
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Antibacterial peptidomimetic and characterization of its efficacy as an antibacterial and biocompatible coating for bioceramic-based bone substitutes†
Sudip
Chakraborty‡
a,
Rajesh
Kuppusamy‡
*ab,
Iman
Roohani
acd,
William R.
Walsh
e,
Mark D. P.
Willcox
b,
Naresh
Kumar
 a and
Renxun
Chen
*a
a and
Renxun
Chen
*a
aSchool of Chemistry, UNSW Sydney, 2052, Kensington, NSW, Australia. E-mail: r.kuppusamy@unsw.edu.au; r.chen@unsw.edu.au; Tel: +61 401426387 Tel: +61 406233203
bSchool of Optometry and Vision Science, UNSW Sydney, 2052, Kensington, NSW, Australia
cCharles Perkins Centre, University of Sydney, Sydney, NSW 2006, Australia
dSchool of Life and Environmental Sciences, University of Sydney, Sydney, NSW 2006, Australia
ePrince of Wales Clinical School, UNSW Medicine, UNSW Sydney, Kensington, NSW, Australia
First published on 8th September 2021
Abstract
Infection of orthopedic devices by pathogenic bacteria, coupled with the development of bacterial resistance against major antibiotics, have caused severe physical and emotional trauma to patients and posed economic challenges to healthcare institutes and governments worldwide. Antimicrobial peptidomimetics have emerged as promising new candidates to fight the rise of bacterial resistance. Conjugation of these peptidomimetics to bone implant materials has become a viable strategy to combat orthopedic device related infections. In this paper, we report on the synthesis of an anthranilamide-based antibacterial peptidomimetic. The compound, which could possibly be acting through the depolarization of bacterial cell membrane, demonstrated a higher toxicity towards S. aureus compared to E. coli. We further demonstrated the ability of this compound to disrupt pre-formed biofilms. Coating of the compound on hydroxyapatite discs was achieved via physical interactions between the charged hydroxyapatite surface and the peptidomimetic. This was confirmed via X-ray photoelectron spectroscopy. The compound imparted antibacterial property to the discs as determined via antibacterial assays and imaging, while rendering the discs mildly cytotoxic towards human fetal osteoblast cells.
Introduction
Infection of orthopedic implants is a major challenge facing healthcare systems around the world.1–3 Infections lead to the failure of implants which necessitates implant revision surgery and, in severe cases, may result in amputation and even mortality.4–6 Management of orthopedic implant related infections (ODRIs) also poses a significant financial burden to health care systems and governments.7 The rate of infection ranges from 0.7% to 4.2% in patients that have undergone elective orthopedic surgery, while for patients with trauma that require complex surgeries, the rate can be as high as 30%.8–12 Nearly two thirds of all ODRIs are caused by Staphylococcus species, of which, Staphylococcus aureus and Staphylococcus epidermis are the most significant. Between 20% to 30% of ODRIs are caused by Staphylococcus aureus.13–17S. aureus can make up a significant proportion of the resident microbiota of the skin, and from there can cause hospital- or community-acquired infections.18–20Treatments of ODRIs have depended heavily on the use of antibiotics. However, one of the major complications with the treatment of ODRIs with traditional antibiotics is the development of bacterial resistance.21 The emergence of methicillin resistant Staphylococcus aureus has posed serious challenges to established therapeutic protocols for ODRIs.22 A decline in the development of new antibiotics has happened in parallel with the acquisition of resistance to established antibiotics.23,24 Just two new antibiotics were developed during 2008–2012.25,26 This decline can be attributed to a decrease in antibiotic research, reduced investment in the development of new antibiotics, and unfavorable government regulations.27–29
These developments have necessitated research into alternative strategies to combat bacterial infection. One class of molecules have gained prominence. These are referred to as antimicrobial peptides (AMPs), and are a diverse group of small proteins, some of which play a key role in the innate immune system of various species.30 One of the primary modes of action of AMPs is through the disruption of bacterial cell membranes. AMPs also exert their influence through the modulation of immune responses and the regulation of inflammation.31 The primary advantages of AMPs over traditional antibiotics are their broad-spectrum activity and lesser susceptibility towards the development of bacterial resistance.32 There has been a significant amount of research on AMP coated bone implants.33–36 Despite a lot of promise, there are certain drawbacks of natural AMPs that hinder their clinical translation. These can include toxicity towards mammalian cells, protease susceptibility,37 sensitivity to changes in pH and salt concentrations,38,39 and high costs of production.40 These shortcomings have inspired research on mimics of AMPs. Our group has designed short molecule peptidomimetics which have significant antibacterial activity.41,42 However, little research has been conducted to demonstrate the application of such peptidomimetics in controlling ODRIs.
Hydroxyapatite (HA) has been used extensively for the fabrication of biomaterials particularly bone substitutes.43 A comparative study of HA based materials with autografts demonstrated increased implant incorporation of the HA graft in a canine model for cervical interbody fusion.44 In another study, hydroxyapatite disc fusion exhibited slightly superior performance over autologous iliac crest interbody fusion in anterior cervical disc surgery.45 HA has been used as a stand-alone material or in combination with other materials and nanoparticles for the fabrication of bone tissue engineering scaffolds.43 On other occasions, HA has been used as coatings and other formulations to improve the performance of metal and polymer-based implants.46–48 Due to its versatility and widespread usage as a material for fabricating bone implants and biomaterials, HA serves as a suitable model material for testing the efficacy of novel antimicrobial peptidomimetics.
In this work, we report the synthesis of a small molecule peptidomimetic and its antibacterial efficacy. We then determine its effectiveness as an antibacterial coating material on hydroxyapatite surface.
Materials and methods
1. Synthesis and characterization of the antimicrobial peptidomimetic
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20) in MHB medium and 200 μL aliquots were dispensed to flat bottom 96-well plate wells (Sarstedt Australia). Biofilm was allowed to grow in the 96-well plate for 24 h, followed by the addition of the compound and further incubation for 24 h. Four different concentrations of the compound; 1×, 2×, 4×, 8× MIC, corresponding to one-fold, two-fold, four-fold, and eight-fold MIC were chosen for this study. Untreated biofilm was used as control. Plates were sealed with self-adhesive microplate sealers (TopSeal-A, PerkinElmer) to allow diffusion of air and to prevent condensation. Biofilms adhered on polystyrene substratum were quantified by crystal violet staining as described previously.50 This experiment was performed in triplicate.
:20) in MHB medium and 200 μL aliquots were dispensed to flat bottom 96-well plate wells (Sarstedt Australia). Biofilm was allowed to grow in the 96-well plate for 24 h, followed by the addition of the compound and further incubation for 24 h. Four different concentrations of the compound; 1×, 2×, 4×, 8× MIC, corresponding to one-fold, two-fold, four-fold, and eight-fold MIC were chosen for this study. Untreated biofilm was used as control. Plates were sealed with self-adhesive microplate sealers (TopSeal-A, PerkinElmer) to allow diffusion of air and to prevent condensation. Biofilms adhered on polystyrene substratum were quantified by crystal violet staining as described previously.50 This experiment was performed in triplicate.
2. Synthesis of HA nanoparticles and discs
3. Synthesis and physical characterization of peptidomimetic coated hydroxyapatite discs
4. In vitro antimicrobial evaluation of peptidomimetic coated discs
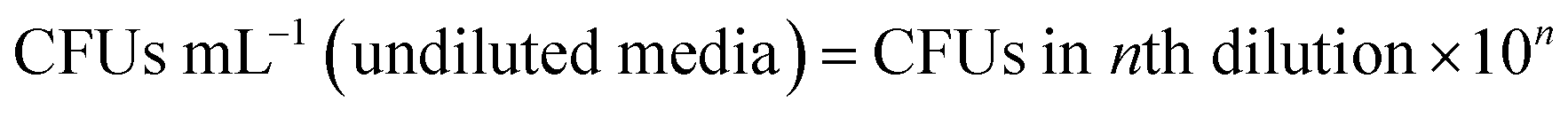 .
.
 .
.
5. In vitro cytotoxicity
where: M1 = molar extinction coefficient (E) of oxidized alamar blue (blue) at 570 nm, M2 = molar extinction coefficient (E) of oxidized alamar, blue (blue) at 570 nm, A1 = absorbance of test wells at 570 nm, A2 = absorbance of test wells at 600 nm, P1 = absorbance of positive growth control well (cells in tissue culture plate) at 570 nm, P2 = absorbance of positive growth control well at 600 nm.

The morphology of the bacteria attached to the discs was observed under SEM. The same protocol as above was followed for fixing the cells. NanoSEM 230, a field-emission scanning electron microscope (FE-SEM) was used for the imaging. The spot size was fixed at 3 and an accelerating voltage of 10 kV was used for the imaging.
Statistics. The bar graphs are plotted as mean ± standard deviation. Unless specified otherwise, all the experiments were performed in triplicates and repeated at least three independent times. The data was analyzed via one way Analysis of Variance (one way ANOVA). A p-value of less than 0.05 was considered to be statistically significant.
Results
1. Synthesis and characterization of novel antimicrobial peptidomimetic for surface coating
We have previously reported amphiphilic anthranilamide compounds as antimicrobial peptidomimetics.41 In this study, we have designed a cationic synthetic peptide for noncovalent, electrostatic binding with HA, aiming to construct an antimicrobial coating. Amongst the different biaryl compounds that were synthesized (data not presented), one compound (6 in Scheme 1) was found to be active against S. aureus (MIC = 3.9 μM) and E. coli (MIC = 31.2 μM). The compound was further investigated to understand its mode of action. As most AMP mimics kill bacteria by disrupting their cytoplasmic membranes, membrane permeability assays are illuminating for analyzing their mode of action.53 The membrane potential-sensitive dye cyanine disC3-5 (3,3′-dipropylthiadicarbocyanine iodide) assay was used to analyze the ability of our compound to depolarize the cytoplasmic membrane of S. aureus and E. coli. Perturbation of the bacterial cell membrane by a membrane disruptive compound leads to the loss of the membrane potential gradient, thereby causing the dye to be released into the medium. The release of the dye subsequently increases the fluorescence intensity of the medium, which can be used as a measure of membrane activity of the compound. The addition of biaryl compound 6 at 1× MIC (3.9 μM) against S. aureus rapidly increased the fluorescence intensity indicating the perturbation of the cell membrane (Fig. 1(a)). Similar results were obtained when the cytoplasmic permeability assay was performed on E. coli as well (Fig. 1(b)). | ||
| Scheme 1 Synthesis of antibacterial peptide mimics for surface coating. | ||
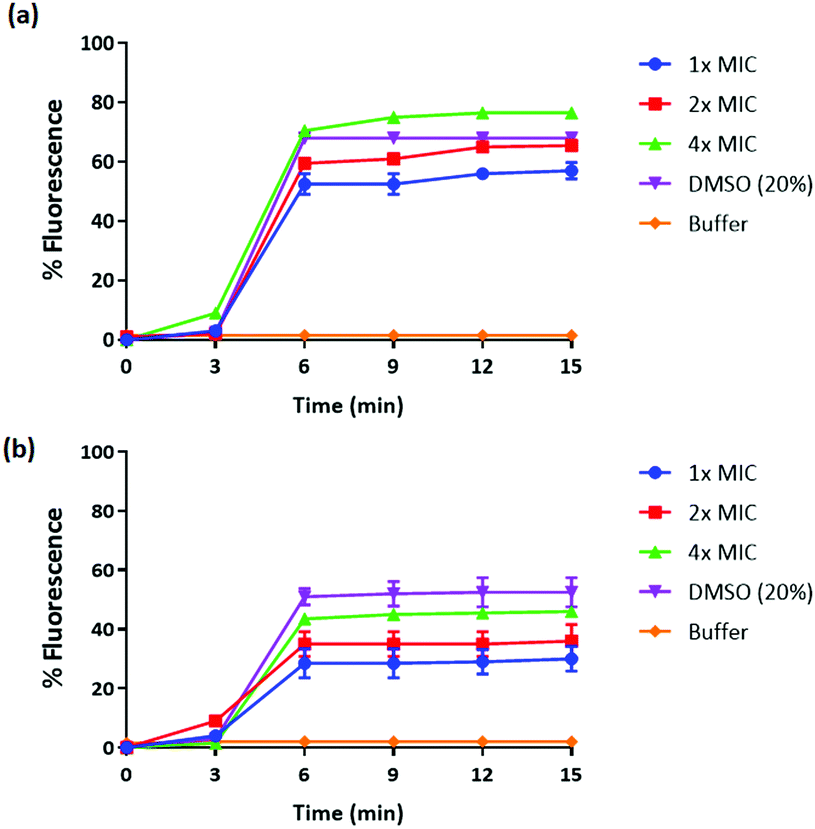 | ||
| Fig. 1 Cytoplasmic membrane depolarization of (a) S. aureus 38 and (b) E. coli K12 by 6, as assessed by release of the membrane potential-sensitive dye DiSC3-(5) measured spectroscopically at 622 nm and 670 nm, as the excitation and emission wavelength respectively. 1×, 2×, and 4× MIC denote one-fold, two-fold, and four-fold MIC, respectively. Data presented as means (± SD) of three independent repeats in triplicate cells 20% DMSO was used as a positive control. | ||
Biofilms have been shown to be 10–1000 times more resistant to conventional antibiotics.54 We tested the ability of our peptidomimetic compound for its ability to disrupt established S. aureus and E. coli biofilms using the crystal violet staining assay. The ability of compound 6 to disrupt the biofilms was measured at increasing concentrations that were multiples of its MIC; 1×, 2×, 4×, and 8×, where × is the MIC of compound 6 (Fig. 2). Compound 6 disrupted around 93% of biofilm at 4× MIC (15.6 μM) concentration. However, the compound did not show biofilm disruption of E. coli even at a concentration of >125 μM, corresponding to 32× MIC.
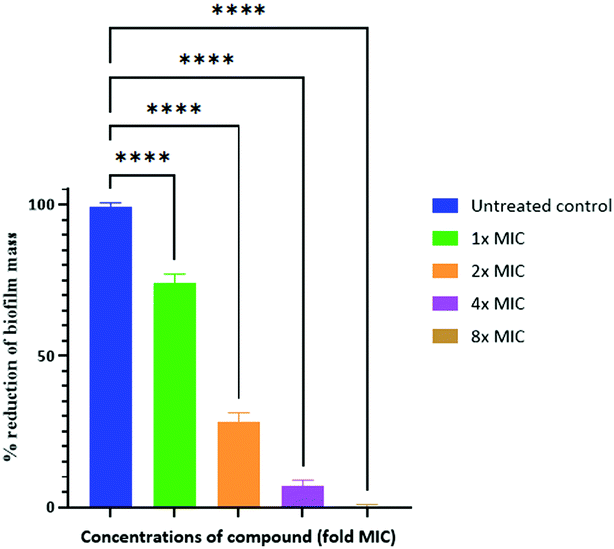 | ||
| Fig. 2 Disruption of established biofilm of S. aureus after 24 h with 1× to 8× MIC concentrations of compound 6. 1×, 2×, 4×, and 8× MIC denote one-fold, two-fold, four-fold and eight-fold MIC, respectively. The positive control represents the preestablished biofilms without any compounds. **** = p-value <0.0001. | ||
The active peptidomimetic compound was further assessed for its activity after attachment on HA discs.
2. Synthesis of HA nanoparticles and discs
 | ||
| Fig. 3 Scanning electron micrographs of (a) un-sintered and (b)–(e), HA nanoparticles sintered at 800, 1000, 1050, and 1100 °C, respectively. | ||
 | ||
| Fig. 4 (a) Sintered HA discs, (b) scanning electron micrograph of HA disc surface. | ||
3. Synthesis and characterization of peptidomimetic coated HA discs
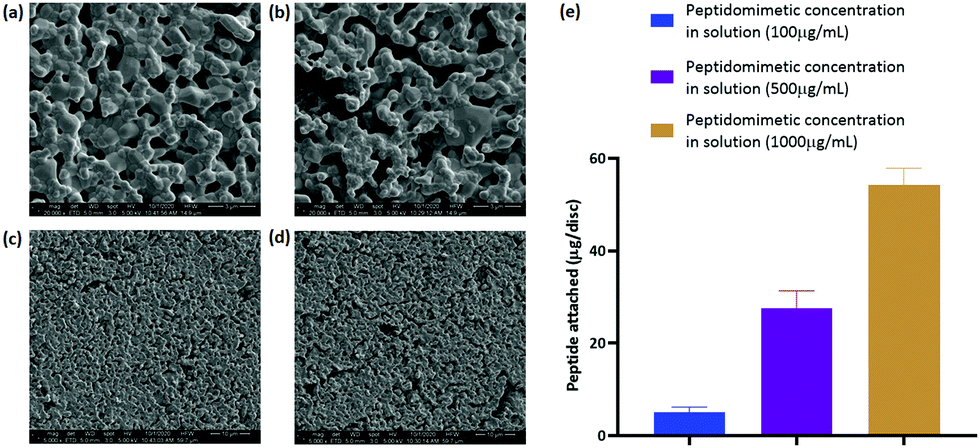 | ||
| Fig. 5 (a) and (c) Scanning electron micrographs of HA discs incubated in water for 24 h and (b) and (d) incubated in 1000 μg mL−1 peptidomimetic solution for 24 h. (e) Quantification of peptidomimetic attachment on HA discs corresponding to three different concentrations of peptidomimetic loading solution. n = 3. | ||
 | ||
| Fig. 6 (a) Surface elemental composition of the uncoated and coated HA discs, (b) ratio of different carbon species on the surface of the discs, (c) surface coverage of nitrogen on the discs. ns = no significance, * = p-value <0.5, **** = p-value <0.0001. n = 3. | ||
 | ||
| Fig. 7 FT-IR spectra of unmodified and modified HA discs. | ||
All the four spectra follow the characteristic spectrum of HA; however, the intensity and sharpness of the peaks decreased with an increase in the amount of bound peptidomimetic.
 | ||
| Fig. 8 RAMAN spectra of unmodified and modified HA discs. | ||
4. Antibacterial adhesion assay
The antibacterial activity of the peptidomimetic coated discs was evaluated in a direct contact antibacterial assay.36 The coated discs demonstrated higher antibacterial activity than the uncoated control discs, exhibiting a 2log reduction in the number of viable bacteria in the media (Fig. 9(a)). However, a significant dose dependent increase in the antibacterial activity of the coated discs was not observed. A trend towards an increasing activity was noticed but the differences between groups with different amounts of loaded peptide were not statistically significant.
 | ||
| Fig. 9 (a) CFUs in supernatant, (b) CFUs on disc surface. ** = p-value <0.01, **** = p-value <0.0001. n = 3. | ||
The number of bacteria attached to the discs was quantified by a slight modification of the protocol of the direct contact antibacterial assay. A reduction in the total number of bacteria attached to the discs was observed between the coated and the uncoated discs (Fig. 9(b)). However, like the case with bacteria in media, no significant difference was observed within the three different coated discs.
The morphology of the attached bacteria was observed under SEM. S.A. 38 exhibited a spherical morphology on all the discs and no structural deformities were visible on the bacteria grown on the coated discs (Fig. 10). The number of bacteria within a field of view was higher in the uncoated disc compared to that on the coated discs.
 | ||
| Fig. 10 SEM micrographs of S. aureus on the surfaces of (a) HA disc, (b)–(d) HA disc soaked in 100, 500, 1000 μg mL−1 peptidomimetic, respectively. (e)–(h) corresponding images of the same samples in the same order at a lower magnification. | ||
5. In vitro cytotoxicity
 | ||
| Fig. 11 Cytotoxicity after (a) 24 h, (b) 72 h of cell culture on discs, (c) side by side comparison of cytotoxicity at 24 and 72 hours, respectively. ns = no significance, ** = p-value <0.01, *** = p-value <0.001, **** = p-value <0.0001. n = 3. | ||
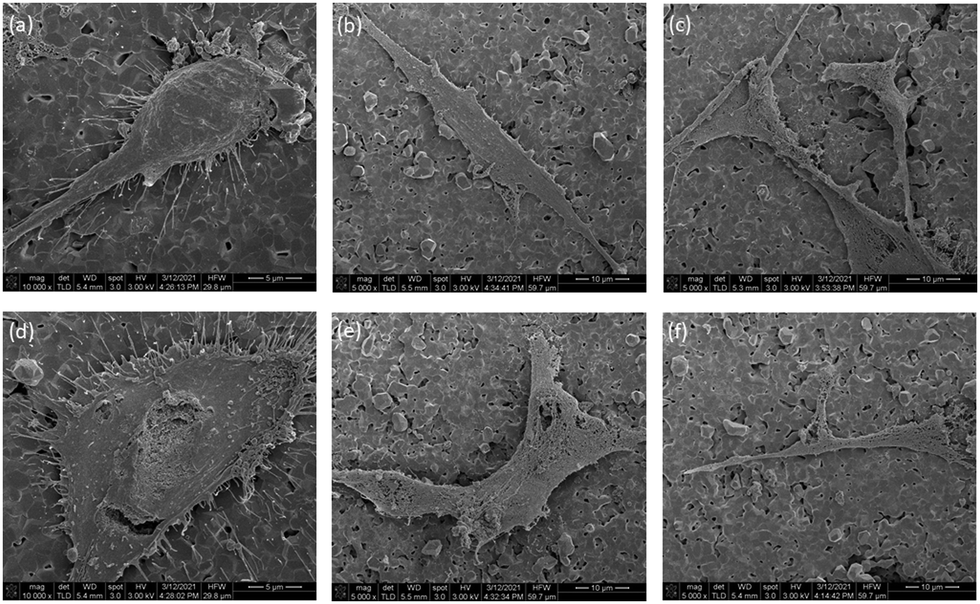 | ||
| Fig. 12 SEM of hFOB on (a)–(c), HA disc, (d)–(f), Ha disc soaked in 100 μg mL−1 peptidomimetic. | ||
Discussion
The prevalence of bacterial infections on orthopedic implants and biomaterials is a major challenge for governments and healthcare institutes around the world. The presence of S. aureus in a significant majority of these infections makes it a major target of therapeutic interventions. However, traditional antibiotics, despite their success, have become ineffectual in treating a lot of infections due to the emergence of antibiotic resistance in bacteria. Novel strategies for treating infections have utilized AMPs and their synthetic analogs. Natural AMPs are not particularly suitable for use in the pharmaceutical industry due to their drawbacks mentioned before. Synthetic mimics of AMPs, also known as antibacterial peptidomimetics, retain crucial structural features of AMPs responsible for their antibacterial activity while overcoming some of their drawbacks.Our group has previously reported amphiphilic anthranilamide compounds as novel antibacterial peptidomimetics.41,42 Inspired by the success of those compounds, our current work describes the synthesis of a novel antibacterial peptidomimetic for potential application as a coating material for orthopedic devices. In this paper, we reported on the synthesis of a new anthranilamide-based peptidomimetic. The compound demonstrated activity against both Gram-positive and Gram-negative bacteria and its MIC against S. aureus 38 strain was significantly lower than its MIC against E. coli K12 strain. One possible common mechanism of action of AMPs and AMP mimics is the disruption of bacterial cell membrane. Membrane disruption is initiated by the attachment of the AMPs and their mimics to the membrane surface which is facilitated by the cationic and amphiphilic nature of these molecules. Our compound demonstrated the ability to destabilize artificial membrane in the cytoplasmic membrane permeability assay, which suggests that membrane disruption is an integral part of mechanism of action for the peptidomimetic. This can be attributed to the cationic and amphiphilic nature of our compound which, as mentioned before, are crucial parameters determining the attachment of antibacterial entities to the surface of bacterial membranes. Furthermore, our compound demonstrated the ability to disrupt pre-formed biofilms. These results suggests that the peptidomimetic is a suitable candidate for the prevention and/or treatment of bacterial infections.
One of the strategies for combatting orthopedic device associated infections is the release of antibacterial agents from the devices in situ. Biomaterials and devices used in orthopedic therapy have been coated with antibacterial entities, both traditional antibiotics and AMPS/peptidomimetics.55–57 Amongst the different coating strategies, direct covalent and non-covalent attachment of antibacterial entities to surfaces have been explored quite extensively.36,58 Non-covalent attachment strategies leverage the charged and/or polar properties of some drugs and material surfaces to form favorable weak interactions between the two. This type of attachment is reversible and the characteristics of the material and the antibacterial entity contributing to their interaction can be modified to tune the strength of attachment and dissociation kinetics. Non-covalent attachment of an antibacterial entity on a biomaterial surface also allows for sustained release of the antibacterial over a prolonged period.
To investigate the utility of the peptidomimetic as antibacterial coating, we evaluated the efficacy of non-covalently attached peptidomimetic coating on HA discs. HA is a biocompatible and osteoconductive material used extensively in repair of bone defects. It serves as a good model material for testing the efficacy of novel antibacterial agents as coatings for biomaterials used in orthopedics. For our studies, we prepared cylindrical HA discs using HA nanoparticles. Cylindrical discs are easy to handle and transport for experimental purposes and the results obtained from them can be generalized to other HA based formulations, provided the surface properties of HA remain intact and play a dominant role in the function of the material. HA nanoparticles with approximately spherical morphology were selected for the formulation of the discs.
HA has a negatively charged surface at physiological pH. Studies have reported on the effect of solution pH on the charge of HA surfaces, and although there is some variability in the data owing to the different sources and fabrication techniques of HA used in these studies, the isoelectric point, and the point of zero charge of HA have been found to be below 7 in most cases. Thus, at pH 7, HA has a net negative charge on its surface, which has been found to be favourable for protein adsorption.59 This surface property makes physical adsorption of charged antibacterial entities a viable route of infection therapy. We took advantage of the cationic nature of our peptidomimetic to bind it physically through electrostatic interaction to the surface of HA discs. The binding was confirmed via XPS. The adsorption of the peptidomimetic on the surface led to an increase in the nitrogen and a specific type of carbon (C–C/C–H) content on the surface. Some of the surface carbon was attributed to nonspecific adsorption of contaminants from the environment (adventitious carbon), however, the proportion of the adventitious carbon within the total carbon species reduced as more peptidomimetic attached to the surface, and the major fraction of the surface carbon was attributed to the peptidomimetic. Binding of the peptidomimetic to the discs did not interfere with the native functional groups present within HA, as was demonstrated via FT-IR and RAMAN spectroscopy. The lack of peaks specific to the peptidomimetic in the FT-IR and RAMAN spectra of the discs could be attributed to the formation of a very thin layer of coating, below the detection limit of the two spectroscopic methods.
We subsequently evaluated the antibacterial activity of the discs. Preliminary evaluation of antibacterial activity was done via a Kirby–Bauer disc diffusion assay (data not shown). A visible zone of inhibition in a lawn of S. aureus surrounding the peptidomimetic coated discs indicated the presence of antibacterial activity in the discs. To characterize the activity further, we went ahead with direct contact antibacterial assays and quantified the decrease in the number of viable S. aureus on the surface of coated discs and their surrounding medium compared to the number of viable S. aureus on uncoated HA disc and its surrounding medium. A significant decrease in the number of viable S. aureus was observed both on the surface of the coated discs and their surrounding medium compared to their uncoated counterparts, indicating the retention of antibacterial activity of the peptidomimetic on HA surface and also its release into the surrounding medium. The magnitude of decrease in viable bacteria in our study is comparable to those described in previous publications with different AMPs and peptidomimetics.36,60 These results look promising and further characterization of the peptidomimetic HA interactions can be performed to optimize their interactions. Simultaneously, structural analogs of the peptidomimetic used in this study can be synthesized and assessed for their efficacy.
In order for satisfactory regeneration of bone, biomaterials and implants used in orthopedic reconstruction should allow the growth of cells (osteoblasts and stem cells) on their surfaces. HA is intrinsically osteoconductive and therefore, provides a favorable environment for osteoblasts to attach and proliferate. Alamar blue assay was performed to assess the cytotoxicity of the coated discs and SEM imaging was performed to visualize the morphology of the cells on the discs. hFOB 1.19 cells were chosen for these studies as it is a widely accepted cell line for modeling the behavior of human osteoblasts. Incorporation of our peptidomimetic rendered the discs cytotoxic in a dose dependent manner. However, the magnitude of the cytotoxicity is within the acceptable range for biomaterials. Furthermore, the discs with the lowest amount of peptidomimetic yielded a cytotoxicity value that wasn’t significantly different to the uncoated HA discs. The morphology of the cells was unaffected by the incorporation of the peptidomimetic as can be inferred from the SEM images. It is plausible that contact with the peptidomimetic coated discs alters the metabolic rate of the cells and this resulted in a reduced viability count. However, the behavior of osteoblasts in contact with these discs needs to be studies further to reach a conclusion on the mechanism of cytotoxicity.
Conclusions
We have successfully designed a novel antibacterial peptidomimetic. The possible mode of action of this compound is via the disruption of the bacterial cell wall. Its antibacterial activity against S. aureus suggests its possible applicability towards the treatment of orthopedic device related infections. By coating the surface of HA discs with the peptidomimetic and then testing the antibacterial efficacy and cytotoxicity of the coated discs, we have determined the propensity of the peptidomimetic to attach to HA via electrostatic interactions and the compound retains its activity after the attachment. This study opens up the opportunity for further exploration into different modes of binding of the compound onto surfaces of medical importance. Future studies are planned to include in vitro release kinetics of the peptidomimetics from HA as well as challenge models in mice and a rabbit distal femoral defect model. The results from this study paves the way for further exploration of different antibacterial peptidomimetics on HA surfaces.Author contributions
N. K., M. W., I. R., R. C., B. W., R. K., and S. C. conceptualized the study, S. C. and R. K. conducted in vitro experiments and analyzed the data, N. K., M. W., I. R., and R. C. analyzed the data, S. C. and R. K. drafted the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the electron microscopy unit and the spectroscopy unit at Mark Wainwright Analytical Centre, UNSW Sydney for providing the SEM imaging, and FT-IR, RAMAN spectroscopy facilities, respectively. Sudip Chakraborty would like to acknowledge the Graduate Research School, UNSW Sydney, for providing him with the Tuition Fee Scholarship for pursuing his research degree. This work was supported by the NHMRC [APP1183597].References
- T. F. Moriarty, R. Kuehl, T. Coenye, W. J. Metsemakers, M. Morgenstern, E. M. Schwarz, M. Riool, S. A. J. Zaat, N. Khana, S. L. Kates and R. G. Richards, EFORT Open Rev., 2016, 1, 89–99 CrossRef PubMed.
- R. Spitzmüller, D. Gümbel, C. Güthoff, S. Zaatreh, A. Klinder, M. Napp, R. Bader, W. Mittelmeier, A. Ekkernkamp, A. Kramer and D. Stengel, BMC Musculoskeletal Disord., 2019, 20, 184 CrossRef PubMed.
- G. Tsaras, D. R. Osmon, T. Mabry, B. Lahr, J. St Sauveur, B. Yawn, R. Kurland and E. F. Berbari, Infect. Control Hosp. Epidemiol., 2012, 33, 1207–1212 CrossRef PubMed.
- A. Patel, G. Pavlou, R. E. Mújica-Mota and A. D. Toms, Bone Joint J., 2015, 97-b, 1076–1081 CrossRef CAS PubMed.
- R. E. Delanois, J. B. Mistry, C. U. Gwam, N. S. Mohamed, U. S. Choksi and M. A. Mont, J. Arthroplasty, 2017, 32, 2663–2668 CrossRef PubMed.
- K. J. Bozic, E. Lau, S. Kurtz, K. Ong, H. Rubash, T. P. Vail and D. J. Berry, J. Bone Jt. Surg., Am. Vol., 2012, 94, 794–800 CrossRef PubMed.
- E. Sullivan, A. Gupta and C. H. Cook, Surg. Infect., 2017, 18, 451–454 CrossRef PubMed.
- M. J. Patzakis and J. Wilkins, Clin. Orthop. Relat. Res., 1989, 36–40 CAS.
- B. Sugarman and E. J. Young, Infect. Dis. Clin. North Am., 1989, 3, 187–198 CrossRef CAS PubMed.
- W. Zimmerli, A. Trampuz and P. E. Ochsner, N. Engl. J. Med., 2004, 351, 1645–1654 CrossRef CAS PubMed.
- M. Saadatian-Elahi, R. Teyssou and P. Vanhems, Int. J. Surg., 2008, 6, 238–245 CrossRef PubMed.
- S. M. Kurtz, E. Lau, J. Schmier, K. L. Ong, K. Zhao and J. Parvizi, J. Arthroplasty, 2008, 23, 984–991 CrossRef PubMed.
- S. Corvec, M. E. Portillo, B. M. Pasticci, O. Borens and A. Trampuz, Int. J. Artif. Organs, 2012, 35, 923–934 CrossRef PubMed.
- J. L. Del Pozo and R. Patel, N. Engl. J. Med., 2009, 361, 787–794 CrossRef CAS PubMed.
- L. Montanaro, P. Speziale, D. Campoccia, S. Ravaioli, I. Cangini, G. Pietrocola, S. Giannini and C. R. Arciola, Future Microbiol., 2011, 6, 1329–1349 CrossRef CAS PubMed.
- A. Trampuz and W. Zimmerli, Injury, 2006, 37(Suppl 2), S59–S66 CrossRef PubMed.
- A. J. Tande and R. Patel, Clin. Microbiol. Rev., 2014, 27, 302–345 CrossRef PubMed.
- E. Klein, D. L. Smith and R. Laxminarayan, Emerg. Infect. Dis. J., 2007, 13, 1840 CrossRef PubMed.
- A. G. Jensen, C. H. Wachmann, K. B. Poulsen, F. Espersen, J. Scheibel, P. Skinhøj and N. Frimodt-Møller, Arch. Intern. Med., 1999, 159, 1437–1444 CrossRef CAS PubMed.
- C.-J. Kim, H.-B. Kim, M.-D. Oh, Y. Kim, A. Kim, S.-H. Oh, K.-H. Song, E. S. Kim, Y. K. Cho, Y. H. Choi, J. Park, B.-N. Kim, N.-J. Kim, K.-H. Kim, E. J. Lee, J.-B. Jun, Y. K. Kim, S. M. Kiem, H. J. Choi, E. J. Choo, K.-M. Sohn, S. Lee, H. H. Chang, J. H. Bang, S. J. Lee, J. H. Lee, S. Y. Park, M. H. Jeon, N. R. Yun and K. S. G. The, BMC Infect. Dis., 2014, 14, 590 CrossRef PubMed.
- C. L. Ventola, P T, 2015, 40, 277–283 Search PubMed.
- D. Teterycz, T. Ferry, D. Lew, R. Stern, M. Assal, P. Hoffmeyer, L. Bernard and I. Uçkay, Int. J. Infect. Dis., 2010, 14, e913–e918 CrossRef PubMed.
- J. Conly and B. Johnston, Can. J. Infect. Dis. Med. Microbiol., 2005, 16, 159–160 CrossRef PubMed.
- M. I. Hutchings, A. W. Truman and B. Wilkinson, Curr. Opin. Microbiol., 2019, 51, 72–80 CrossRef CAS PubMed.
- B. Li and T. J. Webster, J. Orthop. Res., 2018, 36, 22–32 Search PubMed.
- H. W. Boucher, G. H. Talbot, D. K. Benjamin, Jr., J. Bradley, R. J. Guidos, R. N. Jones, B. E. Murray, R. A. Bonomo, D. Gilbert and A. Infectious Diseases Society of, Clin. Infect. Dis., 2013, 56, 1685–1694 CrossRef PubMed.
- J. A. DiMasi, H. G. Grabowski and R. W. Hansen, J. Health Econ., 2016, 47, 20–33 CrossRef PubMed.
- L. J. Piddock, Lancet Infect. Dis., 2012, 12, 249–253 CrossRef PubMed.
- J. G. Bartlett, D. N. Gilbert and B. Spellberg, Clin. Infect. Dis., 2013, 56, 1445–1450 CrossRef CAS PubMed.
- M. Magana, M. Pushpanathan, A. L. Santos, L. Leanse, M. Fernandez, A. Ioannidis, M. A. Giulianotti, Y. Apidianakis, S. Bradfute, A. L. Ferguson, A. Cherkasov, M. N. Seleem, C. Pinilla, C. de la Fuente-Nunez, T. Lazaridis, T. Dai, R. A. Houghten, R. E. W. Hancock and G. P. Tegos, Lancet Infect. Dis., 2020, 20, e216–e230 CrossRef CAS PubMed.
- N. Mookherjee and R. E. Hancock, Cell. Mol. Life Sci., 2007, 64, 922–933 CrossRef CAS PubMed.
- R. E. Hancock and D. S. Chapple, Antimicrob. Agents Chemother., 1999, 43, 1317–1323 CrossRef CAS PubMed.
- M. Kazemzadeh-Narbat, J. Kindrachuk, K. Duan, H. Jenssen, R. E. W. Hancock and R. Wang, Biomaterials, 2010, 31, 9519–9526 CrossRef CAS PubMed.
- M. Kazemzadeh-Narbat, S. Noordin, B. A. Masri, D. S. Garbuz, C. P. Duncan, R. E. W. Hancock and R. Wang, J. Biomed. Mater. Res., Part B, 2012, 100B, 1344–1352 CrossRef CAS PubMed.
- M. Kazemzadeh-Narbat, B. F. L. Lai, C. Ding, J. N. Kizhakkedathu, R. E. W. Hancock and R. Wang, Biomaterials, 2013, 34, 5969–5977 CrossRef CAS PubMed.
- L. Townsend, R. L. Williams, O. Anuforom, M. R. Berwick, F. Halstead, E. Hughes, A. Stamboulis, B. Oppenheim, J. Gough, L. Grover, R. A. H. Scott, M. Webber, A. F. A. Peacock, A. Belli, A. Logan and F. de Cogan, J. R. Soc., Interface, 2017, 14, 20160657 CrossRef PubMed.
- A. Rozek, J.-P. S. Powers, C. L. Friedrich and R. E. W. Hancock, Biochemistry, 2003, 42, 14130–14138 CrossRef CAS PubMed.
- I. H. Lee, Y. Cho and R. I. Lehrer, Infect. Immun., 1997, 65, 2898–2903 CrossRef CAS PubMed.
- T. Rydlo, S. Rotem and A. Mor, Antimicrob. Agents Chemother., 2006, 50, 490 CrossRef CAS PubMed.
- A. K. Marr, W. J. Gooderham and R. E. Hancock, Curr. Opin. Pharmacol., 2006, 6, 468–472 CrossRef CAS PubMed.
- R. Kuppusamy, M. Yasir, E. Yee, M. Willcox, D. S. Black and N. Kumar, Org. Biomol. Chem., 2018, 16, 5871–5888 RSC.
- R. Kuppusamy, M. Yasir, T. Berry, C. G. Cranfield, S. Nizalapur, E. Yee, O. Kimyon, A. Taunk, K. K. K. Ho, B. Cornell, M. Manefield, M. Willcox, D. S. Black and N. Kumar, Eur. J. Med. Chem., 2018, 143, 1702–1722 CrossRef CAS PubMed.
- Z. Bal, T. Kaito, F. Korkusuz and H. Yoshikawa, Emergent Mater., 2019, 3, 521–544 CrossRef.
- S. D. Cook, J. E. Dalton, E. H. Tan, W. V. Tejeiro, M. J. Young and T. S. Whitecloud, 3rd, Spine, 1994, 19, 1856–1866 CrossRef CAS PubMed.
- H. J. Senter, R. Kortyna and W. R. Kemp, Neurosurgery, 1989, 25, 39–43 CrossRef CAS PubMed.
- S. Allegrini, Jr., E. Rumpel, E. Kauschke, J. Fanghanel and B. Konig, Jr., Ann. Anat., 2006, 188, 143–151 CrossRef PubMed.
- W. S. W. Harun, R. I. M. Asri, A. B. Sulong, S. A. C. Ghani and Z. Ghazalli, Hydroxyapatite – Advances in Composite Nanomaterials, Biomedical Applications and Its Technological Facets, 2018 DOI:10.5772/intechopen.71063 ch. 5.
- C. J. Oosterbos, H. Vogely, M. W. Nijhof, A. Fleer, A. J. Verbout, A. J. Tonino and W. J. Dhert, J. Biomed. Mater. Res., 2002, 60, 339–347 CrossRef CAS PubMed.
- M. Wu, E. Maier, R. Benz and R. E. W. Hancock, Biochemistry, 1999, 38, 7235–7242 CrossRef CAS PubMed.
- G. A. O'Toole, J. Vis. Exp., 2011, 2437, DOI:10.3791/2437.
- M. Balouiri, M. Sadiki and S. K. Ibnsouda, J. Pharm. Anal., 2016, 6, 71–79 CrossRef PubMed.
- L. Deng, Y. Deng and K. Xie, Colloids Surf., B, 2017, 160, 483–492 CrossRef CAS PubMed.
- B. Mojsoska and H. Jenssen, Pharmaceuticals, 2015, 8, 366–415 CrossRef CAS PubMed.
- N. Høiby, T. Bjarnsholt, M. Givskov, S. Molin and O. Ciofu, Int. J. Antimicrob. Agents, 2010, 35, 322–332 CrossRef PubMed.
- Y. He, Y. Jin, X. Ying, Q. Wu, S. Yao, Y. Li, H. Liu, G. Ma and X. Wang, Regener. Biomater., 2020, 7, 515–525 CrossRef CAS PubMed.
- L. Chen, L. Shao, F. Wang, Y. Huang and F. Gao, RSC Adv., 2019, 9, 10494–10507 RSC.
- Z. Ye, X. Zhu, I. Mutreja, S. K. Boda, N. G. Fischer, A. Zhang, C. Lui, Y. Qi and C. Aparicio, Bioact. Mater., 2021, 6, 2250–2260 CrossRef CAS PubMed.
- C. T. Johnson and A. J. García, Ann. Biomed. Eng., 2015, 43, 515–528 CrossRef PubMed.
- J. R. Sharpe, R. L. Sammons and P. M. Marquis, Biomaterials, 1997, 18, 471–476 CrossRef CAS PubMed.
- D. M. Ibrahim, E. S. Sani, A. M. Soliman, N. Zandi, E. Mostafavi, A. M. Youssef, N. K. Allam and N. Annabi, ACS Appl. Bio Mater., 2020, 3, 3313–3325 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis route and details of each step is provided along with supporting 1H and 13C NMR data. See DOI: 10.1039/d1ma00648g |
| ‡ Equal first authors-Sudip Chakraborty and Rajesh Kuppusamy both contributed equally to the manuscript. |
| This journal is © The Royal Society of Chemistry 2021 |