C double bond on vinyl dithioether, allowing the control of the reactivity of the product. Disulfide–yne photochemistry could be applied on surface, resulting a surface with similar properties as the surface produced by thiol–yne chemistry, but with hidden reactivity. Further modification can be performed on this surface using thiols or disulfides. Disulfide–yne reaction is highly compatible and complemental to the existed thiol–yne toolbox, offering the flexibility and reversibility that is lacked in thiol–yne chemistry. We expect a wide application for this chemistry and look forward for further investigations in this field.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research is supported by the National Key R&D Program of China (No. 2017YFA0700500). X. Du thanks the Fundamental Research Funding from Jiangsu Province (BK20170662). X. Du thanks the Southeast University-Nanjing Medical University Cooperative Research Project (2242018K3DN17) for funding support.References
- A. Massi and D. Nanni, Org. Biomol. Chem., 2012, 10, 3791–3807 RSC.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed. Engl., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- A. B. Lowe, Polymer, 2014, 55, 5517–5549 CrossRef CAS.
- A. B. Lowe, C. E. Hoyle and C. N. Bowman, J. Mater. Chem., 2010, 20, 4745 RSC.
- J. V. Burykina, N. S. Shlapakov, E. G. Gordeev, B. König and V. P. Ananikov, Chem. Sci., 2020, 11, 10061–10070 RSC.
- G. Chen, J. Kumar, A. Gregory and M. H. Stenzel, Chem. Commun., 2009, 12, 6291–6293 RSC.
- L. Li, D. Zahner, Y. Su, C. Gruen, G. Davidson and P. a Levkin, Biomaterials, 2012, 33, 8160–8166 CrossRef CAS PubMed.
- E. Lallana, F. Fernandez-Trillo, A. Sousa-Herves, R. Riguera and E. Fernandez-Megia, Pharm. Res., 2012, 29, 902–921 CrossRef CAS PubMed.
- J. G. Ray, J. T. Ly and D. a. Savin, Polym. Chem., 2011, 2, 1536 RSC.
- Z. Zhang, L. Yin, Y. Xu, R. Tong, Y. Lu, J. Ren and J. Cheng, Biomacromolecules, 2012, 13, 3456–3462 CrossRef CAS PubMed.
- E. Paruli, T. Griesser, F. Merlier, C. Gonzato and K. Haupt, Polym. Chem., 2019, 10, 4732–4739 RSC.
- Y. Jiang, J. Chen, C. Deng, E. J. Suuronen and Z. Zhong, Biomaterials, 2014, 35, 4969–4985 CrossRef CAS PubMed.
- L. J. Macdougall, M. M. Pérez-Madrigal, M. C. Arno and A. P. Dove, Biomacromolecules, 2018, 19, 1378–1388 CrossRef CAS PubMed.
- H. Ma, A. S. Caldwell, M. A. Azagarsamy, A. Gonzalez Rodriguez and K. S. Anseth, Biomaterials, 2020, 255, 120205 CrossRef CAS PubMed.
- M. Li, A. P. Dove and V. X. Truong, Angew. Chem., Int. Ed., 2020, 59, 2284–2288 CrossRef CAS PubMed.
- D. Konkolewicz, A. Gray-Weale and S. Perrier, J. Am. Chem. Soc., 2009, 131, 18075–18077 CrossRef CAS PubMed.
- B. D. Fairbanks, T. F. Scott, C. J. Kloxin, K. S. Anseth and C. N. Bowman, Macromolecules, 2009, 42, 211–217 CrossRef CAS PubMed.
- K. Kutonova, B. Ebenhoch, L. Graf Von Reventlow, S. Heißler, L. Rothmann, S. Bräse and A. Colsmann, J. Mater. Chem. C, 2020, 8, 16498–16505 RSC.
- A. B. Cook and S. Perrier, Adv. Funct. Mater., 2020, 30, 1901001 CrossRef CAS.
- R. M. Hensarling, V. A. Doughty, J. W. Chan and D. L. Patton, J. Am. Chem. Soc., 2009, 131, 14673–14675 CrossRef CAS PubMed.
- S. B. Rahane, R. M. Hensarling, B. J. Sparks, C. M. Stafford and D. L. Patton, J. Mater. Chem., 2012, 22, 932 RSC.
- D. Meziane, A. Barras, A. Kromka, J. Houdkova, R. Boukherroub and S. Szunerits, Anal. Chem., 2012, 194–200 CrossRef CAS PubMed.
- W. Feng, L. Li, E. Ueda, J. Li, S. Heißler, A. Welle, O. Trapp and P. a. Levkin, Adv. Mater. Interfaces, 2014, 1, 1400269 CrossRef.
- I. Roppolo, F. Frascella, M. Gastaldi, M. Castellino, B. Ciubini, C. Barolo, L. Scaltrito, C. Nicosia, M. Zanetti and A. Chiappone, Polym. Chem., 2019, 10, 5950–5958 RSC.
- C. Lanz, M. Schlötter, N. Klinkenberg, P. Besirske and S. Polarz, Angew. Chem., Int. Ed., 2020, 59, 8902–8906 CrossRef CAS PubMed.
- E. I. Heiba and R. M. Dessau, J. Org. Chem., 1967, 32, 3837–3840 CrossRef CAS.
- P. A. Of, D. Diselenide, T. O. Allenes, A. Ogawa, M. Sekiguchi, N. Kambe, N. Sonoda and A. Chemistry, Tetrahedron Lett., 1990, 31, 5931–5934 CrossRef.
- T. Mitamura, K. Iwata and A. Ogawa, Org. Lett., 2009, 11, 3422–3424 CrossRef CAS PubMed.
- T. Shirai, S. Kawaguchi, A. Nomoto and A. Ogawa, Tetrahedron Lett., 2008, 49, 4043–4046 CrossRef CAS.
- A. Ogawa, K. Yokoyama, R. Obayashi, L.-B. Han, N. Kambe and N. Sonoda, Tetrahedron, 1993, 49, 1177–1188 CrossRef CAS.
- A. Ogawa, R. Obayashi, M. Doi, N. Sonoda and T. Hirao, J. Org. Chem., 1998, 3263, 4277–4281 CrossRef.
- A. Ogawa, H. Yokoyama, K. Yokoyama, T. Masawaki and N. Kambe, J. Org. Chem., 1991, 2, 5721–5723 CrossRef.
- N. Yamagiwa, Y. Suto and Y. Torisawa, Bioorg. Med. Chem. Lett., 2007, 17, 6197–6201 CrossRef CAS PubMed.
- N. R. Gandavarapu, M. A. Azagarsamy and K. S. Anseth, Adv. Mater., 2014, 26, 2521–2526 CrossRef CAS PubMed.
- W. Feng, L. Li, E. Ueda, J. Li, S. Heißler, A. Welle, O. Trapp and P. a. Levkin, Adv. Mater. Interfaces, 2014, 1, 1400269 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra02262h |
| ‡ Y Li and S Li contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |