
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLight-driven selective aerobic oxidation of (iso)quinoliniums and related heterocycles†
Meimei Zhou‡
a,
Keyang Yu‡b,
Jianxin Liuc,
Weimei Shic,
Yingming Pan a,
Haitao Tanga,
Xiangjun Peng*b,
Qian Liu*b and
Hengshan Wang*a
a,
Haitao Tanga,
Xiangjun Peng*b,
Qian Liu*b and
Hengshan Wang*a
aState Key Laboratory for Chemistry and Molecular Engineering of Medicinal Resources, School of Chemistry and Pharmaceutical Sciences of Guangxi Normal University, Guilin 541004, People's Republic of China. E-mail: whengshan@163.com
bKey Laboratory of Prevention and Treatment of Cardiovascular and Cerebrovascular Diseases of Ministry of Education, Gannan Medical University, Ganzhou 341000, People's Republic of China. E-mail: pengxiangjun99@163.com; 18907979876@126.com
cSchool of Pharmaceutical Science, Gannan Medical University, Ganzhou, Jiangxi 341000, P. R. China
First published on 4th May 2021
Abstract
Selective C1–H/C4–H carbonylation of N-methylene iminium salts, catalyzed by visible-light photoredox and oxygen in the air, has been reported. A ruthenium complex acts as a chemical switch to conduct two different reaction pathways and to afford two different kinds of products. In the absence of the ruthenium complex, the Csp2–H bonds adjacent to the nitrogen atoms are oxidized to α-lactams by the N-methyleneiminium substrates themselves as photosensitizers. In the presence of the ruthenium complex, the oxidation reaction site of quinoliniums is switched to the C4 region, resulting in the formation of 4-quinolones. The use of two transformations directly introduces oxygen into the nitrogen heterocyclic skeletons under an air atmosphere.
Introduction
The biomimetic characteristics of isoquinolone and quinolone rings, reminiscent of the rigidly fixed skeleton of N-heterocycles, frequently emerge in many natural products and pharmaceuticals.1 For example, the alkaloids doryanine and sarcomejine have been isolated from Cryptocarya chinensis (Lauraceae) and Sarcomelicope megistophylla (Rutaceae), respectively.2 The isoquinolone scaffold is widely found as the core structure in topoisomerase I (Top I) inhibitors.3 Elvitegravir (EVG), which contains the elementary unit of the corresponding quinolone, is a potent inhibitor of human immunodeficiency virus type 1 (HIV-1) integrase (Fig. 1).4 Due to their diverse biological activities, tremendous effort has been devoted to the effective synthesis of (iso)quinolones. The traditional methods usually require stoichiometric amounts of K3Fe(CN)6 as an oxidant, and this inevitably produces a considerable number of toxic by-products such as K4Fe(CN)6.5 Other methods, such as Conrad–Limpach and Niementowski reactions, have proven to be powerful tactics using the condensation of amines with carboxyl derivatives followed by cyclization to generate quinolone analogs.6 However, most of these syntheses suffer from harsh reaction conditions (high temperature, Eaton reagent, etc.), poor conversion of the substrates and the use of excessive oxidants or additives. Consequently, considerable efforts have been devoted to establishing mild, environmentally friendly and sustainable procedures for these valuable scaffolds.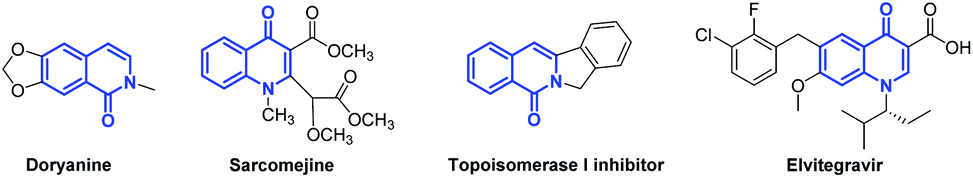 | ||
| Fig. 1 Representative examples of natural products and pharmaceuticals containing the (iso)quinolone structural motif. | ||
Using O2 (or ambient air) as an oxidant is the most ideal methodology for direct oxygenation.7 Moreover, visible-light- promoted aerobic oxidations have been demonstrated in the synthesis of biologically active molecules in green and environmentally friendly conditions.8 Recently, Fu et al. successfully achieved the visible-light mediated ortho C–H oxidation of N-alkyliminiums using Eosin Y as the photocatalyst and air as the oxidant, obtaining isoquinoline-1-ones (Scheme 1a).9 Deng and co-authors highlighted the photocatalytic oxidation and subsequent cyclization protocol to transform indoles to quinolones.10 The attractive properties of these photocatalytic reactions were their mild reaction conditions, low catalyst loading, ubiquity and versatility functionality of directing C–H activation with O2 (or air). In our designed method, the isoquinolinium salts (1) formed excited isoquinolinium salts (1*) under visible-light irradiation without other photosensitizers,11 and then underwent photoinduced electron transfer (PET) to transform the α-carbon radicals (A), which were captured by O2 to obtain isoquinolones (2) (Scheme 1b). This process sets the stage for carbonylation to the oxidation onto the C1-position of isoquinoliniums, and another successful strategy that obtains quinoliniums by para(C4)-selective oxidation will significantly broaden the potential of remote site-selective functionalization. Herein, we delineate the successful realization of different reactive sites in (iso)quinoliniums and related heterocycles through visible-light sources, orchestrating a Ru-photocatalyst, which controls the ortho- or para-position of oxidation.
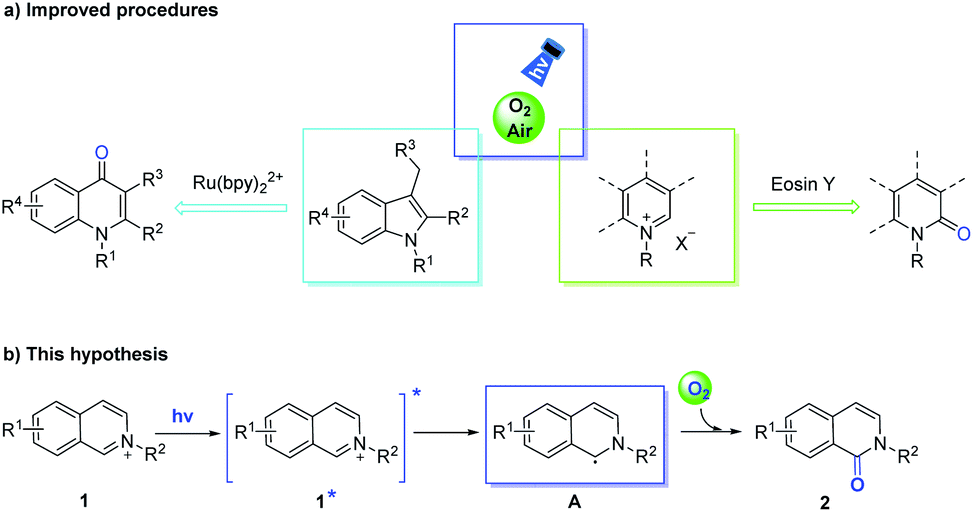 | ||
| Scheme 1 The representative photocatalysis of oxidative approaches to the construction of N-substituted isoquinolin-1(2H)-ones and 4-quinolones (a), and our hypothesis (b). | ||
Experimental section
General methods
Unless otherwise specified, commercial reagents and solvents were used without further purification. All manipulations were performed in air at room temperature. For flash column chromatography, 100–200 mesh and 300–400 mesh silica gels were used by eluting with petroleum ether/ethyl acetate. 1H and 13C spectra were recorded on a 400 MHz spectrometer. The chemical shifts were reported in ppm. 1H NMR spectra were referenced to DMSO (2.50 ppm) or CDCl3 (7.26 ppm), and 13C NMR spectra were referenced to DMSO (39.5 ppm) or CDCl3 (77.0 ppm). Peak multiplicities were designated by the following abbreviations: s, singlet; d, doublet; t, triplet; m, multiplet, and J is the coupling constant in Hz. HRMS spectra were recorded with a Micromass QTOF2 Quadrupole/time-of-flight tandem mass spectrometer using electron spray ionization.The synthesis of halogenated N-methylene iminiums (1a as the example)
A solution of isoquinoline (5 mmol), benzyl bromide (10 mmol) and CH3CN (15 mL) was refluxed for 12 hours and cooled to room temperature. After the addition of ethyl acetate, isoquinoline salt was precipitated rapidly into a solid, and this was then filtered and washed with ethyl acetate to obtain a white solid 1a. The volatile solvents were removed by vacuum evaporation.The synthesis of α-lactams
A solution of isoquinoline (5 mmol), halogenated N-methylene iminiums 1 (0.2 mmol), and DABCO (0.4 mmol), was added to a 10 mL quartz tube in CH3CN (2 mL), and then the mixture was irradiated with green LEDs (15 W) (approximately 1.5 cm away from the light source) at room temperature in an air atmosphere. After complete conversion of the substrate (monitored by TLC), the reaction mixture was diluted with 20 mL of EtOAc and the solution was filtered by flash chromatography (petroleum ether/ethyl acetate 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). The filtrate was evaporated by rotary evaporator and the residue was purified by silica gel column chromatography to give the desired product 2.
:1). The filtrate was evaporated by rotary evaporator and the residue was purified by silica gel column chromatography to give the desired product 2.
The synthesis of 4-quinolones
Halogenated quinoliniums 1ag–1aq (0.2 mmol), DABCO (0.4 mmol), and Ru(bpy)3Cl2 (0.004 mmol) were added to a 10 mL quartz tube in CH3CN (2 mL), and then the mixture was irradiated with blue LEDs (15 W) (approximately 1.5 cm away from the light source) at room temperature in an air atmosphere. After complete conversion of the substrate (monitored by TLC), the reaction mixture was diluted with 20 mL of EtOAc, and the solution was filtered by flash chromatography (petroleum ether/ethyl acetate 2:1). The filtrate was evaporated by rotary evaporator and the residue was purified by silica gel column chromatography to give the desired product 3.
In vitro cardiovascular activities
The MOVAS (mouse aorta vascular smooth muscle cells), HUVEC (human umbilical vein endothelial cells) and AC16 (human cardiomyocytes cells) cell lines were all obtained from the Institute of Biochemistry and Cell Biology, China Academy of Sciences. The cells were cultured in DMEM (Sigma), which was supplemented with 10% fetal bovine serum in a humidified atmosphere of 5% CO2/95% air at 37 °C. All compounds were dissolved in phosphate buffered saline (PBS) with 1% DMSO to obtain various concentrations in 96-well plates, whilst control wells contained supplemented media with 1% DMSO, and the wells continued to be incubated for 48 h at 37 °C in a 5% CO2 atmosphere. MTT (5 mg mL−1) was added into the wells and the plates were incubated at 37 °C for 4 h. The MTT assay was stopped by adding dimethyl sulfoxide (100 μL per well) and mixing for 10 min vigorously before measuring the absorbance at 490 nm in a multi-well plate reader. The cytotoxicity was estimated based on the percentage cell survival in a dose dependent manner relative to the negative control. The final IC50 (drug concentration that kills 50% of the cells) values were calculated by the Bliss method. All target compounds were tested on MOVAS, HUVEC and AC16 cell lines, respectively.Results and discussion
We started to investigate the reactivity of N-benzylisoquinolinium 1a as a model substrate in an air atmosphere and at room temperature (Table 1; more details in the ESI†). The optimal reaction was achieved with substrate 1a as the photosensitizer, 1,4-diazobicyclo[2.2.2]octane (DABCO) as a base, and air as an oxidant in CH3CN under the irradiation of 15 W blue light-emitting diodes (LEDs), thereby affording the carbonylation product 2a (entry 1). Inhibition of the reactivity was observed in the presence of other solvents (entries 2 and 3). Other bases, such as Et3N and KOtBu, were also effective for this C–H oxidation transformation, but in inferior yields (entries 4 and 5). There was a negative effect on the reaction with the addition of the photocatalyst Ru(bpy)3Cl2 or without light (entries 6 and 7). The experiments showed that no desired product was formed in the absence of base, revealing the crucial role of the base in this reaction (entry 8).
|
|||
|---|---|---|---|
| Entry | Substrate | Variation from standard conditions | 2ab (%) |
| a Reaction conditions: N-methylene iminiums 1 (0.2 mmol), DABCO (2.0 equiv.), LED lamp as light source, room temperature, under air atmosphere.b Isolated yield. | |||
| 1 | 1a | None | 87 |
| 2 | 1a | DMF instead of CH3CN | 22 |
| 3 | 1a | THF instead of CH3CN | 58 |
| 4 | 1a | Et3N instead of DABCO | 54 |
| 5 | 1a | KOtBu instead of DABCO | 67 |
| 6 | 1a | Addition 2 mol% Ru(bpy)3Cl2 | 63 |
| 7 | 1a | Without light | 21 |
| 8 | 1a | Without base | NR |

According to the readily available optimization conditions, we significantly extended the scope of ortho-selective oxidation to synthesize α-lactams. As shown in Scheme 2, isoquinolinium chloride (X = Cl) showed good reaction activity and the target product 2a was obtained in 85% yield. Moderate yields were realized when the pyridine ring contained Me, Cl and Br functional groups (2b–2d). Substituting the benzene ring with electron-donating groups, such as methyl and methoxy groups, and with electron-withdrawing groups, such as chloro- and nitro-groups, proceeded smoothly to give isoquinolones 2e–2i in 75–88% yields. N-Benzylquinolinium was transformed into the corresponding quinoline-2-one 2j. It is worth noting that heteroarenium derivatives, such as quinazolinium, quinoxalinium, benzothiazolium and 3-methoxypyridinium, were produced smoothly by our standard protocol as products 2k–2n. Substituent R represents the phenyl group, such as the various electron-donating groups (Me, tBu, OMe, and Ph) and electron-withdrawing groups (Cl, NO2, CN, CF3, and F), that can be transformed smoothly to obtain corresponding products (2o–2z). Many functional groups, including benzyl and alkyl, were well tolerated (2aa–2ad). Among them, isoquinolinium halides (X = Cl and I) also proceeded in good yields (2q and 2ab). In addition, the heterocyclic substrates were well tolerated in the oxidative reactions, and target products 2ae and 2af were obtained with yields of 87% and 83%, respectively.
 | ||
| Scheme 2 The ortho C–H selective oxidation reaction using N-methylene iminium salts.a Reaction conditions: N-methylene iminiums 1 (0.2 mmol), DABCO (2.0 equiv.), blue LED (15 W), room temperature, under air atmosphere. b Isolated yield. | ||
When 3-methylquinolinium (1ag) instead of N-benzylisoquinolinium bromide (1a) was applied to this system, 4-quinolone 3a was successfully achieved in a 68% yield using the Ru-catalyst (Table 2, entry 1). Further screening of the reactions could not be effectively performed with other photocatalysts, solvents and bases (entries 2–4). No aerobic oxidation occurred in the absence of light or base, demonstrating that these components were essential factors (entries 5 and 6).
|
|||
|---|---|---|---|
| Entry | Substrate | Variation from standard conditions | 3ab (%) |
| a Reaction conditions: N-methylene iminiums 1 (0.2 mmol), DABCO (2.0 equiv.), Ru(bpy)3Cl2 (2.0 mol%), LED lamp as light source, room temperature, under air atmosphere.b Isolated yield. | |||
| 1 | 1ag | None | 68 |
| 2 | 1ag | Eosin Y instead of Ru(bpy)3Cl2 | NR |
| 3 | 1ag | 1,4-Dioxane instead of CH3CN | 33 |
| 4 | 1ag | Et3N instead of DABCO | NR |
| 5 | 1ag | Without light | Trace |
| 6 | 1ag | Without base | NR |

Subsequently, we assessed the reaction of quinolinium substrates with a ruthenium catalyst, and this protocol straightforwardly photocatalyzed the carbonylation of the Csp2−H bond to afford the desired products with excellent C4-regiocontrol, as illustrated in Scheme 3. The quinoline ring bearing various groups, such as methyl, chloro- and bromo-groups, were successfully engaged in the current photocatalytic oxidation reaction (3b–3d). The benzene ring with different electronic groups, connected to the nitrogen atom, maintained selectivity for the C4-site (3e–3g). The substrate with the benzyl group was still suitable for the reaction, and the product 3h was obtained in a 75% yield. Other alkyl quinoliniums, such as cyclohexane, n-propyl, and vinyl, were also well-tolerated and provided the corresponding products (3i–3k) in 78%–80% yields. Thiophene quinolinium was tolerated to give the desired product 3l in a good yield.
 | ||
| Scheme 3 The C4-selective oxidation reaction using N-benzylquinolinium salts. a Reaction conditions: quinoliniums 1ag–1ar (0.2 mmol), DABCO (2.0 equiv.), Ru(bpy)3Cl2 (2.0 mol%), blue LED (15 W), ambient temperature, under air atmosphere. b Isolated yield. | ||
Having established our developed methods and scope for a wide range of applications, we investigated the proposed mechanism. Isotope experiments indicated that the oxygen atom in the target products was mainly derived from oxygen in the atmosphere (Scheme 4a). Only a trace amount of isoquinolinone 2a was observed in an argon atmosphere. Under the optimal reaction conditions, the reaction was clearly inhibited by the addition of 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) or 2,6-di-tert-butyl-4-methylphenol (BHT), indicating that the conversion could have undergone a free radical process. It is well known that N-functionalized onium salts act as photosensitizers.11 Hence, the excited state A was formed under continuous visible light irradiation. Subsequently, the nitrogen-centered cation A could undergo photoinduced electron transfer to generate the α-amino radical B (trapped by TEMPO),12 and DABCO was simultaneously oxidized to DABCO+ (trapped by TEMPO). The radical B was coupled with oxygen in the air to produce the peroxide radical C.13 The other route for radical B involved being oxidized to the ground state isoquinolinium 1a, while O2 was reduced to O2, and so the photocatalytic cycle was completed. Intermediate C continued to form the carbon radical D (trapped by TEMPO) through hydrogen atom transfer (HAT), and then D was oxidized to the oxygen cation E by DABCO+, and was then simultaneously reduced to DABCO. Finally, treatment of the cation E with NaOH and Br− afforded the desired adduct 2a (Scheme 4b).14
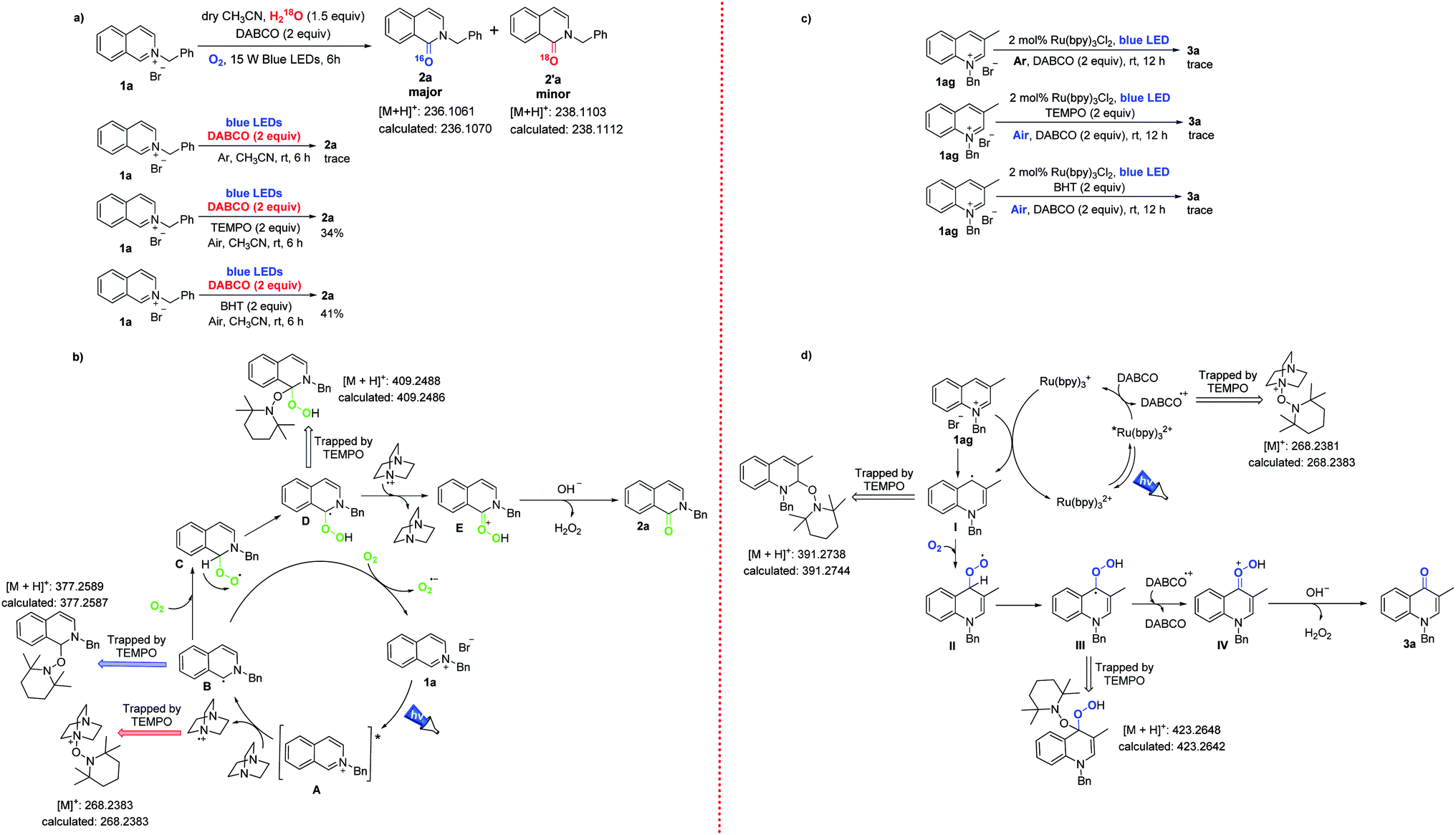 | ||
| Scheme 4 Control experiments (a and c) and the proposed mechanism (b and d). | ||
Similarly, a trace amount of 4-quinolone 3a was observed in an argon atmosphere (Scheme 4c). The reaction was inhibited by adding the free radical inhibitors TEMPO or BHT under the optimal reaction conditions, indicating that the conversion involves a radical process. Based on these control experiments, we speculated the following mechanism (Scheme 4d). Photoactivated Ru(II)* could be reduced to Ru(I) species by DABCO. The redox potential of Ru(bpy)3Cl2 was E1/2 Ru(II)*/Ru(I) = 0.77 V vs. SCE in CH3CN,15 and this was higher than E1/2 DABCO = 0.6 V vs. SCE.16 Therefore, the photoredox reactions could occur spontaneously between Ru(bpy)3Cl2 and DABCO. 3-Methylquinolinium (1ag) could accept a single electron from the Ru(I) species to form the C4-site quinoline radical I,11b,17 which was coupled with oxygen to form the peroxy radical II. The C4-position radical III was generated from II, which was promoted by a hydrogen atom transfer (HAT) process. This was similar to the above oxidation of isoquinoliniums, in that intermediate III was oxidized to cation IV by DABCO+, (radical cation) and then reacted with base to remove H2O2 to obtain target product 3a.
The in vitro cardiovascular activities of all the synthesized compounds were tested on several cell lines, including MOVAS (mouse aorta vascular smooth muscle cells), HUVEC (human umbilical vein endothelial cells) and AC16 (human cardiomyocytes cells). As reported in Table 3, the biological activities were expressed as IC50 values. The table shows that two products demonstrated moderate activities of MOVAS cells with IC50 values of 59.14 ± 2.31 and 94.93 ± 4.57, respectively. The results showed that these products only had potential effects on mouse aorta vascular smooth muscle cells. Unfortunately, the products did not inhibit the activity of HUVEC and AC16 cells.
Conclusions
In summary, we have developed the selective oxidation of Csp2–H functionalization by employing oxygen under light-driven conditions. Clearly, the ruthenium complex was used as a chemical switch to trigger two different oxidation pathways of the N-onium salts. In the absence of the Ru-complex, the N-functionalized onium ions were irradiated from the α-amino radicals of tertiary amines to the corresponding amides using air as an oxidant. On the other hand, the quinoliniums underwent single electron transfer to form the C4-site quinoline radical using the Ru(bpy)3Cl2 catalyst, and this was shown to be smoothly carried out by the formation of 4-quinolones. The outstanding advantages of these transformations are that carbonylation at different sites of the N-methyleneiminiums could be directly oxidized to obtain diverse ketones under air and at room temperature.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the National Natural Science Foundation of China (21977021 and 81760626), the Ministry of Education Innovation Team Fund (IRT_16R15, 2016GXNSFGA380005), and the Natural Science Foundation of Guangxi Province (AB17292075) and Guangxi Funds for Distinguished Experts.Notes and references
- (a) M. J. Fisher, B. P. Gunn, S. Um and J. A. Jakubowski, Tetrahedron Lett., 1997, 38, 5747–5750 CrossRef CAS; (b) H. Huse and M. Whiteley, Chem. Rev., 2011, 111, 152–159 CrossRef CAS PubMed; (c) D. B. Khadka and W. J. Cho, Bioorg. Med. Chem., 2011, 19, 724–734 CrossRef CAS PubMed; (d) Z. Jin, Nat. Prod. Rep., 2013, 30, 849–868 RSC; (e) B.-R. Kang, S. Li, S. Mao, Y.-X. Cao and S.-Q. Zhang, Bioorg. Med. Chem., 2015, 25, 5808–5812 CrossRef CAS PubMed; (f) Y.-M. Ma, K. Qiao, Y. Kong, M.-Y. Li, L.-X. Guo, Z. Miao and C. Fan, Nat. Prod. Res., 2017, 31, 951–958 CrossRef CAS PubMed; (g) J. Drogosz-Stachowicz, A. Długosz-Pokorska, K. Gach-Janczak, A. Jaskulska, T. Janecki and A. Janecka, Chem. Biol. Interact., 2020, 320, 109005 CrossRef CAS PubMed.
- (a) N. Fokialakis, P. Magiatis, A. L. Skaltsounis, F. Tillequin and T. Sévenet, J. Nat. Prod., 2000, 63, 1004–1005 CrossRef CAS PubMed; (b) T. S. Wu and F. W. Lin, J. Nat. Prod., 2001, 64, 1404–1407 CrossRef CAS PubMed; (c) S. Mitaku, N. Fokialakis, P. Magiatis and F. Tillequin, Fitoterapia, 2007, 78, 169–170 CrossRef CAS PubMed; (d) P. H. Coombes, E. M. Mwangi, B. K. Peters, N. R. Crouch and D. A. Mulholland, Biochem. Syst. Ecol., 2009, 37, 494–496 CrossRef CAS; (e) J.-Y. Cho, S.-H. Bae, H.-K. Kim, M.-L. Lee, Y.-S. Choi, J.-Y. Cho, S.-H. Bae, H.-K. Kim, M.-L. Lee, Y.-S. Choi, B.-R. Jin, H. J. Lee, H. Y. Jeong, Y. G. Lee and J.-H. Moon, J. Agric. Food Chem., 2015, 63, 3587–3592 CrossRef CAS PubMed.
- (a) A. Morrell, S. Antony, G. Kohlhagen, Y. Pommier and M. Cushman, J. Med. Chem., 2006, 49, 7740–7753 CrossRef CAS PubMed; (b) H. T. My Van and W. J. Cho, Bioorg. Med. Chem., 2009, 19, 2551–2554 CrossRef PubMed.
- (a) M. Sato, H. Kawakami, T. Motomura, H. Aramaki, T. Matsuda, M. Yamashita, Y. Ito, Y. Matsuzaki, K. Yamataka, S. Ikeda and H. Shinkai, J. Med. Chem., 2009, 52, 4869–4882 CrossRef CAS PubMed; (b) S. Ramanathan, A. A. Mathias, P. German and B. P. Kearney, Clin. Pharmacokinet., 2011, 50, 229–244 CrossRef CAS PubMed.
- (a) I. M. Khalil, D. Barker and B. R. Copp, J. Org. Chem., 2016, 81, 282–289 CrossRef CAS PubMed; (b) T. T. Fan-Chiang, H. K. Wang and J.-C. Hsieh, Tetrahedron, 2016, 72, 5640–5645 CrossRef CAS.
- (a) J. K. Son, S. I. Kim and Y. Jahng, Heterocycles, 2001, 55, 1981–1986 CrossRef CAS; (b) D. Zewge, C. Y. Chen, C. Deer, P. G. Dormer and D. L. Hughes, J. Org. Chem., 2007, 72, 4276–4279 CrossRef CAS PubMed; (c) J. C. Brouet, S. Gu, N. P. Peet and J. D. Williams, Synth. Commun., 2009, 39, 1563–1569 CrossRef CAS PubMed.
- (a) A. A. Ghogare and A. Greer, Chem. Rev., 2016, 116, 9994–10034 CrossRef CAS PubMed; (b) Y.-F. Liang and N. Jiao, Acc. Chem. Res., 2017, 50, 1640–1653 CrossRef CAS PubMed; (c) Y. Zhang, W. Schilling and S. Das, ChemSusChem, 2019, 12, 2898–2910 CrossRef CAS PubMed; (d) W. Schilling, Y. Zhang, P. K. Sahoo, S. K. Sarkar, S. Gandhi, H. W. Roesky and S. Das, Green Chem., 2021, 23, 379–387 RSC.
- (a) Q. Liu and L. Z. Wu, Natl. Sci. Rev., 2017, 4, 359–380 CrossRef CAS; (b) A. Motaleb, A. Bera and P. Maity, Org. Biomol. Chem., 2018, 16, 5081–5085 RSC; (c) X. Zhang, K. P. Rakesh, L. Ravindar and H. L. Qin, Green Chem., 2018, 20, 4790–4833 RSC; (d) Y. Zhou, W. Liu, Z. Xing, J. Guan, Z. Song and Y. Peng, Org. Chem. Front., 2020, 7, 2405–2413 RSC; (e) W. Schilling, Y. Zhang, D. Riemer and S. Das, Chem.–Eur. J., 2020, 26, 390–395 CrossRef CAS PubMed; (f) Y. Zhou, W. Liu, Z. Xing, J. Guan, Z. Song and Y. Peng, Org. Chem. Front., 2020, 7, 2405–2413 RSC.
- Y. Jin, L. Ou, H. Yang and H. Fu, J. Am. Chem. Soc., 2017, 139, 14237–14243 CrossRef CAS PubMed.
- X. Ji, D. Li, Z. Wang, M. Tan, H. Huang and G. J. Deng, Asian J. Org. Chem., 2018, 7, 711–714 CrossRef CAS.
- (a) P. Hewavitharanage, E. O. Danilov and D. C. Neckers, J. Org. Chem., 2005, 70, 10653–10659 CrossRef CAS PubMed; (b) K. Ohkubo, T. Kobayashi and S. Fukuzumi, Angew. Chem., Int. Ed., 2011, 50, 8652–8655 CrossRef CAS PubMed; (c) J. P. Dinnocenzo, P. B. Merkel and S. Farid, J. Phys. Chem. A, 2017, 121, 7903–7909 CrossRef CAS PubMed.
- (a) Y. L. Chow, W. C. Danen, S. F. Nelsen and D. H. Rosenblatt, Chem. Rev., 1978, 78, 243–274 CrossRef CAS; (b) U. C. Yoon and P. S. Mariano, Acc. Chem. Res., 1992, 25, 233–240 CrossRef CAS; (c) X. Zhang, S.-R. Yeh, S. Hong, M. Freccero, A. Albini, D. Falvey and P. S. Mariano, J. Am. Chem. Soc., 1994, 116, 4211–4220 CrossRef CAS; (d) P. Renaud and L. Giraud, Synthesis, 1996, 1996, 913–926 CrossRef.
- (a) Z. Zhang, J. Su, Z. Zha and Z. Wang, Chem. Commun., 2013, 49, 8982–8984 RSC; (b) Q. B. Zhang, Y. L. Ban, D. G. Zhou, P. P. Zhou, L. Z. Wu and Q. Liu, Org. Lett., 2016, 18, 5256–5259 CrossRef CAS PubMed; (c) Y. Zhang, D. Riemer, W. Schilling, J. Kollmann and S. Das, ACS Catal., 2018, 8, 6659–6664 CrossRef CAS.
- (a) N. Suzuki, Y. Kazui, T. Tsukamoto, M. Kato and Y. Izawa, Bull. Chem. Soc. Jpn., 1983, 56, 1519–1521 CrossRef CAS; (b) J. Motoyoshiya, H. Inoue, Y. Takaguchi and H. Aoyama, Heteroat. Chem., 2002, 13, 252–257 CrossRef CAS.
- (a) C. Creutz and N. Sutin, Inorg. Chem., 1976, 15, 496–499 CrossRef CAS; (b) Y. Matsubara, K. Koga, A. Kobayashi, H. Konno, K. Sakamoto, T. Morimoto and O. Ishitani, J. Am. Chem. Soc., 2010, 132, 10547–10552 CrossRef CAS PubMed; (c) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC.
- S. F. Nelsen and P. J. Hintz, J. Am. Chem. Soc., 1972, 94, 7114–7117 CrossRef CAS.
- (a) Y. Su, L. Zhang and N. Jiao, Org. Lett., 2011, 13, 2168–2171 CrossRef CAS PubMed; (b) R. R. Kotha, J. J. Nash and H. I. Kenttämaa, Eur. J. Org. Chem., 2017, 2017, 1407–1412 CrossRef CAS; (c) D. Ding, H. Jiang, X. Ma, J. J. Nash and H. I. Kenttämaa, J. Org. Chem., 2020, 85, 8415–8428 CrossRef CAS PubMed; (d) J. M. Ganley, P. R. D. Murray and R. R. Knowles, ACS Catal., 2020, 10, 11712–11738 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H, 19F and 13C NMR spectra of compounds 2 and 3. See DOI: 10.1039/d1ra01226f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |