
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA stable ring-expanded NHC-supported copper boryl and its reactivity towards heterocumulenes†
Thomas M.
Horsley Downie
 ,
Rex S. C.
Charman
,
Jonathan W.
Hall
,
Mary F.
Mahon
,
John P.
Lowe
and
David J.
Liptrot
*
,
Rex S. C.
Charman
,
Jonathan W.
Hall
,
Mary F.
Mahon
,
John P.
Lowe
and
David J.
Liptrot
*
Department of Chemistry, University of Bath. Claverton Down, Bath, BA2 7AY, UK. E-mail: dl260@bath.ac.uk
First published on 26th October 2021
Abstract
Reaction of bis(pinacolato)diboron with (6-Dipp)CuOtBu generates a ring-expanded N-heterocyclic carbene supported copper(I) boryl, (6-Dipp)CuBpin. This compound showed remarkable stability and was characterised by NMR spectroscopy and X-ray crystallography. (6-Dipp)CuBpin readily dechalcogenated a range of heterocumulenes such as CO2, isocyanates and isothiocyanates to yield (6-Dipp)CuXBpin (X = O, S). In the case of CO2 catalytic reduction to CO is viable in the presence of excess bis(pinacolato)diboron. In contrast, in the case of iso(thio)cyanates, the isocyanide byproduct of dechalcogenation reacted with (6-Dipp)CuBpin to generate a copper(I) borylimidinate, (6-Dipp)CuC(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NR)Bpin, which went on to react with heterocumulenes. This off-cycle reactivity gives selective access to a range of novel boron-containing heterocycles bonded to copper, but precludes catalytic reactivity.
NR)Bpin, which went on to react with heterocumulenes. This off-cycle reactivity gives selective access to a range of novel boron-containing heterocycles bonded to copper, but precludes catalytic reactivity.
Nucleophilic boron fragments are increasingly important “umpolong” reagents applied in the generation of synthetically useful boron-element bonds.1–8 Copper(I) boryls are, by far, their pre-eminent class, emerging from chemical curiosities to become important synthetic intermediates in the generation of a huge number of new, boron-containing organic molecules.9–15 Synthetic access to copper boryls has principally relied upon two synthetic routes; salt metathesis reactions of isolable boryl anions with copper halides16–18 and Cu–O/B–B σ-bond metathesis. Whilst the former route is limited by the extremely small number of R2BM (M = Li, Na, K) fragments thus reported, the latter has emerged as a generalisable route to copper boryl systems under both stoichiometric and catalytic regimes.13,14
Despite this significant interest, well-defined compounds containing explicit Cu–BR2 bonds are still sparse.9,12,19–23 Although the first of these species to be crystallographically characterised, two coordinate copper(I) boryls are an even more rarefied sub-class of these molecules. In 2006 Sadighi reported the synthesis of (IPr)CuBpin (IPr = 1,3-bis-(2,6-diisopropylphenyl)-imidazol-2-ylidene; pin = (OCMe2)2) from (IPr)CuOtBu and pin2B2.9 This has been followed by a small number of more recent reports of related monomeric (NHC)CuBR2 species.22–24 (IPr)CuBpin was shown to readily reduce CO2 to CO, with concomitant formation of (IPr)CuOBpin. This species could then be converted back to (IPr)CuBpin via its reaction with bis(pinacolato)diboron, resulting in a net catalytic deoxygenation of CO2 to carbon monoxide and (pinB)2O.9,25 This remarkable transformation belongs to the broader class of dechalcogenation of heterocumulenes which, under homogeneous conditions, was previously restricted to the stoichiometric application of a limited number of reactive phosphorus compounds and electrophilic metals.26–30
The breakdown pathways of copper boryls to copper clusters and boryl radicals have been intensively investigated, and these studies imply that the Cu–B bond is inherently thermolabile.21,22 Nevertheless, the steric demands of the ancillary ligands both bonded to copper and around the boron centre have been shown to significantly influence the stability of these bonds. For example, having found its IPr analogue to be unstable, Kleeberg and co-workers were able to synthesise (IDipp*)CuBneop (neop = (OCH2)2CMe2), supported by the extremely bulky 5-membered NHC, IDipp* (1,3-bis(2,6-(diphenylmethyl)-4-methylphenyl)imidazol-2-ylidene).31 Ring-expanded NHCs are a class of ligands that have shown enhanced steric demand relative to their 5-membered analogues, with a consequent ability to enhance the stability of ligated fragments and isolate otherwise inaccessible reaction intermediates.32–34 To our knowledge, no such exploration of RE-NHC supported copper boryls has yet been described.
Herein we report the synthesis of a copper boryl supported by a RE-NHC, 6-Dipp (Scheme 1). This compound shows significantly enhanced stability relative to its 5-membered NHC supported analogue, but shares a similar reactivity profile; it deoxygenates CO2 to generate CO. This reaction can be also rendered catalytic in the presence of an excess of diboron via a Cu–O/B–B σ-bond metathesis. Whilst stoichiometric CX bond cleavage reactivity can be extended to other heterocumulenes such as isocyanates, isothiocyanates and carbodiimides, catalytic turnover for these systems was not observed. The high stability imparted by the 6-Dipp ligand facilitates isolation of reaction intermediates and off-cycle products which provide insight into these distinct sets of reactivity.
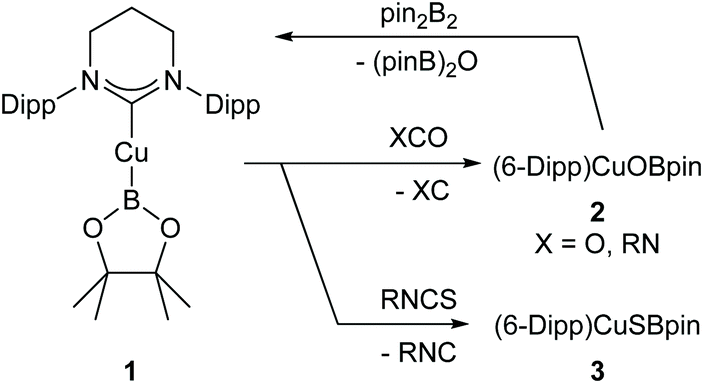 | ||
| Scheme 1 Dechalcogenation of heterocumulenes by (6-Dipp)CuBpin. | ||
Synthesis of (6-Dipp)CuBpin, 1, and its reduction of CO2
In an initial reaction, (6-Dipp)CuOtBu was added to an equimolar amount of bis(pinacolato)diboron in C6D6 (see Fig. S1 and S2†), with a number of new resonances observable after 30 minutes. After 18 hours, growth of these resonances in the 1H NMR spectrum implied the formation of a single 6-Dipp supported species and in the 11B NMR spectrum showed a sharp singlet at 21.4 ppm attributed to pinBOtBu, alongside a broad resonance centred on 41.2 ppm (FWHM = 700 Hz), similar to the 11B peak observed for (IPr)CuBpin (41.7 ppm). Repetition of this reaction in toluene, followed by diffusion of hexane into the resultant solution provided material suitable for single crystal X-ray crystallography which confirmed the formation of (6-Dipp)CuBpin, 1 (Fig. 1).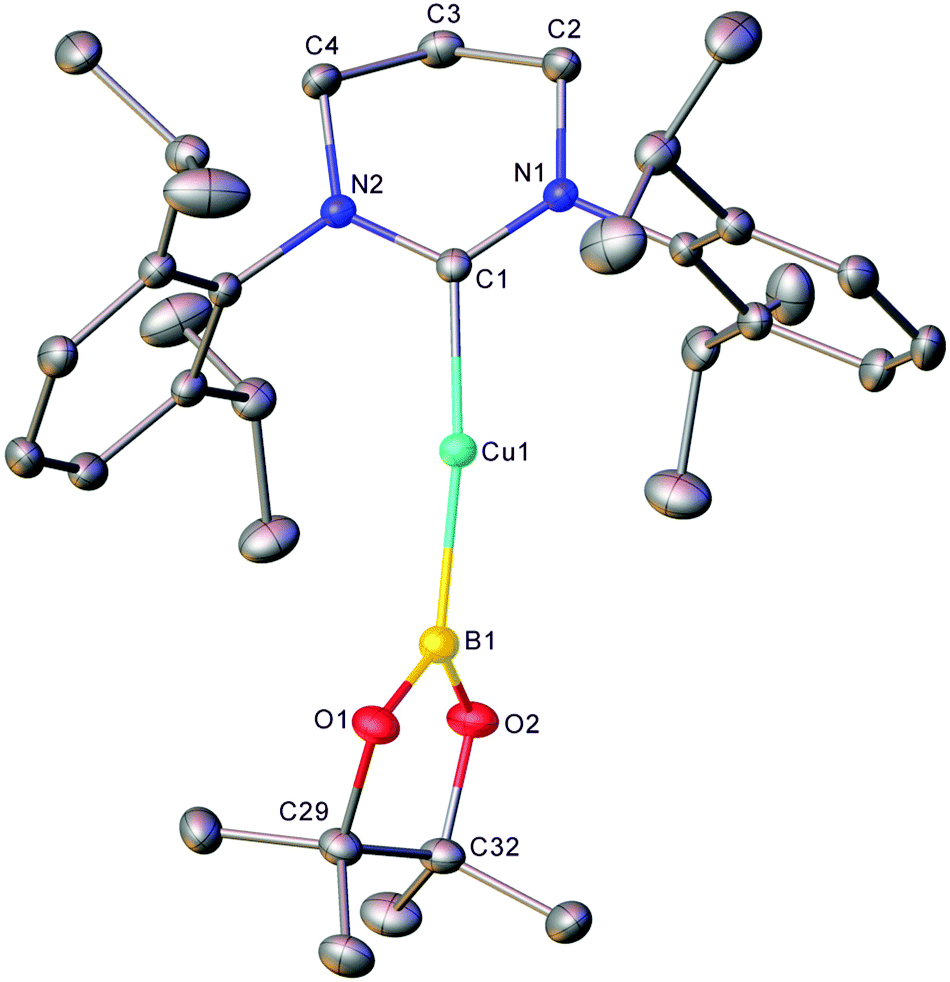 | ||
| Fig. 1 Molecular structure (30% probability ellipsoids) of compound 1. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (°) C1–Cu1 1.9587(12); Cu1–B1 2.0017(15); B1–O1 1.3977(18); B1–O2 1.3928(18); C1–Cu1–B1 164.24(6); Cu1–B1–O1 129.85(11); Cu1–B1–O2 121.08(10); O1–B1–O2 108.32(11); ∠(N,C,N,Cu)/(O,B,O,Cu) 70.41(5). | ||
Compound 1 could be synthesised on a preparative scale using the methodology described for (IPr)CuBpin, and isolated as a brown powder. Unlike its 5-membered analogue, C6D6 solutions of which decomposed to elemental copper and unidentified byproducts within days at room temperature,9 (6-Dipp)CuBpin appears to be essentially indefinitely stable in both the solid- and solution state. A C6D6 solution of this compound evidenced only very limited decomposition over the course of weeks, as assessed by 11B NMR spectroscopy (see Fig. S3 and S4†).
The metric parameters for the C1–Cu1 (1.9587(12) Å) and Cu1–B1 (2.0017(15) Å) distance, as well as the C1–Cu1–B1 angle (164.24(6)°) (Fig. 1) are commensurate with those previously reported for two-coordinate NHC copper(I) boryls: (IPr)CuBpin9 C–Cu 1.937(2) Å, Cu–B 2.002(3) Å, C–Cu–B 168.1(2)°; and (IDipp*)CuBneop31 C–Cu 1.935(2) Å, Cu–B 2.020(2) Å, C–Cu–B 177.36(8)°. The interplanar angle at copper ∠(N,C,N,Cu)/(O,B,O,Cu) of 70.41° is much closer to that of (IDipp*)CuBneop (82.6°) which showed a similarly enhanced stability profile relative to (IPr)CuBpin (40.0°), reflecting the consequences of enhanced steric protection offered by the IDipp* and 6-Dipp ligands.
The high stability of 1 facilitates the exploration of its chemistry by allowing well-defined stoichiometric investigations of its reactions with various substrates (vide infra). It may, however, have profound effects on its reactivity by decreasing access to the copper centre relative to less sterically encumbered complexes. In order to compare it to (IPr)CuBpin, a C6D6 solution of pin2B2, in the presence of 10 mol% (6-Dipp)CuOtBu, was exposed to an atmosphere of 13CO2. NMR spectroscopic analysis indicated catalytic deoxygenation analogous to that reported by Sadighi and co-workers for (IPr)CuOtBu.9 Over the course of several days, the peak associated with pin2B2 in the 11B NMR spectrum attenuated, with a concurrent growth of the peak attributed to (pinB)2O (see Fig. S5†). Interrogation of the 13C NMR spectrum indicated the formation of a resonance at 184.8 ppm attributed to 13CO (see Fig. S6†). Complete consumption of pin2B2 occurred after 96 hours of reaction at room temperature, a significant decrease in rate relative to (IPr)CuOtBu where 2 mol% of the catalyst completed the same transformation in 20 hours. This diminished rate could originate from variations in the experimental procedure (solvent, concentration, pressure) but is more likely to reflect the increased steric demand of the 6-Dipp ligand versus its 5-membered cousin, IPr.
Dechalcogenation reactivity of (6-Dipp)CuBpin, 1, towards other heterocumulenes
The generality of this transformation to other heterocumulenes was then explored through the reaction of 1 with isocyanates and isothiocyanates. Initially, 1 generated in situ from (6-Dipp)CuOtBu and pin2B2 in C6D6 was exposed to one equivalent of isopropyl isocyanate (see Fig. S7 and S8†). After mixing, the 11B NMR no longer contained the resonance associated with 1, but direct observation of resonances associated with a new B–O containing species were hindered by the residual pinBOtBu present in the reaction mixture. Overnight, colourless crystalline material precipitated from the reaction mixture and heating of the resultant suspension followed by slow cooling yielded colourless blocks which, via single crystal X-ray crystallography, were found to be (6-Dipp)CuOBpin, 2 (Fig. 2). The bond lengths and angles in 2 are similar to those reported for (IPr)CuOBpin9 and the 11B NMR spectroscopic resonance of 21.2 ppm (FWHM = 650 Hz) also corresponds closely.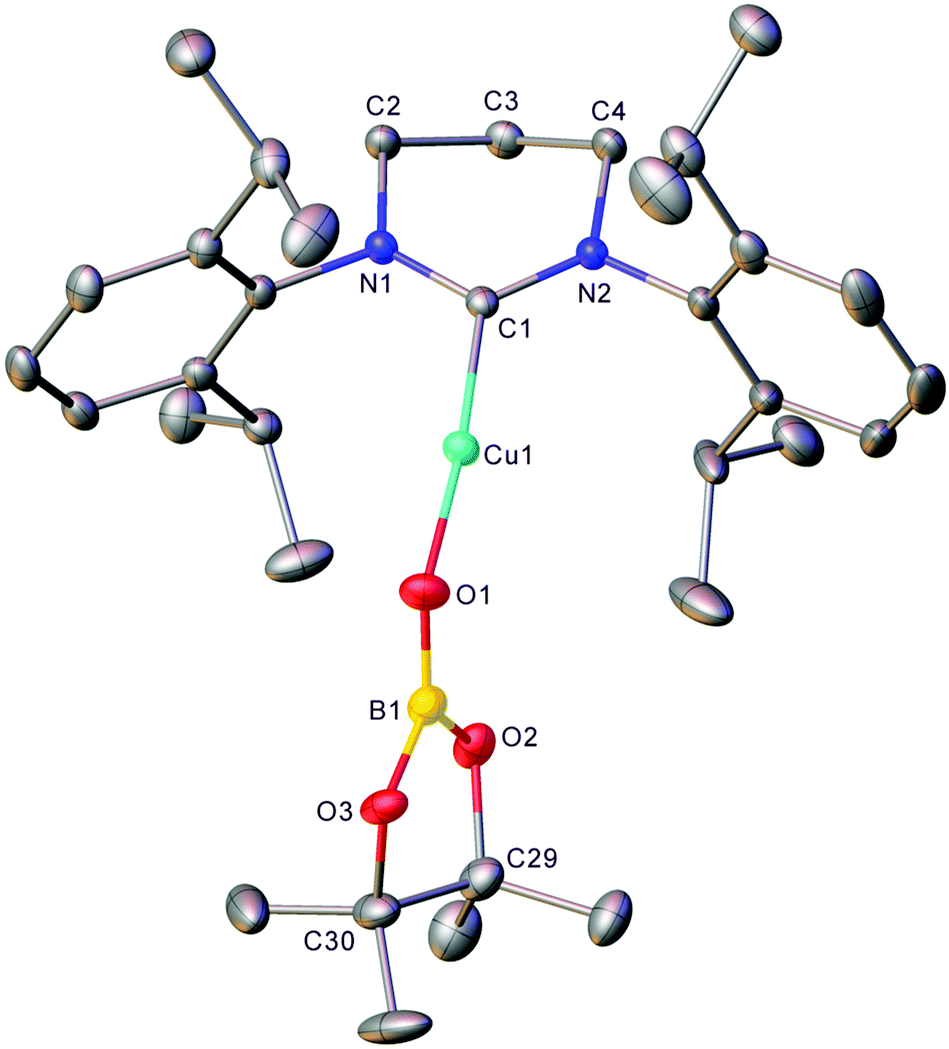 | ||
| Fig. 2 Molecular structure (30% probability ellipsoids) of compound 2. Hydrogen atoms and the minor disordered component are omitted for clarity. Selected bond lengths (Å) and angles (°) C1–Cu1 1.8834(14); Cu1–O1 1.8320(11); O1–B1 1.308(2); B1–O2 1.416(2); B1–O3 1.394(2); C1–Cu1–O1 173.87(6); Cu1–O1–B1 128.24(11); O1–B1–O2 127.08(16); O1–B1–O3 123.16(16); O2–B1–O3 109.74(17). | ||
Extension of this chemistry to isothiocyanates proceeded in a similar fashion; 1, generated in situ as above, was reacted with one equivalent of tBuNCS (see Fig. S9 and S10†). The 11B NMR spectrum of this reaction mixture contained a new resonance at ca. 34 ppm, as well as residual pinBOtBu. Overnight, colourless material precipitated from this solution which was found to be suitable for single-crystal X-ray crystallography. The results of this analysis indicate that dechalcogenation can be extended to isothiocyanates to yield 3, (6-Dipp)CuSBpin (Fig. 3).
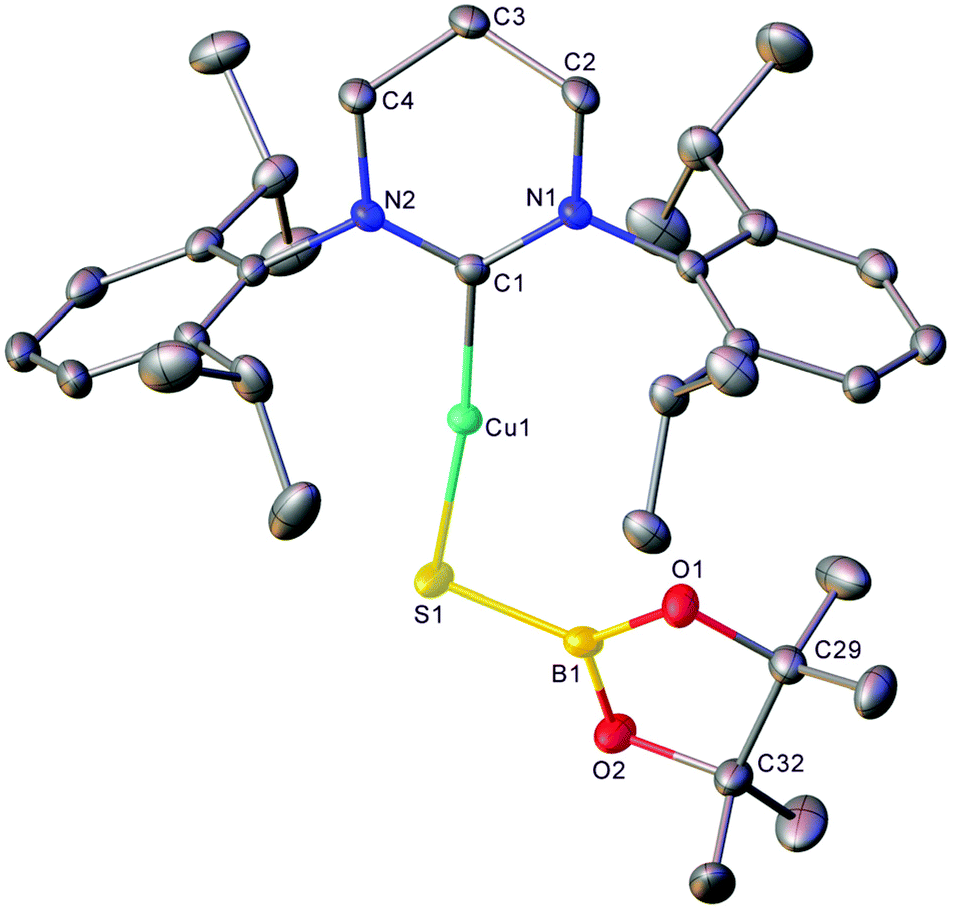 | ||
| Fig. 3 Molecular structure (30% probability ellipsoids) of compound 3. Solvent and hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (°) C1–Cu1 1.9038(17); Cu1–S1 2.1426(5); S1–B1 1.788(2); B1–O1 1.373(2); B1–O2 1.381(3); C1–Cu1–S1 172.81(5); Cu1–S1–B1 101.43(7); S1–B1–O1 125.45(16); S1–B1–O2 123.06(15); O1–B1–O2 111.48(18). | ||
Compound 3 is one of only a limited number of structurally characterised borylthiolates, only two of which contain a terminal SBpin moiety and which display similar S–B bond lengths and M–S–B angles.29,30 Its 11B NMR spectrum contains a single resonance at 34.3 ppm (FWHM = 220 Hz), which is comparable to other SBpin-containing compounds30,35–37
The formation of (6-Dipp)CuXBpin (X = O, 2; S, 3) hinted at the potential of (6-Dipp)CuBpin to dechalcogenate a wide range of heterocumulenes to yield isocyanides, although attempts to confirm the presence of these side products via NMR spectroscopy and mass spectrometry proved fruitless. Nevertheless, it was hoped that 2 and 3 would react with pin2B2 to reform 1 and facilitate such reactivity under catalytic conditions. The reaction of either tBuNCO or tBuNCS with an equimolar amount of bis(pinacolato)diboron in the presence of 10 mol% of (6-Dipp)CuOtBu, however, provided no evidence of catalytic activity. During many of the reactions between isocyanates and 1, resonances around 10–5 ppm in the 11B NMR spectrum were observed. It was proposed that such peaks might represent off-cycle copper containing species which were precluding catalytic turnover.
Stoichiometric mechanistic investigations into heterocumulene dechalcogenation by (6-Dipp)CuBpin, 1
In order to gain insight into the identity of these species, reactions between isolated 1 and phenyl isocyanate were investigated whilst varying the stoichiometry of the reactants.Reaction of 1 with two equivalents of PhNCO in toluene resulted in the precipitation of material overnight which was analysed by X-ray crystallography (Fig. 4).
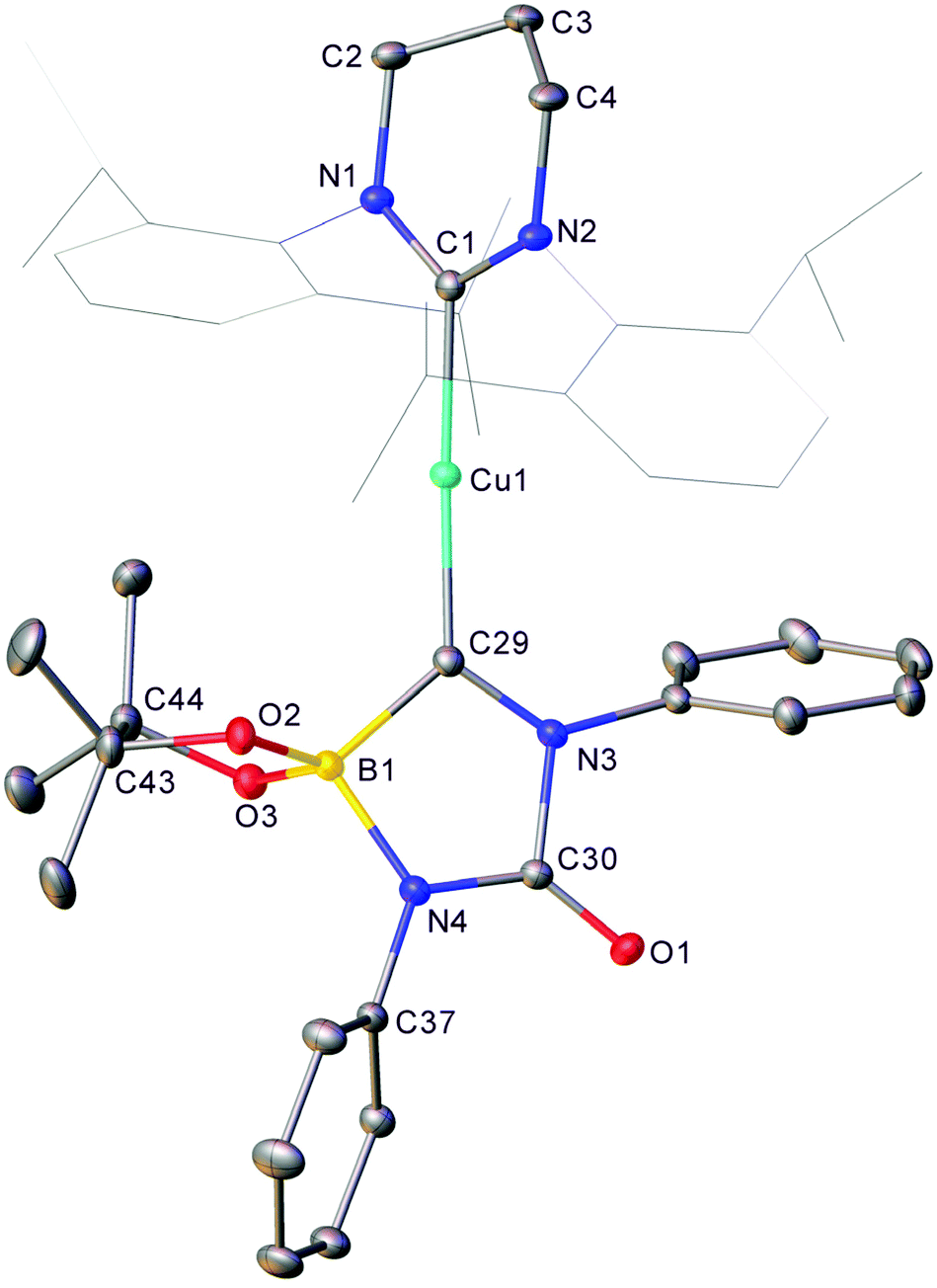 | ||
| Fig. 4 Molecular structure (30% probability ellipsoids) of compound 4. Solvent and hydrogen atoms are omitted for clarity. Wireframe view has been employed for Dipp groups, also for visual simplicity. Selected bond lengths (Å) and angles (°) C1–Cu1 1.9530(13); Cu1–C29 1.9163(13); C29–N3 1.3130(17); N3–C30 1.4912(15); C30–N4 1.3238(17); N4–B1 1.5772(17); B1–C29 1.6693(18); C30–O1 1.2124(16); B1–C29–N3 104.86(10); C29–N3–C30 115.84(10); N3–C30–N4 107.03(10); C30–N4–B1 112.46(10); N4–B1–C29 98.10(10). | ||
The product of this reaction, 4, comprises a 6-Dipp supported copper fragment bonded to a 5-membered C–N–C–N–B ring. Interrogation of the C29–N3 distance in this ring (1.3130(17) Å) implies significant double bond character which, alongside the other bond length and angle data, indicates that this ring exists in the form of a zwitterionic analogue of a boraimidazolinone (Scheme 2).
 | ||
| Scheme 2 The proposed mechanism for the formation of 4 and 5via FLP activation of phenyl isocyanate by a copper(I) boraniminomethanide, 6. | ||
4 can be considered to originate from the reaction of PhNCO, PhNC and 1. The formation of this species was surprising but did provide a hint as to why catalytic turnover for deoxygenation of isocyanates was not observed; the isocyanide, produced from reduction of the isocyanate by 1, intercepts any 1 present in the solution precluding turnover. Whilst under equimolar conditions such a step must produce a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of 2 and 4, under a catalytic regime where 2 is rapidly re-converted to 1 in the presence of excess isocyanate, rapid and complete off-cycle sequestration of any copper species can be envisioned. We propose that such off-cycle reactivity does not occur in the case of catalytic CO2 reduction because of the gaseous, and less reactive, nature of CO in comparison to PhNC.
:1 mixture of 2 and 4, under a catalytic regime where 2 is rapidly re-converted to 1 in the presence of excess isocyanate, rapid and complete off-cycle sequestration of any copper species can be envisioned. We propose that such off-cycle reactivity does not occur in the case of catalytic CO2 reduction because of the gaseous, and less reactive, nature of CO in comparison to PhNC.
The formation of 4 is proposed to proceed via the insertion of PhNC, generated from the reaction of 1 with half an equivalent PhNCO, into the Cu–B bond of the remaining half equivalent of 1 (Scheme 2). The resultant boraniminomethanide contains a Lewis acidic boron centre in the vicinity of a Lewis basic nitrogen atom, generating a potentially reactive system reminiscent of an intramolecular frustrated Lewis pair (FLP) in the form of a copper(I) boryliminomethanide. This intermediate species readily reacts with the residual isocyanate to generate 4.
Figueroa and co-workers reported similar reactivity of a related boryliminomethane (Cy2BC(H)NAr, Ar = 2,6-bis(2,6-diisopropylphenyl)phenyl) which was found to react similarly with CO2. In their hands the pinacol analogue, pinBC(H)NAr was unreactive, which the authors propose was a result of its low Lewis acidity at boron.38 A related, but structurally distinct copper-containing Lewis pair has also been implicated in CO2 activation reactivity by Hou and co-workers.15
In order to probe this mechanistic hypothesis, the equimolar reaction between 1 and the commercially available isocyanide, tBuNC, was attempted in C6D6 (see Fig. S11 and S12†). The formation of a resonance at 28.3 ppm (FWHM = 610 Hz) in the 11B NMR spectrum was taken to imply such reactivity furnished the boraniminomethanide, (6-Dipp)CuC(NtBu)Bpin (Scheme 2). Addition of one equivalent of phenyl isocyanate to this reaction mixture resulted in precipitation of colourless material overnight which was suitable for X-ray crystallography (Fig. 5).
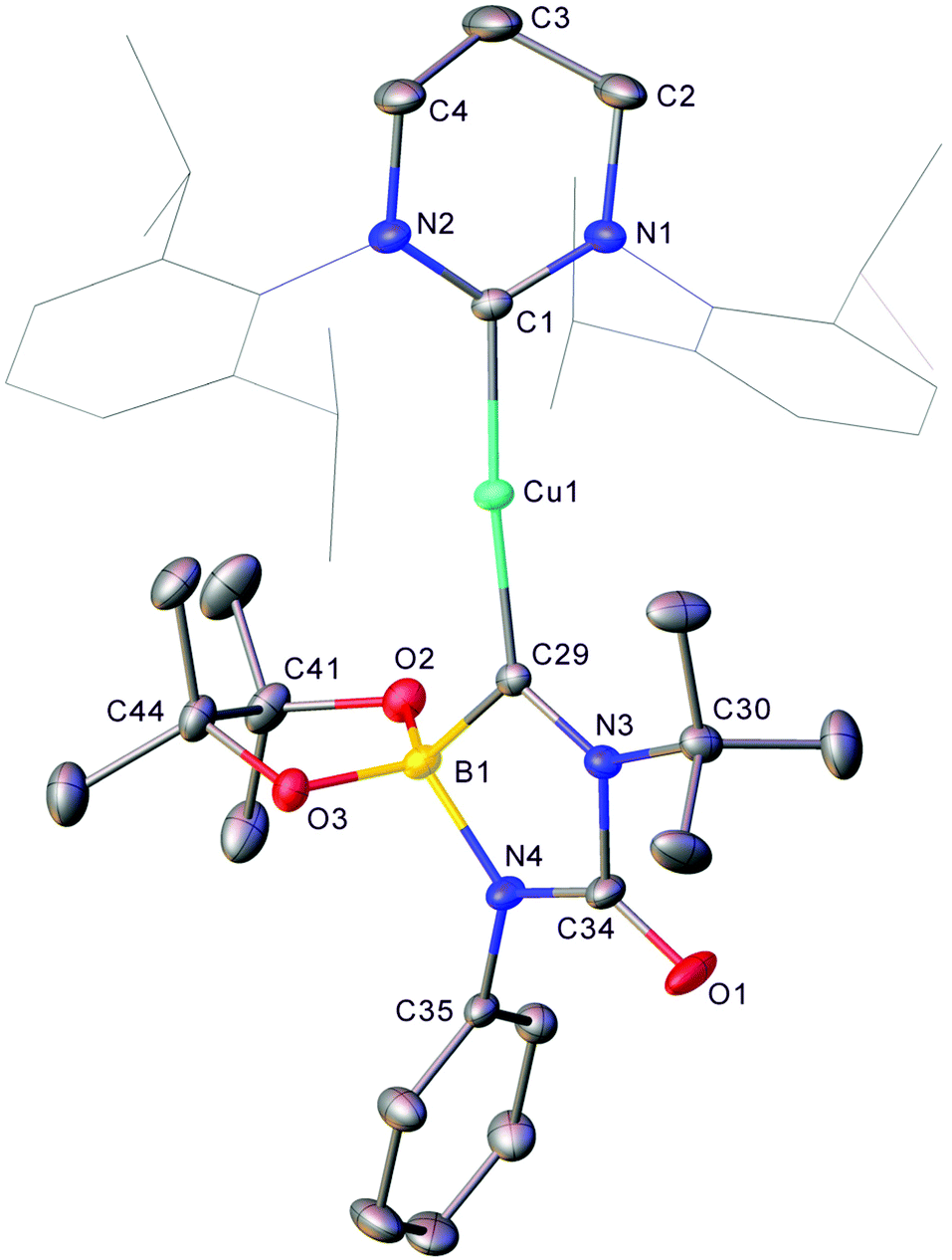 | ||
| Fig. 5 Molecular structure (30% probability ellipsoids) of compound 5. Hydrogen atoms and the minor disordered component are omitted for clarity. Wireframe view has been employed for Dipp groups, also for visual simplicity. Selected bond lengths (Å) and angles (°) C1–Cu1 1.955(2); Cu1–C29 1.925(2); C29–N3 1.314(3); N3–C34 1.493(3); C34–N4 1.315(3); N4–B1 1.574(3); B1–C29 1.656(3); C34–O1 1.209(3); B1–C29–N3 106.83(18); C29–N3–C34 113.96(18); N3–C34–N4 107.73(19); C34–N4–B1 113.30(18); N4–B1–C29 97.61(17). | ||
The product, 5, is the mixed phenyl/tert-butyl analogue of 4 and provides strong experimental support for the mechanistic hypothesis. No significant structural deviations from that of 4 are observed for this minor variation in ancillary R groups.
In contrast to the reactions with isocyanates, no unexpected resonances were observed in the borate region of the 11B NMR spectrum during catalytic attempts to dethiolate tert-butyl isothiocyanate which reflected only the presence of pin2B2 and pinBOtBu generated from activation of the catalyst. The presence of 3 could not be confirmed as its 11B NMR spectroscopic peak overlaps that of pin2B2. Nevertheless, when an equimolar mixture of previously isolated 1 and tBuNCS were reacted in C6D6, the 11B NMR spectrum reflected the presence of 3 and the copper(I) boraniminomethanide, 6 (see Fig. S13†). Fractional crystallisation from this solution provided analytically pure samples of 3 and 6. Although its solid state structure could not be confirmed, reaction of isolated 6 with PhNCS in toluene provided, after filtration, crystalline material found to be compound 7 (Fig. 6).
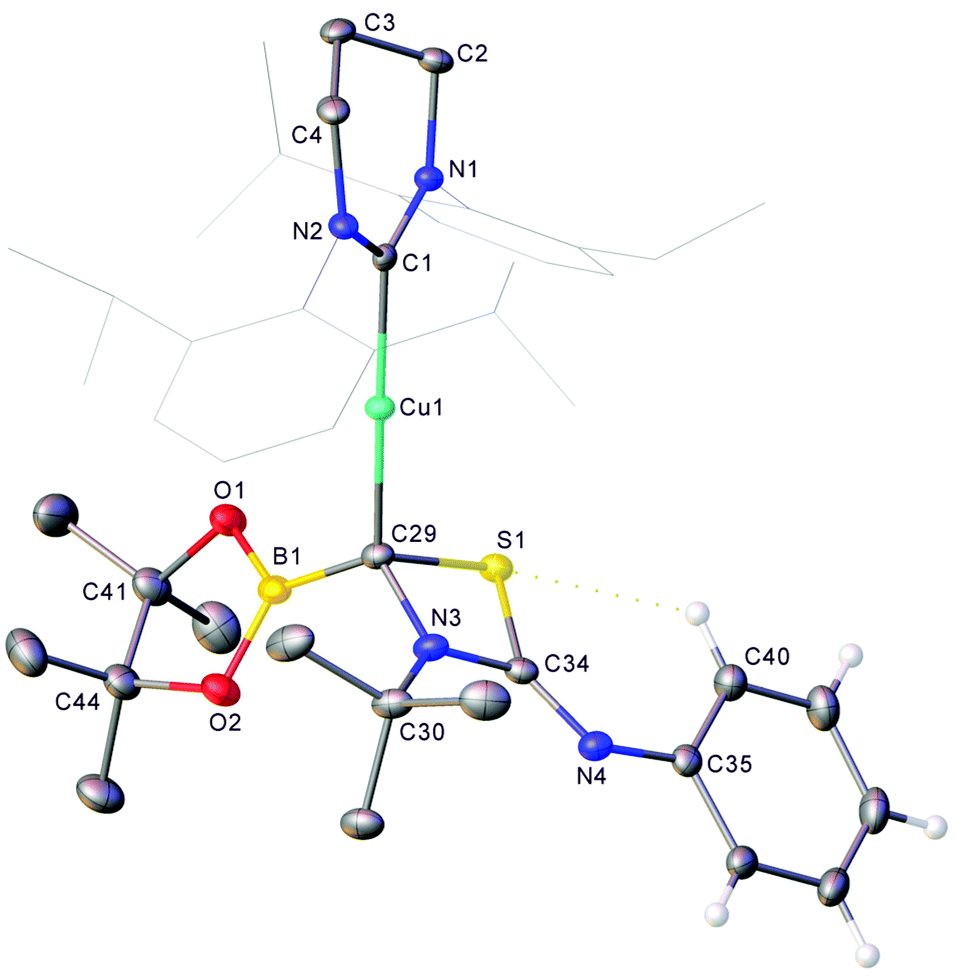 | ||
| Fig. 6 Molecular structure (30% probability ellipsoids) of compound 7. Hydrogen atoms are omitted for clarity, with the exceptions of those pertaining to the phenyl ring based on C35. Wireframe view has been employed for Dipp groups, also for visual simplicity. Selected bond lengths (Å) and angles (°) C1–Cu1 1.9271(12); Cu1–C29 1.9430(13); C29–B1 1.555(2); C29–S1 1.8740(13); S1–C34 1.7856(13); C34–N3 1.3454(18); N3–C29 1.4944(17); C34–N4 1.2958(18); C29–S1–C34 74.77(6); S1–C34–N3 95.37(9); C34–N3–C29 102.91(10); N3–C29–S1 86.96(8). | ||
Compound 7 comprises a 6-Dipp supported copper(I) centre bonded to a 4-membered C–S–C–N ring. Examination of the metric parameters associated with this structure imply a bonding motif with a conjugated N3–C34–N4 fragment but otherwise localised bonds. C29 is bonded to four different substituents and consequently chiral, although the material crystallises in the centrosymmetric P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group as a racemic mixture. The structure of 7 can once again be rationalised as the product of activation of a heterocumulene by the boraniminomethanide, 6, albeit through the electrophilic imine carbon instead of the Bpin fragment. The formation of this net [2 + 2] cycloaddition product likely reflects the weak SC bond in the isothiocyanate starting material, and the limited thiophilicity of boron.
space group as a racemic mixture. The structure of 7 can once again be rationalised as the product of activation of a heterocumulene by the boraniminomethanide, 6, albeit through the electrophilic imine carbon instead of the Bpin fragment. The formation of this net [2 + 2] cycloaddition product likely reflects the weak SC bond in the isothiocyanate starting material, and the limited thiophilicity of boron.
Whilst this off-cycle reactivity could act as a sink for any copper present under catalytic conditions, attempts to react isolated 3 with pin2B2 were unsuccessful (see Fig. S14†), implying that whilst isocyanide-centred reactivity may be present, a major stumbling block to dethiolation is the inaccessibility of Cu–S/B–B metathesis. This contrast to the activity of the boroxide analogue can most likely be attributed to the softer nature of copper(I), and a consequent affinity for the borylthiolate in 3.
Reactivity of (6-Dipp)CuBpin, 1, towards isopropyl carbodiimide
Despite such reactivity being confined to a stoichiometric regime, it does add to the limited pool of dechalcogenations of heterocumulenes. In order to probe the reactivity of Cu–B bonds towards other CX bond cleavages, the reaction of 1 and diisopropyl carbodiimide was investigated. When iPrNCNiPr was added to 1 in C6D6 complete attenuation of the peak at 41.2 ppm in the 11B NMR spectrum was observed over the course of 9 days alongside a colour change to yellow. The formation of two new resonances in the 11B NMR at 25.6 and 8.0 ppm was interpreted as a promising sign that CN bond cleavage may have occurred. The resultant solution was filtered, and hexane allowed to diffuse into it overnight resulting in the formation of yellow crystalline material. Single crystal X-ray diffraction analysis of this material (8) revealed it to be analogue of 4 where the exocyclic oxygen atom has been exchanged for an NiPr moiety (Fig. 7).
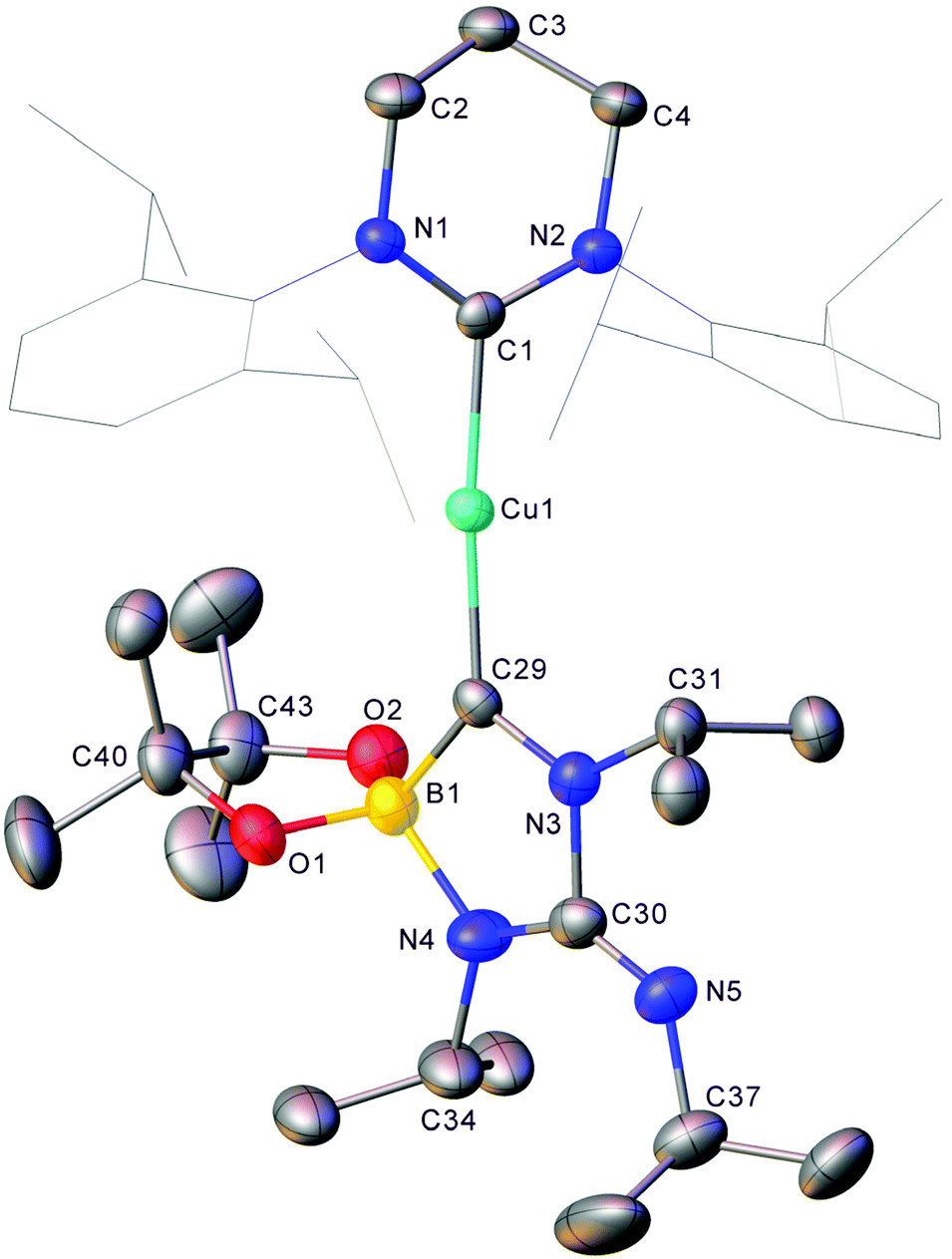 | ||
| Fig. 7 Molecular structure (30% probability ellipsoids) of compound 8. Hydrogen atoms and the minor disordered component are omitted for clarity. Wireframe view has been employed for Dipp groups, also for visual simplicity. Selected bond lengths (Å) and angles (°) C1–Cu1 1.957(4); Cu1–C29 1.941(4); C29–N3 1.336(5); N3–C30 1.492(6); C30–N4 1.361(6); N4–B1 1.563(7); B1–C29 1.640(6); C30–N5 1.283(6); B1–C29–N3 105.4(4); C29–N3–C30 115.7(4); N3–C30–N4 106.2(4); C30–N4–B1 112.2(4); N4–B1–C29 100.0(4). | ||
Compound 8 shares a similar cyclic C–N–C–N–B core with 4 and 5, but has an exocyclic CN bond, reflecting its nature as the product of the reaction of diisopropyl carbodiimide by a copper boraniminomethanide. 11B NMR spectroscopic analysis of compound 8 indicated a single resonance at 8.0 ppm, and its formation should be concomitant with that of (6-Dipp)CuN(iPr)Bpin to which the peak at 25.6 ppm in the 11B NMR spectrum of the formation of compound 9 (Fig. 8) can be attributed based on literature data of comparable compounds.30,39
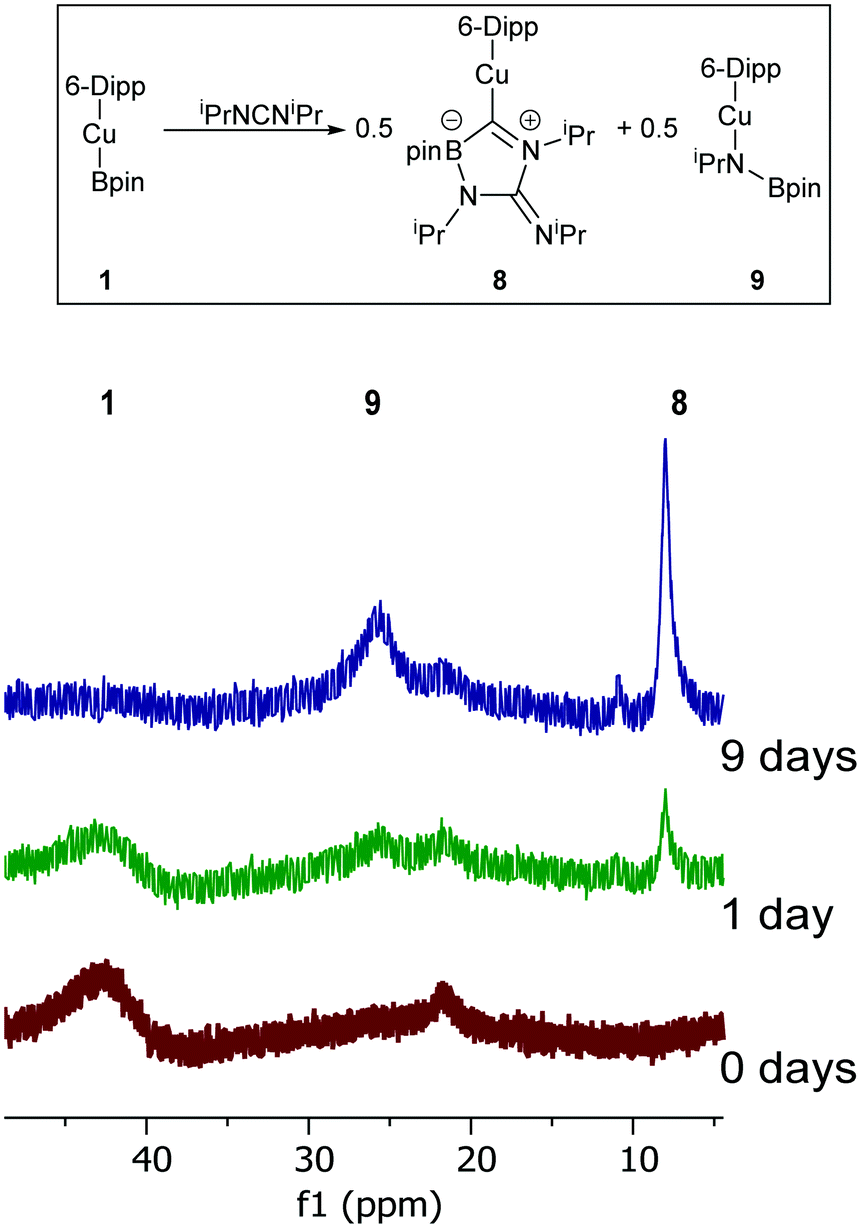 | ||
| Fig. 8 A proposed reaction scheme and 11B NMR spectra (ppm) for the reaction between 1 and iPrNCNiPr over 9 days. | ||
Conclusions
In conclusion, we have shown that the application of a RE-NHC ligand on copper can profoundly enhance the stability of a copper(I) boryl compared to corresponding 5-membered NHCs. The generation of a stable copper(I) boryl system, 1, allowed us to explore the chemistry of the highly reactive Cu–B bond towards a range of heterocumulenes. We were, thus, able to show that the previously reported deoxygenation reactivity of the copper-boryl moiety could be extended to a variety of CX bonds, including the CO bond in isocyanates, the CS bond in isothiocyanates and the CN in carbodiimides. Whilst we do not expect the mechanism of this bond cleavage to deviate significantly from that calculated for CO bond cleavage in carbon dioxide by (NHC)CuBpin,25 the promiscuity of the isonitrile side-products of each of these reactions results in the clean assembly of a range of unprecedented ring systems bonded to copper, the formation of which was probed mechanistically via stepwise stoichiometric reactions. Although currently confined to a stoichiometric regime, these ring systems comprise a number of novel structural motifs whose formation can be attributed to a mode of reactivity at the copper centre which has the potential for exploitation in productive catalysis, an area we continue to explore. We expect this work to contribute to the ever-growing suite of productive transformations reliant on Cu–B bond containing intermediates.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
DJL thanks the Royal Society for the support of a University Research Fellowship. We acknowledge financial support from the EPSRC Centre for Doctoral Training in Catalysis (EP/L016443/1) for JWH.Notes and references
- R. D. Dewhurst, E. C. Neeve, H. Braunschweig and T. B. Marder, Chem. Commun., 2015, 51, 9594–9607 RSC.
- E. C. Neeve, S. J. Geier, I. A. I. Mkhalid, S. A. Westcott and T. B. Marder, Chem. Rev., 2016, 116, 9091–9161 CrossRef CAS PubMed.
- A.-F. Pécharman, M. S. Hill, C. L. McMullin and M. F. Mahon, Angew. Chem., Int. Ed., 2017, 56, 16363–16366 CrossRef PubMed.
- A.-F. Pécharman, A. L. Colebatch, M. S. Hill, C. L. McMullin, M. F. Mahon and C. Weetman, Nat. Commun., 2017, 8, 15022 CrossRef PubMed.
- A.-F. Pécharman, M. S. Hill and M. F. Mahon, Dalton Trans., 2018, 47, 7300–7305 RSC.
- A.-F. Pécharman, M. S. Hill, C. L. McMullin and M. F. Mahon, Organometallics, 2018, 37, 4457–4464 CrossRef.
- A.-F. Pécharman, N. A. Rajabi, M. S. Hill, C. L. McMullin and M. F. Mahon, Chem. Commun., 2019, 55, 9035–9038 RSC.
- H. Shere, M. S. Hill, A.-F. Pécharman and M. F. Mahon, Dalton Trans., 2021, 50, 1283–1292 RSC.
- D. S. Laitar, P. Müller and J. P. Sadighi, J. Am. Chem. Soc., 2005, 127, 17196–17197 CrossRef CAS PubMed.
- D. S. Laitar, E. Y. Tsui and J. P. Sadighi, J. Am. Chem. Soc., 2006, 128, 11036–11037 CrossRef CAS PubMed.
- L. Zhang, J. Cheng, B. Carry and Z. Hou, J. Am. Chem. Soc., 2012, 134, 14314–14317 CrossRef CAS PubMed.
- K. Semba, M. Shinomiya, T. Fujihara, J. Terao and Y. Tsuji, Chem. – Eur. J., 2013, 19, 7125–7132 CrossRef CAS PubMed.
- T. Fujihara, K. Semba, J. Terao and Y. Tsuji, Catal. Sci. Technol., 2014, 4, 1699–1709 RSC.
- D. Hemming, R. Fritzemeier, S. A. Westcott, W. L. Santos and P. G. Steel, Chem. Soc. Rev., 2018, 47, 7477–7494 RSC.
- Z. Li, L. Zhang, M. Nishiura, G. Luo, Y. Luo and Z. Hou, J. Am. Chem. Soc., 2020, 142, 1966–1974 CrossRef CAS PubMed.
- Y. Segawa, M. Yamashita and K. Nozaki, Angew. Chem., Int. Ed., 2007, 46, 6710–6713 CrossRef CAS PubMed.
- T. Kajiwara, T. Terabayashi, M. Yamashita and K. Nozaki, Angew. Chem., Int. Ed., 2008, 47, 6606–6610 CrossRef CAS PubMed.
- Y. Okuno, M. Yamashita and K. Nozaki, Angew. Chem., Int. Ed., 2011, 50, 920–923 CrossRef CAS PubMed.
- C. Borner and C. Kleeberg, Eur. J. Inorg. Chem., 2014, 2014, 2486–2489 CrossRef CAS.
- C. M. Wyss, J. Bitting, J. Bacsa, T. G. Gray and J. P. Sadighi, Organometallics, 2016, 35, 71–74 CrossRef CAS.
- C. Borner, L. Anders, K. Brandhorst and C. Kleeberg, Organometallics, 2017, 36, 4687–4690 CrossRef CAS.
- C. Kleeberg and C. Borner, Organometallics, 2018, 37, 4136–4146 CrossRef CAS.
- W. Drescher and C. Kleeberg, Inorg. Chem., 2019, 58, 8215–8229 CrossRef PubMed.
- C. Kleeberg, L. Dang, Z. Lin and T. B. Marder, Angew. Chem., Int. Ed., 2009, 48, 5350–5354 CrossRef CAS PubMed.
- H. Zhao, Z. Lin and T. B. Marder, J. Am. Chem. Soc., 2006, 128, 15637–15643 CrossRef CAS PubMed.
- J. E. Baldwin, J. C. Bottaro, P. D. Riordan and A. E. Derome, J. Chem. Soc., Chem. Commun., 1982, 942–943 RSC.
- L. Yun-Shan, B. Mei-Zhi, Z. Zhi-Hua, T. Ken and F. Yuzo, Chem. Lett., 1992, 21, 1143–1144 CrossRef.
- S. V. Ley and S. J. Taylor, Bioorg. Med. Chem. Lett., 2002, 12, 1813–1816 CrossRef CAS PubMed.
- T. Ohnishi, H. Seino, M. Hidai and Y. Mizobe, J. Organomet. Chem., 2005, 690, 1140–1146 CrossRef CAS.
- S. I. Kalläne, T. Braun, M. Teltewskoi, B. Braun, R. Herrmann and R. Laubenstein, Chem. Commun., 2015, 51, 14613–14616 RSC.
- W. Drescher, C. Borner and C. Kleeberg, New J. Chem., 2021, 45, 14957–14964 RSC.
- L. Jie, S. Wang-xing and L. Xiang-rong, Curr. Org. Chem., 2012, 16, 2879–2891 CrossRef.
- A. J. Jordan, C. M. Wyss, J. Bacsa and J. P. Sadighi, Organometallics, 2016, 35, 613–616 CrossRef CAS.
- T. M. Horsley Downie, J. W. Hall, T. P. Collier Finn, D. J. Liptrot, J. P. Lowe, M. F. Mahon, C. L. McMullin and M. K. Whittlesey, Chem. Commun., 2020, 56, 13359–13362 RSC.
- C. A. G. Carter, C. M. Vogels, D. J. Harrison, M. K. J. Gagnon, D. W. Norman, R. F. Langler, R. T. Baker and S. A. Westcott, Organometallics, 2001, 20, 2130–2132 CrossRef CAS.
- M. A. Esteruelas, A. M. López, M. Mora and E. Oñate, Chem. Commun., 2013, 49, 7543–7545 RSC.
- J. A. Fernández-Salas, S. Manzini and S. P. Nolan, Chem. Commun., 2013, 49, 5829–5831 RSC.
- B. R. Barnett, C. E. Moore, A. L. Rheingold and J. S. Figueroa, Chem. Commun., 2015, 51, 541–544 RSC.
- D. J. Liptrot, M. S. Hill, M. F. Mahon and A. S. S. Wilson, Angew. Chem., Int. Ed., 2015, 54, 13362–13365 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: General synthetic experimental details, NMR spectra, X-ray analysis of compounds 1–5, 7 and 8. CCDC 2099327–2099333. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1dt03540a |
| This journal is © The Royal Society of Chemistry 2021 |