
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePreparation of 3,5-diarylsubstituted 5-hydroxy-1,5-dihydro-2H-pyrrol-2-ones via base-assisted cyclization of 3-cyanoketones†
Nicolai A. Aksenov *a,
Dmitrii A. Aksenova,
Igor A. Kurenkova,
Alexander V. Aksenova,
Anton A. Skomorokhova,
Lidiya A. Pritykoa and
Michael Rubin*ab
*a,
Dmitrii A. Aksenova,
Igor A. Kurenkova,
Alexander V. Aksenova,
Anton A. Skomorokhova,
Lidiya A. Pritykoa and
Michael Rubin*ab
aDepartment of Chemistry, North Caucasus Federal University, 1a Pushkin St., Stavropol 355009, Russian Federation. E-mail: aaksenov@ncfu.ru
bDepartment of Chemistry, University of Kansas, 1567 Irving Hill Rd, Lawrence, KS 66045-7582, USA. E-mail: mrubin@ku.edu; Tel: +1-785-864-5071
First published on 30th April 2021
Abstract
A convenient preparative method is developed allowing for expeditious assembly of 3,5-diarylsubstituted 5-hydroxy-1,5-dihydro-2H-pyrrol-2-ones from routinely available inexpensive synthetic precursors. These compounds could not be prepared via the previously known protocols, as 2-aminofuran derivatives were produced instead.
Introduction
Isomers of tetramic acid heterocyclic core of 3,5-disubstituted 5-hydroxy-1,5-dihydro-2H-pyrrol-2-one are highly attractive for modern medicinal chemistry. Compounds possessing this structural unit are found in nature and often demonstrate a wide array of important biological activities. For example, an array of alkaloids ianthellidones A–F, isolated from Australian marine sponges of Ianthella genus showed promising anti-cancer activity.1,2 Antimicrobial agents myceliothermophins A–D, isolated from fungus Myceliophthora thermophila3 were targets for synthetic exercises4 and subject for mechanistic investigations.5 Recently, a new wave of interest in these compounds was raised stimulated by their newly discovered cytotoxic activity (Fig. 1).6 Tryptophan–polyketide hybrid, codinaeopsin, isolated from endophytic fungus Codinaeopsis gonytrichoides demonstrated potent anti-malarial properties7 and was also a target for total synthesis.8 Rollipyrrole, a representative of propentdyopents typical for animals, but highly unusual for plants, was isolated from magnolia Rollinia mucosa (Fig. 1).9 Alkaloid cannabisin L originally isolated from seeds of black henbane (Hyoscamus niger)10 was also recently discovered in devil's trumpet plants (Datura metel).11,12 Finally, Penicillenol D, an alkaloid with an ample anti-tumor effect was isolated from marine-derived fungus Trichoderma citrinoviride (Fig. 1).13 A typical synthetic approach to these nitrogen-based heterocycles involves the reaction of five-membered lactone precursors with ammonia14–17 or amines,18,19 reductive cyclization of nitroolefins with 1,3-diketones,20 or various aldol-type cyclocondensations.21–24 In application to the synthesis of 3,5-diarylsubstituted 5-hydroxy-1,5-dihydro-2H-pyrrol-2-one, just a handful of synthetic approaches provided decent results,23,25 while most of the described protocols rely on the utilization of precursors that are quite exotic. Herein, we report a novel synthesis of 3,5-diarylsubstituted 5-hydroxy-1,5-dihydro-2H-pyrrol-2-ones via unusual base-assisted cyclization of readily available 3-cyanoketones (Scheme 1).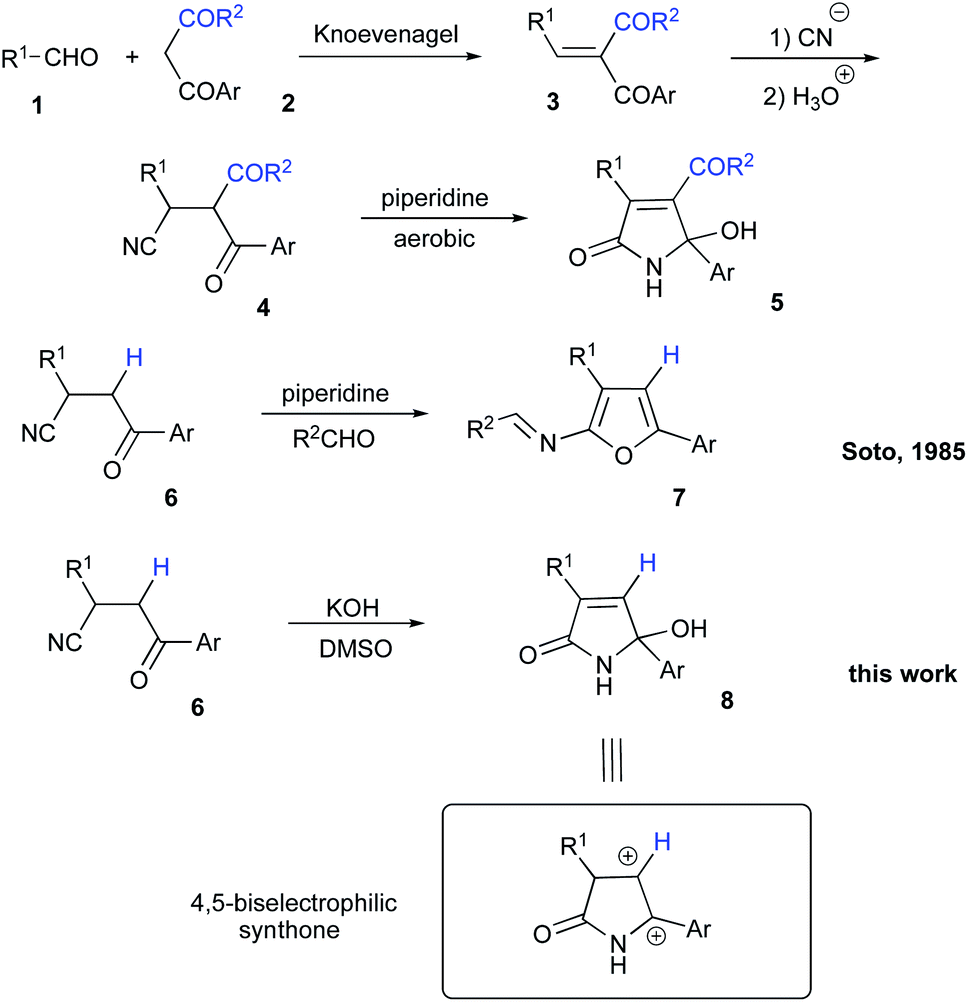 | ||
| Scheme 1 Alternative pathways for the cyclization of 3-cyanoketones. | ||
 | ||
| Fig. 1 Naturally occurring biologically active 5-hydroxy-1,5-dihydro-2H-pyrrol-2-ones. | ||
Results and discussion
During our research involving nitrogen-based heterocyclic compounds26–28 we got very enthusiastic about the possibility of employing synthetic equivalents of bis-electrophilic 3,4-dihydro-2H-pyrrol-1-ium-4-ylium synthon for expeditious assembly of bicyclic and tricyclic heterocyclic scaffolds. Although the structure of 5-hydroxy-1,5-dihydro-2H-pyrrol-2-ones 8 seems to be a suitable synthetic equivalent, lengthy and laborious synthetic approaches to these molecules rendered this idea cost-prohibitive and much less exciting. In 1985, Soto and co-workers reported on a very appealing strategy allowing for the preparation of very similar cyclic compound 5 from readily available and very inexpensive precursors.29 Their method involved Knoevenagel condensation between aldehyde 1 and 1,3-dicarbonyl compound 2 to obtain conjugate olefin 3, which was then subjected to hydrocyanation to provide nitrile 4. Next, piperidine-assisted oxidative cyclization was carried out to furnish 5 (Scheme 2).29 It should be pointed out, that this method is specific for the cyclization of dicarbonyl compounds 4 and can provide only heterocyclic products 5 with requisite acyl substituent at C-4 (Scheme 2). This substitution pattern is not suitable for our purposes, since this electron-withdrawing group would potentially revert the polarization of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond. So, we opted for the preparation of the lactam 8, non-substituted at this position. However, an attempt to involve mono-carbonyl precursor 6 into the reaction with piperidine did not provide any reactivity at room temperature (Table 1, entry 1). Upon heating, a different cyclization was triggered resulting in the formation of unstable 2-aminofuran, which could be isolated in a form of imine 7 (Scheme 2 and Table 1, entry 2).29 It should be pointed out, that the same result can be achieved under acidic conditions in the presence of polyphosphoric acid, which was demonstrated in our recent report.30
C double bond. So, we opted for the preparation of the lactam 8, non-substituted at this position. However, an attempt to involve mono-carbonyl precursor 6 into the reaction with piperidine did not provide any reactivity at room temperature (Table 1, entry 1). Upon heating, a different cyclization was triggered resulting in the formation of unstable 2-aminofuran, which could be isolated in a form of imine 7 (Scheme 2 and Table 1, entry 2).29 It should be pointed out, that the same result can be achieved under acidic conditions in the presence of polyphosphoric acid, which was demonstrated in our recent report.30
 | ||
| Scheme 2 Mechanistic rationales for the featured transformation. | ||
|
||||
|---|---|---|---|---|
| # | Base | Oxidant/solvent | Time, h (temp., °C) | Yielda, % |
| a All test reactions were performed on 0.5 mmol scales. NMR yields are provided unless specified otherwise.b Forms 7a (R1 = R2 = Ar = Ph) in the presence of benzaldehyde.c Reaction was carried out under argon atmosphere.d 300 μL of water was added to improve the solubility of the base.e Organic materials were poorly soluble in this mixture. | ||||
| 1 | Pyridine | Air/EtOH | 48 (20) | NR |
| 2 | Pyridine | Air/EtOH | 48 (100) | Decompositionb |
| 3 | KOH | Air/DMF–water | 72 (20) | 32% + 64% of 6a |
| 4 | KOH | DMF–waterc | 4 (20) | NR |
| 5 | KOH | H2O2–urea/DMF | 1.5 (20) | 29 |
| 6 | KOH | H2O2–urea/MeOH | 1.5 (20) | 40 |
| 7 | KOH (2 equiv.) | DMSO (0.2 mL)d,e | 0.5 (20) | 48 |
| 8 | KOH (4 equiv.) | DMSO (0.5 mL)d,e | 1.0 (20) | 71 |
| 9 | KOH (4 equiv.) | DMSO (1 mL)d | 0.67 (20) | 85 |
| 10 | KOH (4 equiv.) | DMSO (2 mL)d | 2 (20) | 72 |
| 11 | KOH (4 equiv.) | DMSO (3 mL)d | 4.5 (20) | 41 |
To gain access to the desired lactam 8 we decided to search for alternative conditions for the cyclization of 6. To this end, we tested KOH as a base in aqueous DMF for the reaction medium. When performed at room temperature in air, this reaction proceeded very sluggishly and provided the desired product in low yield, and was isolated along with unreacted starting material (entry 3). It should be stressed that the presence of the oxidant is crucial for this cyclization, which does not proceed at all under an inert atmosphere (entry 4). In an attempt to facilitate the oxidation step, we tested reactions in the presence of hydrogen peroxide/urea complex taking DMF or methanol as solvents. In both cases reaction proceeded much faster, going to completion within 1.5 h and affording 29 and 40% of lactam 8a, respectively (entries 5 and 6).
Next, we tried to employ DMSO as an oxidant. This reaction was carried out in the presence of water as a co-solvent to improve the solubility of the base in the reaction mixture. The initial attempt involved 2 equiv. of KOH and 0.2 mL of DMSO to trigger a rather quick reaction, which provided, however, only marginal yield (entry 7). To facilitate the reaction, amounts of both base and DMSO were increased, and the yield was greatly improved (entry 8). However, organic materials were still quite poorly soluble in this combination of solvents, so we decided to increase the concentration of DMSO. In a mixture of water/DMSO 0.3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, the observed rate of the reaction hits the maximum. The reaction was complete in 40 min at room temperature, affording 85% NMR yield, which translated into a 72% isolated yield of purified material (entry 9). Further increase in concentration of DMSO proved detrimental. The rate of the reaction slowed down, and the yields dropped quite noticeably (entries 10 and 11).
:1, the observed rate of the reaction hits the maximum. The reaction was complete in 40 min at room temperature, affording 85% NMR yield, which translated into a 72% isolated yield of purified material (entry 9). Further increase in concentration of DMSO proved detrimental. The rate of the reaction slowed down, and the yields dropped quite noticeably (entries 10 and 11).
With optimized conditions in hand, we proceeded to perform the reaction on a 1 mmol scale to isolate the product 8a in 72% yield (Table 2, entry 1). The reaction proved very general, and a series of 3,5-diarylsubstituted 5-hydroxy-1,5-dihydro-2H-pyrrol-2-ones were easily obtained in good to high isolated yields under the same conditions (Table 2). It was demonstrated, that phenyl groups with different alkyl, alkoxy, and halide substituents as well as 2-naphthyl and 2,3-dihydrobenzo[b][1,4]dioxin-6-yl moieties are very well tolerated in the featured transformation.
|
||||
|---|---|---|---|---|
| 6, 8 | R1 | R2 | Yielda, % | |
| a Isolated yields of purified materials are provided. | ||||
| 1 | 6a, 8a | Ph | Ph | 72 |
| 2 | 6b, 8b | p-MeC6H4 | Ph | 71 |
| 3 | 6c, 8c | p-EtC6H4 | Ph | 65 |
| 4 | 6d, 8d | p-MeOC6H4 | Ph | 88 |
| 5 | 6e, 8e | o-MeOC6H4 | Ph | 70 |
| 6 | 6f, 8f | p-Me2NC6H4 | Ph | 68 |
| 7 | 6g, 8g | p-FC6H4 | Ph | 71 |
| 8 | 6h, 8h | o-FC6H4 | Ph | 66 |
| 9 | 6i, 8i | p-ClC6H4 | Ph | 68 |
| 10 | 6j, 8j | o-ClC6H4 | Ph | 76 |
| 11 | 6k, 8k | p-BrC6H4 | Ph | 59 |
| 12 | 6l, 8l | Ph | p-MeOC6H4 | 77 |
| 13 | 6m, 8m | Ph | p-ClC6H4 | 59 |
| 14 | 6n, 8n | Ph | 2-Naphthyl | 77 |
| 15 | 6o, 8o | Ph | 2,3-Dihydrobenzo[b][1,4]dioxin-6-yl | 61 |
| 16 | 6p, 8p | p-MeOC6H4 | p-MeOC6H4 | 64 |
The formation of the 1,5-dihydro-2H-pyrrol-2-one ring in the reaction of ketonitrile 6e (entry 5) was unambiguously confirmed by single-crystal X-ray analysis of the product 8e (Fig. 2).
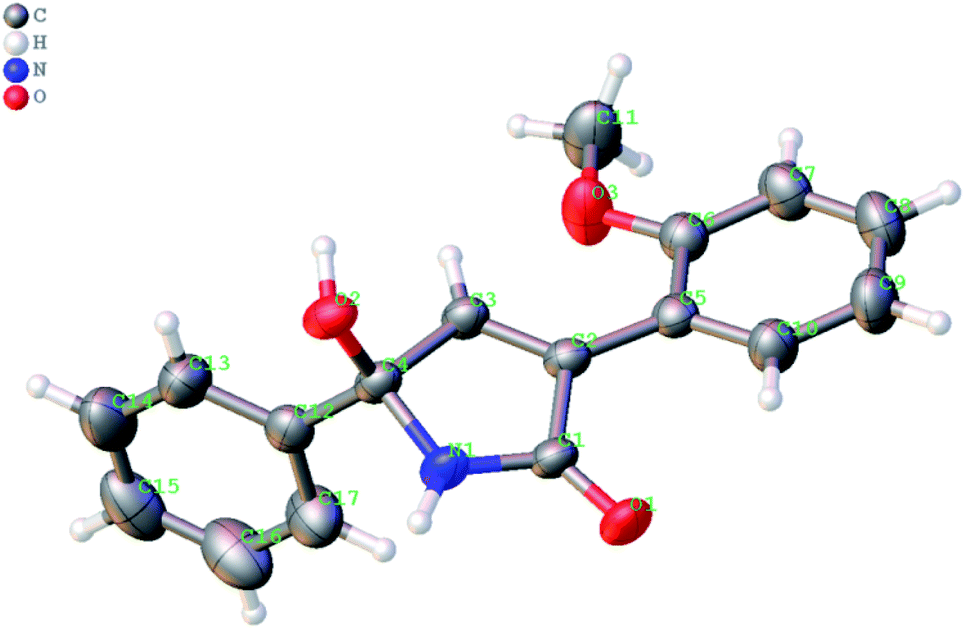 | ||
| Fig. 2 X-ray structure of 5-hydroxy-3-(2-methoxyphenyl)-5-phenyl-1,5-dihydro-2H-pyrrol-2-one 8e (the thermal ellipsoids are shown at 50% probability) (CCDC #2069260). | ||
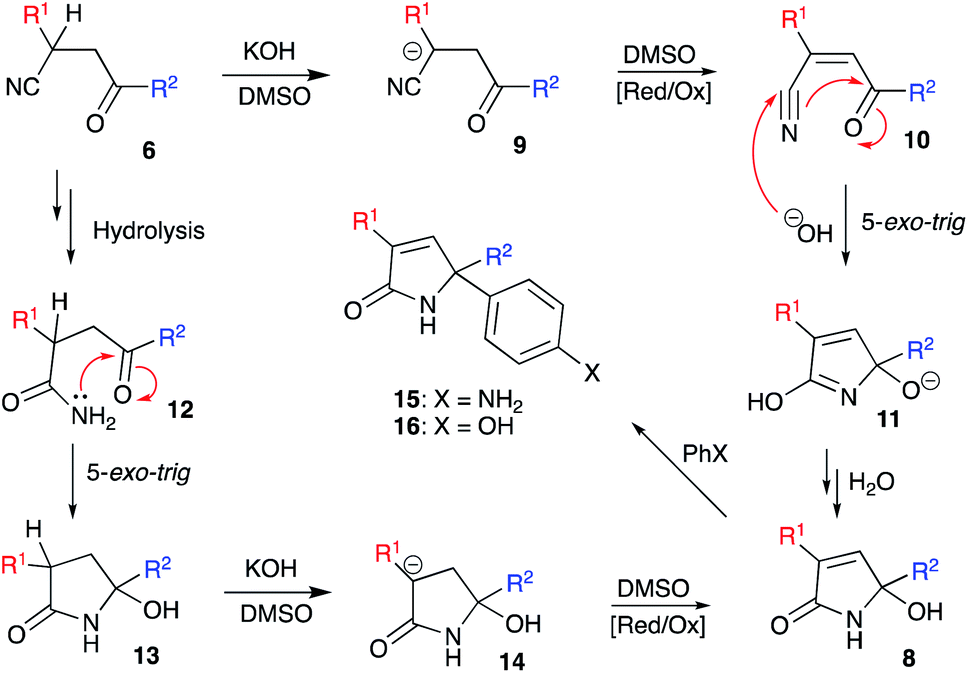
We envision the described transformation to proceed via the following mechanistic pathway. Initial base-assisted cleavage of acidic α-CH-bond of nitrile would lead to the formation of anionic moiety 9, which should be susceptible to oxidation in the presence of DMSO. The resulting acrylonitrile 10 would then experience a nucleophilic attack with hydroxide species triggering subsequent 5-exo-trig cyclization to afford 5-hydroxy-2H-pyrrol-2-olate 11. Re-protonation of this species, followed by tautomerization of the imidic acid entity into lactam function would finally provide product 8 (Scheme 2). An alternative pathway would include initial hydrolysis of nitrile function in 6 to afford primary amide 12, which could undergo a subsequent 5-exo-trig cyclization providing lactam 13. Deprotonation of α-CH-bond would lead to the formation of anionic intermediate 14, which would be further oxidized into 1,5-dihydro-2H-pyrrol-2-one 8 with DMSO. The latter rationale, however, seems less likely, as in the absence of DMSO as an oxidizing agent we failed to detect the formation of intermediate 12 or 13. The hydrolysis step did not proceed and nitrile function remained unchanged. This suggests that the oxidation process should proceed prior to cyclization.
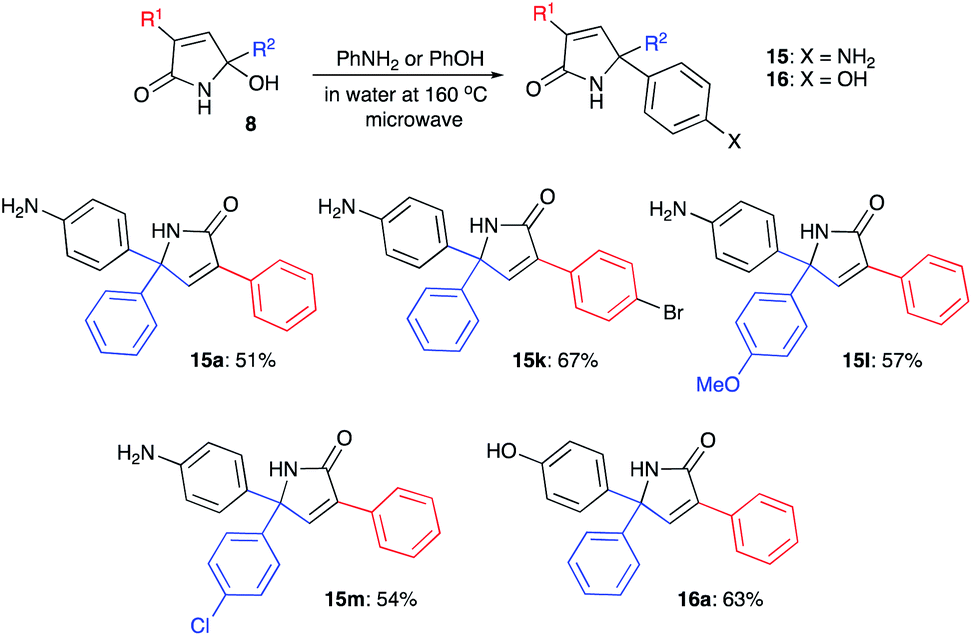
Next, we evaluated the possibility of further increasing the molecular complexity of the synthesized scaffolds by targeting the introduction of a third aryl substituent to obtain products 15, 16 (Scheme 2). To this end, we tested the SEAr reaction between 1,5-dihydro-2H-pyrrol-2-ones and electron-rich aromatic compounds, such as aniline and phenol. The reactions were carried out in a sealed tube upon heating at 160 °C with microwave irradiation (Scheme 3). Gratifyingly, all the tested reactions proceeded smoothly affording compounds 15a, k–m and 16a in moderate to good yields (Scheme 3). Successful installation of the new aniline moiety at C-5 was unambiguously confirmed by the single-crystal X-ray diffraction analysis of compound 15k re-crystallized from benzene (Fig. 3).
 | ||
| Scheme 3 Further modification of 1,5-dihydro-2H-pyrrol-2-ones via electrophilic aromatic substitution. | ||
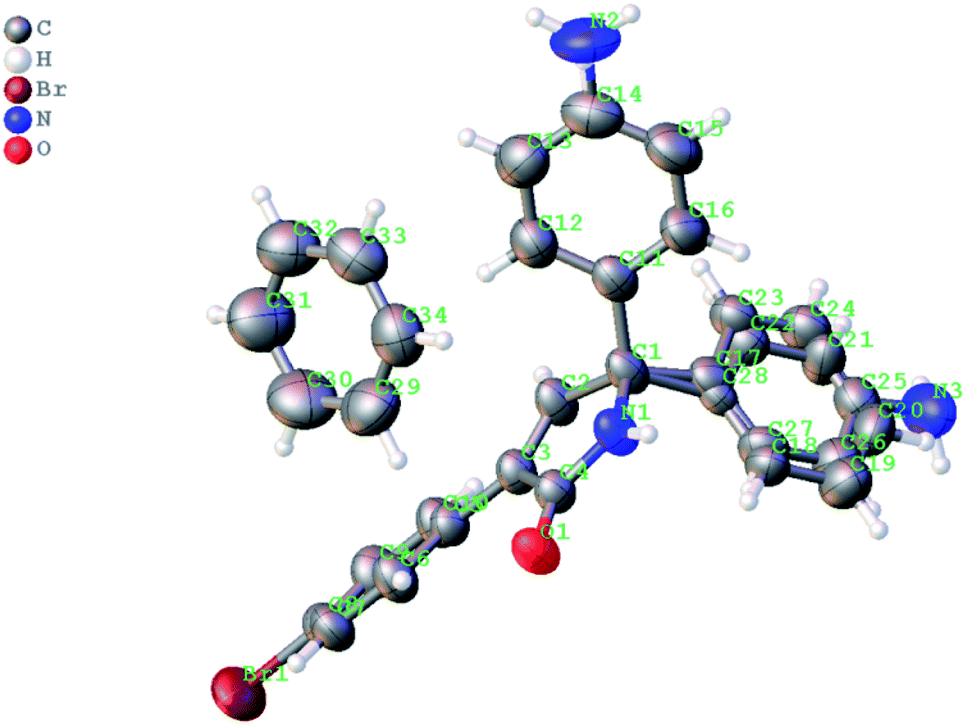 | ||
| Fig. 3 X-ray structure of 5-(4-aminophenyl)-3-(4-bromophenyl)-5-phenyl-1,5-dihydro-2H-pyrrol-2-one 15k as solvate with benzene. Two enantiomers of 15k are disordered in the crystalline lattice and their overlay is shown (the thermal ellipsoids are shown at 50% probability) (CCDC #2069203). | ||
As suggested by the reviewer of this paper, we also evaluated the possibility of lowering the reaction temperature employing Brønsted acid catalysis. To this end, reactions of phenol with lactam 8a were carried out in the presence of 20 mol% of HClO4 (70% aqueous, the 0.60 M reaction mixture was heated in nitromethane at 70 °C for 1 h)31,32 or MeSO3H33 (0.60 M reaction mixture was heated in toluene at 70 °C for 1 h). Upsettingly, these reactions afforded only marginal yields of the corresponding product 16a, 46% and 48%, respectively. The reaction of 8a with aniline carried out in the presence of MeSO3H under the same reaction conditions was accompanied by significant decomposition and allowed for the isolation of 15a in very low yield (12%).
Conclusion
In conclusion, a novel synthetic method allowing for highly efficient preparation of formerly poorly-accessible 3,5-diarylsubstituted 5-hydroxy-1,5-dihydro-2H-pyrrol-2-ones was developed. The method relies on the employment of routinely available and very inexpensive precursors and provides good yields of the target products. Following the modification of the prepared structures via thermally-induced SEAr reaction with aniline or phenol allowed for the introduction of the third aryl substituent at this heterocyclic scaffold, which seems attractive for building diverse libraries for drug discovery. Evaluation of biological activities of the synthesized compounds and further exploration of their reactivity as bis-electrophiles is currently underway in our laboratories.Experimental part
General information
1H and 13C NMR spectra were recorded on a Bruker Avance-III spectrometer (400 or 100 MHz, respectively) equipped with a BBO probe in CDCl3 or DMSO-d6, using TMS as an internal standard. High-resolution mass spectra were obtained using a Bruker Maxis spectrometer (electrospray ionization, MeCN solution, using HCO2Na–HCO2H for calibration). Melting points were measured with a Stuart SMP30 apparatus. All reactions were performed in oven-dried flasks equipped with reflux condensers and magnetic stir bars. All reactions were followed by thin-layer chromatography (TLC) using ALUGRAM Xtra SIL G UV254 plates, which were visualized under UV light (254 nm), with hexane/EtOAc mixtures as eluent. Nitriles 6c,34 6h, 6n,34 6o, 6p35 were synthesized according to the protocols provided below from the corresponding chalcones. All other nitriles were prepared according to known procedures and their physical and spectral properties were identical to those described in the literature.36 Reagents and solvents were purchased from commercial vendors and used as received. Abbreviation “PE” is used for light petroleum ether employed as an eluent for preparative chromatography.2-(4-Ethylphenyl)-4-oxo-4-phenylbutanenitrile (6c)34
Use of well-ventilated fume hood is necessary as the release of hydrogen cyanide occurs during the reaction. In a 25 mL round bottom flask equipped with magnetic stir bar (E)-3-(4-ethylphenyl)-1-phenylprop-2-en-1-one37 (472 mg, 2.00 mmol), KCN (195 mg, 3 mmol), EtOH (5 mL), H2O (0.3 mL) AcOH (114 μL, 120 mg, 2.00 mmol) were placed, closed with reflux condenser and allowed to stir under reflux for 2 h (TLC control). After the consumption of the starting material, the reaction mixture was cooled to room temperature and the precipitated product was collected by vacuum filtration, washed twice with H2O. Colorless solid, mp 114.3–115.4 °C (EtOH); yield 420 mg (1.6 mmol, 80%). Rf = 0.49, EtOAc/hexane (1:4, v/v). 1H NMR (400 MHz, CDCl3) δ 8.00–7.85 (m, 2H), 7.63–7.56 (m, 1H), 7.52–7.42 (m, 2H), 7.38–7.30 (m, 2H), 7.26–7.19 (m, 2H), 4.54 (dd, J = 7.9, 6.0 Hz, 1H), 3.72 (dd, J = 17.9, 8.0 Hz, 1H), 3.50 (dd, J = 18.0, 6.0 Hz, 1H), 2.65 (q, J = 7.6 Hz, 2H), 1.23 (t, J = 7.6 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 194.9, 144.7, 135.9, 134.0, 132.6, 120.0 (2C), 128.9 (2C), 128.2 (2C), 127.6 (2C), 120.9, 44.7, 31.7, 28.6, 15.6. FTIR (ZnSe) ν (cm−1): 2969, 2241, 1769, 1675, 1595, 1511, 1443, 1352, 1246, 1053, 998; HRMS (ES TOF, m/z) calcd for C18H17NNaO+ ([M + Na]+): 286.1202, found 286.1200 (0.9 ppm).
2-(2-Fluorophenyl)-4-oxo-4-phenylbutanenitrile (6h)
Product 6h was obtained via the method described for 6c employing (E)-3-(2-fluorophenyl)-1-phenylprop-2-en-1-one38 (452 mg, 2.00 mmol), and purified by recrystallization from ethanol. Colorless solid, mp 125.3–126.4 °C (EtOH); yield 385 mg (1.52 mmol, 76%). Rf = 0.51, EtOAc/hexane (1:4, v/v). 1H NMR (400 MHz, chloroform-d) δ 7.99–7.91 (m, 2H), 7.65–7.52 (m, 2H), 7.52–7.43 (m, 2H), 7.42–7.30 (m, 1H), 7.21 (td, J = 7.6, 1.2 Hz, 1H), 7.12 (ddd, J = 10.3, 8.2, 1.2 Hz, 1H), 4.75 (dd, J = 8.5, 5.4 Hz, 1H), 3.74 (dd, J = 18.0, 8.5 Hz, 1H), 3.55 (dd, J = 18.0, 5.3 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ 194.6, 160.1 (d, J = 248.2 Hz), 135.7, 134.1, 130.6 (d, J = 8.4 Hz), 129.8 (d, J = 3.2 Hz), 129.0 (2C), 128.2 (2C), 125.1 (d, J = 3.7 Hz), 122.4 (d, J = 13.6 Hz), 119.6, 116.3 (d, J = 21.0 Hz), 42.6 (d, J = 1.6 Hz), 26.8 (d, J = 3.1 Hz). FTIR (ZnSe) ν (cm−1): 2242, 1677, 1595, 1496, 1451, 1410, 1357, 1304, 1246, 1212, 1000; HRMS (ES TOF) calcd for C16H12FNNaO+ ([M + Na]+): 276.0795, found 276.0795 (0.0 ppm).
4-(Naphthalen-2-yl)-4-oxo-2-phenylbutanenitrile (6n)
Product 6n was obtained via the method described for 6c, employing (E)-1-(naphthalen-2-yl)-3-phenylprop-2-en-1-one39 (516 mg, 2.00 mmol), and purified by recrystallization from ethanol. Colorless solid, mp 109.6–110.4 °C (EtOH), lit40 mp 128 °C (EtOH); yield 496 mg (1.74 mmol, 87%). Rf = 0.54, EtOAc/hexane (1:4, v/v). 1H NMR (400 MHz, chloroform-d) δ 8.42 (s, 1H), 8.00 (dd, J = 8.7, 1.8 Hz, 1H), 7.94 (d, J = 8.1 Hz, 1H), 7.91–7.85 (m, 2H), 7.62 (ddd, J = 8.2, 7.0, 1.3 Hz, 1H), 7.56 (ddd, J = 8.0, 6.9, 1.2 Hz, 1H), 7.51–7.45 (m, 2H), 7.44–7.38 (m, 2H), 7.38–7.32 (m, 1H), 4.63 (dd, J = 8.0, 5.9 Hz, 1H), 3.87 (dd, J = 17.8, 8.0 Hz, 1H), 3.65 (dd, J = 17.8, 6.0 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ 194.7, 136.0, 135.5, 133.1, 132.5, 130.2, 129.7, 129.4 (2C), 129.1, 128.9, 128.5, 128.0, 127.7 (2C), 127.2, 123.6, 120.8, 44.7, 32.2. FTIR (ZnSe) ν (cm−1): 2241, 1680, 1624, 1595, 1455, 1405, 1366, 1258, 1173, 1128, 1017; HRMS (ES TOF, m/z) calcd for C20H15NNaO+ ([M + Na]+): 308.1046, found 308.1042 (1.1 ppm).
4-(2,3-Dihydrobenzo[b][1,4]dioxin-6-yl)-4-oxo-2-phenylbutanenitrile (6o)
Product 6o was obtained via the method described in literature employing (E)-1-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)-3-phenylprop-2-en-1-one40 (532 mg, 2.00 mmol), and purified by recrystallization from ethanol. White solid, mp 108.9–109.4 °C (EtOH); yield 410 mg (1.4 mmol, 70%). Rf = 0.27, EtOAc/hexane (1:4, v/v). 1H NMR (400 MHz, chloroform-d) δ 7.53–7.29 (m, 7H), 6.90 (d, J = 8.2 Hz, 1H), 4.55 (dd, J = 7.9, 6.0 Hz, 1H), 4.36–4.23 (m, 4H), 3.64 (dd, J = 17.7, 7.9 Hz, 1H), 3.42 (dd, J = 17.7, 6.1 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ 193.1, 148.8, 143.6, 135.5, 129.7, 129.4 (2C), 128.5, 127.6 (2C), 122.4, 120.9, 117.8, 117.6, 64.8, 64.2, 44.3, 32.1. FTIR (ZnSe) ν (cm−1): 2241, 1665, 1602, 1501, 1431, 1345, 1284, 1260, 1166, 1135, 1063; HRMS (ES TOF, m/z) calcd for C18H15NNaO3+ ([M + Na]+): 316.0944, found 316.0948 (−1.1 ppm).
2,4-Bis(4-methoxyphenyl)-4-oxobutanenitrile (6p)35
Product 6p was obtained via the method described for 6c employing (E)-1,3-bis(4-methoxyphenyl)prop-2-en-1-one41 (536 mg, 2.00 mmol), and purified by recrystallization from ethanol. White crystals, mp 112.6–113.3 °C (EtOH); yield 0.354 mg (1.20 mmol, 60%). Rf = 0.21, EtOAc/hexane (1:1, v/v). 1H NMR (400 MHz, CDCl3) δ 7.94–7.85 (m, 2H), 7.39–7.30 (m, 2H), 6.97–6.86 (m, 4H), 4.52 (dd, J = 7.7, 6.3 Hz, 1H), 3.87 (s, 3H), 3.80 (s, 3H), 3.63 (dd, J = 17.6, 7.6 Hz, 1H), 3.43 (dd, J = 17.6, 6.4 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ 193.3, 164.2, 159.6, 130.6 (2C), 129.0 (2C), 128.8, 127.5, 121.2, 114.7 (2C), 114.1 (2C), 55.7, 55.5, 44.3, 31.3. FTIR (ZnSe) ν (cm−1): 2246, 1675, 1598, 1516, 1361, 1306, 1248, 1219, 1164, 1022; HRMS (ES TOF, m/z) calcd for C18H17NNaO3+ ([M + Na]+): 318.1101, found 318.1104 (−1.0 ppm).
5-Hydroxy-3,5-diphenyl-1,5-dihydro-2H-pyrrol-2-one (8a)
In a 5 mL vial equipped with a magnetic stirring bar, KOH (224 mg, 4 mmol) was dissolved in H2O (0.6 mL), followed by the addition of 4-oxo-2,4-diphenylbutanenitrile36 (235 mg, 1 mmol) and DMSO (2 mL). The reaction mixture was allowed to stir at r.t. for approximately 1 hour. After the consumption of all the starting compound, the reaction mixture was diluted with water (15 mL), extracted with ethyl acetate (4 × 15 mL), washed with water (2 × 15 mL) and purified by column chromatography (gradient: EtOAc/PE 1:3–1:1). The fractions were concentrated on a rotary evaporator. Compounds can also be purified by recrystallization from a suitable solvent. White solid, mp 234.1–236.1 °C (EtOAc); yield 181 mg (0.72 mmol, 72%). Rf = 0.31, EtOAc/hexane (1:2, v/v), Rf = 0.42, EtOAc/hexane (1:1, v/v). 1H NMR (400 MHz, DMSO-d6) δ 9.17–9.05 (m, 1H), 8.05–7.90 (m, 2H), 7.59–7.47 (m, 2H), 7.46–7.24 (m, 7H), 6.63 (s, 1H). 13C NMR (101 MHz, DMSO-d6) δ 170.8, 145.8, 140.8, 131.8, 131.15, 128.5, 128.3 (2C), 128.3 (2C), 127.9, 127.2 (2C), 125.7 (2C), 86.1. FTIR (ZnSe) ν (cm−1): 3242, 2960, 1767, 1617, 1605, 1501, 1463, 1447, 1433, 1363, 1234, 1204; HRMS (ES TOF, m/z) calcd for C16H13NNaO2+ ([M + Na]+): 274.0838, found 274.0839 (−0.2 ppm).
5-Hydroxy-5-phenyl-3-(p-tolyl)-1,5-dihydro-2H-pyrrol-2-one (8b)
Product 8b was obtained via the method described for compound 8a employing 4-oxo-4-phenyl-2-(p-tolyl)butanenitrile36 (249 mg, 1.00 mmol), and purified by column chromatography (gradient: EtOAc/PE 1:2–2:1) or recrystallization from ethanol. Reaction time: 1 hour. Yellow solid, mp 161.6–163.8 °C (EtOH); yield 188 mg (0.71 mmol, 71%). Rf = 0.45, EtOAc/hexane (1:1, v/v). 1H NMR (400 MHz, DMSO-d6) δ 9.09 (s, 1H), 7.86 (dd, J = 8.3, 2.2 Hz, 2H), 7.56–7.49 (m, 2H), 7.40–7.29 (m, 4H), 7.20 (d, J = 7.9 Hz, 2H), 6.60 (d, J = 2.3 Hz, 1H), 2.31 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 171.0, 144.8, 141.0, 138.0, 131.6, 128.9 (2C), 128.3, 128.2 (2C), 127.8, 127.1 (2C), 125.7 (2C), 86.0, 20.9. FTIR (ZnSe) ν (cm−1): 3268, 2362, 1672, 1508, 1489, 1426, 1207, 1087, 1058, 976; HRMS (ES TOF, m/z) calcd for C17H15NNaO2+ ([M + Na]+): 288.0995, found 288.0989 (1.9 ppm).
3-(4-Ethylphenyl)-5-hydroxy-5-phenyl-1,5-dihydro-2H-pyrrol-2-one (8c)
Product 8c was obtained via the method described for compound 8a, employing 2-(4-ethylphenyl)-4-oxo-4-phenylbutanenitrile (263 mg, 1.00 mmol), and purified by column chromatography (gradient: EtOAc/PE 1:2–2:1) or recrystallization from ethanol. White solid, mp 164.4–165.2 °C (EtOH); yield 181 mg (0.65 mmol, 65%). Rf = 0.23, EtOAc/hexane (1:2, v/v), Rf = 0.58, EtOAc/hexane (1:1, v/v). 1H NMR (400 MHz, DMSO-d6) δ 9.09 (s, 1H), 7.90–7.83 (m, 2H), 7.56–7.48 (m, 2H), 7.41–7.26 (m, 4H), 7.22 (d, J = 8.1 Hz, 2H), 6.60 (s, 1H), 2.61 (q, J = 7.6 Hz, 2H), 1.17 (t, J = 7.6 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 171.0, 144.9, 144.3, 141.0, 131.8, 128.6, 128.2 (2C), 127.8, 127.7 (2C), 127.2 (2C), 125.7 (2C), 86.1, 28.0, 15.5. FTIR (ZnSe) ν (cm−1): 3258, 1672, 1511, 1489, 1414, 1231, 1202, 1087, 1058, 976; HRMS (ES TOF, m/z) calcd for C18H17NNaO2+ ([M + Na]+): 302.1151, found 302.1149 (0.9 ppm).
5-Hydroxy-3-(4-methoxyphenyl)-5-phenyl-1,5-dihydro-2H-pyrrol-2-one (8d)
Product 8d was obtained via the method described for compound 8a, employing 2-(4-methoxyphenyl)-4-oxo-4-phenylbutanenitrile36 (265 mg, 1.00 mmol), and purified by column chromatography (gradient: EtOAc/PE 1:2–2:1) or recrystallization from ethanol. White solid, mp 163.4–164.1 °C (EtOH); yield 247 mg (0.88 mmol, 88%). Rf = 0.15, EtOAc/hexane (1:2, v/v), Rf = 0.5, EtOAc/hexane (2:1, v/v). 1H NMR (400 MHz, DMSO-d6) δ 9.07 (d, J = 1.8 Hz, 1H), 7.97–7.90 (m, 2H), 7.55–7.48 (m, 2H), 7.40–7.34 (m, 2H), 7.33–7.28 (m, 1H), 7.27 (d, J = 1.7 Hz, 1H), 6.98–6.90 (m, 2H), 6.56 (s, 1H), 3.77 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 171.1, 159.5, 143.6, 141.1, 131.2, 128.6 (2C), 128.2 (2C), 127.8, 125.7 (2C), 123.6, 113.8 (2C), 86.0, 55.1. FTIR (ZnSe) ν (cm−1): 3297, 1699, 1667, 1446, 1255, 1048, 966; HRMS (ES TOF, m/z) calcd for C17H15NNaO3+ ([M + Na]+): 304.0944, found 304.0944 (−0.3 ppm).
5-Hydroxy-3-(2-methoxyphenyl)-5-phenyl-1,5-dihydro-2H-pyrrol-2-one (8e)
Product 8e was obtained via the method described for compound 8a, employing 2-(2-methoxyphenyl)-4-oxo-4-phenylbutanenitrile36 (265 mg, 1.00 mmol), and purified by column chromatography (gradient: EtOAc/PE 1:2–2:1) or recrystallization from ethanol. Reaction time: 1 h. Colorless crystals, mp 193.9–195.4 °C (EtOH); yield 0.196 g (0.7 mmol, 70%). Rf = 0.61, EtOAc/hexane (1:1, v/v). 1H NMR (400 MHz, DMSO-d6) δ 9.05 (s, 1H), 8.22 (dt, J = 7.7, 1.6 Hz, 1H), 7.51 (d, J = 8.3 Hz, 2H), 7.42–7.27 (m, 5H), 7.07 (dd, J = 8.5, 1.1 Hz, 1H), 6.98 (td, J = 7.5, 1.1 Hz, 1H), 6.59 (s, 1H), 3.80 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 171.4, 157.9, 148.7, 141.2, 129.6, 129.6, 128.3 (2C), 127.8, 127.8, 125.7 (2C), 119.9, 119.6, 111.2, 86.1, 55.5. FTIR (ZnSe) ν (cm−1): 3268, 3195, 1670, 1614, 1489, 1407, 1243, 1216, 1045; HRMS (ES TOF, m/z) calcd for C17H15NNaO3+ ([M + Na]+): 304.0944, found 304.0951 (2.2 ppm).
3-(4-(Dimethylamino)phenyl)-5-hydroxy-5-phenyl-1,5-dihydro-2H-pyrrol-2-one (8f)
Product 8f was obtained via the method described for compound 8a 2-(4-(dimethylamino)phenyl)-4-oxo-4-phenylbutanenitrile36 (278 mg, 1.00 mmol), and purified by column chromatography (gradient: EtOAc/PE 1:2–2:1) or recrystallization from ethanol. Reaction time: 1.5 h. Colorless solid, mp 270.9–273.3 °C (EtOH); yield 200 mg (0.68 mmol, 68%). Rf = 0.55, EtOAc/hexane (1:1, v/v). 1H NMR (400 MHz, DMSO-d6) δ 8.97 (s, 1H), 7.83 (d, J = 8.3 Hz, 2H), 7.50 (d, J = 8.1 Hz, 2H), 7.39–7.24 (m, 3H), 7.10 (s, 1H), 6.69 (d, J = 8.5 Hz, 2H), 6.49 (d, J = 1.2 Hz, 1H), 2.92 (s, 6H). 13C NMR (101 MHz, DMSO-d6) δ 172.0, 150.7, 142.0, 141.5, 132.1, 128.6 (2C), 128.5 (2C), 128.1, 126.2 (2C), 119.2, 112.1 (2C), 86.5, 40.3 (2C). FTIR (ZnSe) ν (cm−1): 3268, 2762, 2540, 2366, 1663, 1612, 1525, 1424, 1359, 1198, 1051; HRMS (ES TOF, m/z) calcd for C18H18N2NaO2+ ([M + Na]+): 317.1260, found 317.1252 (2.7 ppm).
3-(4-Fluorophenyl)-5-hydroxy-5-phenyl-1,5-dihydro-2H-pyrrol-2-one (8g)
Product 8g was obtained via the method described for compound 8a employing 2-(4-fluorophenyl)-4-oxo-4-phenylbutanenitrile36 (253 mg, 1.00 mmol), and purified by column chromatography (gradient: EtOAc/PE 1:2–2:1) or recrystallization from ethanol. White solid, mp 186.1–187.1 °C (EtOH); yield 191 mg (0.71 mmol, 71%). Rf = 0.26, EtOAc/hexane (1:2, v/v). 1H NMR (400 MHz, DMSO-d6) δ 9.16 (s, 1H), 8.08–7.98 (m, 2H), 7.54–7.50 (m, 2H), 7.43 (d, J = 1.7 Hz, 1H), 7.41–7.34 (m, 2H), 7.34–7.29 (m, 1H), 7.26–7.14 (m, 2H), 6.64 (s, 1H). 13C NMR (101 MHz, DMSO-d6) δ 170.8, 162.2 (d, J = 246.1 Hz), 145.6 (d, J = 1.9 Hz), 140.8, 130.7, 129.4 (2C, d, J = 8.1 Hz), 128.3 (2C), 127.9, 127.7 (d, J = 3.2 Hz), 125.7 (2C), 115.3 (2C, d, J = 21.3 Hz), 86.1. FTIR (ZnSe) ν (cm−1): 3253, 1771, 1704, 1672, 1499, 1414, 1229, 1159, 1051; HRMS (ES TOF, m/z) calcd for C16H12FNNaO2+ ([M + Na]+): 292.0744, found 292.038 (2.1 ppm).
3-(2-Fluorophenyl)-5-hydroxy-5-phenyl-1,5-dihydro-2H-pyrrol-2-one (8h)
Product 8h was obtained via the method described for compound 8a employing 2-(2-fluorophenyl)-4-oxo-4-phenylbutanenitrile (253 mg, 1.00 mmol), and purified by column chromatography (gradient: EtOAc/PE 1:2–2:1) or recrystallization from ethanol. White solid, mp 183.0–184.0 °C (EtOH); yield 177 mg (0.66 mmol, 66%). Rf = 0.25, EtOAc/hexane (1:2, v/v). 0.62 (EtOAc/hexane, 2:1). 1H NMR (400 MHz, DMSO-d6) δ 9.23 (s, 1H), 8.13 (td, J = 7.7, 1.9 Hz, 1H), 7.53 (dd, J = 7.5, 1.8 Hz, 2H), 7.48–7.22 (m, 7H), 6.73 (s, 1H). 13C NMR (101 MHz, DMSO-d6) δ 170.3, 160.3 (d, J = 249.7 Hz), 149.5 (d, J = 9.2 Hz), 140.6, 130.4 (d, J = 8.6 Hz), 130.2 (d, J = 2.9 Hz), 128.3 (2C), 128.0, 126.5 (d, J = 1.6 Hz), 125.7 (2C), 124.2 (d, J = 3.4 Hz), 118.9 (d, J = 12.3 Hz), 115.7 (d, J = 21.9 Hz), 86.5. FTIR (ZnSe) ν (cm−1): 3253, 1672, 1489, 1446, 1224, 1053, 971; HRMS (ES TOF, m/z) calcd for C16H12FNNaO2+ ([M + Na]+): 292.0744, found 292.0744 (0.2 ppm).
3-(4-Chlorophenyl)-5-hydroxy-5-phenyl-1,5-dihydro-2H-pyrrol-2-one (8i)
Product 8i was obtained via the method described for compound 8a employing 2-(4-chlorophenyl)-4-oxo-4-phenylbutanenitrile36 (269 mg, 1.00 mmol), and purified by column chromatography (gradient: EtOAc:PE 1:2–2:1) or recrystallization from ethanol. Reaction time: 1 h. Colorless solid, mp 148.1–148.7 °C (EtOH); yield 194 mg (0.68 mmol, 68%). Rf = 0.40, EtOAc/hexane (1:1, v/v). 1H NMR (400 MHz, DMSO-d6) δ 9.18 (d, J = 1.8 Hz, 1H), 8.07–7.97 (m, 2H), 7.51 (dd, J = 8.1, 1.6 Hz, 3H), 7.49–7.43 (m, 2H), 7.42–7.26 (m, 3H), 6.66 (s, 1H). 13C NMR (101 MHz, DMSO-d6) δ 170.6, 146.4, 140.6, 133.2, 130.5, 130.0, 128.9 (2C), 128.4 (2C), 128.3 (2C), 128.0, 125.8 (2C), 86.1. FTIR (ZnSe) ν (cm−1): 3292, 1708, 1670, 1487, 1455, 1417, 1200, 1089, 1051; HRMS (ES TOF, m/z) calcd for C16H12ClNNaO2+ ([M + Na]+): 308.0449, found 308.0446 (0.8 ppm).
3-(2-Chlorophenyl)-5-hydroxy-5-phenyl-1,5-dihydro-2H-pyrrol-2-one (8j)
Product 8j was obtained via the method described for compound 8a, employing 2-(2-chlorophenyl)-4-oxo-4-phenylbutanenitrile36 (269 mg, 1.00 mmol), and purified by column chromatography (gradient: EtOAc/PE 1:2–2:1). This product is unstable upon heating above 60 °C. Yellowish amorphous solid; yield 217 mg (0.76 mmol, 76%). Rf = 0.15, EtOAc/hexane (1:2, v/v), Rf = 0.53, EtOAc/hexane (2:1, v/v). 1H NMR (400 MHz, DMSO-d6) δ 9.15 (s, 1H), 7.63–7.48 (m, 4H), 7.44–7.36 (m, 4H), 7.37–7.29 (m, 1H), 7.22 (d, J = 1.6 Hz, 1H), 6.75 (s, 1H). 13C NMR (101 MHz, DMSO-d6) δ 170.0, 149.7, 140.7, 132.6, 132.1, 131.4, 130.2, 129.9, 129.7, 128.3 (2C), 128.0, 126.9, 125.7 (2C), 86.8. FTIR (ZnSe) ν (cm−1): 3330, 1696, 1663, 1448, 1255, 1058, 1036, 756; HRMS (ES TOF, m/z) calcd for C16H12ClNNaO2+ ([M + Na]+): 308.0449, found 308.0449 (−0.2 ppm).
3-(4-Bromophenyl)-5-hydroxy-5-phenyl-1,5-dihydro-2H-pyrrol-2-one (8k)
Product 8k was obtained via the method described for compound 8a, employing 2-(4-bromophenyl)-4-oxo-4-phenylbutanenitrile36 (313 mg, 1.00 mmol), and purified by column chromatography (gradient: EtOAc/PE 1:2–2:1) or recrystallization from benzene. Reaction time: 2 hours. Colorless solid, mp 152.6–155.2 °C (benzene); yield 194 mg (0.59 mmol, 59%). Rf = 0.54, EtOAc/hexane (1:1, v/v). 1H NMR (400 MHz, DMSO-d6) δ 9.19 (s, 1H), 7.95 (d, J = 8.2 Hz, 2H), 7.59 (d, J = 8.3 Hz, 2H), 7.55–7.46 (m, 3H), 7.37–7.27 (m, 3H), 6.66 (s, 1H). 13C NMR (101 MHz, DMSO-d6) δ 170.5, 146.5, 140.6, 131.3 (2C), 130.6, 130.4, 129.2 (2C), 128.3 (2C), 128.0, 125.8 (2C), 121.9, 86.1. FTIR (ZnSe) ν (cm−1): 3248, 1663, 1612, 1487, 1451, 1424, 1210, 1055, 1010, 970; HRMS (ES TOF, m/z) calcd for C16H12BrNNaO2+ ([M + Na]+): 351.9944, found 351.9952 (−2.5 ppm).
5-Hydroxy-5-(4-methoxyphenyl)-3-phenyl-1,5-dihydro-2H-pyrrol-2-one (8l)
Product 8l was obtained via the method described for compound 8a, employing 4-(4-methoxyphenyl)-4-oxo-2-phenylbutanenitrile35 (265 mg, 1.00 mmol), and purified by column chromatography (gradient: EtOAc/PE 1:2–2:1) or recrystallization from ethanol. Colorless solid, mp 176.8–179.0 °C (ethanol); yield 216 mg (0.77 mmol, 77%). Rf = 0.22, EtOAc/hexane (1:2, v/v), Rf = 0.7, EtOAc/hexane (1:1, v/v). 1H NMR (400 MHz, DMSO-d6) δ 9.07 (s, 1H), 7.94 (d, J = 7.6 Hz, 2H), 7.57–7.27 (m, 6H), 6.93 (d, J = 8.3 Hz, 2H), 6.53 (s, 1H), 3.75 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 170.8, 159.0, 146.0, 132.7, 131.4, 131.2, 128.5, 128.3 (2C), 127.2 (2C), 127.0 (2C), 113.6 (2C), 85.9, 55.2. FTIR (ZnSe) ν (cm−1): 3354, 3258, 1703, 1662, 1515, 1419, 1243, 1183, 1062; HRMS (ES TOF, m/z) calcd for C17H15NNaO3+ ([M + Na]+): 304.0944, found 304.0941 (1.1 ppm).
5-(4-Chlorophenyl)-5-hydroxy-3-phenyl-1,5-dihydro-2H-pyrrol-2-one (8m)
Product 8m was obtained via the method described for compound 8a, employing 4-(4-chlorophenyl)-4-oxo-2-phenylbutanenitrile36 (269 mg, 1.00 mmol), and purified by column chromatography (gradient: EtOAc/PE 1:3–1:1) or recrystallization from benzene. White solid, mp 157.9–159.6 °C (benzene); yield 168 mg (0.59 mmol, 59%). Rf = 0.33, EtOAc/hexane (1:2, v/v). 1H NMR (400 MHz, DMSO-d6) δ 9.17 (d, J = 1.8 Hz, 1H), 8.01–7.91 (m, 2H), 7.56–7.50 (m, 2H), 7.48–7.32 (m, 6H), 6.75 (s, 1H). 13C NMR (101 MHz, DMSO-d6) δ 170.8, 145.4, 140.0, 132.6, 132.1, 131.0, 128.6, 128.3 (2C), 128.3 (2C), 127.8 (2C), 127.3 (2C), 85.7. FTIR (ZnSe) ν (cm−1): 3354, 3258, 1768, 1698, 1667, 1414, 1243, 1062; HRMS (ES TOF, m/z) calcd for C16H12ClNNaO2+ ([M + Na]+): 308.0449, found 308.0440 (3.0 ppm).
5-Hydroxy-5-(naphthalen-2-yl)-3-phenyl-1,5-dihydro-2H-pyrrol-2-one (8n)
Product 8n was obtained via the method described for compound 8a, employing 4-(naphthalen-2-yl)-4-oxo-2-phenylbutanenitrile34 (285 mg, 1.00 mmol), and purified by column chromatography (gradient: EtOAc/PE 1:3–1:1) or recrystallization from ethanol. White solid, mp 175.9–177.9 °C (ethanol); yield 232 g (0.77 mmol, 77%). Rf = 0.33, EtOAc/hexane (1:2, v/v). 1H NMR (400 MHz, DMSO-d6) δ 9.27 (s, 1H), 8.14 (s, 1H), 8.09–7.80 (m, 5H), 7.59 (d, J = 8.7 Hz, 1H), 7.56–7.48 (m, 3H), 7.46–7.26 (m, 3H), 6.82 (s, 1H). 13C NMR (101 MHz, DMSO-d6) δ 170.9, 145.7, 138.3, 132.7, 132.6, 132.2, 131.2, 128.6, 128.4 (2C), 128.1, 127.9, 127.5, 127.3 (2C), 126.3, 126.3, 124.3, 124.2, 86.2. FTIR (ZnSe) ν (cm−1): 3258, 1768, 1665, 1624, 1424, 1231, 1043; HRMS (ES TOF, m/z) calcd for C20H15NNaO2+ ([M + Na]+): 324.0995, found 324.1003 (−2.4 ppm).
5-(2,3-Dihydrobenzo[b][1,4]dioxin-6-yl)-5-hydroxy-3-phenyl-1,5-dihydro-2H-pyrrol-2-one (8o)
Product 8o was obtained via the method described for compound 8a, employing 4-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)-4-oxo-2-phenylbutanenitrile (293 mg, 1.00 mmol), and purified by column chromatography (gradient/EtOAc:PE 1:3–1:1) or recrystallization from ethanol. Reaction time: 1 h. White solid, mp 161.8–164.6 °C (EtOH); yield 0.189 g (0.61 mmol, 61%). Rf = 0.58, EtOAc/hexane (1:1, v/v). 1H NMR (400 MHz, DMSO-d6) δ 9.04 (s, 1H), 7.93 (d, J = 7.2 Hz, 2H), 7.43–7.30 (m, 4H), 6.99 (d, J = 2.1 Hz, 1H), 6.94 (dd, J = 8.4, 2.2 Hz, 1H), 6.84 (d, J = 8.4 Hz, 1H), 6.53 (s, 1H), 4.22 (s, 4H). 13C NMR (101 MHz, DMSO-d6) δ 170.8, 145.8, 143.1, 143.0, 133.9, 131.5, 131.2, 128.5, 128.3 (2C), 127.2 (2C), 118.6, 116.8, 114.7, 85.7, 64.1, 64.1. FTIR (ZnSe) ν (cm−1): 3248, 1667, 1498, 1431, 1313, 1277, 1253, 1060, 1000; HRMS (ES TOF, m/z) calcd for C18H15NNaO4+ ([M + Na]+): 332.0893, found 332.0895 (−0.4 ppm).
5-Hydroxy-3,5-bis(4-methoxyphenyl)-1,5-dihydro-2H-pyrrol-2-one (8p)
Product 8p was obtained via the method described for compound 8a, employing 2,4-bis(4-methoxyphenyl)-4-oxobutanenitrile35 (295 mg, 1.00 mmol), and purified by column chromatography (gradient: EtOAc/PE 1:2–2:1) or recrystallization from ethanol. Reaction time: 40 min. Yellow solid, mp 141.2–143.1 °C (EtOH); yield 199 mg (0.64 mmol, 64%). Rf = 0.50, EtOAc/hexane (1:1, v/v). 1H NMR (400 MHz, DMSO-d6) δ 9.00 (d, J = 1.8 Hz, 1H), 7.98–7.90 (m, 2H), 7.47–7.38 (m, 2H), 7.24 (d, J = 1.7 Hz, 1H), 7.01–6.86 (m, 4H), 6.46 (s, 1H), 3.77 (s, 3H), 3.74 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 171.1, 159.4, 158.9, 143.7, 133.0, 130.8, 128.5 (2C), 127.0 (2C), 123.7, 113.7 (2C), 113.5 (2C), 85.8, 55.1, 55.1. FTIR (ZnSe) ν (cm−1): 3335, 3244, 1704, 1602, 1506, 1417, 1304, 1251, 1183, 1067; HRMS (ES TOF, m/z) calcd for C18H17NNaO4+ ([M + Na]+): 334.1050, found 334.1048 (0.4 ppm).
5-(4-Aminophenyl)-3,5-diphenyl-1,5-dihydro-2H-pyrrol-2-one (15a)
5-Hydroxy-3,5-diphenyl-1,5-dihydro-2H-pyrrol-2-one (251 mg, 1.00 mmol) and aniline (186 mg, 2.00 mmol) were placed in a glass vial G10 Anton PAAR, equipped with a magnetic stirring bar heated in a boiling water bath for 1 min to homogenize the reaction mass. Then the vial was placed in an Anton PAAR microwave oven and heated to 160 °C for 40 min (temperature control using an infrared sensor). Next, the mixture was transferred from the reactor to chromatography column (gradient: EtOAc/PE 1:3–2:1) for anilines and (gradient: EtOAc/PE 1:4–1:2) for phenols. The fractions were concentrated on a rotary evaporator.
Anilines can also be purified by dissolving in 20% HCl, rinsing with EtOAc, neutralizing with NaHCO3 and subsequent extraction with CH2Cl2 (4 × 15 mL) followed by evaporation on a rotary evaporator.
Yellowish transparent solid, mp 119.5–122.1 °C; yield 143 mg (0.44 mmol, 44%). Rf = 0.36, EtOAc/hexane (1:1, v/v). 1H NMR (400 MHz, DMSO-d6) δ 9.59 (d, J = 2.0 Hz, 1H), 8.20 (d, J = 1.9 Hz, 1H), 8.04–7.99 (m, 2H), 7.45–7.21 (m, 8H), 7.06–6.92 (m, 2H), 6.58–6.46 (m, 2H), 5.10 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 170.6, 147.9, 147.8, 143.0, 131.6, 131.5, 128.9, 128.3 (5C), 127.5 (2C), 127.1, 126.9 (2C), 126.7 (2C), 113.5 (2C), 68.0. FTIR (ZnSe) ν (cm−1): 3380, 3253, 2998, 1771, 1679, 1516, 1369, 1241, 1048; HRMS (ES TOF, m/z) calcd for C22H18N2NaO+ ([M + Na]+): 349.1311, found 349.1307 (1.2 ppm).
5-(4-Aminophenyl)-3-(4-bromophenyl)-5-phenyl-1,5-dihydro-2H-pyrrol-2-one (15k)
Product 15k was obtained via the method described for compound 15a, employing 3-(4-bromophenyl)-5-hydroxy-5-phenyl-1,5-dihydro-2H-pyrrol-2-one (329 mg, 1.00 mmol) and aniline (186 mg, 2.00 mmol). Additional purification can be achieved by recrystallization from ethanol. Colorless solid, mp 247.0–249.5 °C (ethanol); yield 271 mg (0.67 mmol, 67%). Rf = 0.36, EtOAc/hexane (1:1, v/v). 1H NMR (400 MHz, DMSO-d6) δ 9.65 (d, J = 1.9 Hz, 1H), 8.29 (d, J = 1.9 Hz, 1H), 8.10–7.86 (m, 2H), 7.66–7.53 (m, 2H), 7.42–7.21 (m, 5H), 7.04–6.93 (m, 2H), 6.59–6.41 (m, 2H), 5.10 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 170.4, 148.5, 148.0, 142.8, 131.3 (2C), 130.8, 130.3, 129.0 (2C), 128.7, 128.3 (2C), 127.5 (2C), 127.2, 126.7 (2C), 121.7, 113.6 (2C), 68.2. FTIR (ZnSe) ν (cm−1): 3431, 3349, 1679, 1619, 1513, 1489, 1294, 1178, 1074; HRMS (ES TOF, m/z) calcd for C22H17BrN2NaO+ ([M + Na]+): 427.0416, found 427.0407 (2.3 ppm).
5-(4-Aminophenyl)-5-(4-methoxyphenyl)-3-phenyl-1,5-dihydro-2H-pyrrol-2-one (15l)
Product 15l was obtained via the method described for compound 15a, employing 5-hydroxy-5-(4-methoxyphenyl)-3-phenyl-1,5-dihydro-2H-pyrrol-2-one (281 mg, 1.00 mmol) and aniline (186 mg, 2.00 mmol). Additional purification can be achieved by recrystallization from ethanol. Colorless solid, mp 256.3–258.6 °C; yield 1.015 g (3.44 mmol, 86%). Rf = 0.25, EtOAc/hexane (1:2, v/v). 1H NMR (400 MHz, DMSO-d6) δ 9.50 (d, J = 2.0 Hz, 1H), 8.14 (d, J = 1.9 Hz, 1H), 8.03–7.95 (m, 2H), 7.43–7.23 (m, 5H), 6.97 (d, J = 8.2 Hz, 2H), 6.89 (d, J = 8.7 Hz, 2H), 6.50 (d, J = 8.2 Hz, 2H), 5.07 (s, 2H), 3.73 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 170.6, 158.2, 148.1, 147.8, 134.8, 131.7, 131.1, 129.1, 128.3 (2C), 128.2, 127.9 (2C), 127.4 (2C), 126.9 (2C), 113.6 (2C), 113.5 (2C), 67.5, 55.1. FTIR (ZnSe) ν (cm−1): 3370, 1773, 1674, 1592, 1513, 1445, 1356, 1248, 1211; HRMS (ES TOF, m/z) calcd for C23H21N2O2+ ([M + H]+): 357.1598, found 357.1588 (2.8 ppm).
5-(4-Aminophenyl)-5-(4-chlorophenyl)-3-phenyl-1,5-dihydro-2H-pyrrol-2-one (15m)
Product 15m was obtained via the method described for compound 15a, employing 5-(4-chlorophenyl)-5-hydroxy-3-phenyl-1,5-dihydro-2H-pyrrol-2-one (285 mg, 1.00 mmol) and aniline (186 mg, 2.00 mmol). Colorless solid, mp 209.0–211.9 °C; yield 166 mg (0.46 mmol, 46%). Rf = 0.36, EtOAc/hexane (1:1, v/v). 1H NMR (400 MHz, DMSO-d6) δ 9.61 (s, 1H), 8.19 (d, J = 1.8 Hz, 1H), 8.07–7.95 (m, 2H), 7.48–7.21 (m, 7H), 6.99 (d, J = 8.4 Hz, 2H), 6.52 (d, J = 8.4 Hz, 2H), 5.13 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 170.6, 148.1, 147.4, 142.0, 131.8, 131.7, 131.5, 128.6 (2C), 128.4, 128.4, 128.3 (2C), 128.3 (2C), 127.4 (2C), 127.0 (2C), 113.6 (2C), 67.6. FTIR (ZnSe) ν (cm−1): 3161, 2997, 1766, 1681, 1513, 1489, 1241, 1091; HRMS (ES TOF, m/z) calcd for C22H17ClN2NaO+ ([M + Na]+): 383.0922, found 383.0911 (2.8 ppm).
5-(4-Hydroxyphenyl)-3,5-diphenyl-1,5-dihydro-2H-pyrrol-2-one (16a)
Product 16a was obtained via the method described for compound 15a, employing 5-hydroxy-3,5-diphenyl-1,5-dihydro-2H-pyrrol-2-one (251 mg, 1.00 mmol) and phenol (188 mg, 1.00 mmol). White solid, mp 138.1–140.6 °C; yield 206 mg (0.63 mmol, 63%). Rf = 0.64, EtOAc/hexane (1:2, v/v). 1H NMR (400 MHz, DMSO-d6) δ 9.69 (d, J = 1.7 Hz, 1H), 9.49 (s, 1H), 8.26 (d, J = 1.9 Hz, 1H), 8.07–7.89 (m, 2H), 7.47–7.23 (m, 8H), 7.21–7.12 (m, 2H), 6.78–6.69 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 170.6, 156.6, 147.6, 142.6, 132.5, 131.8, 131.5, 128.4 (3C), 128.3 (2C), 128.0 (2C), 127.2, 127.0 (2C), 126.7 (2C), 115.1 (2C), 67.9. FTIR (ZnSe) ν (cm−1): 3344, 3176, 1679, 1513, 1492, 1359, 1241, 1173, 1096; HRMS (ES TOF, m/z) calcd for C22H17NNaO2+ ([M + Na]+): 350.1151, found 350.1139 (3.5 ppm).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Russian Science Foundation (grant #19-73-00091).Notes and references
- X.-C. Huang, X. Xiao, Y.-K. Zhang, T. T. Talele, A. A. Salim, Z.-S. Chen and R. J. Capon, Mar. Drugs, 2014, 12, 3818–3837 CrossRef CAS PubMed.
- H. Zhang, M. M. Conte, X.-C. Huang, Z. Khalil and R. J. Capon, Org. Biomol. Chem., 2012, 10, 2656–2663 RSC.
- Y.-L. Yang, C.-P. Lu, M.-Y. Chen, K.-Y. Chen, Y.-C. Wu and S.-H. Wu, Chem.–Eur. J., 2007, 13, 6985–6991 CrossRef CAS PubMed.
- N. Shionozaki, T. Yamaguchi, H. Kitano, M. Tomizawa, K. Makino and H. Uchiro, Tetrahedron Lett., 2012, 53, 5167–5170 CrossRef CAS.
- L. Li, P. Yu, M.-C. Tang, Y. Zou, S.-S. Gao, Y.-S. Hung, M. Zhao, K. Watanabe, K. N. Houk and Y. Tang, J. Am. Chem. Soc., 2016, 138, 15837–15840 CrossRef CAS PubMed.
- Y.-L. Gao, M.-L. Zhang, X. Wang, H.-D. Zhang, J.-Z. Huang and L. Li, Nat. Prod. Res., 2019 DOI:10.1080/14786419.2019.1641810.
- R. Kontnik and J. Clardy, Org. Lett., 2008, 10, 4149–4151 CrossRef CAS PubMed.
- M. Ramanathan, C.-J. Tan, W.-J. Chang, H.-H. G. Tsai and D.-R. Hou, Org. Biomol. Chem., 2013, 11, 3846–3854 RSC.
- R. Y. Kuo, F. R. Chang and Y. C. Wu, Tetrahedron Lett., 2001, 42, 7907–7909 CrossRef CAS.
- W.-N. Zhang, J.-G. Luo and L.-Y. Kong, J. Agric. Food Chem., 2012, 60, 1682–1687 CrossRef CAS PubMed.
- Y. S. Hui, L. Yan, W. Qi, S. Y. Ping, G. Wei, L. Yuan, Y. B. You and K. H. Xue, Heterocycles, 2020, 100, 568–584 CrossRef CAS.
- B.-y. Yang, Y. Liu, X. Wang, Y.-g. Xia, Q.-h. Wang and H.-x. Kuang, Zhongcaoyao, 2013, 44, 1877–1880 CAS.
- X. Hu, M.-W. Gong, W.-W. Zhang, Q.-H. Zheng, Q.-Y. Liu, L. Chen and Q.-Q. Zhang, Heterocycles, 2014, 89, 189–196 CrossRef CAS.
- B. Almohaywi, T. T. Yu, G. Iskander, D. S. H. Chan, K. K. K. Ho, S. Rice, D. S. Black, R. Griffith and N. Kumar, Bioorg. Med. Chem. Lett., 2019, 29, 1054–1059 CrossRef CAS PubMed.
- S. Mykhaylychenko, D. Harakat, G. Dupas, Y. G. Shermolovich and J.-P. Bouillon, J. Fluorine Chem., 2009, 130, 418–427 CrossRef CAS.
- S. Seo and M. C. Willis, Org. Lett., 2017, 19, 4556–4559 CrossRef CAS PubMed.
- R. Shiraki, A. Sumino, K.-i. Tadano and S. Ogawa, Tetrahedron Lett., 1995, 36, 5551–5554 CrossRef CAS.
- W.-K. Goh, C. R. Gardner, K. V. G. Chandra Sekhar, N. N. Biswas, S. Nizalapur, S. A. Rice, M. Willcox, D. S. Black and N. Kumar, Bioorg. Med. Chem., 2015, 23, 7366–7377 CrossRef CAS PubMed.
- W. K. Goh, G. Iskander, D. S. Black and N. Kumar, Tetrahedron Lett., 2007, 48, 2287–2290 CrossRef CAS.
- M.-Y. Wu, K. Li, N. Wang, T. He and X.-Q. Yu, Synthesis, 2011, 1831–1839 CAS.
- R. Adhikari, D. A. Jones, A. J. Liepa and R. H. Nearn, Aust. J. Chem., 2005, 58, 882–890 CrossRef CAS.
- W. Birru, R. T. Fernley, L. D. Graham, J. Grusovin, R. J. Hill, A. Hofmann, L. Howell, P. J. James, K. E. Jarvis, W. M. Johnson, D. A. Jones, C. Leitner, A. J. Liepa, G. O. Lovrecz, L. Lu, R. H. Nearn, B. J. O'Driscoll, T. Phan, M. Pollard, K. A. Turner and D. A. Winkler, Bioorg. Med. Chem., 2010, 18, 5647–5660 CrossRef CAS PubMed.
- C. Li, K. Jiang, Q. Ouyang, T.-Y. Liu and Y.-C. Chen, Org. Lett., 2016, 18, 2738–2741 CrossRef CAS PubMed.
- H. Roeber and K. Hartke, Chem. Ber., 1975, 108, 3262–3270 CrossRef CAS.
- F.-M. Liao, Z.-Y. Cao, J.-S. Yu and J. Zhou, Angew. Chem., Int. Ed., 2017, 56, 2459–2463 CrossRef CAS PubMed.
- A. V. Aksenov, V. Khamraev, N. A. Aksenov, N. K. Kirilov, D. A. Domenyuk, V. A. Zelensky and M. Rubin, RSC Adv., 2019, 9, 6636–6642 RSC.
- A. V. Aksenov, D. A. Aksenov, N. A. Arutiunov, N. A. Aksenov, E. V. Aleksandrova, Z. Zhao, L. Du, A. Kornienko and M. Rubin, J. Org. Chem., 2019, 84, 7123–7137 CrossRef CAS PubMed.
- A. V. Aksenov, D. A. Aksenov, G. D. Griaznov, N. A. Aksenov, L. G. Voskressensky and M. Rubin, Org. Biomol. Chem., 2018, 16, 4325–4332 RSC.
- J. A. Ciller, C. Seoane and J. L. Soto, Liebigs Ann. Chem., 1985, 51–57 CrossRef CAS.
- N. A. Aksenov, D. A. Aksenov, A. A. Skomorokhov, L. A. Prityko, A. V. Aksenov, G. D. Griaznov and M. Rubin, J. Org. Chem., 2020, 85, 12128–12146 CrossRef PubMed.
- L. Chen, F. Zhou, T.-D. Shi and J. Zhou, J. Org. Chem., 2012, 77, 4354–4362 CrossRef CAS PubMed.
- L. Chen and J. Zhou, Chem.–Asian J., 2012, 7, 2510–2515 CrossRef CAS PubMed.
- D. Glavac, N. Topolovcan and M. Gredicak, J. Org. Chem., 2020, 85, 14253–14261 CrossRef CAS PubMed.
- M. S. Hidayetulla, R. C. Shah and T. S. Wheeler, J. Chem. Soc., 1941, 111–112, 10.1039/JR9410000111.
- Z. Li and J. Yin, Chin. J. Chem., 2017, 35, 1179–1184 CrossRef CAS.
- S. Lin, Y. Wei and F. Liang, Chem. Commun., 2012, 48, 9879–9881 RSC.
- A. Stroba, F. Schaeffer, V. Hindie, L. Lopez-Garcia, I. Adrian, W. Froehner, R. W. Hartmann, R. M. Biondi and M. Engel, J. Med. Chem., 2009, 52, 4683–4693 CrossRef CAS PubMed.
- G. Bist, N. T. Pun, T. B. T. Magar, A. Shrestha, H. J. Oh, A. Khakurel, P.-H. Park and E.-S. Lee, Bioorg. Med. Chem. Lett., 2017, 27, 1205–1209 CrossRef CAS PubMed.
- P. Shaykhutdinova and M. Oestreich, Org. Lett., 2018, 20, 7029–7033 CrossRef CAS PubMed.
- Y.-S. Yang, B. Yang, Y. Zou, G. Li and H.-L. Zhu, Bioorg. Med. Chem., 2016, 24, 3052–3061 CrossRef CAS PubMed.
- R. Kumar, P. Sharma, A. Shard, D. K. Tewary, G. Nadda and A. K. Sinha, Med. Chem. Res., 2012, 21, 922–931 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2069203 and 2069260. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra02279b |
| This journal is © The Royal Society of Chemistry 2021 |