
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
(Hydroxy)apatite on cement: insights into a new surface treatment†
Ronald J.
Turner
 *a,
Pieter
Bots
*a,
Alan
Richardson
b,
Paul A.
Bingham
c,
Alex
Scrimshire
c,
Andrew
Brown
d,
Mark
S’Ari
d,
John
Harrington
d,
Susan A.
Cumberland
a,
Joanna C.
Renshaw
a,
Matthew J.
Baker
e,
Paul R.
Edwards
f,
Cerys
Jenkins
g and
Andrea
Hamilton
*a
*a,
Pieter
Bots
*a,
Alan
Richardson
b,
Paul A.
Bingham
c,
Alex
Scrimshire
c,
Andrew
Brown
d,
Mark
S’Ari
d,
John
Harrington
d,
Susan A.
Cumberland
a,
Joanna C.
Renshaw
a,
Matthew J.
Baker
e,
Paul R.
Edwards
f,
Cerys
Jenkins
g and
Andrea
Hamilton
*a
aDepartment of Civil and Environmental Engineering, University of Strathclyde, Glasgow, G1 1XJ, UK. E-mail: andrea.hamilton@strath.ac.uk; ronald.turner@strath.ac.uk; pieter.bots@strath.ac.uk
bDepartment of Mechanical and Construction Engineering, University of Northumbria, Newcastle Upon Tyne, NE1 8ST, UK
cDepartment of Engineering and Mathematics, Sheffield Hallam University, Sheffield, S1 1WB, UK
dSchool of Chemical and Process Engineering, University of Leeds, Leeds, LS2 9JT, UK
eDepartment of Pure and Applied Chemistry, University of Strathclyde, Glasgow, G1 1BX, UK
fDepartment of Physics, SUPA, University of Strathclyde, Glasgow, G4 0NG, UK
gDepartment of Physics, Swansea University, Swansea, SA2 8PP, UK
First published on 12th July 2021
Abstract
(Hydroxy)apatite (HAp) [Ca10(PO4)6(OH)2], has emerging potential as a cement coating material, with applications in environmental remediation, nuclear waste storage and architectural preservation. In these low temperature environments and when precipitating from aqueous solution on to a porous substrate, the crystal size, nucleation sites and modified surface properties created are key to designing the most effective coating. In this study we show that bacterial (biogenic) or chemical (abiotic) syntheses on to Portland cement alter these critical performance parameters. We identify that the most significant difference between these two methods is the rate of pH change of the solution during synthesis, as this alters the surface properties and layer structure of HAp formed on cement. We show that iron present in Portland cement is not incorporated into the HAp structure; that formation of nanoparticulate/nanocrystalline HAp begins in the top 20–50 μm of the cement pore structure; and that a slow pH rise in the deposition solution controlled by bacteria metabolic activity leads to a rougher and more hydrophilic HAp coating compared to the abiotic synthesis. The results present the possibility of tailoring the surface topography and hydrophilicity of (hydroxy)apatite coated cement.
Introduction
The generation of hydrophobic surface coatings for concretes has been of significant recent research interest.1–4 There is an increasing need for such barrier coatings due to the corrosive effects of water and chloride ingress on cementitious building materials.4,5 Calcium phosphate phases, in particular (hydroxy)apatite (HAp, Ca10(PO4)6(OH)2), have been proposed as an effective conservation treatment for calcitic building materials such as limestone and marble.6–8 The mechanisms of apatite formation on cement have received little investigation to date, and the applied properties of these apatites remains under studied. The presented work addresses this knowledge gap, through a thorough investigation of two different treatment methods to form apatite (biogenic, and abiotic) with the aim of investigating its formation as a surface treatment, not a mechanical consolidant. Apatite may provide two potential benefits as a surface treatment versus alternative methods of coating or repairing cement.The first potential benefit is that apatite has a lower solubility in the built environment compared to calcite, which could increase the durability of the resulting coating. The apatite produced in this work is closer to the hydroxyapatite end member, based on XRD and FTIR analysis, than either of the chloro- or fluoro-apatite end members and therefore we label it HAp but acknowledge it is not pure stoichiometric HAp. Furthermore, many naturally occurring forms of HAp, such as human tooth enamel,9 and bone10 contain some CO32− incorporated within the structure. If this incorporation occurs within the built environment it would be undesirable for an architectural coating because it may increase HAp solubility,10 however acid resistance testing has shown HAp layers on marble to be an effective protective surface barrier.11 Therefore HAp surface coatings could be a viable treatment strategy in architectural conservation.
A second important benefit of the apatite family mineral phases (Ca10(PO4)6(F, Cl, OH)2) is the ability to incorporate cations and anions into their structure.12,13 HAp has also demonstrated considerable potential as a sorbent in contaminated environments. Biogenically produced, poorly crystalline HAp, is shown to be more effective against remobilisation of radionuclide analogues such as strontium14–16 than commercially produced nanoparticles of HAp, and a better sorbent of Co2+ in saline solutions (90% seawater) than natural zeolite.15 We therefore consider HAp has a potential as a physical barrier to water ingress and a chemical barrier to radionuclide transport, when applied to cementitious substrates.
Portland cement is widely used as a building material and as a waste form for low level radioactive waste and backfill within geodisposal facilities.17–21 Here, we investigate the aqueous solution based formation of HAp on Portland cement22 (CEM II/A-LL) to determine the mechanisms of formation and its surface engineering properties. Pseudomonas fluorescens was selected for use as the HAp-generating strain based on past observations of its capacity to generate calcium phosphate minerals,23 combined with the relative ease with which it can be cultured. Here, we compare HAp formed with and without Pseudomonas fluorescens, to develop a conceptual model of the process, and show that in both cases the deposited layer begins as nanocrystals in pores of the top ∼50 μm of the cement, creating an altered layer. Subsequently, a thin, smooth HAp layer (Ra = 1.67 μm, sessile drop water contact angle = 85°) formed on top of the altered layer under abiotic conditions, and large well faceted HAp crystals (Ra = 8.68 μm, sessile drop water contact angle = 52°) precipitated when formation was aided by microbial metabolic activity.
Materials and methods
Cement substrate preparation
Cement blocks (5 × 2 × 1 cm) were prepared from 'Multicem 32,5R (CEM-II/A-LL)', Hanson Heidelberg cement powder (500 g) with deionised water (200 g) to form a paste. After mixing for 15 minutes at low speed using a rotary mixer, the resulting cement paste was cast into silicone moulds and left to set for 24 hours at 100% RH and 20 °C. Samples were then removed from their moulds and cured in a Ca(OH)2 saturated solution to minimise carbonation. Curing was carried out at 20 °C for at least 28 days. After curing, the weight of each block was recorded (20 g each), then all samples were vacuum packed prior to further treatment to minimise cement carbonation.Phosphate treatments
All cement samples were placed in an autoclaved, phosphate amended lysogeny broth (LB), which consisted of 20 g L−1 LB (Lennox), (Sigma-Aldrich), 9.4 g L−1 KH2PO4 (0.0691 mol L−1, Sigma-Aldrich), 2.2 g L−1 K2HPO4 (0.0126 mol L−1, Fluka). During microbial treatment the cement samples were equilibrated with 100 mL autoclaved phosphate amended LB for 1 day, prior to the addition of a 1 mL inoculum of Pseudomonas fluorescens SBW25 (OD600 = 0.1) grown in regular LB, to induce the formation of HAp. For direct comparison with the biogenic system, abiotic experiments were conducted to understand the role of bacteria in HAp production. During abiotic treatment, HAp formation was induced through the addition of 10 mL 1 M NaOH to 90 mL phosphate-amended LB, instead of adding P. fluorescens. Treatment progress was monitored visually (e.g. the colour of the cement samples) and by measuring the pH of the phosphate amended LB. After ageing for 21 days in the LB the cement samples had changed colour (from dark grey to murky white) and the pH had stabilised in both treatments. At this point the cement samples were removed from the phosphate amended LB, washed in deionised water under mild agitation and vacuum sealed for storage prior to further analysis.To induce HAp formation as a control, without the presence of a cement substrate, 0.1 g Ca(OH)2 was added as a source of Ca ions instead of cement, to 100 mL of phosphate amended LB and autoclaved. All flasks were incubated for 20 days at ambient temperature with continual shaking (Yellowline OS2 shaker, speed setting 2) inside a fume cabinet. After 20 days the precipitate was collected from an aliquot of 10 mL by centrifuging for 20 minutes at 8000 × g and 20 °C. The supernatant was decanted and the collected solid was suspended in 50 mL deionised water to wash the precipitate and re-centrifuged to collect the solid. The control precipitate was dried at 37 °C for 24 hours and stored at ambient temperature in a sealed centrifuge tube prior to further analyses.
Composition and speciation
HAp was removed from the surface of the cement samples using diamond-coated files to produce a fine powder for compositional analysis using ICP-OES (inductively coupled plasma-optical emission spectroscopy), CHN (carbon–hydrogen–nitrogen), TOC (total organic carbon), FT-IR (Fourier transform infra-red spectroscopy) and Mössbauer spectroscopy. All samples were vacuum-packed for storage prior to analysis, to minimise carbonation.To quantify Ca, Si, Fe, and P content, 10 mg of powdered HAp from the cement treatments and the abiotic precipitate were dissolved in 10 mL of concentrated trace-element grade hydrochloric acid (2 mL) and nitric acid (8 mL) via microwave digestion using a MARSXpress microwave digestion system (CEM Corporation). Samples were diluted 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 in ultrapure water prior to analysis using an iCAP 7000 Plus series ICP-OES (Thermo-Fisher). To quantify the total and organic carbon (CHN and TOC respectively) content of the HAp, 5 mg of each sample was mixed with 5 mL sulphurous acid (TOC only). Further sulphurous acid was added until effervescence was observed to stop. Untreated and acidified samples were then freeze-dried and weighed into tin capsules for CHN and TOC analysis respectively, using an Exeter CE440 Elemental Analyser.
:10 in ultrapure water prior to analysis using an iCAP 7000 Plus series ICP-OES (Thermo-Fisher). To quantify the total and organic carbon (CHN and TOC respectively) content of the HAp, 5 mg of each sample was mixed with 5 mL sulphurous acid (TOC only). Further sulphurous acid was added until effervescence was observed to stop. Untreated and acidified samples were then freeze-dried and weighed into tin capsules for CHN and TOC analysis respectively, using an Exeter CE440 Elemental Analyser.
X-ray diffraction (XRD) was carried out using a Bruker D8 Advance diffractometer on intact cement samples and powder from the abiotic precipitate; these samples were washed in deionised water and vacuum sealed for storage prior to analyses. During the XRD analyses, the step size used was 0.01° 2θ, count time was 4 s per step, a Göbel mirror was used on the primary beam path and only the powder sample was rotated. A silicon standard was analysed to determine instrumental peak broadening, using the same instrument settings as the samples. For quantification of the dominant crystalline phases (HAp and calcite), Rietveld refinements were performed using TOPAS (Bruker).
For FT-IR analysis, the powdered samples were transferred onto the diamond ATR element of the Spectrum Two FT-IR spectrometer (PerkinElmer, USA). Background spectra were taken before each measurement and automatically subtracted from the sample spectra using Spectrum (PE) software. Spectral repeats were taken of three sub-samples and averaged to produce the final FT-IR spectra representative of each sample.
57Fe Mössbauer spectroscopy was used to investigate the oxidation state and coordination environment of Fe within surface scrapings from samples of ordinary Portland cement and the abiotic and biogenic treatments. Mössbauer spectra were collected at 293 K using a constant acceleration spectrometer with a 25 mCi 57Co source in a Rh matrix. Absorbers were prepared from ground samples mixed with graphite powder to ensure a Mössbauer thickness t < 1. Spectra were measured in the velocity range ±12 mm s−1 relative to α-Fe and were fitted using the Recoil analysis software package. Two broadened Lorentzian paramagnetic doublets were fitted to the resultant spectra. It was assumed for the purposes of fitting that the recoil-free fraction ratio f(Fe3+)/f(Fe2+) = 1.0. Spectral measurements were carried out for 2 weeks per sample. Data collection was halted after 2 weeks, because beyond 2 weeks an unfeasible (>1 month) additional collection time would be required to produce a statistically significant improvement in spectral signal-to-noise ratio.
Imaging
Samples for electron probe microanalysis (EPMA) were embedded in epoxy resin, sectioned and polished by Petrolab Ltd (Cornwall, UK). The resin-embedded polished cement samples were coated with Ir to improve the electrical conductivity for electron imaging. EPMA analyses were performed using a JEOL JXA-8530F field-emission EPMA with four integrated wavelength-dispersive X-ray (WDX) spectrometers. Beam conditions were 20 kV and 40 nA defocused to a 1 μm spot, with a 50 ms per pixel sampling time and 1 μm per step stage scan.For transmission electron microscopy (TEM), thin lamellae of the samples (approx. 100 nm thick) were prepared via the in situ lift-out method using a FEI Helios G4 CX DualBeam high resolution monochromated, field emission gun, scanning electron microscope (FEG-SEM) with precise focused ion beam (FIB). The samples were initially sputter-coated with a 30 nm thick Pt/Pd layer to make the samples conductive for SEM imaging. After transferring the cement blocks to the dual beam microscope, 500 nm of electron beam platinum (Pt) was deposited (at 5 kV, 6.4 nA for the electron source) on the surface of the target area. This was followed by a second Pt layer (1 μm) using the FIB (at 30 kV, 80 pA for the liquid Ga ion source). A bulk lamella was initially cut (by the FIB at 30 kV, 47 nA), before a final cut-out was performed (at 30 kV, 79 nA). Thinning and polishing of the lamellae to electron translucency was performed with a final polish/clean using a gentle ion beam (5 kV, 41 pA). The lamellae were attached, using Ion Beam Pt, to a Cu FIB lift-out grid (Omniprobe, USA) mounted within the SEM chamber (in situ) and then stored under vacuum before and during transport to and from the TEM. The resulting FIB-SEM lift-outs allow TEM imaging perpendicular to the surface of the cement samples, enabling analysis of the HAp layer and interface at this surface. Bright field TEM, high-angle annular dark-field-scanning transmission electron microscopy (HAADF-STEM) images and energy dispersive X-ray (EDX) spectra and maps were recorded using an FEI Titan(3) Themis G2 STEM operating at 300 kV with an FEI Super-X 4-detector EDX system, a Gatan One-View CCD and a Gatan Quantum 965 ER imaging filter. HAp and cement are both susceptible to alteration under electron irradiation.24 Therefore probe currents and electron doses (dwell times) were limited to ensure that no significant changes in morphology or crystal structure were observed during data acquisition, either imaging or during mapping; the latter for <15 min per region of interest.
Surface properties
The surface topography of the cement substrate and treated samples were mapped using focus-variation microscopy (Alicona 4G infinite focus scanner). Contact angle measurements were made on identical samples, thoroughly rinsed with DI water and dried under a stream of nitrogen gas. Three individual 20 μL droplets of deionised water were spotted onto the surface and images were captured using a Nikon digital camera. Droplet contact angle measurement was performed using Fiji and the LBADSA plugin.25Results and discussion
Impacts of treatment on the composition, speciation and distribution of mineral and cement phases
The evolution of pH in the phosphate amended LB is summarised in Fig. 1. Prior to any amendments, the pH of phosphate amended LB was approximately 6.2. This shows that during the biogenic treatment, pH increased from ∼6.2 to ∼8.5, while the adjusted pH in the abiotic treatment remained stable at ∼9, as shown before in previous work.22 Between the treatments, there are two main differences: (1) a starting pH of 6 or 9 in the biogenic and abiotic treatments, respectively, and (2) the presence or absence of microbial metabolic activity. Preliminary PHREEQC models show that due to the high phosphate buffer capacity (81.7 mM PO43−), the pH should remain stable, even with the dissolution of alkaline phases within the cement samples (e.g. Ca(OH)2). Thus, it is likely that microbial metabolic activity was the cause of pH increase during the biogenic treatment, e.g. from the formation of ammonia as a by-product of microbial proteolysis.26,27 Therefore stable pH during the abiotic treatment is likely due to the absence of microbial metabolic activity and the elevated initial pH.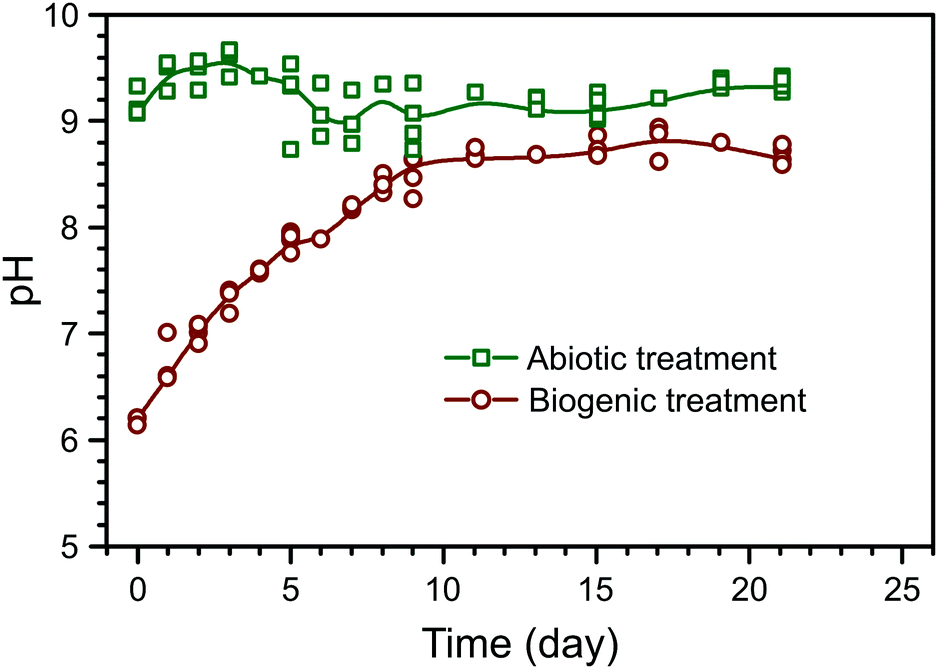 | ||
| Fig. 1 Summary of pH evolution during the abiotic and biogenic treatment of the cement blocks; the coloured lines represent a B-spline through the median values of parallel experiments and are intended as a guide to the eye only. | ||
The composition of the control HAp precipitate and powdered surface scrapings from the cement sample after biogenic and abiotic treatments are summarised in Table 1. In the control HAp precipitate, Ca and P are 48.3 wt% and 21.9 wt% respectively, corresponding to a molar Ca/P ratio of 1.70, which is close to the expected value of 1.67–1.68 from stoichiometric HAp.28,29 Similarly, powdered surface scrapings from the biogenic treatment samples had 37.3 and 16.8 wt% Ca and P respectively, and hence a molar Ca/P ratio of 1.72. The abiotic treatment, surface scrapings had 28.4 and 3.1 wt% Ca and P respectively, which corresponds to a molar Ca/P ratio of 7.13. This is more than 4 times higher compared to the control precipitate and other reported values at 1.38–1.76 for natural, stoichiometric, or carbonated HAp.29–31 However it is clear that HAp does not account for all Ca in the samples and the XRD results show calcite is present in the coated cement samples from both treatments (Fig. 2). In addition, C–S–H is not detected by XRD but is certainly present (EPMA results) and will also contribute to Ca measured by ICP-OES on digested surface scrapings. Si and Fe were detected within the scrapings, also confirming the presence of cement phases (Table 1). Inorganic carbon was found in all samples, ranging from 0.65 wt% in the control HAp precipitate to 5.32 wt% in scrapings from the cement substrate after the abiotic treatment. Finally, within all samples, organic carbon was also detected. Organic carbon is lower in both cement samples compared to the control HAp precipitate. For samples where no microbes have been added, it probably comes from the growth media used.
| Control precipitate (wt%) | Biogenic treatment (wt%) | Abiotic treatment (wt%) | |
|---|---|---|---|
| Ca | 48.3 (+9.58, −10.5) | 37.3 (+6.35, −6.95) | 28.4 (+5.62, −8.28) |
| Fe | 0.04 (+0.03, −0.01) | 1.21 (+0.20, −0.27) | 1.57 (+0.37, −0.49) |
| P | 21.9 (+8.41, −5.29) | 16.8 (+2.15, −3.44) | 3.1 (+0.15, −0.87) |
| Si | 0.07 (+0.01, −0.01) | 1.99 (+ 0.31, −0.07) | 2.03 (+1.59, −0.18) |
| Total carbon | 1.56 (+0.08, −0.09) | 3.68 (+0.38, −0.19) | 5.67 (+0.54, −0.03) |
| Organic carbon | 0.91 (+0.00, −0.01) | 0.50 (+0.24, −0.03) | 0.35 (+0.16, −0.02) |
| Inorganic carbon | 0.65 (+0.08, −0.08) | 3.18 (+0.41, −0.43) | 5.32 (+0.38, −0.01) |
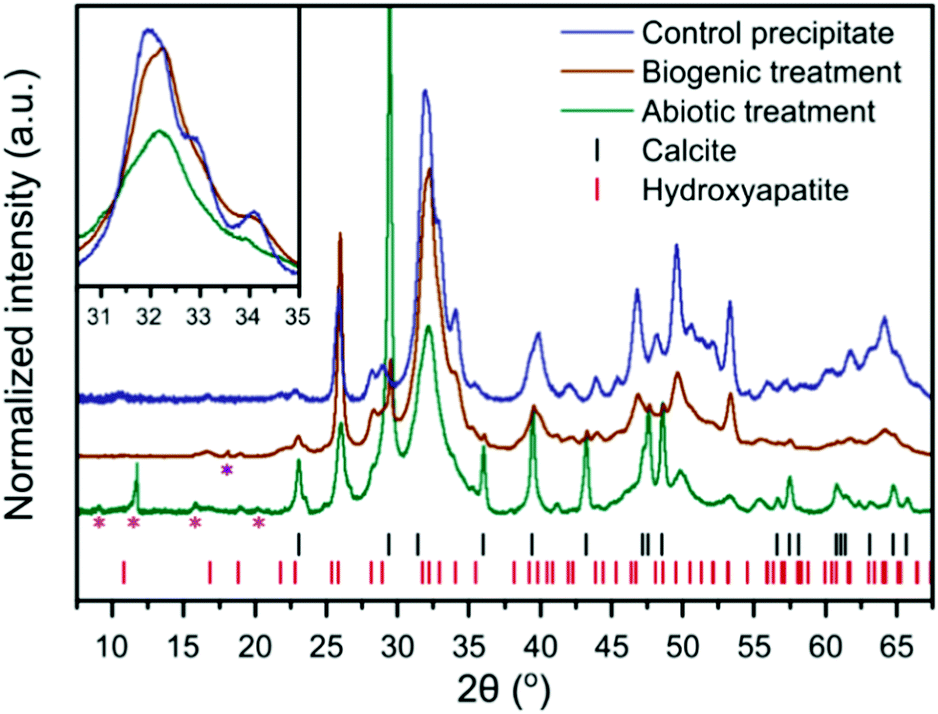 | ||
| Fig. 2 XRD patterns of the control HAp precipitate and the intact surface of the abiotically and biogenically treated cement blocks. Shown are stick plots for the reflection positions of HAp and calcite, in red and black respectively. Magenta asterisks represent minor quantitites of cement phases, ettringite (ICSD 16045)32 and monocarboaluminate (ICSD 59327),33 but there are insufficient isolated reflections to unequivocably confirm identification. The purple asterisk represents a portlandite reflection (ICSD 15471).34 These minor reflections do not interfere with the main HAp and calcite reflections, thus were not included within the refinements to quantify the dominant crystalline phases (i.e. HAp and calcite). Inset is a higher magnification of the region from 30–35° 2θ, without intensity offset to emphasise reflection broadening. | ||
XRD of the control HAp precipitate and both biogenic and abiotic coatings on the intact cement samples after treatment are plotted in Fig. 2. On precipitation from aqueous solution and at pH > 6, octacalcium phosphate (OCP, Ca8H2(PO4)6·5H2O) and dicalcium phosphate dihydrate (DCPD, brushite, CaHPO4·2H2O) are the main other non-apatite crystalline phases which could form.35 OCP has been formed along with HAp on marble.36 In the work presented here, we did not observe the formation of brushite or OCP. All Bragg peaks in the XRD of control HAp precipitate can be assigned to HAp, suggesting that inorganic carbon in that sample (Table 1) may be carbonate incorporated into the HAp structure37,38 rather than precipitated as calcite. In contrast, the analysed cement blocks showed significant calcite reflections, accounting for 24.1 wt% calcite in the sample after abiotic treatment and 3.6 wt% calcite after the biogenic treatment. This demonstrates that the difference in inorganic carbon content (Table 1) of surface scrapings between the 2 coatings was due to differences in the calcite content. This is likely responsible for the higher Ca/P ratios compared to the control precipitate, particularly for the abiotic treated sample which had the highest calcite content. There is a clear difference in the FWHM (Full Width-Half Maximum) of the HAp reflections with the broadest peaks present in the abiotic control. This is most obvious at 30–35° 2θ, where at least three distinct HAp reflections are identifiable in the control HAp precipitate sample, yet appear merged in the abiotic treated cement sample (see inset Fig. 2). This suggests variations in the crystallite size of HAp between the two treatments of the cement samples and the control HAp precipitate, as confirmed later by TEM (Fig. 6).
The FT-IR spectra are plotted in Fig. 3. In these spectra, HAp is identified from the PO43− group vibrations which are dominant at ∼560 and 1020 cm−1 and include bands at 471, (559–561), 601, 962, (1020–1025) and a shoulder at 1087 cm−1.39 Broad water vibrations (3200–3600 cm−1) are also characteristic of HAp.39 CO32− vibrational bands were identified at 873, 1469–1454 and 1420–1417 cm−1, which have been observed previously in FT-IR studies as typical for both carbonated HAp and calcite alone.39,40 These bands are stronger in the abiotic coating, consistent with the higher percentage of calcite detected by XRD. Additional vibrational bands at 1652–1640 cm−1 can be assigned to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and N–H related to organic matter. They show little variation in intensity between samples, confirming that organic carbon is present in all samples, as shown in Table 1.
O and N–H related to organic matter. They show little variation in intensity between samples, confirming that organic carbon is present in all samples, as shown in Table 1.
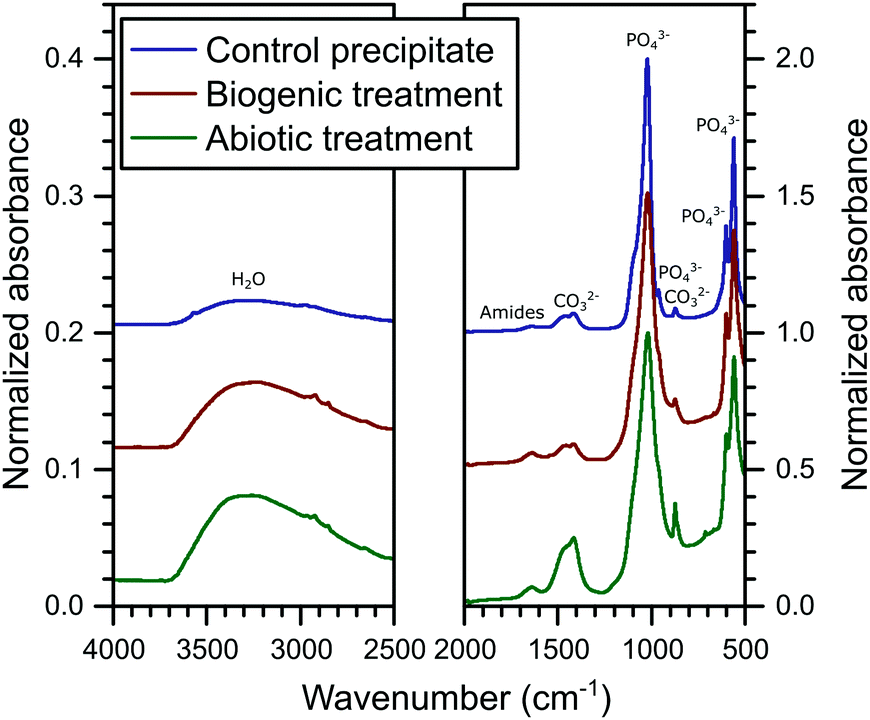 | ||
| Fig. 3 Summary of the FTIR spectra collected on the powders from the control HAp precipitate and the cement samples after the abiotic and biogenic treatments; including annotation of the dominant vibrational bands. | ||
The 57Fe Mössbauer spectra and their respective fits are plotted in Fig. 4. All 57Fe Mössbauer spectra could be fitted with two doublets (D1: centre shift: 0.26–0.42 mm s−1, quadrupole splitting: 0.41–0.49 mm s−1, and D2: centre shift: 0.23–0.45 mm s−1, quadrupole splitting: 1.21–1.61 mm s−1, Table 2). These doublets (within experimental uncertainties and taking into account the low signal-to-noise ratio) indicate the existence of at least two different paramagnetic Fe3+ sites and that all of the observed Fe is present as ferric Fe in all three of the cement samples.41–45 The hyperfine parameters from fitting the Mössbauer spectra associated with the cement samples are all fully consistent with values for cement from Harchand et al. (D1: centre shift: 0.278 mm s−1, quadrupole splitting: 0.428 mm s−1, and D2: centre shift: 0.285 mm s−1, quadrupole splitting: 1.667 mm s−1, Table 2).41 Doublet D2, with the greater quadrupole splitting (QS), is consistent with 6-fold coordinated Fe3+, and doublet D1, with the smaller QS, is consistent with 4-fold coordinated Fe3+.41
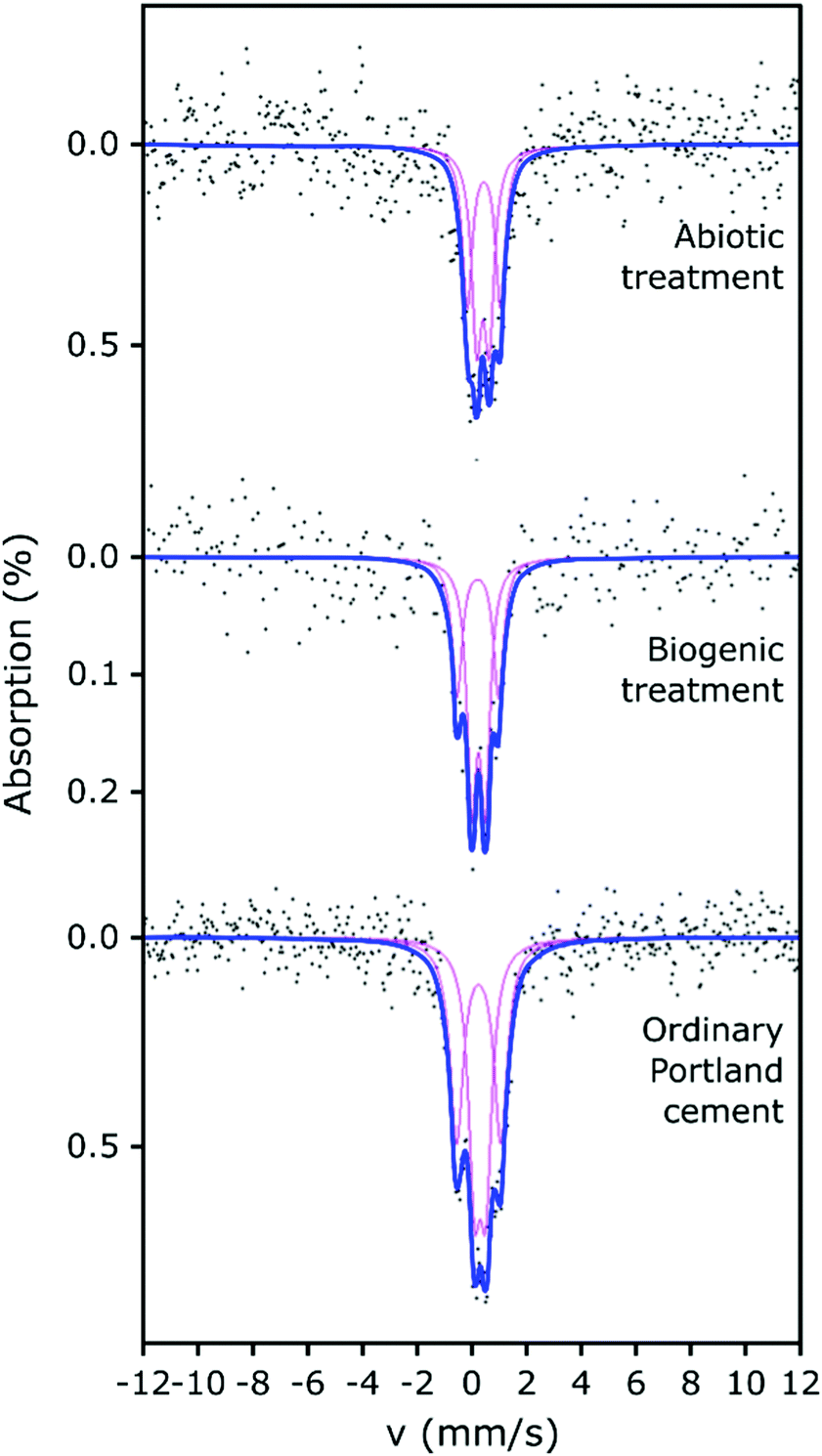 | ||
| Fig. 4 Fitted Mössbauer spectra of abiotic HAp coating on Portland cement (top), biogenic HAp coating on Portland cement (middle), and Portland cement (lower). Points are the collected data, bold blue lines are the overall fit to the data and magenta lines are the fitted doublet components. | ||
| Sample/description | Doublet | Centre shift (mm s−1) | Quadrupole splitting (mm s−1) | HWHM linewidth (mm s−1) | Area (%) |
|---|---|---|---|---|---|
| a Reported linewidths are consistent with FWHM (Full Width-Half Maximum) linewidths, therefore dividing these reported linewidths by a factor of two enables direct comparison with our HWHM linewidths reported here. b No FWHM was reported by Rea et al.48 | |||||
| Hydrated ordinary Portland cement | D1 | 0.32(3) | 0.41(6) | 0.27(8) | 50 |
| D2 | 0.26(4) | 1.61(5) | 0.30(6) | 50 | |
| Biogenic treated cement sample | D1 | 0.26(5) | 0.49(1) | 0.21(1) | 61 |
| D2 | 0.23(1) | 1.52(2) | 0.23(5) | 39 | |
| Abiotic treated cement sample | D1 | 0.42(4) | 0.46(9) | 0.22 | 53 |
| D2 | 0.45(3) | 1.21(1) | 0.22 | 47 | |
| Fe-doped HAp (Jiang et al.)47 | D1 | 0.23 | 1.72 | 0.35a | 6 |
| D2 | 0.22 | 1.14 | 0.40a | 18 | |
| D3 | 0.22 | 0.69 | 0.40a | 18 | |
| Leached ordinary Portland cement paste surface (Faucon et al.)45 | D1 | 0.34 | 1.50 | 0.27 | 16 |
| D2 | 0.39 | 0.86 | 0.23 | 53 | |
| D3 | 0.31 | 0.49 | 0.17 | 31 | |
| Hydrated ordinary Portland cement (Harchand et al.)41 | D1 | 0.278 | 0.428 | 0.495a | 18 |
| D2 | 0.285 | 1.667 | 0.495a | 82 | |
| Ferrihydrite (Rea et al.)48 | D1 | 0.37(1) | 0.60(1) | —b | 27(6) |
| D2 | 0.36(2) | 1.02(2) | —b | 73(6) | |
A significant proportion of the Fe in hydrated OPC resides in the calcium aluminoferrite phases and its hydration products. Multiple researchers41–45 have shown that room-temperature 57Fe Mössbauer spectroscopy of hydrated OPC produces complex spectra with 3 or more components, due to the hydration products, including Fe substituted phases such as ettringite, monosulphoaluminate, C–S–H, hydrogarnet and partly-unresolved magnetic components.43–45 However, given the low signal-to-noise ratio obtained for our Mössbauer spectra, plus the absence of any weakly- (or strongly-) magnetic signals (which would give rise to spectral components at more positive and negative velocities than were observed), fitting with 2 doublets has been considered sufficiently robust to enable any significant differences arising between the samples to be identified.
Ferric iron could be expected to be present in Fe-substituted HAp produced under aerobic conditions.46 However, the fitted parameters for the two doublets from the biogenic treated cement sample (with HAp known to be present) are similar to those obtained for the untreated cement sample and the differences between the two samples could arguably have arisen from uncertainties caused by the limited signal-to-noise ratio. Therefore, it is not possible to demonstrate significant differences between the fitted Mössbauer parameters for the biogenic sample and the untreated cement. However, we note that the fitted parameters (CS, QS) to the spectrum from the cement sample after the biogenic treatment are mostly (3 out of 4 parameters), lower than those for cement. This suggests that the average Fe3+ environments in the biogenic HAp may be slightly less distorted and slightly closer to cubic symmetry than in cement, and/or that small differences arise in the partitioning of Fe between the different hydration products. The spectra of these cement samples vary significantly in terms of their absorption intensities, with the biogenic treated cement sample having lower absorption and therefore lower iron content than the cement sample, which is consistent with the formation of HAp. This indicates that observable Fe was diluted in the prepared surface scrapings from the biogenic treated cement sample due to the formation of HAp.
The abiotic treated cement sample presents a Mössbauer spectrum and fitted parameters that are slightly different to those of the cement and biogenic treated cement spectra (Fig. 4 and Table 2). The CS values for both doublets in the abiotic treated cement sample are higher than for the other two samples, which indicates a slightly lower s-electron density at the Fe nuclei. The QS values are largely similar to the untreated and biogenic treated cement samples, one 4-fold and one 6-fold coordinated ferric Fe site, again consistent with literature.41–45 However, the higher CS values for the abiotic treated cement sample are consistent with the view that the average Fe3+ coordination number is higher than in the other two samples and consequently there may be small differences in the partitioning of iron between the hydrated phases containing Fe.41–45 The overall absorption intensity for the abiotic HAp samples is similar to that of the untreated cement, indicating a higher Fe content than the biogenic HAp sample. This suggests that the HAp layer covering the cement is thinner in the abiotic sample than in the biogenic sample as it is diluting the Fe signal less, which is confirmed by EPMA imaging (as described below, Fig. 5).
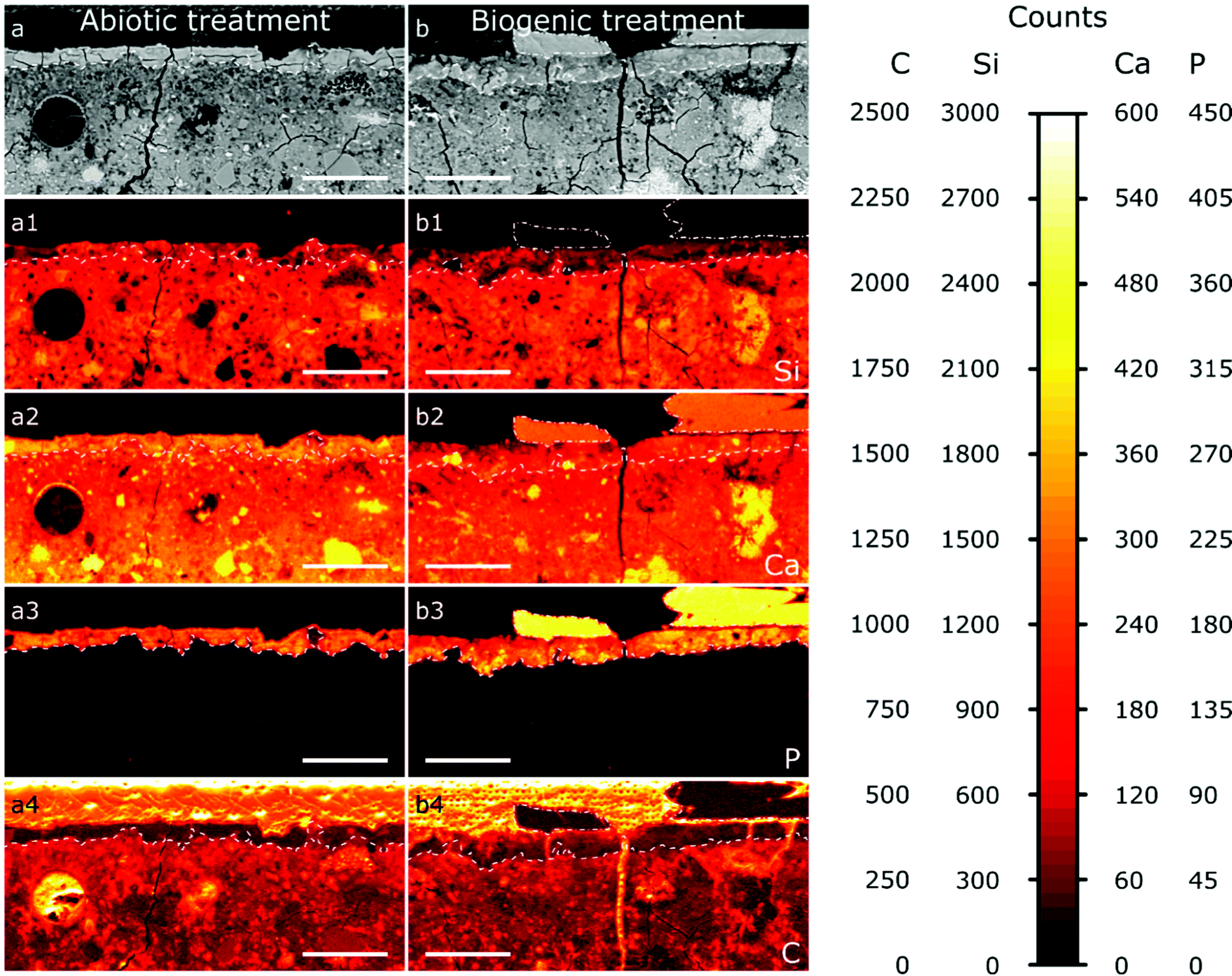 | ||
| Fig. 5 A selection of the EPMA-WDS maps of the abiotic sample (a) and the biogenic sample (b); a and b are the backscatter electron images, (a1–a4 and b1–b4) are the elemental maps (WDS) for Si (a1 and b1), Ca (a2 and b2), P (a3 and b3), and C (a4 and b4). The dashed lines demarcate surface layers within the cement blocks with a high P signal; the dash dotted lines outline large areas on top of the cement layers with a high P and high Ca signal. The scale bar represents 100 μm. | ||
It is important to note that none of the spectra presented support the presence of Fe-doped natural apatite, as reported by Jiang et al.47 Fe in the structure of HAp would be present in highly distorted polyhedra that would generate Mössbauer absorption at approximately −0.65 mm s−1 and 2.8 mm s−1. Rather, the similarities of the fitted doublets (in all analysed samples) with the doublets in hydrated ordinary Portland cement41 and (to a lesser extent) with the doublets in ferrihydrite48 (a hydration product of calcium aluminoferrite) suggests that the formation of HAp during the two treatments did not extensively impact on the original Fe speciation in the cement samples.
The results from EPMA-WDS mapping on polished sections of the treated cement samples embedded in resin are summarised in Fig. 5. In both abiotic and biogenic samples there is a bright relatively homogenous layer within the backscattered electron images at the top 20–50 μm of the cement blocks, identified by dashed lines in the images. This layer is distinct compared to lower into the treated samples, so we assume that this is an altered/new layer that arose due to the respective treatments. This altered layer in the biogenic treated sample is less bright compared to the abiotic treated cement sample, suggesting an overall lower atomic mass compared to the abiotic treated cement sample. On top of this altered layer are (a) large, well faceted crystals, present only in the biogenic treated cement sample (Fig. 5b), shown with a dot-dash outline and (b) a thin, smooth layer, 1–2 μm thick, on the abiotic treated cement sample (Fig. 5a and Fig. S2, ESI†). Deeper into the sample cross section, the (altered) top layer is followed by a heterogeneous dark blotchy layer which brightens at around 100 μm depth into the cement blocks. This indicates that the atomic mass of the mineral phases in the cement block are lower closer to the (altered) top layer than deeper in the cement samples, possibly indicating a more porous region right below the altered layer, with this porosity decreasing around 100 μm into the cement samples. This could be due to either dissolution of cement phases close to the interface or settling of denser phases during sample curing.
The wide distribution of Si in the sample, likely from C–S–H, represents the extent of the original cement block (Fig. 5a1 and b1). It shows that the altered/new layer formed within the pores of the top 20–50 μm of the cement samples. The lower Si counts in this 20–50 μm layer show HAp is intermixed with silicate (cement) phases, confirming pore filling. In both samples the top 20–50 μm layer contains significant amounts of P (Fig. 5a3 and b3), while deeper in the cement samples no P is found; this is consistent with HAp formation in the top, altered layer and not deeper into the sample. Consequently, we do not anticipate any change in the mechanical properties of the entire sample as a result of this coating, further work will investigate the mechanical properties of the HAp filled layer. The large faceted crystals on top of the biogenic sample (Fig. 5b3) have very high P counts, representing HAp, as no other P-rich phases were detected by XRD (Fig. 2). P counts in the faceted crystals on top of the biogenic treated cement sample are higher than in the altered layer, showing a higher wt% of HAp in the large particles compared to the altered layers of the cement samples, as expected.
Ca is distributed throughout the cement samples and within the faceted crystals on top of the biogenic treated cement sample. In the altered layer (dashed line Fig. 5a2 and b2), Ca and Si are elevated in the abiotic sample compared to the biogenic. This suggests more C–S–H present in the altered layer of the abiotic sample than the biogenic sample, corresponding with lower Si in the same layer in the biogenic sample.
Carbon counts are elevated outside of the cement and HAp coating from the resin used to make the polished sections (Fig. 5a4 and b4). Several fractures are visible throughout the samples (Fig. 5a and b). However, these cracks are likely to be perturbations occurring post experiment as a result of sample preparation. Few of these fractures have elevated C counts, while most fractures have low C counts (Fig. 5a4 and b4), suggesting that most fractures formed after embedding the samples in the resin and therefore occur during sample preparation and polishing.
Thin FIB-SEM lift-out cross-sections (c. 100 nm thick) of the top 5–10 μm (representing the top of the altered layers) of the treated cement surfaces were imaged using TEM (Fig. 6), and for comparison a sample of the control HAp precipitate was also imaged. The selected area electron diffraction (SAED) pattern (Fig. 6a1) of the control HAp precipitate matches the plotted standard for HAp,49 confirming its presence in the field of view. Additionally, the control HAp precipitate presents various crystal sizes and morphologies (Fig. 6a and b), possibly representing different stages within the nucleation and growth process.52–55
 | ||
| Fig. 6 Summary of the TEM images on the control precipitate (a and b), and the FIB lift-outs of the abiotic treated cement sample (c to f) and the biogenic treated cement sample (g–k). Control HAp precipitate: (a) overview image of the control HAp precipitate with respective SAED pattern (a1) plotted with a HAp reference reflections of half circles (LHS) where the thickness of the lines are proportional to the intensity of the respective Bragg reflections49 and (b) a HR image of the area demarcated by the black square in a. Abiotic treated cement sample: (c) overview image of the FIB lift-out with (d) higher resolution image on the top 1–2 μm top layer identified in the EPMA-BSE image (Fig. 5a) including a respective SAED pattern (d1) plotted with a HAp reference.49 (e) Higher resolution image of the interface between the top 1–2 μm and the cement sample including a respective SAED pattern (e1) plotted with a HAp reference49 and (f) a higher resolution image of an area with elevated calcium (ESI,† Fig. S3b) including a respective SAED pattern (f1) plotted with a calcite reference.50 Biogenic treated cement sample: (c) overview image of the FIB lift-out with (h) a HR/lattice image of a calcium rich crystal (Fig. S3i (ESI†) with a respective SAED pattern (h1), (i) a higher resolution image of an iron and silicon rich region (Fig. S3l and m, ESI†) with a respective SAED pattern (i1) plotted with a HAp reference49 in white half circles and a 2-line ferrihydrite reference51 in black. (j) A higher resolution image of a calcium and silicon rich region (Fig. S3i and j, ESI†) with a respective SAED pattern (j1) plotted with a HAp reference.49 (k) a higher resolution image of a calcium and phosphorus region (Fig. S3i and j, ESI†) plotted with a respective SAED pattern (k1) plotted with a HAp reference. Solid shapes in the overview images represent the areas where the high(er) resolution images were taken. | ||
In the TEM images of the abiotic treated cement sample (Fig. 6c–f) the 1–2 μm top layer identified in the EPMA images (Fig. 5a, and Fig. S2, ESI†) is confirmed (Fig. 5c and d). The layer is composed of densely packed crystals with a platelet morphology of ∼5 nm thick and ∼0.5 μm long which have high Ca and P contents (Fig. 6c, d and Fig. S3b and c, ESI†). The SAED pattern of this 1–2 μm thick layer with platelet crystals (Fig. 6d1) matches the HAp standard49 but the Bragg diffraction rings are not complete, particularly the 002 reflection, showing some preferred orientation. This 1–2 μm thick layer of densely packed HAp crystals could possibly have prevented resin entering the HAp-cement altered layer below, as observed in the carbon EPMA analyses (Fig. 5a4).
Below the top 1–2 μm thick platy layer (Fig. 6c and e) we see regions with high Si, and with high Ca and P (Fig. S3b, c and e, ESI†). Furthermore, the SAED pattern captured from this area (Fig. 6e1) shows relatively diffuse Bragg circles matching HAp. Combined EDX and SAED shows that nanoparticulate HAp coexists with C–S–H in this region, confirming that HAp is intermixed with cement phases in the altered layer (Fig. 5a). Further down, in the abiotic treated cement sample, an area with a high Ca content was observed (Fig. 6f) with a morphology distinct from the other regions visible in the abiotic treated cement sample (Fig. 6c). Additionally, the SAED pattern of this near single crystal region (Fig. 6f1) shows several Bragg spots which match the position for calcite,56,57 suggesting a relatively large calcite crystal.
The TEM image of the biogenic treated cement sample (Fig. 6g–k) shows a more rugged surface compared to the abiotic sample (Fig. 6g). Additionally, no identifiable layer of oriented HAp platelets, as observed for the abiotic treated cement sample, is detected. The larger crystals on top of the altered layer, detected by EPMA, were not visible in the lift out taken for TEM analysis.
We identify an area near the surface with high calcium (Fig. S3i, ESI†) and lattice fringes (Fig. 6h). These lattice fringes and the respective single crystal SAED pattern (Fig. 6h1) match the crystallographic information of portlandite (Ca(OH)2),58 which was also detected by XRD.
The higher resolution images shown in Fig. 6i, j and k all show no clearly defined crystal morphological features, suggesting that these areas throughout the altered layer (Fig. 6g, i, j and k) consist of amorphous material and/or nanoparticles. However, the composition in these images is distinct from each other (Fig. S3i–n, ESI†). Fig. 6i contains elevated Fe and Si, Fig. 6j elevated Si, and Fig. 6k elevated P. Additionally, the corresponding SAED patterns (Fig. 6i1, j1 and k1) have a broad diffuse scattering band, showing no strongly crystalline structure. For the area with elevated P (Fig. 6k), this diffuse scattering band matches the expected position of the dominant Bragg rings for HAp (Fig. 6k1).49,59 We thus suggest that this area is dominated by a nanocrystalline calcium phosphate phase similar to HAp. The diffuse scattering band in the SAED for the area with high Si (Fig. 6j1), is observed at higher angles compared to what would be expected from HAp. Thus, we tentatively assign this to C–S–H. Finally, the diffuse scattering band for the area with high Fe and Si (Fig. 6i1) appears at a position expected for both 2-line ferrihydrite (a nanocrystalline, iron oxyhydroxide phase) and HAp. Due to the Fe (and Si) content in this area (Fig. 6i and Fig. S3l, m, ESI†) we tentatively assign the dominant phase in this area to ferrihydrite, potentially a silica rich ferrihydrite as identified by Kinsela et al.60 but cannot exclude the presence of another Fe rich poorly crystalline calcium aluminoferrite hydration product such as hydrogarnet. P could also be detected in this region with high Fe and Si (Fig. 6i and Fig. S3j, l, m, ESI†). This could be due to adsorption of phosphate to these iron oxide phases or to the presence of a nanocrystalline calcium phosphate phase similar to HAp.
In summary, a number of HAp morphologies are observed in the abiotic and biogenic treated cement samples. After abiotic treatment, the cement sample has a densely packed 1–2 μm thick layer of platelets (Fig. 6c and d) at the surface, with nanoparticulate HAp intermixed with cement phases immediately below (Fig. 6c and e). In the biogenic treated samples, there are large faceted HAp crystals at the surface (Fig. 5b) with nanoparticulate HAp – cement intermixed below (Fig. 6g and k). Combined, these provide insights into the XRD results (Fig. 2). First, this shows that the XRD patterns (Fig. 2) reveal multiple HAp phases in the altered layer and above. Second, the increase in the FWHM of the Bragg peaks in the XRD patterns (Fig. 2) of HAp in the treated cement samples compared to the control HAp precipitate is due to the presence of nanoparticulate HAp intermixed with the cement phases.
Treatment impact on the cement surface properties
The topography of the HAp coatings and untreated cement samples were examined using focus-variation microscopy, and results are given in Table 3. Ra is a measurement of surface roughness, representing the arithmetical mean height deviation of the surface. Ra was lowest for the abiotic treated cement sample (1.67 μm), compared to the untreated cement sample (4.00 μm) in Table 3, showing that formation of the densely packed HAp platelets reduced surface roughness. The biogenic treated cement sample had the highest Ra at 8.68 μm, due to formation of the large faceted HAp crystals on the surface of the cement during this treatment. Similarly, Rq (root mean squared roughness) is also a measure of surface roughness, however the Rq measurement is more sensitive to the surface maxima and minima. The abiotic treated cement sample was the lowest of all samples measured (2.27 μm) and the biogenic treated cement sample the highest (11.19 μm) while the untreated cement sample was not statistically different to the biogenic treated sample (6.85 μm). This shows that the flattest surface was created by the abiotic treatment, in contrast with the biogenic treatment, which fits with the EPMA images. Measurements of Rsk (skewness) provide a statistical description of the symmetry and shape of features on the cement surfaces, respectively (Table 3).61 The negative Rsk value for the untreated cement sample (−2.98) indicates a surface with more pores (or cracks) than peaks.61Rsk values for the biogenic (0.57) and abiotic treated cement samples (−0.39) were statistically similar but both were distinct from the untreated cement. This demonstrates the effect of both treatments in producing HAp that filled or blocked surface cement pores.28,61 The biogenic treated cement sample had a positive Rsk value, indicating relatively high peaks and shallow valleys61 due to large crystals deposited on the surface. Rsk was close to 0 for the abiotic sample suggesting a relatively flat surface,28 and confirming the smoothing effect of the densely packed HAp platelets on the cement surface.| Sample | R a [μm] | R q [μm] | R sk (−) |
|---|---|---|---|
| The roughness average (Ra), root-mean-squared roughness (Rq), and skewness (Rsk) were measured using focus-variation microscopy. Values are the median of 3 scans of different areas of the same sample. The ±distance from the median is given in brackets. The permuted Brunner–Munzel test was used to pair-wise evaluate the samples for statistical significance (p value < 0.1). Biogenic treated and untreated cement samples that are not statistically different are marked A; similarly, abiotic and biogenic samples that are not statistically different are marked B. | |||
| Biogenic treated cement sample | 8.68 (+0.89, −3.35) | 11.19 (+0.79, −4.16)A | 0.57 (+0.23, −0.17)B |
| Abiotic treated cement sample | 1.67 (+0.12, −0.20) | 2.27 (+0.13, −0.26) | −0.39 (+0.52, −0.36)B |
| Untreated cement | 4.00 (+0.34, −1.11) | 6.85 (+0.40, −2.08)A | −2.98 (+ 0.97, −0.31) |
Hydrophobicity of the coatings was assessed by measuring the contact angle of water droplets on the surface, similar to biologically treated concretes in previous studies.3 These are summarised in Fig. 7. The abiotic treated cement sample had the highest contact angle (∼85°), compared to the biogenic treated sample (∼52°) and the untreated cement sample (∼38° ). This shows that the abiotic treatment had the largest impact on contact angle. As no differences were observed in the total organic carbon in any of the samples, it is likely that the roughness (due to the large faceted HAp crystals) in the biogenic treated cement sample impacted on the contact angle measurements62 but this requires further exploration. However, it should be noted that none of the treatments resulted in a truly hydrophobic surface (>90°).
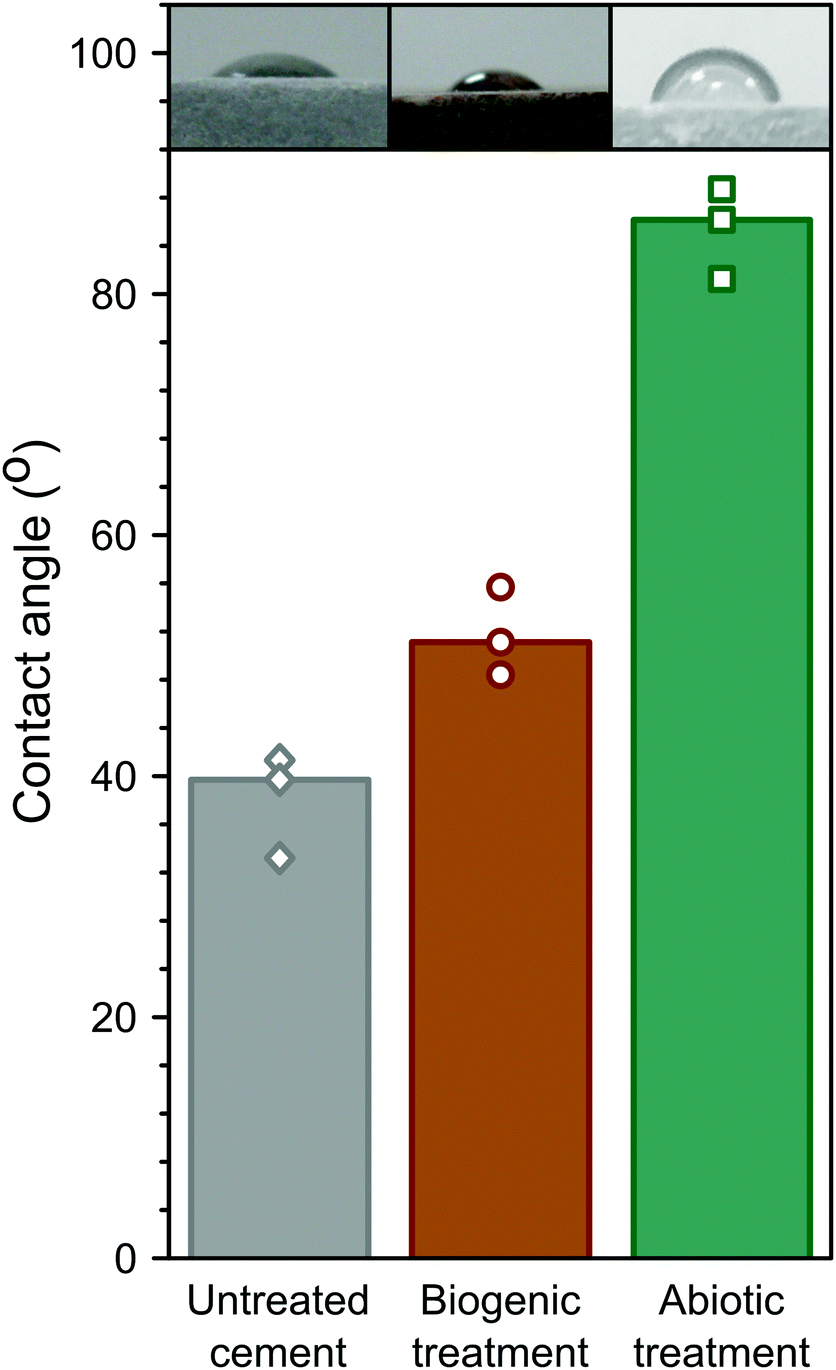 | ||
| Fig. 7 Summary of the contact angle measurements for untreated cement; and biogenic and abiotic treated cement samples. The bars represent the median values and the symbols the measured values. The inset images are examples of those used to measure contact angle. | ||
Conceptual model on the formation of HAp phases during the treatment
Based on pH evolution in the phosphate amended LB (Fig. 1), complementary qualitative PHREEQC modelling63 (Fig. S4, ESI†), mineralogical composition (Fig. 2), chemical composition (Table 1) and imaging analyses (Fig. 5 and 6), we have developed a conceptual model of the treatments. This is visualised in Fig. 8, and provides a schematic representation of the major processes that occurred during the two treatments.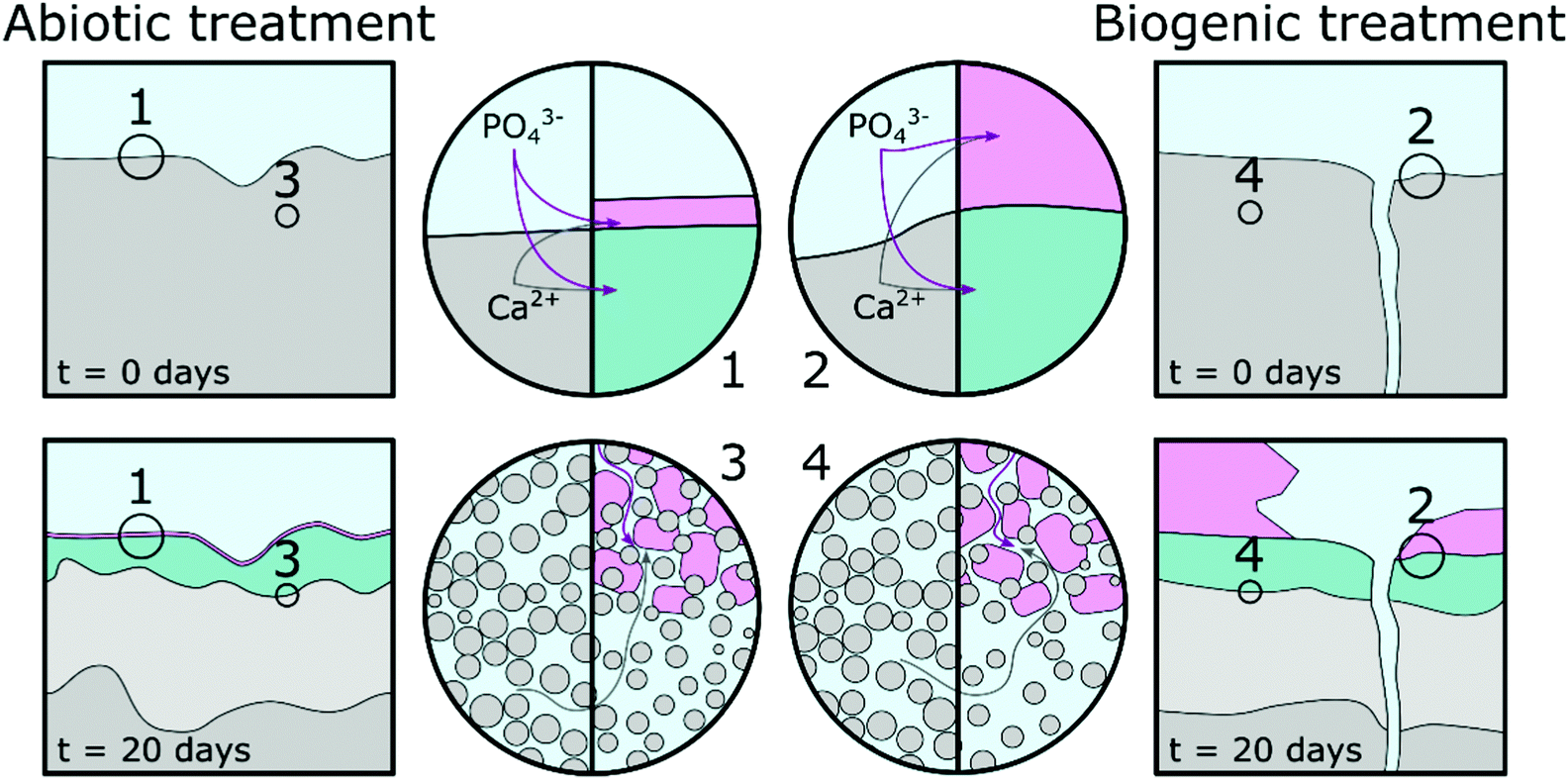 | ||
| Fig. 8 Schematic representation of a proposed conceptual model; grey represents the unaltered cement, light grey represents altered cement where calcium rich phases dissolved, pink represents hydroxyapatite phases and green represents cement phases intermixed with hydroxyapatite phases. | ||
The similarity of the Mössbauer spectra, and their respective fits (Fig. 4 and Table 2), shows that neither of the treatments impacted on the Fe mineralogy in the cement samples.41,64,65 Calcite, detected in the TEM (Fig. 6), XRD (Fig. 2) and EPMA (Fig. 5) results throughout the cement samples, combined with no calcite found in the precipitate control, suggests all calcite originated from the cement powder (CEM II/A-LL) and/or portlandite carbonation.
The main impact of treatment on the top 20–50 μm of the cement substrate is dissolution of Ca-rich cement phases, providing aqueous Ca2+ for HAp formation as indicated by the lower density region in the cement samples (Fig. 5, Fig. 8 highlight 3 and 4, and light grey areas in Fig. 8). The following stages are described: (1) PO43− from phosphate amended LB diffusing into the surface region pores of the cement samples where it precipitates with Ca2+ as a poorly crystalline calcium phosphate phase intermixed with cement phases. From the angle position of the diffuse scattering rings in the SAED image (Fig. 6e1 and k1) we propose that this calcium phosphate phase is similar to a nanocrystalline HAp forming throughout the top 20–50 μm of the cement samples (Fig. 5 and 6, Fig. 8 highlight 3 and 4, and green areas in Fig. 8). Interestingly, qualitative PHREEQC modelling shows that diffusion of LB into cement pore water (Fig. S4e–h, ESI†) has little effect on pH, HAp supersaturation, and therefore the mass of HAp precipitated. This is because only a trace amount of PO43− from the LB is required to produce a supersaturated solution (SI > 0) with respect to HAp in cement equilibrated pore water. While HAp supersaturation from diffusion of cement pore water into the LB (Fig. S4a–d, ESI†) is strongly affected by starting pH. (2) Ca2+ also diffuses into the PO43− amended LB, which induces precipitation of HAp on the surface of the cement samples (Fig. 5, Fig. 8 highlight 1 and 2 and the pink regions in Fig. 8). During biogenic treatment this forms as (isolated) large, well faceted HAp crystals, while in the abiotic treatment this forms as a 1–2 μm thick continuous layer of oriented HAp platelets (Fig. 5 and 6, 8 highlight 1 and 2, and pink areas in Fig. 8). The reasoning for this difference is discussed below.
We found no difference in the quantity of organic carbon in any of the samples, showing that no microbial matter was intermixed with any of the newly formed phases on the cement surfaces. Therefore, we propose that P. fluorescens present in the biogenic treatment only indirectly impacted on differences between the abiotic and biogenic treatments. We suggest this occurred via inducing gradual increase in pH from ∼6 to ∼8.5 (Fig. 1), rather than a constant elevated pH of ∼9. To understand the impacts of pH variation, we performed complementary qualitative PHREEQC modelling (Fig. S4, ESI†)63 on cement pore water by bringing water in equilibrium with portlandite, C–S–H and calcite (the dominant calcium phases present in hydrated CEM II/A-LL) into contact with phosphate amended LB (Fig. S4a–d, ESI†) and vice versa (Fig. S4e–h, ESI†). Results show that at the initial phosphate concentration in solution (81.7 mM PO43−), the pH of the LB should be stable, irrespective of OH− diffusion from cement pore water. This supports the pH measurement shown in Fig. 1 where microbial activity generates a slow pH rise, while during the abiotic treatment the pH is stable. The PHREEQC model shows furthermore that at equal amounts of diffusion from the cement porewater into the phosphate amended LB, at pH 9 the HAp saturation index is about 10 times higher compared to at pH 6 (Fig. S4a–d, ESI†). Diffusion of only trace amounts of Ca2+ (∼0.2 μM) from the cement equilibrated porewater is required in the phosphate amended LB to produce supersaturation with respect to HAp. This rapid supersaturation could then have aided widespread nucleation of HAp at the cement-solution interface during the abiotic treatment, resulting in the formation of a 1–2 μm thick layer of HAp platelets with preferred orientation (Fig. 6c, d, and 8 highlight 1). Furthermore, significantly more Ca2+ is required (0.1 mM) in the phosphate amended LB at pH 6 (during the initial stage of the biogenic treatment) for supersaturation of HAp, compared to at pH 9 in the abiotic treatment (Fig. S4c, ESI†). This suggests that during the initial stages of treatment, Ca2+ could accumulate within the phosphate amended LB until supersaturation was reached, through the continual mixing of pore water Ca2+ into LB, or through the microbially induced increase in the pH. We suggest that this slower process in reaching supersaturation with respect to HAp caused less widespread nucleation of HAp and slower growth of the HAp crystals. This in turn enabled the formation of large well faceted crystals as observed on the surface of the biogenic treated cement sample (Fig. 5b and 8 highlight 2). Finally, the PHREEQC models show that during ingress of the lower pH phosphate amended LB (during the biogenic treatment), additional cement phases dissolved (Fig. S4e and g, ESI†) inducing a higher porosity in the top 20–50 μm of the biogenic compared to the abiotic treated sample (Fig. 5 and 8 highlight 3 and 4). The PHREEQC model suggests that this did not impact on the quantity of HAp formed in the altered layer (Fig. S4g, ESI†); conversely this did likely provide additional Ca2+ to diffuse from the altered layer into the phosphate amended LB for the formation of the large faceted HAp crystals.
Conclusions
Through combined imaging, spectroscopy, diffraction and pH, analyses we have developed a conceptual model of the overall process and mechanisms behind the formation of HAp on Portland cement, and of the observed surface alterations induced through abiotic and biogenic cement treatments. We show that a phosphate amended LB impacts on the cement samples by inducing the formation of distinct calcium phosphate phases: (1) nanocrystalline HAp intermixed with cement phases throughout the top 20–50 μm (for both treatments); (2) larger (individual) crystalline HAp phases (sparsely) covering the surface of the cement samples (for the biogenic treatment); and (3) a thin, smooth layer of crystalline plate-like HAp showing preferred orientation and (nearly) full coverage (for the abiotic treatment). We determined that the presence of P. fluorescens impacted on the crystallinity and morphology of the HAp phases formed indirectly by inducing a gradual pH rise in the LB. This promoted slower growth and led to large crystals deposited on the uppermost surface. Using topography and contact angle measurement, we additionally show that the two different treatments had a significant impact on surface roughness and hydrophobicity. Most notably, the formation of a continuous 1–2 μm thick layer of platelet HAp nanoparticles, led to a smoother surface compared to the untreated cement sample, while the formation of large faceted crystals during the biogenic treatment increased surface roughness. In both cases, Rsk measurements show that the treatments cover the surface. These observations establish a direct link between mechanism of formation and surface properties.The biogenic and abiotic treatment methods described in this manuscript provide new insights into the mechanisms for generating surface coatings with various surface properties (including roughness and hydrophobicity) on cement-based materials. Here, we observed that both treatment methods produced complete coverage of nanoparticulate HAp phases intermixed with cement phases through the top 20–50 μm of the cement samples. Such coatings could be expected to reduce the rate of deterioration in real-world applications. The abiotic treatment produced a smoother, more hydrophobic surface composed of ∼5 nm thick platelets of HAp (Fig. 6d), better suited to protecting building materials from rainfall. This treatment produced particles with a greater surface area (due to the nanoparticulate nature of the HAp platelets, Fig. 6c and d) and may be preferable for applications involving the uptake and/or immobilization of contaminants and fission products.14–16,66 Finally, the properties of both the abiotic and biogenic coatings should be compared against those of previously reported non-biological methods;67 we suggest that based on our presented conceptual model (Fig. 8) the treatment (including application time), and resulting surface properties (e.g. roughness, hydrophobicity; Table 3 and Fig. 7) could be refined for their envisaged final applications.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors gratefully acknowledge the Engineering and Physical Sciences Research Council (UKRI-EPSRC) for funding the research as a Doctoral Training Partnership. We acknowledge support from the Leeds spoke of the Henry Royce Institute, the UK's national body for advanced materials research and innovation, which enabled external access to FIB sample preparation and transmission electron microscopy in LEMAS, Leeds Electron Microscopy And Spectroscopy centre. Thanks to Dr Tanya Peshkur and Mara Knapp for their assistance with the microwave digestion and ICP-OES analyses and Dr Bart Craenen for assistance with statistics.References
- H. S. Wong, R. Barakat, A. Alhilali, M. Saleh and C. R. Cheeseman, Cem. Concr. Res., 2015, 70, 9–20 CrossRef CAS.
- S. Muzenski, I. Flores-Vivian and K. Sobolev, Constr. Build. Mater., 2015, 81, 291–297 CrossRef.
- S. Grumbein, D. Minev, M. Tallawi, K. Boettcher, F. Prade, F. Pfeiffer, C. U. Grosse and O. Lieleg, Adv. Mater., 2016, 8138–8143 CrossRef CAS PubMed.
- D. Barnat-Hunek and P. Smarzewski, Constr. Build. Mater., 2016, 102, 367–377 CrossRef CAS.
- S. Ahmad, Cem. Concr. Compos., 2003, 25, 459–471 CrossRef CAS.
- E. Sassoni and E. Franzoni, Built Herit. 2013 Monit. Conserv. Manag., 2013, pp. 1287–1295.
- E. Sassoni, G. Graziani and E. Franzoni, Constr. Build. Mater., 2016, 102, 931–942 CrossRef CAS.
- E. Sassoni, S. Naidu and G. W. Scherer, J. Cult. Herit., 2011, 12, 346–355 CrossRef.
- J. Reyes-Gasga, E. L. Martinez-Pinerio and É. F. Bres, J. Microsc., 2012, 248, 102–109 CrossRef CAS PubMed.
- H. Pan and B. W. Darvell, Cryst. Growth Des., 2010, 10, 845–850 CrossRef CAS.
- G. Graziani, E. Sassoni, E. Franzoni and G. W. Scherer, Appl. Surf. Sci., 2016, 368, 241–257 CrossRef CAS.
- T. Wen, X. Wu, M. Liu, Z. Xing, X. Wang and A. W. Xu, Dalton Trans., 2014, 43, 7464–7472 RSC.
- S. Yan, D. Yang, S. Chen, J. Wen, W. He, S. Ji, Y. Xia, Y. Wang, L. Zhou and Y. Li, Dalton Trans., 2020, 49, 2578–2588 RSC.
- S. Handley-Sidhu, J. A. Hriljac, M. O. Cuthbert, J. C. Renshaw, R. A. D. Pattrick, J. M. Charnock, B. Stolpe, J. R. Lead, S. Baker and L. E. Macaskie, Environ. Sci. Technol., 2014, 48, 6891–6898 CrossRef CAS PubMed.
- S. Handley-Sidhu, T. K. Mullan, Q. Grail, M. Albadarneh, T. Ohnuki and L. E. MacAskie, Sci. Rep., 2016, 6, 4–11 CrossRef.
- S. Handley-Sidhu, J. C. Renshaw, S. Moriyama, B. Stolpe, C. Mennan, S. Bagheriasl, P. Yong, A. Stamboulis, M. Paterson-Beedle, K. Sasaki, R. A. D. Pattrick, J. R. Lead and L. E. MacAskie, Environ. Sci. Technol., 2011, 45, 6985–6990 CrossRef CAS.
- NDA, Geological Disposal: An Overview of the Generic Disposal System Safety Case, NDA/RWMD/010, 2010.
- J. Wragg, A. E. Milodowski and R. P. Shaw, Br. Geol. Surv. Comm. Rep., 2014, 26 Search PubMed.
- A. J. Williamson, K. Morris, G. T. W. Law, A. Rizoulis, J. M. Charnock and J. R. Lloyd, Environ. Sci. Technol., 2014, 48(22), 13549–13556 CrossRef CAS PubMed.
- P. B. Bamforth, G. M. N. Baston, J. A. Berry, F. P. Glasser, T. G. Heath, C. P. Jackson, D. Savage and S. W. Swanton, Cement Materials for Use as Backfill, Sealing and Structural Materials in Geological Disposal Concepts, A Review of Current Status, Serco Report, SERCO/005125/001 Issue 3, 2012.
- NDA, Geological Disposal: Summary of generic designs, NDA/RWMD/054, 2010.
- R. J. Turner, J. C. Renshaw and A. Hamilton, ACS Appl. Mater. Interfaces, 2017, 9, 31401–31410 CrossRef CAS PubMed.
- M. R. Fishman, K. Giglio, D. Fay and M. J. Filiatrault, Sci. Rep., 2018, 8, 10156 CrossRef PubMed.
- M. Bilton, S. Milne and A. Brown, Open J. Inorg. Non-Met. Mater., 2012, 2, 1–10 CAS.
- A. F. Stalder, T. Melchior, M. Müller, D. Sage, T. Blu and M. Unser, Colloids Surf., A, 2010, 364, 72–81 CrossRef CAS.
- A. J. Richardson, N. McKain and R. J. Wallace, BMC Microbiol., 2013, 13, 1 CrossRef.
- L. Meng, Y. Zhang, H. Liu, S. Zhao, J. Wang and N. Zheng, Front. Microbiol., 2017, 8, 1–7 Search PubMed.
- M. A. Surmeneva, A. Vladescu, R. A. Surmenev, C. M. Pantilimon, M. Braic and C. M. Cotrut, RSC Adv., 2016, 6, 87665–87674 RSC.
- Y. Sekine, R. Motokawa, N. Kozai, T. Ohnuki, D. Matsumura, T. Tsuji, R. Kawasaki and K. Akiyoshi, Sci. Rep., 2017, 7, 1–8 CrossRef CAS PubMed.
- I. R. Gibson and W. Bonfield, J. Biomed. Mater. Res., 2001, 59, 697–708 CrossRef PubMed.
- H. Wang, J.-K. Lee, A. Moursi and J. J. Lannutti, J. Biomed. Mater. Res., Part A, 2003, 67, 599–608 CrossRef PubMed.
- A. E. Moore and H. F. W. Taylor, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1970, 26, 386–393 CrossRef CAS.
- M. François, G. Renaudin and O. Evrard, Acta Crystallogr., Sect. C: Struct. Chem., 1998, 54, 1214–1217 CrossRef.
- H. E. Petch, Acta Crystallogr., 1961, 14, 950–957 CrossRef CAS.
- S. V. Dorozhkin, Biomatter, 2011, 1, 121–164 CrossRef PubMed.
- S. Naidu and G. W. Scherer, J. Colloid Interface Sci., 2014, 435, 128–137 CrossRef CAS PubMed.
- M. E. Fleet, Carbonated Hydroxyapatite: Materials, Synthesis, and Applications, Pan Stanford Publishing, 2014 Search PubMed.
- W. Kurdowski, Cement and Concrete Chemistry, 2014 Search PubMed.
- I. Rehman and W. Bonfield, J. Mater. Sci.: Mater. Med., 1997, 8, 1–4 CrossRef CAS PubMed.
- F. B. Reig, J. V. G. Adelantado and M. C. M. Moya Moreno, Talanta, 2002, 58, 811–821 CrossRef CAS PubMed.
- K. S. Harchand, Vishwamittar and K. Chandra, Cem. Concr. Res., 1980, 10, 243–252 CrossRef CAS.
- J. M. Fortune and J. M. D. Coey, Cem. Concr. Res., 1983, 13, 696–702 CrossRef CAS.
- K. S. Harchand, R. Kumar, K. Chandra and Vishwamittar, Cem. Concr. Res., 1984, 14, 170–176 CrossRef CAS.
- M. Sharma, Vishwamittar, K. S. Harchand and D. Raj, Cem. Concr. Res., 1991, 21, 484–488 CrossRef CAS.
- P. Faucon, P. Bonville, F. Adenott, J. F. Jacquinot and J. Virlett, Adv. Cem. Res., 1997, 9, 99–104 CrossRef CAS.
- Y. Li, J. Widodo, S. Lim and C. P. Ooi, J. Mater. Sci., 2012, 47, 754–763 CrossRef CAS.
- M. Jiang, J. Terra, A. M. Rossi, M. A. Morales, E. M. Baggio Saitovitch and D. E. Ellis, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 66, 224107 CrossRef.
- B. A. Rea, J. A. Davis and G. A. Waychunas, Clays Clay Miner., 1994, 42, 23–34 CrossRef CAS.
- A. T. Sänger and W. Kuhs, Z. Kristallogr., 1992, 199, 123–148 CrossRef.
- S. Markgraf and R. Reeder, Am. Mineral., 1985, 70, 590–600 CAS.
- F. M. Michel, L. Ehm, S. M. Antao, P. L. Lee, P. J. Chupas, G. Liu, D. R. Strongin, M. A. A. Schoonen, B. L. Phillips and J. B. Parise, Science, 2007, 316, 1726–1729 CrossRef CAS PubMed.
- M. Yu, L. Wang, W. Zhang and B. Ganss, ACS Appl. Nano Mater., 2019, 2, 7566–7576 CrossRef CAS.
- D. V. Krogstad, D. Wang and S. Lin-Gibson, Biomacromolecules, 2017, 18, 3106–3113 CrossRef CAS PubMed.
- S. Yang, J. Chen, Z. Wang, H. Zhang and Q. Zhang, Mater. Lett., 2013, 96, 177–180 CrossRef CAS.
- A. E. S. Van Driessche, L. G. Benning, J. D. Rodriguez-Blanco, M. Ossorio, P. Bots and J. M. García-Ruiz, Science, 2012, 336, 69 LP–72 LP CrossRef PubMed.
- J. Jiang, Q. Zheng, D. Hou, Y. Yan, H. Chen, W. She, S. Wu, D. Guo and W. Sun, Phys. Chem. Chem. Phys., 2018, 20, 14174–14181 RSC.
- K. Sato, Y. Kumagai, T. Kogure, K. Watari and J. Tanaka, J. Ceram. Soc. Jpn., 2006, 114, 754–759 CrossRef CAS.
- R. Iizuka, T. Yagi, K. Komatsu, H. Gotou, T. Tsuchiya, K. Kusaba and H. Kagi, Am. Mineral., 2013, 98, 1421–1428 CrossRef CAS.
- J. M. Hughes, M. Cameron and K. D. Crowley, Am. Mineral., 1989, 74, 870–876 CAS.
- A. S. Kinsela, A. M. Jones, M. W. Bligh, A. N. Pham, R. N. Collins, J. J. Harrison, K. L. Wilsher, T. E. Payne and T. D. Waite, Environ. Sci. Technol., 2016, 50, 11663–11671 CrossRef CAS PubMed.
- A. Mendoza-Arnau, M. F. Vallecillo-Capilla, M. A. Cabrerizo-Vilchez and J. I. Rosales-Leal, Med. Oral. Patol. Oral. Cir. Bucal, 2016, 21, e631–e636 CAS.
- Y. Abe, Y. Okazaki, K. Hiasa, K. Yasuda, K. Nogami, W. Mizumachi and I. Hirata, Biomed. Res. Int., 2013, 1–9, DOI:10.1155/2013/626452.
- D. L. Parkhurst and C. A. J. Appelo, U.S. Geological Survey Techniques and Methods, Book 6, chap. A43, 2013, p. 497 Search PubMed.
- A. Crumbie, G. Walenta and T. Füllmann, Cem. Concr. Res., 2006, 36, 1542–1547 CrossRef CAS.
- F. Guirado and S. Galí, Cem. Concr. Res., 2006, 36, 2021–2032 CrossRef CAS.
- K. Sasaki and T. Goto, Ceram. Int., 2014, 40, 11649–11656 CrossRef CAS.
- E. Sassoni and E. Franzoni, Constr. Build. Mater., 2020, 245, 118409 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ma00320h |
| This journal is © The Royal Society of Chemistry 2021 |