
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInsights into the naphthalenide-driven synthesis and reactivity of zerovalent iron nanoparticles†
Andreas
Reiß
,
Carsten
Donsbach
and
Claus
Feldmann
 *
*
Institut für Anorganische Chemie, Karlsruhe Institute of Technology (KIT), Engesserstrasse 15, D-76131 Karlsruhe, Germany. E-mail: claus.feldmann@kit.edu
First published on 14th October 2021
Abstract
The chemical and thermal stability of alkali metal naphthalenides as powerful reducing agents are examined, including the type of alkali metal ([LiNaph] and [NaNaph]), the type of solvent (THF, DME), the temperature (−30 to +50 °C), and the time of storage (0 to 12 hours). The stability and concentration of [LiNaph]/[NaNaph] are quantified via UV-Vis spectroscopy and the Lambert–Beer law. As a result, the solutions of [LiNaph] in THF at low temperature turn out to be most stable. The decomposition can be related to a reductive polymerization of the solvent. The most stable [LiNaph] solutions in THF are exemplarily used to prepare reactive zerovalent iron nanoparticles, 2.3 ± 0.3 nm in size, by reduction of FeCl3 in THF. Finally, the influence of [LiNaph] and/or remains of the starting materials and solvents upon controlled oxidation of the as-prepared Fe(0) nanoparticles with iodine in the presence of selected ligands is evaluated and results in four novel, single-crystalline iron compounds ([FeI2(MeOH)2], ([MePPh3][FeI3(Ph3P)])4·PPh3·6C7H8, [FeI2(PPh3)2], and [FeI2(18-crown-6)]). Accordingly, reactive Fe(0) nanoparticles can be obtained in the liquid phase via [LiNaph]-driven reduction and instantaneously reacted to give new compounds without remains of the initial reduction (e.g. LiCl, naphthalene, and THF).
Introduction
Alkali metal naphthalenides, which were first described by Berthelot in 1868,1 are well-known as powerful reducing agents.2 In inorganic synthesis, for instance, they were used in main-group chemistry to establish element-element multiple bonds (e.g. B![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) B)3 or to realize low-valence compounds (e.g. stannylenes and germylenes).4 They were also used in organic chemistry to reduce nitrobenzenes, sulfonate esters, or epoxides.5 Alkali metal naphthalenides were also already used to prepare nanoparticles of zerovalent main-group elements (e.g., B, Si, Ge)6 and transition metals (e.g., Co, Cu, Pd, Pt, Pt3Sn, Ag, Au).7 We have also used alkali metal naphthalenides to obtain various reactive base metals such as Ti(0), Mo(0), W(0), or Zn(0).8 Very recently, we could also prepare all rare-earth metals in the form of nanoparticles.9
B)3 or to realize low-valence compounds (e.g. stannylenes and germylenes).4 They were also used in organic chemistry to reduce nitrobenzenes, sulfonate esters, or epoxides.5 Alkali metal naphthalenides were also already used to prepare nanoparticles of zerovalent main-group elements (e.g., B, Si, Ge)6 and transition metals (e.g., Co, Cu, Pd, Pt, Pt3Sn, Ag, Au).7 We have also used alkali metal naphthalenides to obtain various reactive base metals such as Ti(0), Mo(0), W(0), or Zn(0).8 Very recently, we could also prepare all rare-earth metals in the form of nanoparticles.9
Most often lithium and sodium naphthalenide ([LiNaph] and [NaNaph]) were applied and prepared by reacting the respective alkali metal with naphthalene in ethers such as 1,2-dimethoxyethane (DME) or tetrahydrofuran (THF).2 As reducing agents, they offer several advantages: (i) the practical handling of [LiNaph]/[NaNaph] solutions is easier as compared to that of lithium/sodium as bulk metals or as compared to the handling of metal carbonyls, (ii) oxide impurities (e.g. originating from surface contamination of bulk alkali metals) can be avoided due to the insolubility of the respective alkali metal compounds in ethers (i.e. hydroxides, oxides, peroxides, and carbonates), (iii) in contrast to solutions of lithium/sodium in liquid ammonia, [LiNaph]/[NaNaph] solutions can be handled under ambient conditions, (iv) the injection of [LiNaph]/[NaNaph] solutions results in an instantaneous reduction, which is optimal for controlling the nucleation and growth of metal nanoparticles.8,9 Finally, [LiNaph]/[NaNaph] have been reported to be even more powerful reducing agents than lithium/sodium itself (E0(Li/Na in liquid NH3) = –2.3 V and E0([LiNaph]/[NaNaph] in THF) = −3.1 V).2 Despite these advantages and although [LiNaph]/[NaNaph] have been intensely used, the stability of these highly reactive reducing agents was barely studied until now.
Taking zerovalent iron nanoparticles as an example, we here examine the chemical and thermal stability of the [LiNaph]/[NaNaph] solutions in regard to optimal and reproducible use. Subsequent to the [LiNaph]/[NaNaph]-driven formation of Fe(0) nanoparticles, selected follow-up reactions were performed in order to verify the influence of remains originating from the reducing agent. As a result, the concentration of [LiNaph]/[NaNaph] can be quantified, parameters to stabilize the reducing agent are identified, and the [LiNaph]/[NaNaph]-driven formation of small-sized Fe(0) nanoparticles is described, which in turn can be used as a starting material to prepare four novel single-crystalline iron compounds (Fig. 1).
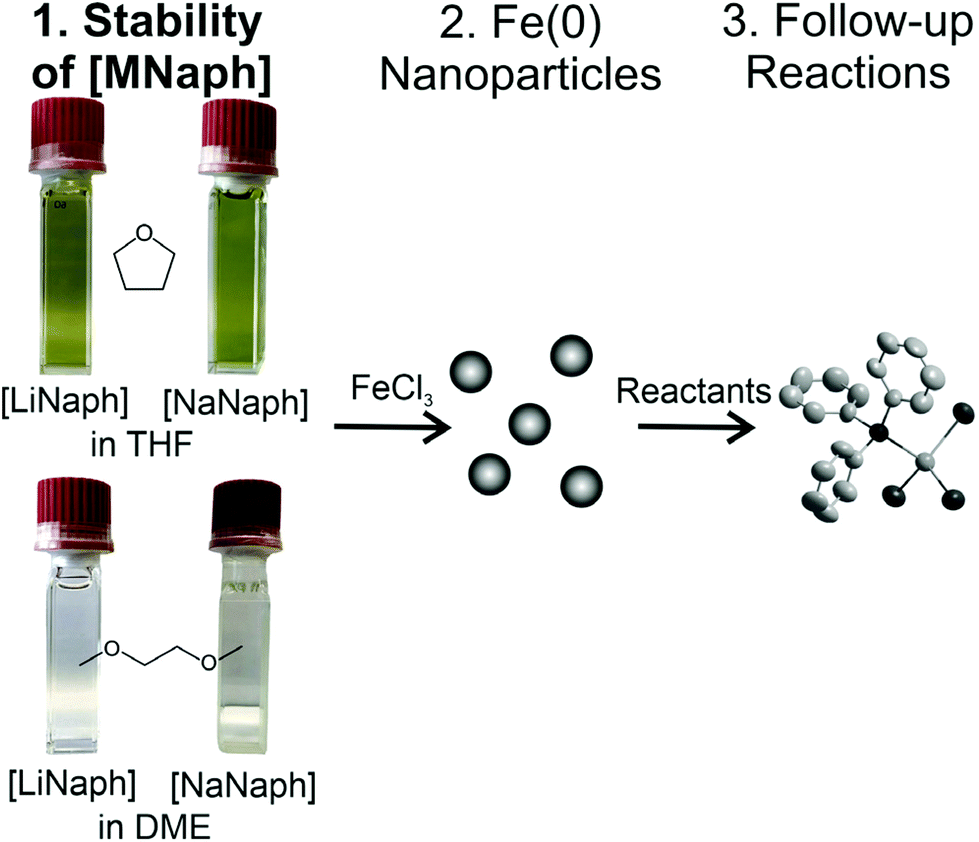 | ||
| Fig. 1 Stability of [LiNaph] and [NaNaph] in THF and DME with photos indicating the discoloration of the as-prepared solutions (0.286 mM of [LiNaph] or [NaNaph]). Moreover, the overall concept with the synthesis of Fe(0) nanoparticles and subsequent follow-up reactions is illustrated. | ||
Results and discussion
Stability of [LiNaph]/[NaNaph] solutions
Since alkali metal naphthalenides are widely used as powerful reducing agents, it is also well-known that the stability of the respective solutions is limited. Because we have frequently used alkali metal naphthalenides as reducing agents ourselves,8,9 it seemed important to us, on the one hand, to examine and to potentially improve the chemical stability, and on the other hand, to quantify the concentration of the [LiNaph]/[NaNaph] solutions used as reducing agents. In regard to the first aspect, we have compared the stability of the [LiNaph] and [NaNaph] solutions in tetrahydrofuran (THF) and dimethoxyethane (DME) as ethers at different temperatures (−30 °C to +50 °C) and over different times of storage (0 to 12 hours). Due to the deep green colour of the [LiNaph]/[NaNaph] solutions, optical spectroscopy (UV-Vis) seemed to be ideal to monitor the stability of the alkali metal naphthalenide solutions and could also allow a quantification of the concentration via the Lambert–Beer law.10 In regard to the selection of the ether, it should be noticed that other conventional ethers, such as diethyl ether or methyl tertiary-butyl ether (MTBE), are less polar than THF/DME, so that the solubility of [LiNaph]/[NaNaph] and of metal salts for follow-up reactions is significantly lower. For other solvents (e.g. DMF and DMSO), the solutions of [LiNaph]/[NaNaph] were already reported not to be stable.11To analyse the [LiNaph] and [NaNaph] solutions in THF or DME, first of all several prerequisites have to be fulfilled. Thus, the preparation of the solutions needs to be performed in a glovebox. Freshly distilled solvents were used throughout. The as-prepared [LiNaph]/[NaNaph] solutions were transferred from the glovebox into gas-tight quartz cuvettes, which then could be analysed by UV-Vis spectroscopy. Due to the intense colour of the [LiNaph]/[NaNaph] solutions, their concentration needs to be limited to ≤0.4 mM to guarantee an absorbance suitable to apply the Lambert–Beer law. When preparing concentration series of [LiNaph]/[NaNaph] (0.1–0.4 mM), we observed that the solutions of [LiNaph] and [NaNaph] in DME showed discoloration on a timescale <1 min (Fig. 1 and ESI: Fig. S1†), whereas the colour of the solutions in THF remained stable (Fig. 1). Here, it needs to be noticed that the discoloration is only visible for diluted solutions (≤0.4 mM). For the [LiNaph]/[NaNaph] solutions with higher concentrations (>0.4 mM), the decomposition of the reducing agent naturally occurs as well but is optically visible only over long periods (i.e. 1–2 hours) due to the intense colour. Based on this first evaluation related to the type of solvent, DME turned out to be less suitable than THF. Therefore, the following more detailed considerations are focused on [LiNaph]/[NaNaph] in THF.
The UV-Vis spectra of the [LiNaph]/[NaNaph] solutions in THF show distinct absorption minima at 411 nm and 435 nm, respectively (Fig. 2a and b), which are suitable to monitor the absorbance and to establish calibration curves. Indeed, a linear correlation of absorbance and concentration was obtained (Fig. 2c and d). Despite this good linear correlation, however, the straight lines do not clearly intersect at the coordinate origin, which is in conflict with the Lambert–Beer law. This finding can be attributed to the decomposition that already occurred prior to the spectroscopic analysis, and which could be initiated by an oxidation caused by humidity, oxygen, or the solvent. To elucidate the underlying effects, we have monitored the absorbance of the [LiNaph]/[NaNaph] solutions in THF (both with 0.286 mM) continuously over a period of 12 hours (Fig. 3). Here, a continuous discoloration (i.e. increase of transmission) was observed. If this behaviour would have been caused by residual moisture in the solvent or on the glass surface of the cuvettes, one would rather expect a fast process that comes to a halt after certain time when all moisture has reacted. Here, it should also be noticed that [NaNaph] not only shows discoloration but also shows a significant colour shift from intense green to light yellow (Fig. 3b and ESI: Fig. S1†). Furthermore, we have evaluated the influence of the temperature on the absorbance of the [LiNaph]/[NaNaph] solutions (0.343 mM [LiNaph], 0.286 mM [NaNaph]). Again, a clear correlation was observed with a slow discoloration at low temperature (after 2 hours at −30 °C) and a significantly faster discoloration at high temperature (after 2 hours at +50 °C) (Fig. 4). The temperature-depending discolouration of the [LiNaph]/[NaNaph] solutions can be followed even with the naked eye (Fig. 4).
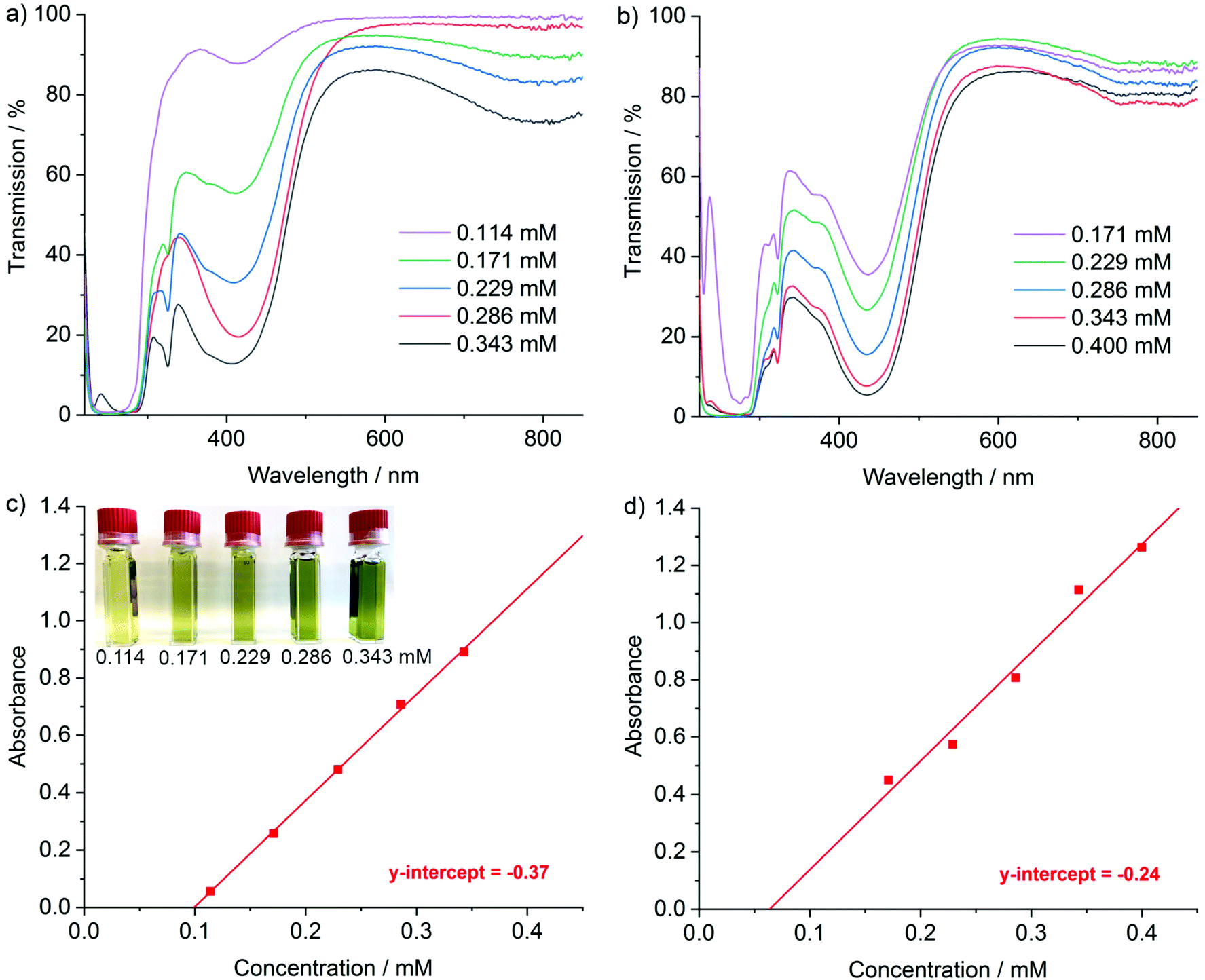 | ||
| Fig. 2 UV/VIS spectra of a concentration series of (a) [LiNaph] in THF and (b) [NaNaph] in THF as well as the resulting calibration lines for (c) [LiNaph] (monitored at 411 nm, with the photo of concentration series) and (d) [NaNaph] (monitored at 435 nm). | ||
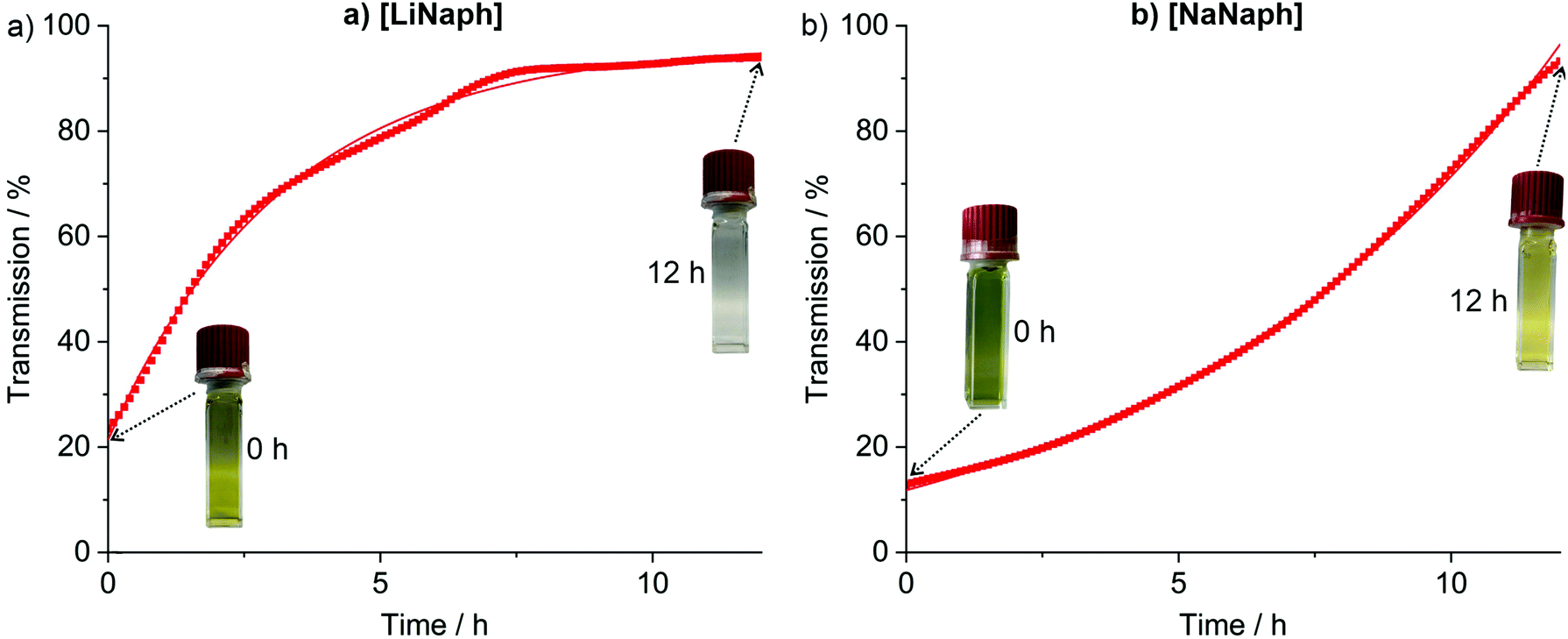 | ||
| Fig. 3 Time-dependent discoloration of (a) [LiNaph] in THF (monitored at 411 nm, 0.343 mM [LiNaph]), and (b) [NaNaph] in THF (monitored at 435 nm, 0.286 mM [NaNaph]) with photos of the respective freshly prepared solutions and the solutions after 12 hours. | ||
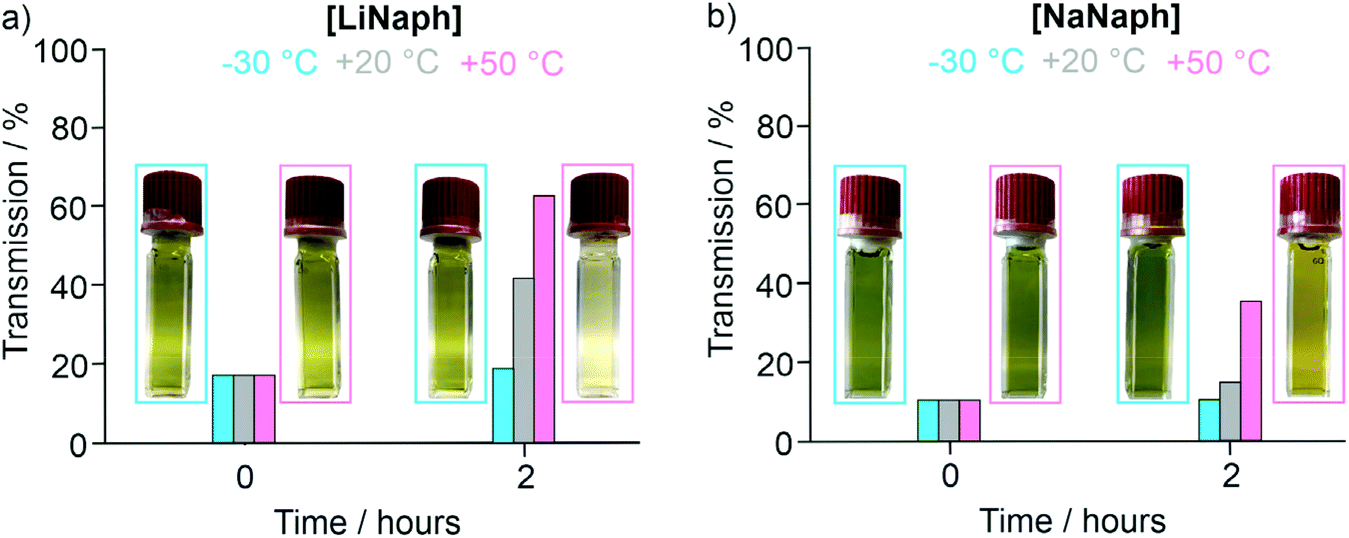 | ||
| Fig. 4 Temperature-dependent discoloration of (a) [LiNaph] in THF (monitored at 411 nm, 0.343 mM [LiNaph]), and (b) [NaNaph] in THF (monitored at 435 nm, 0.286 mM [NaNaph]) with photos for the visual comparison of the [LiNaph]/[NaNaph] solutions in THF at −30 and +50 °C. | ||
Based on the aforementioned effects of chemical stability as well as time- and temperature-depending stability, the [LiNaph] solutions in THF can be concluded to be most stable, especially when stored at low temperature (−30 °C). The experimentally observed slow but continuous discoloration of the [LiNaph]/[NaNaph] solutions, moreover, points to a reaction with THF as the most probable decomposition process. This view is corroborated when calculating the period in time related to the mismatch between the coordinate origin and the intersection of the calibration curve with the x-axis (Fig. 2c and d). Thus, a time slot of 30 minutes can be deduced, which is in good agreement with the real time needed for the preparation of the [LiNaph] solutions in the glovebox prior to recording of UV-Vis spectra. In the case of [NaNaph], such an estimation is not reliable due to the aforementioned time-dependent shift of the optical absorption (Fig. 3b and ESI: Fig. S1†).
In order to validate the influence of THF, finally, a concentrated [LiNaph] solution (80 mM) was stirred at 50 °C until complete discoloration (Fig. 5a). This forced decomposition was accompanied by a slow formation of a colourless precipitate with quantities suitable for separation and analytical characterization. Thus, the precipitate was separated by centrifugation after complete discoloration. Thereafter, the supernatant solution was analysed by 13C nuclear magnetic resonance (13C NMR) spectroscopy, which points to naphthalene and THF only (Fig. 5b). The colourless precipitate was insoluble in common solvents and therefore analysed with Fourier-transform infrared (FT-IR) spectroscopy. The resulting spectra are very similar to those of THF (Fig. 5c). In particular, the deformation oscillation of the THF ring at 1068 cm−1 confirms the molecule backbone to be present and intact. In addition, elemental analysis (EA) of the precipitate shows a C![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H:O ratio of 4:5:1, which is also comparable to that of THF (C:H:O = 4:8:1), except for a lower hydrogen content. These results – including the insolubility and the similarity of IR/EA with pure THF – point to the polymerization of THF.12 In fact, such polymerization of ethers is known, for instance, for the reaction with alkaline earth metals.13 Taken together, the stability of the [LiNaph]/[LiNaph] solutions in ethers is obviously limited by the reduction and polymerization of the solvent. The polymerization is a continuous process, whose speed depends on the type of alkali metal, the type of solvent and the temperature. The [LiNaph] solutions in THF turn out to be most stable, especially, if handled and stored at low temperatures (−30 °C).
:H:O ratio of 4:5:1, which is also comparable to that of THF (C:H:O = 4:8:1), except for a lower hydrogen content. These results – including the insolubility and the similarity of IR/EA with pure THF – point to the polymerization of THF.12 In fact, such polymerization of ethers is known, for instance, for the reaction with alkaline earth metals.13 Taken together, the stability of the [LiNaph]/[LiNaph] solutions in ethers is obviously limited by the reduction and polymerization of the solvent. The polymerization is a continuous process, whose speed depends on the type of alkali metal, the type of solvent and the temperature. The [LiNaph] solutions in THF turn out to be most stable, especially, if handled and stored at low temperatures (−30 °C).
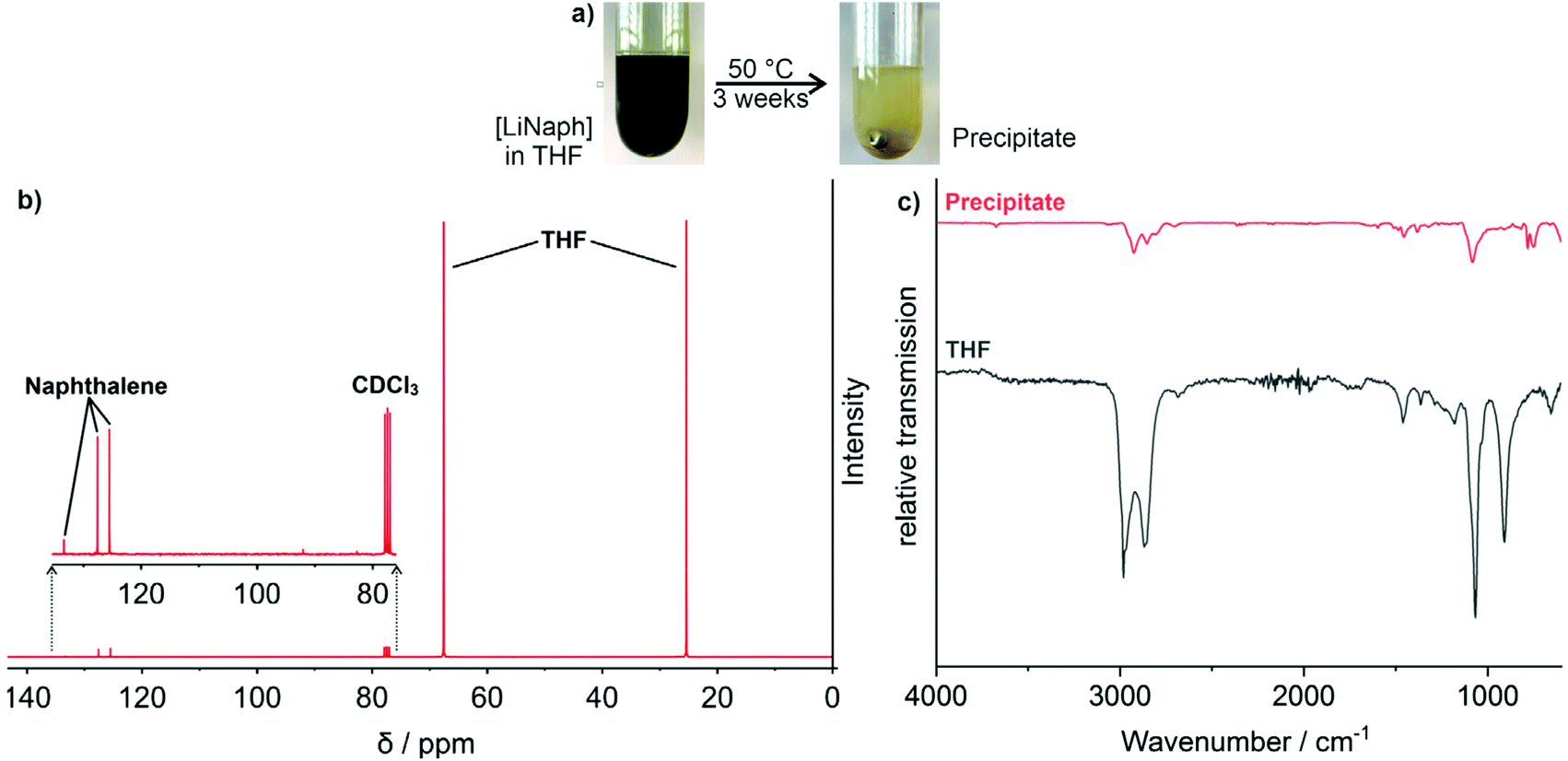 | ||
| Fig. 5 Reaction of [LiNaph] with THF: (a) photos of the as-prepared [LiNaph] in THF (80 mM) and after 3 weeks at 50 °C, (b) 13C-NMR spectrum of the colourless precipitate (in CDCl3), and (c) FT-IR spectrum of the colourless precipitate (with THF as a reference). | ||
Synthesis of Fe(0) nanoparticles
With the knowledge on the stability of [LiNaph] and [NaNaph] in THF or DME, we have exemplarily used the [LiNaph] solutions in THF to obtain Fe(0) nanoparticles (Fig. 6a). First of all, a solution of FeCl3 in THF was prepared. The [LiNaph] solution was injected into the FeCl3 solution at room temperature. The instantaneous reduction and formation of Fe(0) nanoparticles were indicated by a rapid colour change of the yellow-green FeCl3 solution to a deep black suspension. The resulting suspension contained 35 mg of Fe(0) in THF (2.3 mg mL−1). The suspension was colloidally stable for several hours. Thereafter, slow precipitation of iron occurred.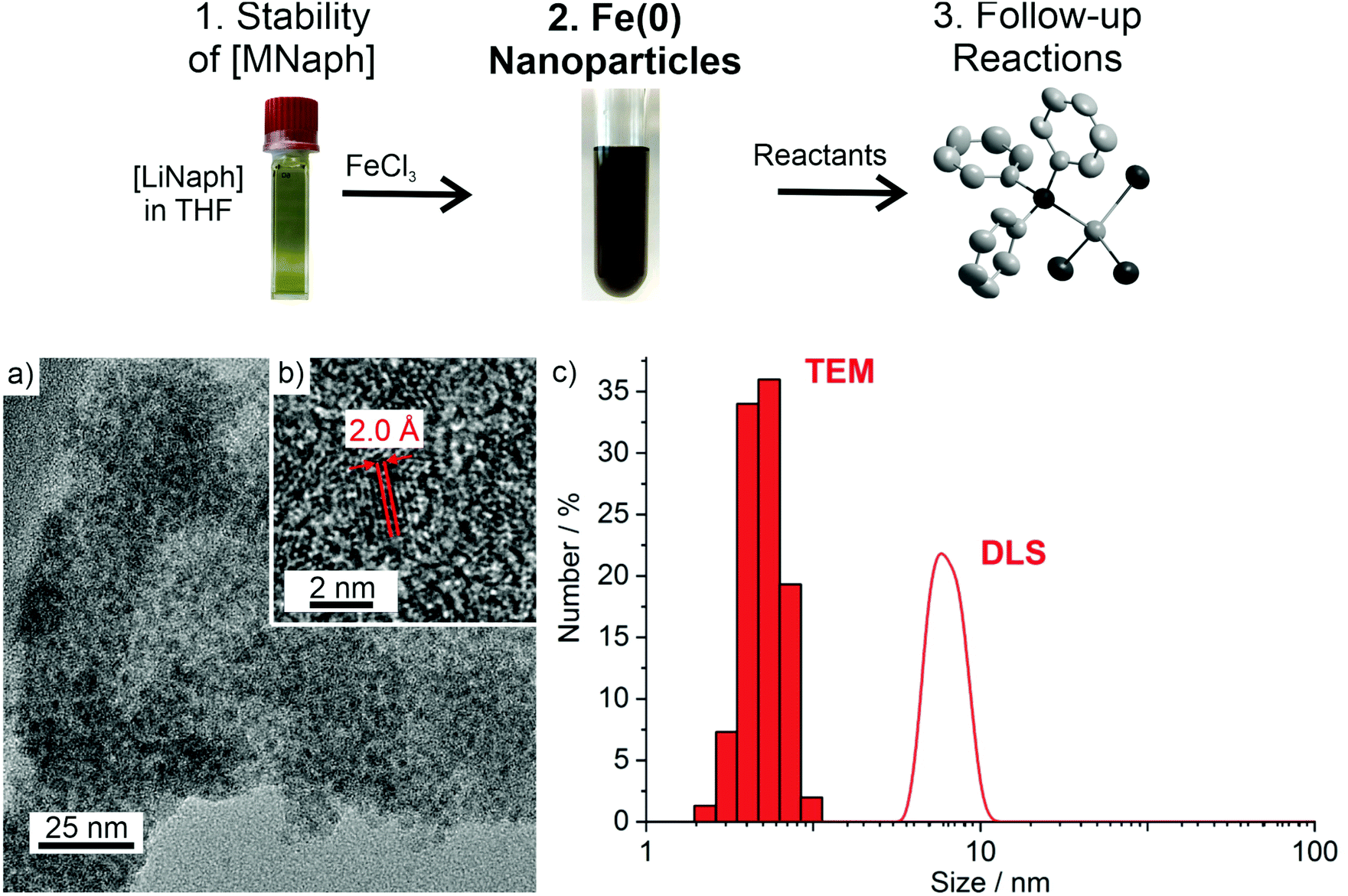 | ||
| Fig. 6 Particle size and size distribution of Fe(0) nanoparticles after the [LiNaph]-based reduction of FeCl3: (a) TEM overview image, (b) HRTEM image, and (c) size distribution according to TEM (statistical evaluation of 150 particles) and DLS (after stabilization with oleylamine in n-heptane. | ||
Transmission electron microscopy (TEM) was performed to examine the size of the as-prepared Fe(0) nanoparticles. Accordingly, the TEM overview images show a great number of non-agglomerated particles with diameters of 1–3 nm at narrow size distribution (Fig. 6a). A statistical evaluation of 150 particles on the TEM images reveals a mean diameter of 2.3 ± 0.3 nm (Fig. 6c). The high-resolution (HR)TEM images confirm the particle diameter and show highly parallel lattice fringes, indicating the crystallinity of the Fe(0) nanoparticles (Fig. 6b). The observed lattice plane distance of 2.0 Å is in good agreement with that of cubic bulk iron (d110: 2.0 Å).14 The presence and purity of zerovalent iron were additionally evidenced via X-ray powder diffraction (XRD) (Fig. 7a). Here, the as-prepared nanoparticles do not show any Bragg peak, which can be attributed to the small particle size and the resulting low scattering power. After sintering at 700 °C, however, Bragg peaks of iron metal are clearly visible. Although the sample is of course not nanosized after sintering, the absence of any other Bragg peaks nevertheless indicates the purity of the as-prepared Fe(0) nanoparticles.
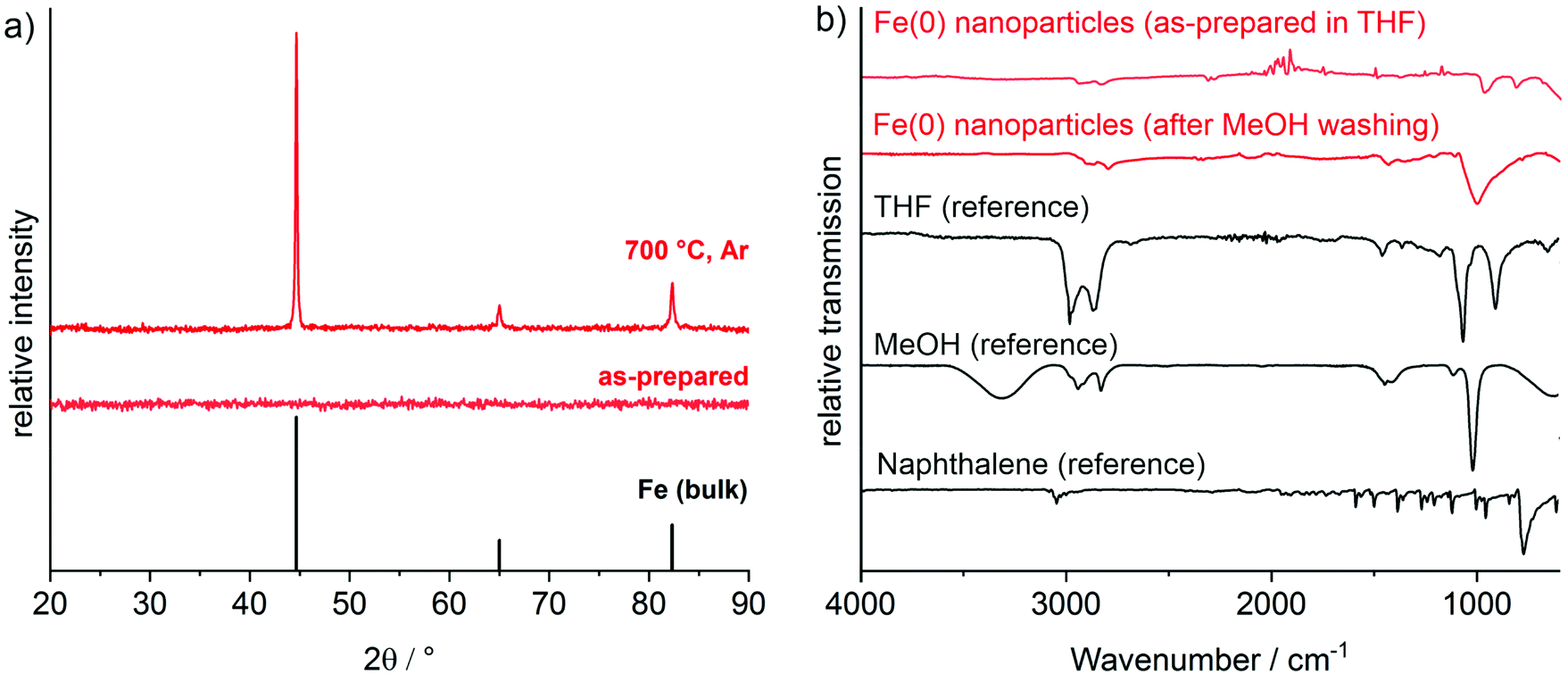 | ||
| Fig. 7 Composition and surface functionalization of the Fe(0) nanoparticles: (a) powder diffractogram (bulk iron as a reference: ICDD-No. 03-065-4899), and (b) FT-IR spectra after synthesis in THF and after washing with MeOH (with the spectra of THF, MeOH and naphthalene as references). | ||
Finally, the surface functionalization and colloidal properties of the as-prepared Fe(0) nanoparticles were studied. First of all, FT-IR spectra were recorded and the as-expected patterns indicate the presence of THF on the particle surface. All vibrations are in accordance with the reference spectra of THF (Fig. 7b). In order to remove LiCl, washing with MeOH as a more polar solvent was performed. Thereafter, the infrared spectra are still dominated by the vibrations of THF. Specifically, the absence of any O–H vibration (3500–3100 cm−1) points to the absence of MeOH showing that MeOH is a suitable solvent for purification subsequent to the reduction (Fig. 7b). Finally, it needs to be noticed that vibrations of naphthalene were neither observed for the as-prepared nor for the MeOH-washed Fe(0) nanoparticles.
In addition to MeOH, we have also treated the as-prepared Fe(0) nanoparticles with oleylamine (OA). OA is a widely applied surface capping agent to functionalize the surface of metal nanoparticles. OA indeed resulted in a long-term stabilization of the as-prepared Fe(0) nanoparticles. Whereas the as-prepared Fe(0) nanoparticles in THF show slow sedimentation after 1 hour, the OA-stabilized Fe(0) nanoparticles in toluene are colloidally stable for months. Dynamic light scattering of such OA-stabilized suspensions shows a mean hydrodynamic diameter of 6.4 ± 1.2 nm at narrow size distribution (Fig. 6c). Taking the size of the OA molecule (1.5–1.8 nm) into account,15 the hydrodynamic diameter is in good agreement with the mean diameter of the as-prepared Fe(0) nanoparticles. As a strong-binding ligand, however, OA is difficult to remove from the particle surface. For follow-up reactions, therefore, the Fe(0) nanoparticle suspensions in THF will be used.
Follow-up reactions
Subsequent to the [LiNaph]-driven synthesis, we have examined if the Fe(0) nanoparticles can be used as a starting material in follow-up reactions. Due to the small size of the Fe(0) nanoparticles, high reactivity can be expected, so that they can be directly reacted in the liquid phase. We were also interested to see if the ingredients of the initial synthesis or their remains (i.e. naphthalene, THF, LiCl) deteriorate the follow-up reactions. To solve this, the as-prepared Fe(0) nanoparticles were washed by redispersion/centrifugation in/from methanol to remove naphthalene and LiCl. Alternatively, the as-prepared Fe(0) nanoparticles were just centrifuged, dried in vacuum at room temperature, and thereafter redispersed in toluene. In regard to the manifold opportunities for follow-up reactions and optional reactants, we have here exemplarily evaluated the oxidation and reactivity of the Fe(0) nanoparticles with iodine in the presence of coordinating ligands such as triphenylphosphane (PPh3) or 18-crown-6. All reactions were performed in the liquid phase near room temperature (20–50 °C). This resulted in four novel iron compounds: [FeI2(MeOH)2] (1), ([MePPh3][FeI3(Ph3P)])4·PPh3·6C7H8 (2), [FeI2(PPh3)2] (3), and [FeI2(18-crown-6)] (4). All of them could be crystallized in the form of suitable single crystals to perform single-crystal structure analysis (Table 1). Beside the higher stability of [LiNaph], it must be noticed that all follow-up reactions are likewise possible with [NaNaph] in THF.| Data | [FeI2(MeOH)2] (1) | ([MePPh3][FeI3(Ph3P)])4·PPh3·6C7H8 (2) | [FeI2(PPh3)2] (3) | [FeI2(18-crown-6)] (4) |
|---|---|---|---|---|
| Sum formula | C2H8FeI2O2 | C52H48.75FeI3P2.25 | C36H30FeI2P2 | C12H24FeI2O6 |
| Crystal system | Triclinic | Hexagonal | Monoclinic | Monoclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P63 (Flack parameter: 0.53(3)) | P21/c | C2/c |
| Lattice parameters | a = 411.9(3) pm | a = 1583.9(1) pm | a = 1939.3(4) pm | a = 1082.8(1) pm |
| b = 628.5(4) pm | b = 1583.9(1) pm | b = 1025.8(1) pm | b = 1203.4(1) pm | |
| c = 783.1(6) pm | c = 4651.3(4) pm | c = 1805.3(4) pm | c = 2836.7(3) pm | |
| α = 91.64(6) ° | ||||
| β = 92.50(6) ° | β = 112.12(2) ° | β = 94.62(1) ° | ||
| γ = 95.76(5) ° | ||||
| Cell volume | V = 201.4(2)×106 pm3 |
V = 10105(1)×106 pm3 |
V = 3327(1)×106 pm3 | V = 3684(1)×106 pm3 |
| Formula units per cell | Z = 1 | Z = 8 | Z = 4 | Z = 8 |
| Calculated density | ρ = 3.081 g cm−3 | ρ = 1.551 g cm−3 | ρ = 1.665 g cm−3 | ρ = 2.069 g cm−3 |
| Measurement limits | −4 ≤ h ≤ 4, −7 ≤ k ≤ 7, −9 ≤ l ≤ 9 | −16 ≤ h ≤ 4, −17 ≤ k ≤ 18, −55 ≤ l ≤ 55 | −19 ≤ h ≤ 23, −12 ≤ k ≤ 11, −21 ≤ l ≤ 21 | −12 ≤ h ≤ 12, −12 ≤ k ≤ 14, −33 ≤ l ≤ 28 |
| Theta range for data collection | 6.52 to 49.97° | 2.97 to 50.00° | 4.53 to 49.99° | 5.07 to 50.00° |
| Linear absorption coefficient | μ = 9.455 mm−1 | μ = 2.235 mm−1 | μ = 2.429 mm−1 | μ = 4.190 mm−1 |
| Number of reflections | 1122 (518 independent) | 20673 (11403 independent) |
15357 (5839 independent) |
9151 (3247 independent) |
| Refinement method | Full-matrix least-squares on F2 for all | |||
| Merging | R int = 0.098 | R int = 0.082 | R int = 0.156 | R int = 0.063 |
| Number of parameters | 38 | 726 | 370 | 190 |
| Residual electron density | 1.12 to −1.16 e−×10−6 pm−3 | 0.44 to −0.45 e−×10−6 pm−3 | 0.55 to −0.46 e−×10−6 pm−3 | 0.64 to −1.20 e−×10−6 pm−3 |
| R1 (I ≥ 2σI) | 0.050 | 0.037 | 0.038 | 0.040 |
| R1 (all data) | 0.080 | 0.102 | 0.127 | 0.062 |
| wR2 (all data) | 0.134 | 0.076 | 0.066 | 0.099 |
| GooF | 0.986 | 0.714 | 0.666 | 0.863 |
[FeI2(MeOH)2] (1) was obtained by reacting the Fe(0) nanoparticles after redispersion/centrifugation in/from methanol with 2-iodo-2-methylpropane (Fig. 8a and ESI: Fig. S2†). Here, 2-iodo-2-methylpropane was selected in regard to an optional formation of metal–carbon bonds. The formation of 1 can be ascribed to the following reaction:
| Fe + 2C4H9I + 2MeOH → [FeI2(MeOH)2] + 2C4H8 + H2 |
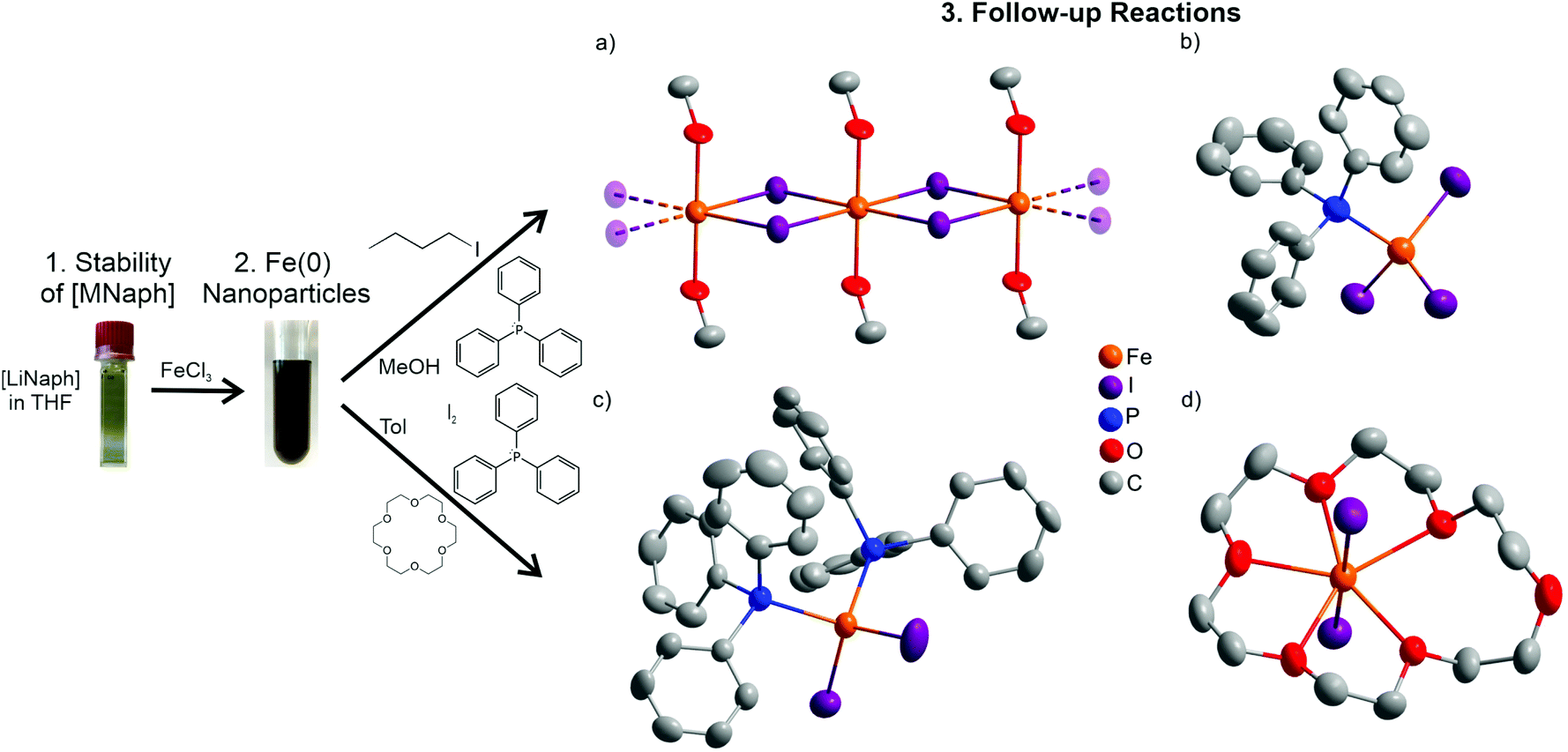 | ||
| Fig. 8 Structures of iron-containing compounds obtained by follow-up reactions with Fe(0) nanoparticles as a starting material: (a) [FeI2(MeOH)2] (1), (b) [FeI3(Ph3P)]− anion in 2, (c) [FeI2(PPh3)2] (3), and (d) [FeI2(18-crown-6)] (4) (H atoms omitted for clarity). | ||
Thus, the reaction was triggered by the decomposition of 2-iodo-2-methylpropane to isobutylene and HI.16 HI is known to decompose at room temperature with the formation of H2 and I2, whereof the latter reacts with the Fe(0) nanoparticles. Interestingly, THF from the initial synthesis of the Fe(0) nanoparticles was obviously completely removed, so that only MeOH remained as the ligand. 1 crystallizes in the triclinic space group P (Table 1). The compound forms linear 1∞[FeI4/2(MeOH)2/1] chains along the crystallographic a axis. Iron is octahedrally coordinated by four corner-bridging iodine atoms and two MeOH ligands located in the trans-position (Fig. 8a). The Fe–I distances (285.3(2)–289.4(2) pm) and the Fe–O distances (209.5(1) pm) are in good agreement with FeI217 and [Fe(H2O)6]2+ (Table 2).18 Interestingly, the Fe(0) nanoparticles did not react with MeOH under the formation of H2, so that the MeOH ligands in 1 were not deprotonated, which was also confirmed by FT-IR spectroscopy (ESI: Fig. S3†).
| Compound | Fe–I | Fe–O | Fe–P |
|---|---|---|---|
| [FeI2(MeOH)2] (1) | 285.3(2)–289.4(2) | 209.5(11) | — |
| ([MePPh3][FeI3(Ph3P)])4·PPh3·6C7H8 (2) | 260.2(1)–262.0(1) | — | 243.1(7)–246.9(7) |
| [FeI2(PPh3)2] (3) | 256.1(1)–257.4(1) | — | 246.9(2)–249.2(2) |
| [FeI2(18-crown-6)] (4) | 2803(1)–280.8(1) | 223.1(5)–249.3(4) | — |
| FeI2 (ref. 17) | 288.5 | — | — |
| [Fe(H2O)62+] (ref. 18) | — | 209.5 | — |
| [FeI4]2− (ref. 20a) | 253.1, 254.0 | — | — |
| [FeCl2(PPh3)2] (ref. 20b) | — | — | 247.6 |
In a second reaction, the Fe(0) nanoparticles were directly reacted in toluene with iodine and in the presence of PPh3 as a ligand after redispersion/centrifugation in/from methanol (Fig. 8b and ESI: Fig. S4†). This reaction results in ([MePPh3][FeI3(Ph3P)])4·PPh3·6C7H8 (2). 2 crystallizes in the chiral hexagonal space group P63 and exhibits [MePPh3]+ cations and [FeI3(Ph3P)]− anions (Table 1 and Fig. 8b). In the anion, Fe+II is tetrahedrally coordinated by iodine (Fe–I: 260.2(12)–262.0(1) pm) and phosphorus (Fe–P: 243.1(7)–246.9(7) pm) (Table 2). Interestingly, PPh3 was methylated to the [MePPh3]+ cation. This methyl group needs to originate from MeOH, so that the following reaction can be postulated for the formation of 2:
| 6Fe + 6I2 + 9PPh3 + 6C7H8 + 4MeOH → ([MePPh3][(Ph3P)FeI3])4·PPh3·6C7H8 + 2Fe(OH)2 |
This reaction is validated by the fact that 2 was only obtained after washing of the as-prepared Fe(0) nanoparticles with MeOH.
[FeI2(PPh3)2] (3) – as the third example – was obtained by the reaction of the Fe(0) nanoparticles with iodine and again PPh3 as a ligand. Since a reaction with MeOH was observed in the cases of 1 and 2, the Fe(0) nanoparticles were not washed by redispersion/centrifugation in/from MeOH but only redispersed in toluene. 3 is a molecular, non-charged complex with tetrahedrally coordinated Fe+II (Fig. 8c and ESI: Fig. S5†). The compound was obtained according to the reaction: Fe + 2PPh3 + I2 → [FeI2(PPh3)2]. [FeI2(PPh3)2] crystallizes in the monoclinic space group P21/c (Table 1). Although the composition of the compound is known,19 this specific reaction and crystal structure were not reported before. 3 exhibits Fe–I (256.1(1), 257.4(1) pm) and Fe–P distances (246.9(2), 249.2(2) pm), which are in accordance with literature data (e.g., [FeI4]2− and [FeCl2(PPh3)2], Table 2).20
Finally, the Fe(0) nanoparticles were reacted with I2 and 18-crown-6 as a ligand in toluene. This results in [FeI2(18-crown-6)] (4) according to the reaction: Fe + I2 + 18-crown-6 → [FeI2(18-crown-6)]. 4 crystallizes in the monoclinic space group C2/c (Table 1). Again, iron was oxidized to Fe+II, which is equatorially coordinated by 18-crown-6 and axially coordinated by two iodine atoms (Fig. 8d and ESI: Fig. S6†). Interestingly, iron is only coordinated by five oxygen atoms of the crown ether, which is uncommon for such strong chelating agents. This finding can be correlated with the mismatch of the cation size of Fe2+ (75 pm)21 and the ring-opening diameter of 18-crown-6 (about 300 pm).22 As a result, a coordination with five shorter Fe–O distances (223.1(5)–249.3(4) pm) and a significantly longer distance to the sixth oxygen atom (446.5(5) pm) of the crown ether is preferred (Table 2). The Fe–I distance (280.5 pm) again corresponds to FeI2 (288.5 pm).17
The reaction of the as-prepared Fe(0) nanoparticles to the compounds 1–4, on the one hand, points to the reactivity of the as-prepared nanoparticles already near room temperature (20–50 °C) and, on the other hand, shows that the Fe(0) nanoparticles can be instantaneously used as a starting material in the liquid phase to obtain new single-crystalline compounds. Moreover, the starting materials, reaction products, and solvents used to prepare the Fe(0) nanoparticles (e.g. [LiNaph], naphthalene, LiCl, and THF) do not necessarily deteriorate follow-up reactions of the nanoparticles. Such reactions, including the temperature range and reaction products, would not be possible with bulk iron and may offer an option for many more reactions with Fe(0) as a starting material in the liquid phase.
Conclusions
Alkali metal naphthalenides are generally well-known as powerful reducing agents and widely used in inorganic, metalorganic and organic chemistry. Therefore, it is all the more surprising that the stability of the respective naphthalenide solutions was rarely examined until now. By comparing the chemical and thermal stability of the most often used [LiNaph] and [NaNaph] depending on the solvent (THF, DME), the temperature (−30 to +50 °C), and the time of storage (up to 12 hours), we could identify [LiNaph] solutions in THF to be most stable, especially when handled and stored at low temperature. Moreover, a reductive polymerization of the solvent turned out to be the major issue to limit the stability of the [LiNaph]/[NaNaph] solutions. Finally, the concentration of the [LiNaph]/[NaNaph] solutions was determined by optical spectroscopy and the Lambert–Beer formalism, which allows a quantification of the reducing reactions.Besides the examination of the chemical and thermal stability of the [LiNaph]/[NaNaph] solutions, we have exemplarily used the reducing agents to obtain reactive Fe(0) nanoparticles by reduction of FeCl3 in THF. The injection of such a powerful reducing agent in the liquid phase in this regard is ideal to control particle nucleation and particle growth and instantaneously resulted in colloidally highly stable suspensions (i.e. 2.3 mg Fe(0) per mL THF) with uniform, small-sized Fe(0) nanoparticles and narrow size distribution (i.e. 2.3 ± 0.3 nm in size). Due to the absence of long-chain, strongly coordinated agents, the as-prepared Fe(0) nanoparticles are highly reactive and can be directly reacted in the liquid phase to obtain new compounds. Exemplarily, the Fe(0) nanoparticles were oxidized by iodine in the presence of different ligands (i.e. 2-iodo-2-methylpropane, PPh3, 18-crown-6), which resulted in [FeI2(MeOH)2], ([MePPh3][FeI3(Ph3P)])4·PPh3·6C7H8, [FeI2(PPh3)2], and [FeI2(18-crown-6)] as novel single-crystalline compounds. Besides the understanding and quantification of [LiNaph]/[NaNaph], the intermediate formation of reactive base nanoparticles as starting materials to obtain new compounds seems interesting. On the one hand, this is one of the first examples of iron metal as a starting material in the form of nanoparticles, and on the other hand, this exemplary study shows that one-pot, liquid-phase reactions with reactive nanoparticles to obtain new single-crystalline compounds are possible. The latter offers numerous options for chemical synthesis.
Experimental section
General aspects
All reactions and sample handling were performed under a dried argon atmosphere using standard Schlenk techniques or gloveboxes.Chemicals
Dimethoxyethane (DME, Sigma-Aldrich, 99%), n-heptane (Seulberger, technical grade), tetrahydrofurane (THF, Seulberger, technical grade), and toluene (Seulberger, 99%) were refluxed over sodium with benzophenone and distilled prior to use. Oleylamine (Acros, 80–90%) was kept for more than 30 days over an activated molecular sieve (4 Å) and then degassed in a vacuum. Methanol (Seulberger, technical grade) was refluxed over magnesium and distilled prior to use. N,N,N′,N′-Tetramethylethylenediamine (TMEDA) (Sigma-Aldrich, 99%) was refluxed and distilled over CaH2. Lithium (Alfa Aesar, 99%) and sodium (Riedel-de-Haën, 99%) were freshly cut under argon prior to use. 18-crown-6 (Sigma-Aldrich, 99%), iodine (Riedel-de-Haën, 99.8%), iron(III) chloride (abcr, 98%), naphthalene (Alfa Aesar, 99%), and triphenylphosphine (PPh3) (Sigma-Aldrich, 99%) were used without further purification.[LiNaph]/[NaNaph] solutions
108.4 mg (0.84 mmol) of naphthalene and either 5.6 mg (0.80 mmol) of lithium or 18.4 mg (0.80 mmol) of sodium were dissolved over 12 h in 10 mL of THF or DME, respectively. The resulting stock solutions were further diluted with the respective solvent to obtain concentration series for spectroscopic analysis. In the case of [LiNaph], a 0.40 mM solution turned out to be too concentrated for spectroscopic analysis. Thus, another sample with a concentration of 0.11 mM was prepared. For [NaNaph], solutions with concentrations between 0.40 mM and 0.17 mM were analysed.Fe(0) nanoparticles
14.0 mg (2.00 mmol) of lithium and 270.0 mg (2.10 mmol) of naphthalene were dissolved in 5 mL of THF over 12 h. In parallel, 108.1 mg (0.67 mmol) of FeCl3 was dissolved in 10 mL of THF. Thereafter, the [LiNaph] solution was injected under vigorous stirring into the FeCl3 solution. The instantaneous reaction was indicated by the change of the yellow-green solution to a deep black suspension. Subsequently, the Fe(0) nanoparticles were separated from the solution via centrifugation. The solid residue was washed two times by redispersion/centrifugation in/from 15 mL of methanol (MeOH) to remove all residual starting materials and LiCl. To use the Fe(0) nanoparticles in follow-up reactions, washing with MeOH was partially avoided in order to exclude a reaction with MeOH during heating. Finally, the Fe(0) nanoparticles were either dried for 30 min in a vacuum to obtain powder samples with a yield of about 90%, or redispersed in a solution of oleylamine in n-heptane to obtain long-term stable suspensions.Follow-up reactions
Analytical techniques
More information related to the analytical methods (including UV/VIS, NMR, TEM, DLS, FT-IR, EA, and XRD with powder diffraction and single-crystal structure analyses) can be found in the ESI.† Further details related to the crystal structures may also be obtained from the joint CCDC/FIZ Karlsruhe deposition service on quoting the depository numbers 2099718 (1), 2099719 (2), 2099720 (3), and 2099721 (4).†Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors are grateful to the Deutsche Forschungsgemeinschaft (DFG) for funding of personnel (NanoMet: FE911/11-1) and TEM equipment (INST 121384/33-1 FUGG).References
- P. E. M. Berthelot, Arch. Pharm. Pharm. Med. Chem., 1868, 268–269 CrossRef.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS PubMed.
- (a) S. Morisako, R. Shang, Y. Yamamoto, H. Matsui and M. Nakano, Angew. Chem., Int. Ed., 2017, 56, 15234–15240 CrossRef CAS PubMed; (b) H. Braunschweig, H. D. Dewhurst, K. Hammond, J. Mies, K. Radacki and A. Vargas, Science, 2012, 336, 1420–1422 CrossRef CAS PubMed; (c) R. C. Fischer and P. P. Power, Chem. Rev., 2010, 110, 3877–3923 CrossRef CAS PubMed.
- (a) C. Mohapatra, L. T. Scharf, T. Scherpf, B. Mallick, K.-S. Feichtner, C. Schwarz and V. H. Gessner, Angew. Chem., Int. Ed., 2019, 58, 7459–7463 CrossRef CAS PubMed; (b) R. E. Jilek, M. Jang, E. D. Smolensky, D. J. Britton and J. E. Ellis, Angew. Chem., Int. Ed., 2008, 47, 8692–8695 CrossRef CAS PubMed; (c) J. E. Ellis, S. R. Frerichs and B. K. Stein, Organometallics, 1993, 12, 1048–1057 CrossRef CAS.
- (a) K. Bannai, T. Tanaka, N. Okamura, A. Hazato, S. Sugiura, K. Manabe, K. Tomimori, Y. Kato, S. Kurozumi and R. Noyori, Tetrahedron, 1990, 46, 6689–6704 CrossRef CAS; (b) N. L. Holy, Can. J. Chem., 1976, 54, 1599–1602 CrossRef CAS; (c) A. Yasuda, H. Yamamoto and H. Nozaki, Tetrahedron Lett., 1976, 17, 2621–2622 CrossRef; (d) J. R. Ganson, S. Schulenberg and W. D. Closson, Tetrahedron Lett., 1970, 50, 4397–4400 CrossRef.
- (a) S. Yao, A. Kostenko, Y. Xiong, A. Ruzicka and M. Driess, J. Am. Chem. Soc., 2020, 142, 12608–12612 CrossRef CAS PubMed; (b) H. W. Chiu, C. N. Chervin and S. M. Kauzlarich, Chem. Mater., 2005, 17, 4858–4864 CrossRef CAS; (c) A. L. Pickering, C. Mitterbauer, N. D. Browning, S. M. Kauzlarich and P. P. Power, Chem. Commun., 2007, 580–582 RSC.
- (a) X. Chen, H. Cao, X. Chen, Y. Du, J. Qi, J. Luo, M. Armbrüster and C. Liang, ACS Appl. Mater. Interfaces, 2020, 12, 18551–18561 CrossRef CAS PubMed; (b) F. Fu, A. M. Martinez-Villacorta, A. Escobar, J. Irigoyen, S. Moya, E. Fouquet, J. Ruiz and D. Astruc, Inorg. Chem. Front., 2017, 4, 2037–2044 RSC; (c) M. Branca, K. Corp, D. Ciuculescu-Pradines, Y. Coppel, P. Lecante and C. Amiens, New J. Chem., 2017, 41, 5960–5966 RSC; (d) M. Schulz-Dobrick, K. V. Sarathy and M. Jansen, J. Am. Chem. Soc., 2005, 127, 12816–12817 CrossRef CAS PubMed.
- (a) A. Egeberg, L.-P. Faden, A. Zimina, J.-D. Grunwaldt, D. Gerthsen and C. Feldmann, Chem. Commun., 2021, 57, 3648–3651 RSC; (b) C. Schöttle, D. E. Doronkin, R. Popescu, D. Gerthsen, J.-D. Grunwaldt and C. Feldmann, Chem. Commun., 2016, 52, 6316–6319 RSC; (c) C. Schöttle, P. Bockstaller, R. Popescu, D. Gerthsen and C. Feldmann, Angew. Chem., Int. Ed., 2015, 54, 9866–9870 CrossRef PubMed.
- D. Bartenbach, O. Wenzel, R. Popescu, L.-P. Faden, A. Reiß, M. Kaiser, A. Zimina, J.-D. Grunwaldt, D. Gerthsen and C. Feldmann, Angew. Chem., Int. Ed., 2021, 60, 17373–11737 CrossRef CAS PubMed.
- T. G. Mayerhöfer and J. Popp, Beer's Law – Why Absorbance Depends (Almost) Linearly on Concentration, ChemPhysChem, 2019, 20, 511–515 CrossRef PubMed.
- A. J. Clancy, J. Melbourne and M. S. P. Shaffer, J. Mater. Chem. A, 2015, 3, 16708–16715 RSC.
- J. R. Sabin, A. Dwivedi, V. Baboo and A. Bajpai, J. Theor. Chem., 2015, 1–11 CAS.
- J. Bhattacharjee, A. Harinath, H. P. Nayek, A. Sarkar and T. K. Panda, Chem. – Eur. J., 2017, 23, 9319–9331 CrossRef CAS PubMed.
- A. T. Gorton, G. Bitsianes and T. L. Joseph, Trans. Metall. Soc. AIME, 1965, 233, 1519 CAS.
- Z. Wang, X. D. Wen, R. Hoffmann, J. S. Son, R. Li, C. C. Fang, D. M. Smilgies and T. Hyeon, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 17119–17124 CrossRef CAS PubMed.
- E. O. Hornig and J. E. Willard, J. Am. Chem. Soc., 1957, 79, 2429–2434 CrossRef CAS.
- A. Ferraria and F. Giorgi, Atti Accad. Naz. Lincei, Rend. Cl. Sci. Fis. Mat., 1929, 10, 522–527 Search PubMed.
- T. K. Sham, J. B. Hastings and M. L. Perlman, J. Am. Chem. Soc., 1980, 102, 5904–5906 CrossRef CAS.
- (a) R. Kliß, R. Hauptmann and G. Henkel, Z. Kristallogr., 1999, 214, 135–136 Search PubMed; (b) L. H. Pignolet, D. Forster and W. D. Horrocks, Inorg. Chem., 1968, 7, 828–830 CrossRef CAS.
- (a) S. Pohl and W. Saak, Z. Anorg. Allg. Chem., 1985, 523, 25–32 CrossRef CAS; (b) O. Seewald, U. Flörke and G. Henkel, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2005, 61, m1829–m1830 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- J. J. Christensen, J. O. Hill and R. M. Izatt, Science, 1971, 174, 459–467 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional data related to the analytical techniques and the unit cells of the title compounds 1–4. CCDC 2099718–2099721. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1dt02523f |
| This journal is © The Royal Society of Chemistry 2021 |