
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Probing catalytic surfaces by correlative scanning photoemission electron microscopy and atom probe tomography†
Kevin
Schweinar
 *a,
Rachel L.
Nicholls
b,
Catherine R.
Rajamathi
b,
Patrick
Zeller
c,
Matteo
Amati
c,
Luca
Gregoratti
c,
Dierk
Raabe
a,
Mark
Greiner
b,
Baptiste
Gault
ad and
Olga
Kasian
ef
*a,
Rachel L.
Nicholls
b,
Catherine R.
Rajamathi
b,
Patrick
Zeller
c,
Matteo
Amati
c,
Luca
Gregoratti
c,
Dierk
Raabe
a,
Mark
Greiner
b,
Baptiste
Gault
ad and
Olga
Kasian
ef
aMax-Planck-Institut für Eisenforschung GmbH, Microstructure Physics and Alloy Design, Düsseldorf, Germany. E-mail: k.schweinar@mpie.de
bMax-Planck-Institut für Chemische Energiekonversion, Heterogeneous Reactions, Mülheim a.d. Ruhr, Germany
cElettra – Sincrotrone Trieste S.C.p.A., Trieste, Italy
dDepartment of Materials, Imperial College London, Royal School of Mines, London, SW7 2AZ, UK
eMax-Planck-Institut für Eisenforschung GmbH, Interface Chemistry and Surface Engineering, Düsseldorf, Germany
fHelmholtz-Zentrum Berlin GmbH, Helmholtz-Institute Erlangen-Nürnberg, 14109 Berlin, Germany
First published on 2nd December 2019
Abstract
The chemical composition and the electronic state of the surface of alloys or mixed oxides with enhanced electrocatalytic properties are usually heterogeneous at the nanoscale. The non-uniform distribution of the potential across their surface affects both activity and stability. Studying such heterogeneities at the relevant length scale is crucial for understanding the relationships between structure and catalytic behaviour. Here, we demonstrate an experimental approach combining scanning photoemission electron microscopy and atom probe tomography performed at identical locations to characterise the surface's structure and oxidation states, and the chemical composition of the surface and sub-surface regions. Showcased on an Ir–Ru thermally grown oxide, an efficient catalyst for the anodic oxygen evolution reaction, the complementary techniques yield consistent results in terms of the determined surface oxidation states and local oxide stoichiometry. Significant chemical heterogeneities in the sputter-deposited Ir–Ru alloy thin films govern the oxide's chemistry, observed after thermal oxidation both laterally and vertically. While the oxide grains have a composition of Ir0.94Ru0.06O2, the composition in the grain boundary region varies from Ir0.70Ru0.30O2 to Ir0.40Ru0.60O2 and eventually to Ir0.75Ru0.25O2 from the top surface into the depth. The influence of such compositional non-uniformities on the catalytic performance of the material is discussed, along with possible engineering levers for the synthesis of more stable and reactive mixed oxides. The proposed method provides a framework for investigating materials of interest in the field of electrocatalysis and beyond.
1. Introduction
Thermally prepared oxides of Ir–Ru are commonly used for catalysis applications, in particular to facilitate the anodic oxygen evolution reaction (OER) in proton exchange membrane water electrolysis (PEMWE) devices.1 These devices convert H2O to molecular H2 that can be used as an energy carrier for the storage of intermittent energy from renewable sources (e.g. wind power, solar power). PEMWEs that perform efficiently and with high longevity will be critical to the development of a sustainable energy economy. The general requirements for materials used in such devices are very demanding, as they must exhibit high electrical conductivity, high chemical activity and long-term stability in harsh electrochemical environments.2 These requirements are particularly critical for anode catalysts that operate in a highly corrosive acidic environment and at high potentials. Among the few active materials that can withstand such conditions are Ir–Ru mixed oxides.3,4 While IrO2 has superior longevity during operation, compared to RuO2, the latter exhibits a higher electrocatalytic activity towards the OER.5 The use of mixed oxides for this reaction is an attempt to combine the strength of both pure oxides. The initial chemical state of the oxide anode material used in an electrolyzer is not only determined by the initial alloy composition and structure, but also by the processes that occur during the preparation procedure. In our proof-of-concept experiments, we closely investigate the local changes in nanoscale chemistry occurring during the thermal oxidation of an Ir–Ru alloy. The presented experiments aim at demonstrating the high potential of combining the utilized spatially resolved methods, to better understand the electrochemical properties of Ir–Ru mixed oxide anodes prepared by this synthesis method.Correlative microscopy approaches, applying different analysis, imaging and diffraction methods to the same portion of material, are increasingly deployed to address fundamental questions pertaining to mechanisms underpinning structure–property relationships. Combining several microscopy and microanalysis techniques on a single specimen allows one to exploit each technique's inherent strengths to provide complementary insights. For materials structured at the nanoscale, a prominent combination of methods is transmission electron microscopy (TEM) and atom probe tomography (APT), allowing one to relate the atomic structure and composition of nanoscale features, such as crystalline defects.6–8 Classically, correlative TEM/APT analyses were conducted on distinct specimens from the same alloy.9–11 Yet, if the features of interest are scarce or heterogeneously distributed, one requires targeted, spatially correlated experiments on the same specimen. Herbig recently reviewed the possibility to carry out a full TEM characterization on APT needle-shaped specimens prior to performing the APT measurement.12 This approach promotes the understanding of solute segregation at crystalline defects and interfaces that impact the macroscopic properties of a material.7,8,13–16 The additional use of scanning electron microscope (SEM) based techniques such as electron backscattered diffraction (EBSD), transmission Kikuchi diffraction (TKD) or electron channeling contrast imaging (ECCI) can be instrumental to guide site-specific preparation of APT specimen.17–20
Developing correlative microscopy approaches amenable to surface-active materials, such as catalysts, has however attracted less attention. The lack of APT studies is partially due to the inherent difficulty of analyzing the top-most layers of the free surface of a material. Yet, the oxidation of metallic bulk alloys used in heterogeneous catalysis was previously the center of some APT studies.21–25 Soon after, nanoparticles (NP) used in real catalysis applications were investigated.26–30 However, the measurement of NPs can introduce further challenges as they typically need to be embedded in a solid matrix material before measurement, potentially introducing a number of measurement artifacts (e.g. ion trajectory aberrations) which complicate the APT data analysis and interpretation.
These attempts demonstrate the growing demand for shedding light on the atomic scale surface chemistry of catalytically active materials. Method development in this field is on-going, but it remains challenging to measure a material's top surface with APT. For catalysts, it is mainly the interface between the gas, liquid and the solid that is catalytically relevant. The surface chemistry of the top-most layers determines how the catalyst engages in a given reaction and how well it performs. The exposure of a catalyst to its service environment, however, will inevitably change its surface structure and composition over time.21,22,24,31–35
Typically, catalytic surfaces are experimentally investigated with surface- and chemical-state sensitive (in situ) methods such as photoemission electron spectroscopy (PES) and X-ray absorption spectroscopy (XAS) techniques. The combination of X-ray photoelectron spectroscopy (XPS) and APT has recently proved to be instrumental in the systematic investigation of the top-most layer of electrochemically grown iridium oxide, an efficient electrocatalyst for the OER.31
Conventional XPS typically lacks the spatial resolution required to resolve confined surface phases or localized chemical state inhomogeneities, that potentially exhibit different catalytic properties. Recently, we complemented XPS with APT to unveil the formation of confined, non-stoichiometric Ir–O species in the near-surface region which gradually transform into IrO2.31 Such confined chemical heterogeneities crucially impact the observed activity and stability of the catalyst in question, and deserve further attention. A fundamental understanding of processes at the atomic scale facilitates the engineering of better catalysts.
Here, we propose an integrated workflow for the investigation of catalytic surfaces by correlative scanning photoemission electron microscopy (SPEM), μ-spot XPS and APT. Thin film catalysts are deposited directly on Si microtip substrates suitable for consecutive spectroscopic and APT measurements at the identical location of a sample. The metallic films can additionally be thermally or electrochemically treated in the desired catalytic environment. The combination of methods with corresponding surface and bulk sensitivity provides fundamental understanding of the complex interplay between surface processes and the underlying microstructure. In our proof-of-concept experiments we show how the proposed workflow can be applied to investigate the change in surface composition and electronic structure by comparing an Ir–Ru alloy catalyst before and after thermal oxidation. We present how the local microstructure controls the growth rate of individual oxide grains and how crystalline defects act as the main diffusional pathways as the oxidation front progresses into the material. These aspects are critical to understand when designing efficient, long-lived catalysts.
2. Experimental section
2.1. Sample preparation
Commercially available Si microtip coupons (Cameca/Ametek, Inc.) were used as a substrate for the deposition of thin films. The doped Si-based conductive coupons feature an array of 22 or 36 pre-fabricated flat top microtips with a diameter of 2 μm (±1 μm) at the top. Fig. 1a schematically depicts an individual as-received microtip. The flat top of each microtip serves as an individual sample for surface spectroscopic measurements.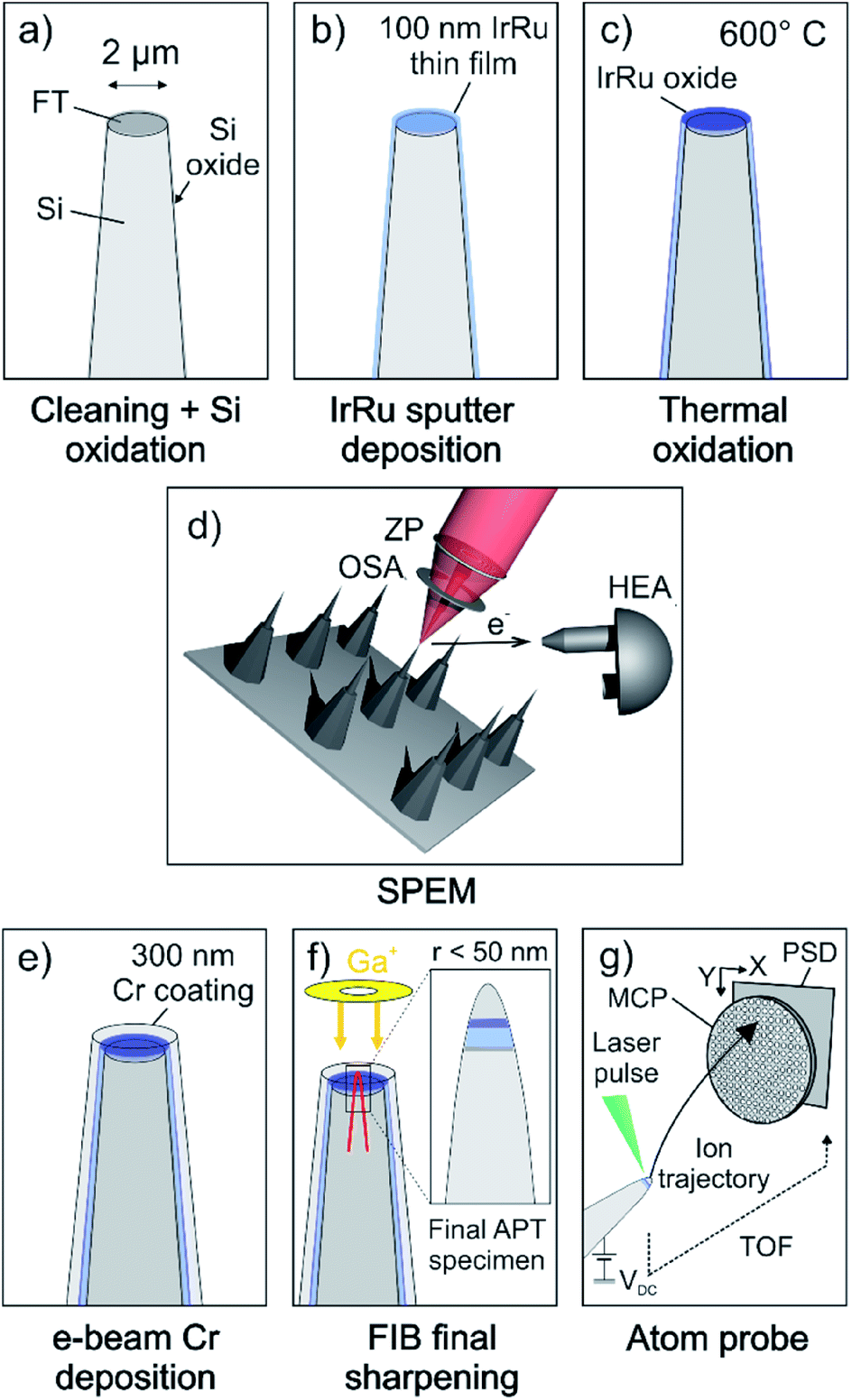 | ||
| Fig. 1 Schematic illustration of the proposed experimental workflow. (a) As-received Si microtips are cleaned by ion-polishing before being oxidized to grow a Si oxide diffusion barrier layer. (b) e-beam deposition of a 5 nm Ti adhesion layer and magnetron sputter co-deposition or Ir–Ru thin films. (c) Thermal oxidation of metallic thin film in air at 600 °C for 5 h. (d) SPEM measurements on top surface of individual microtips. (e) e-beam deposition of 300 nm thick Cr coating as surface protection towards air and enabling the measurement of top-most surface layers with APT. (f) Specimens are sharpened to needle-shaped APT specimen using a FIB-SEM. Inset on the right shows final APT specimen with a remaining Cr coating on top of the Ir–Ru thin film close to the apex of the needle. (g) APT measurement in laser pulsing mode. | ||
Prior to the deposition of thin films, the coupons were ion-polished to remove residual contaminants and oxides introduced during fabrication of the substrates. The rotating substrates were tilted by 30° and cleaned by a broad Ar beam in a precision etching and coating system (PECS, Gatan Inc., USA) for 5 min at an ion energy of 2 keV and current density of 32 μA cm−2.
To prevent the out-diffusion of Si into the thin films during the thermal treatment, we oxidized the Si coupon prior to deposition (Fig. 1a). The oxidation was performed in a near-ambient pressure XPS (NAP-XPS) system enabling the in situ spectroscopic monitoring of the oxidation progress. The coupon was oxidized in 5 mbar O2 at 1000 °C for 4 hours leading to an oxide thickness >10 nm.
Ir–Ru alloy thin films were deposited by magnetron sputter co-deposition (BesTec GmbH, Germany) of Ir and Ru (Fig. 1b). The base vacuum before deposition was 2 × 10−6 Pa. To increase the adhesion of thin films to the substrate, first a 5 nm thick Ti adhesion layer was deposited by e-beam evaporation of high-purity Ti (99.999%). The 3 inch targets of Ir (99.9%, Evochem, Germany) and Ru (99.99%, Evochem, Germany) were pre-cleaned by sputtering against closed shutters prior to deposition. Ar (99.999%) was used as the sputter gas and the chamber pressure was regulated to 0.5 Pa at room temperature. Deposition was performed at constant substrate rotation of 20 rpm during 60 minutes. The sputtering powers of 100 W and 50 W were applied to the Ir and Ru targets, respectively. The thickness of the coating was approximately 100 nm.
While one sample remained in the as-deposited state for further investigation, a second sample was annealed in air at 600 °C for 5 hours (Fig. 1c). This procedure was previously shown to produce oxide films with thicknesses of 30 nm and 15 nm for pure Ir and Ru thin films, respectively,5 which is beyond the probing depth of a conventional lab-source XPS (IrO2 ∼ 5.2 nm, RuO2 ∼ 3.0 nm, with a Al Kα X-ray emission source with a photon energy of 1486.6 eV). Hence, no metallic contribution to the Ir 4f and Ru 3d core levels can be detected anymore.
2.2. Scanning photoemission electron microcopy
The spectroscopy experiments were conducted with the SPEM instrument hosted at the ESCA microscopy beamline at the Elettra synchrotron facility in Trieste, Italy. The instrument can be operated in either (1) the μ-spot spectroscopy mode in which XPS spectra of selected spots on the sample can be collected or (2) the spectro-imaging mode. In imaging mode, the surface is scanned during illumination from a focused beam. At any point (pixel), photoelectrons of a given kinetic energy window are collected resulting in a map, showing the elemental or chemical distribution on the sample's surface.36,37 The analyzer hosts a 48 channel detector determining the energy range/resolution of the spectrum collected at every pixel of the map.38 For image processing, channels containing the peak- and background part were selected and their intensity ratio (i.e. intensity on-peak/intensity off-peak) is plotted as a new map representing a pure chemical contrast map with a removed topographical influence.This instrument is equipped with a Fresnel-type lens (zone plate) and an order selection aperture to demagnify the incident monochromatic X-ray beam (Fig. 1d). Spot sizes down to 120 nm can be achieved, resulting in a lateral resolution of up to 50 nm in imaging mode. The sample surface is mounted perpendicular to the incident X-ray beam. The hemispherical electron analyzer (HEA), collecting the emitted photoelectrons, is mounted at a 30° angle with respect to the probed surface, increasing the surface sensitivity by a factor of two, compared to a HEA perpendicular to the sample surface.
The spatial resolution of SPEM enables the targeted characterization of the chemical and electronic properties of the thin film catalysts on the flat top of the prepared samples (Fig. 1d). The measurements were performed at a photon energy of approx. hν = 650 eV. Binding energies are calibrated using an Au reference sample, assigning the Au 4f7/2 signal to a binding energy of 84.0 eV.
2.3. Atom probe tomography
After the samples were transported back to Germany, a protective Cr layer of 300 nm thickness was deposited on the surface by electron-beam physical vapor deposition (Fig. 1e) prior to sharpening of the specimens. Cr has proven to be a suitable coating to protect the surface of Ir-based materials.31 The chamber pressure during Cr deposition was better than 0.5 Pa (at room temperature). Needle-shaped APT specimens were then prepared by means of annular milling using a FEI Helios 600 Nanolab dual beam focused-ion beam/scanning electron microscope (FIB/SEM), as detailed in ref. 39. To measure across the Cr/Ir–Ru(-oxide) interface, and hence probe the first few atomic layers of the surface of the catalyst, special care was taken to ensure the protective Cr coating remains on top of the specimen, and ends up at the very top of the final APT specimen, as schematically shown in Fig. 1f. The APT experiments were performed on a CAMECA LEAP 5000XR reflectron-fitted instrument equipped with an ultraviolet laser with a spot size of approx. 2 μm and a wavelength of 355 nm. The ion detection efficiency of this microscope is reported to be 52%. Data was acquired in laser pulsing mode at a specimen temperature of 60 K, with an average target detection rate of 0.005 atoms per laser pulse, a pulsing rate of 125 kHz and a laser pulse energy of 80 pJ (Fig. 1g). The APT data was analyzed with the commercial software IVAS 3.8.2.3. Results
3.1. Spectromicroscopy
Photoemission spectromicroscopy was used to investigate the chemical state of the top surface of individual microtips under UHV conditions. The microscope collects spectra in a snapshot mode which substantially reduces the beam exposure time per pixel.38 This prevents radiation damage on the sample's surface that may occur in the standard μ-spot XPS mode, particularly on beam-sensitive materials such as oxides (this will be discussed in the ESI, Fig. S1†). The upper row of Fig. 2a and b shows the raw SPEM maps of the Ir 4f, Ru 3d and O 1s core level lines. The geometry of the sample limits the area available for analysis. For example, the region left of the top surface appears dark in each image because the microtip blocks the photoelectrons (PE), preventing them from reaching the analyzer (shadowing effect) whilst the right shank of the tip, facing the analyzer, appears with a higher intensity.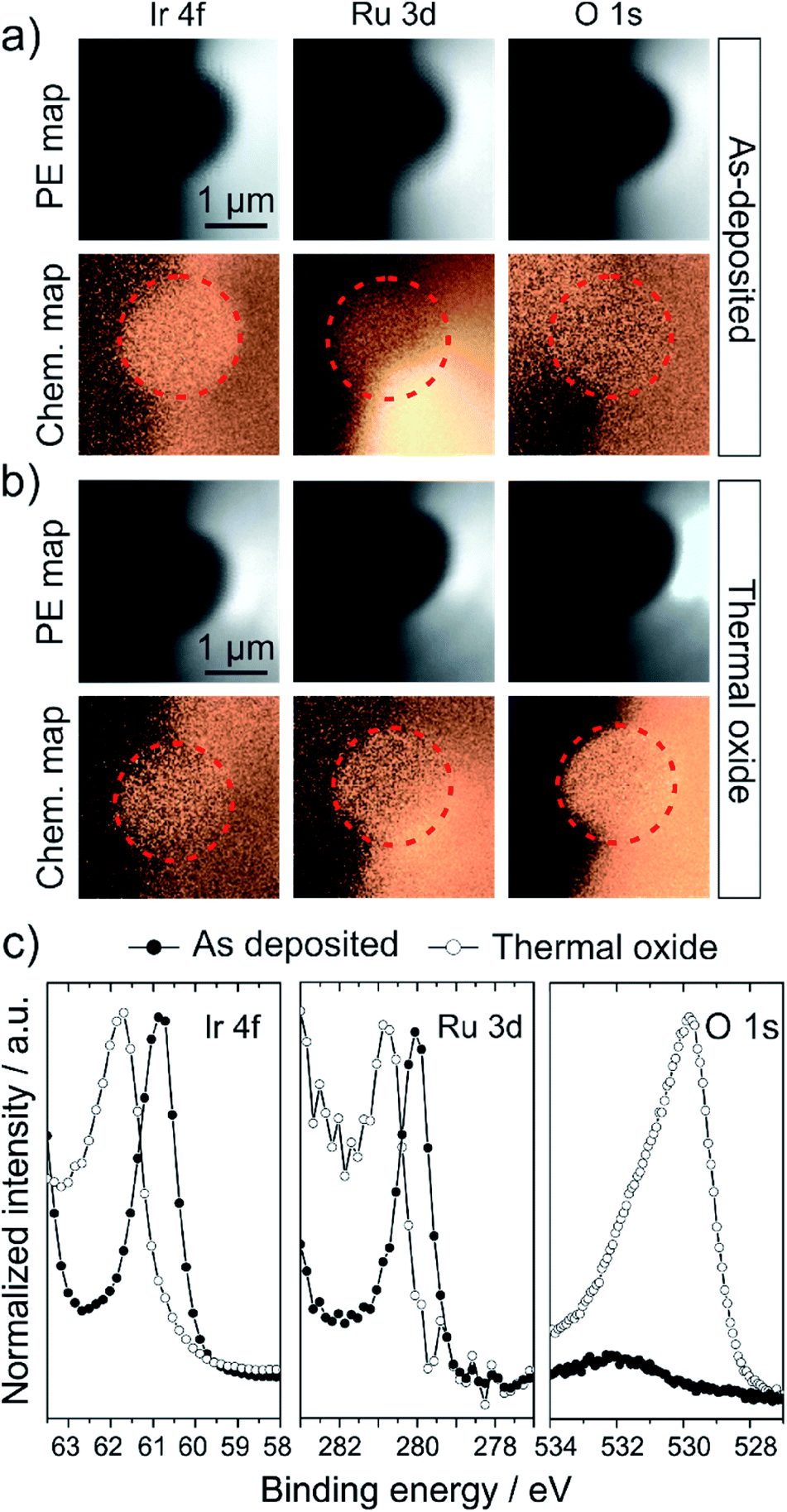 | ||
| Fig. 2 (a and b) Comparison of photoelectron (PE) and corresponding chemical maps of the as-deposited (a) and thermal oxide (b) microtip surface obtained by SPEM. Upper row of each sample shows the raw PE maps of the Ir 4f, Ru 3d and O 1s core levels. Lower row in each case compares the chemical contrast maps of the corresponding core level line, obtained by peak/background division of the raw PE signal as shown in the PE maps. Results indicate no significant lateral chemical heterogeneity on the surface in either of the specimens. (c) Comparison of the core level spectra of the Ir 4f, Ru 3d and O 1s levels of the as-deposited (filled symbols) and thermally oxidized (hollow symbols) sample extracted from the PE maps in (a) and (b). The spectra are normalized for a better comparison (Ir 4f of thermal oxide increased by a factor of 2.8, Ru 3d of thermal oxide by a factor of 3.8). Note that the presented O 1s spectra were collected in the μ-spot XPS mode and show no qualitative disagreement with the spectra from images. The metal core level binding energies shift towards higher binding energies and broaden upon oxidation. The O 1s spectrum of the oxidized sample indicates the presence of several oxygen species on the surface, however the peak is dominated by a species with a binding energy of 529.8 eV, consistent with the binding energy of IrO2 lattice oxygen. | ||
An image processing procedure was applied (peak/background; see Experimental section) to remove PE intensity differences caused by surface topology, and to provide a chemical contrast map. However, the chemical contrast maps in the lower row in each case of Fig. 2a and b show that the intrinsic intensity differences given by the tip geometry cannot always be fully accounted for. Additionally, the Ru 3d maps generally suffer from high variations due to a low signal intensity resulting from the low absolute Ru content on the surface. Hence, the analysis of the SPEM data should be restricted to the flat top surface, marked with a red circle in the chemical maps. The inspection of these areas reveals that in both cases, the as-deposited alloy and the thermal oxide, no lateral chemical heterogeneity developed on the probed length scale.
From these areas, we extracted XPS spectra from the Ir 4f7/2, Ru 3d5/2 and O 1s levels (Fig. 2c). Given the chemical uniformity of the surface, we averaged the spectroscopic signal of a maximum number of pixels belonging to the top surface in order to improve the signal-to-noise ratio in the resulting spectra. The data thereby represents an average over the entire surface of the sample. The currently available 48-channel electron detector on the SPEM instrument allows for the collection of only one peak of the Ir 4f and Ru 3d doublets with a reasonable spectral energy resolution,38 which is however sufficient to extract the qualitative information required herein.
The formation of an oxide is evident by the shift of the Ir 4f7/2 binding energy from 60.9 eV, corresponding to a metallic Ir0, to 61.8 eV commonly assigned to an IrIV oxidation state.32,40,41 This observation is in line with the binding energy shift of the Ru 3d5/2 line from 280.0 eV to 280.8 eV, indicative of the oxidation of Ru0 to RuIV.32,42,43 The O 1s spectrum of the as-deposited sample only shows a minor contribution from adsorbed O-species such as OH, H2O and CO species at binding energies >530 eV, while oxide lattice oxygen is absent. In contrast to that, the O 1s spectrum of the thermal oxide is dominated by the lattice oxygen contribution at a binding energy of 529.8 eV. This is consistent with reported binding energies of IrO2 or Ir–Ru mixed alloys with compositions below 25 at% Ru.32,44 Again, the adsorption or incorporation of additional oxygen species such as OH groups, H2O or carbonate species lead to overlapping oxygen peaks, resulting in the presented O 1s spectrum.
3.2. Atom probe tomography
The atom probe tomography reconstruction of a specimen prepared from the as-deposited sample, the top surface of which had been investigated with SPEM is shown in Fig. S2a.† A view on the cross section of a 5 nm thick slice through the 3D reconstruction (Fig. S2b†) reveals the presence of oxygen and carbon species on the top surface of the sample. Exposure of the sample to air leads to the formation of a native oxide layer on the surface and the deposition of carbon species. The high-density photon beam used during the SPEM measurements, further stimulated the deposition of carbon on the surface. The composition profile through the cross section reveals an average carbon content of up to 10 at% on the surface (see Fig. S3a† for full non-normalized composition profiles). Even though adsorbed carbon species on the surface of an APT specimen can locally alter the evaporation field of surface atoms and hence change the point of evaporation in the evaporation sequence, the unintentionally deposited carbon can also be used as a marker to locate the top surface of the specimen in an APT reconstruction. Note that all compositional profiles presented below start at an artificially defined distance of 0 nm, representing the position of the identified physical surface of the sample identified by means of adsorbed carbon species.The reported distances in the compositional profiles, calculated by means of an iso-composition surface (referred to as a proximity histogram or proxigram45), are relative to the position of the iso-surface. For convenience, the presented proxigrams also start at the identified physical surface (similar to common composition profiles) and hence do not start at 0 nm.
The reported compositions are normalized to Ir + Ru + O = 100 at% for a better comparability of the data (for complete profiles see Fig. S3†). The composition of the as deposited alloy is determined from a composition profile calculated as a function of the distance to an iso-composition surface with a threshold of 5 at% Ir (i.e. proximity histogram45 or proxigram) shown in Fig. S3c.†
The calculation of a proxigram, from an iso-composition surface with an arbitrarily defined composition threshold in the reconstructed volume, is a convenient means to extract an average composition as a function of the analysed depth.
The composition measured is Ir0.92Ru0.08, corresponding to an Ir![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ru ratio of 11.3.
:Ru ratio of 11.3.
Note that nanocrystalline materials rarely reveal sufficient retained crystallographic information to allow for accurate calibration of the tomographic reconstruction.46 The depth (z) values should hence be regarded as qualitative. Consequently, we focus on trends for data interpretation. Furthermore, the calculated iso-surface may not represent the actual specimen's surface, but rather, as described above, we utilize compositional indicators (i.e. adsorbed C species) to identify the position of the physical surface. While this approach is well suited to obtain an overall impression of the compositional changes that occurred during oxidation, it fails to capture the local chemical heterogeneities that may be of catalytic relevance. Hence, a more detailed analysis is required.
Fig. 3a presents a 3D reconstruction of an as-deposited sample with the same average bulk composition than the sample presented in Fig. S2.† Grain boundary regions are highlighted as Ru iso-density surfaces making use of the higher density of Ru atoms in the grain boundary regions. Profile 1, shown in Fig. 3b, shows that the composition along a single columnar grain remains approximately constant along the depth at an average composition of Ir0.92Ru0.08, consistent with the average composition (see Table S1†). Within the grain boundary regions (profiles 2 and 3 in Fig. 3b and c), the Ru content is generally higher than in the grains with an average composition of Ir0.78Ru0.22. This composition is, however, much less uniform than within the grains. There are regions that exhibit a much higher Ru content, with compositions of Ir0.72Ru0.28 (profile 2) or even up to Ir0.55Ru0.45 (profile 3).
 | ||
| Fig. 3 APT composition profiles along intragranular and grain boundary regions within the bulk of an as-deposited sample. (a) 3D reconstruction of evaporated volume. Grain boundary regions are highlighted by means of Ru iso-density surfaces. (b) Intragranular composition profile exhibiting a more or less uniform composition within the bulk grains of the metallic alloy. (c and d) Grain boundary region composition profiles revealing a non-uniform Ru content along these interfaces. | ||
The atom probe reconstructions of the thermal oxide specimen are shown in Fig. 4a and b (further cross sections in Fig. S4†). The qualitative inspection immediately reveals two key attributes: (1) the 3D volume comprises several grains (I–III in Fig. 4c) and (2) the non-uniform distribution of IrOx and RuOx species (see technical note on the formation of molecular species in APT in Fig. S2†) is similar to the distribution of Ir and Ru species in the as-prepared sample. This also leads to a varying average oxygen content. The large grain in the center of the volume (II) exhibits a higher average oxygen content of 68 at%, while the grain on the right-hand side in Fig. 4a and b (III) only has an average oxygen content of 17 at% (for further discussion see Fig. S4†). In the bottom of the tomograph, the oxide–metal interface can be observed. Further qualitative inspection of the cross sections through the volume in Fig. S4c and d† reveals that Ru-containing species tend to appear in close proximity to the grain boundary between two distinct oxidized grains.
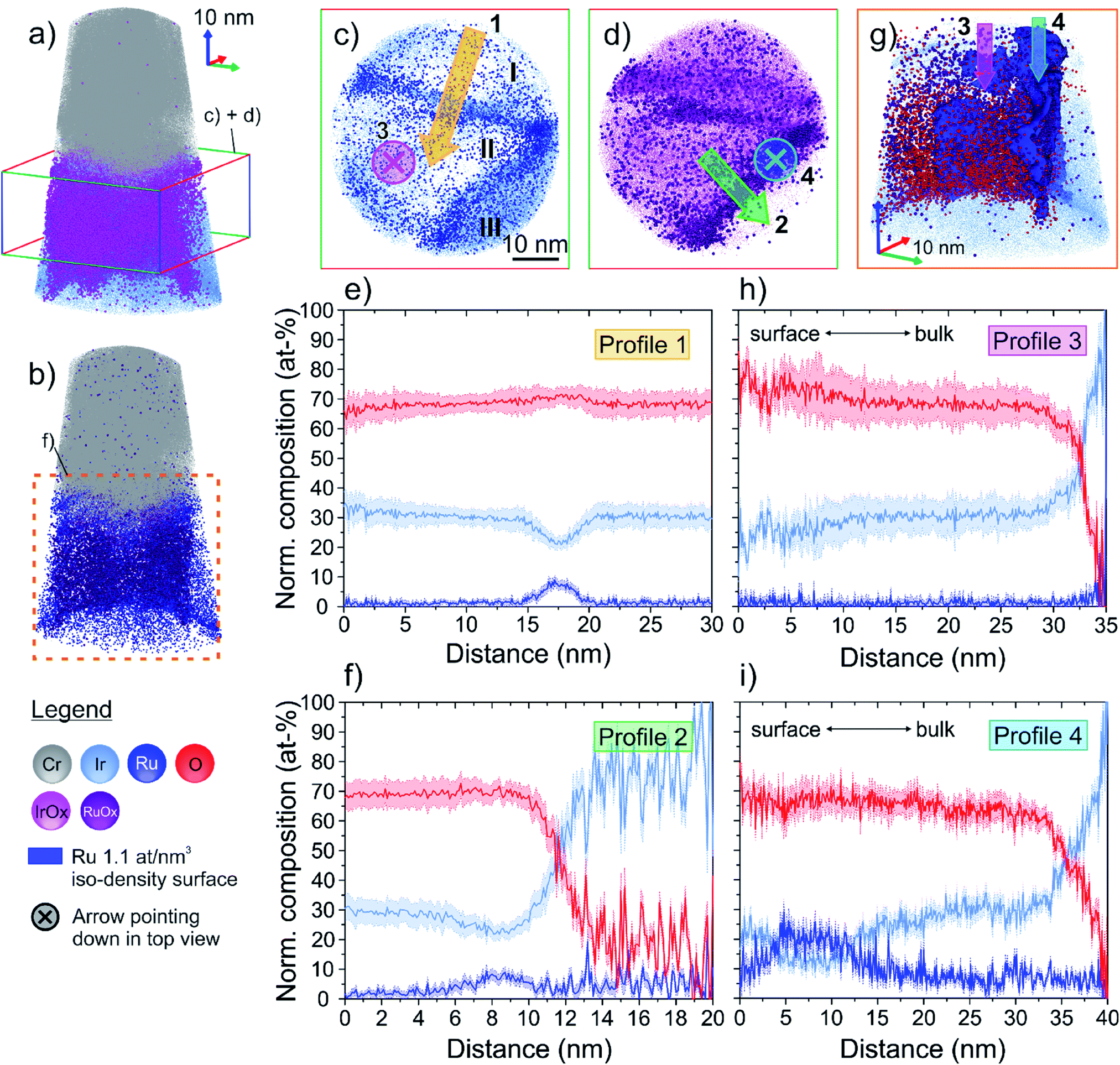 | ||
| Fig. 4 (a and b) 3D APT reconstruction of the thermally oxidized sample. The volume is comprised of several grains that exhibit different oxygen contents. The qualitative inspection reveals a heterogeneous distribution of IrOx and RuOx species within the volume. Coloured boxes indicate the regions illustrated in the top-down view images in (c) and (d), and in the close-up in (g), respectively. (c) and (d) are top-down view elemental maps of a 25 nm thick slice through the 3D volume (perpendicular to z-axis), as indicated in (a). The arrows indicate direction of the composition profiles in (e), (f), (h) and (i). The roman numerals I–III depict different grains. Two distinct grain boundaries (GB) can be identified in the atom maps by local accumulation (high density regions) of Ru, IrOx and RuOx species, respectively. (e) Profile 1 shows the composition across one of the identified GBs. While the composition in both bulk oxide grains are essentially the same, the GB exhibits an elevated Ru content. (f) Profile 2 shows the composition across the second identified GB ranging from the central oxide grain into the less oxidized grain in the bottom right in (c) and (d). Again, the GB reveals similar Ru enrichment and simultaneous local Ir depletion similar to the GB probed with profile 1. (g) Close-up of the 3D reconstruction marked in (b). GB regions are illustrated as Ru iso-density surface. Qualitatively, one can see the higher oxygen content in the central oxide grain, as compared to the less oxidized grain presented in profile 2. (h) Profile 3 shows a vertical composition profile within the central oxide grain. The Ir:Ru ratio remains approximately constant along the profile while the oxygen content increases marginally towards the surface. (i) Profile 4 is measured within the Ru iso-density surface. It evidences the very pronounced chemical inhomogeneities occurring along this microstructural defect, similar to what was previously observed in grain boundaries of the as-prepared sample (Fig. 3). | ||
To investigate the average chemical composition of the volume along the z-axis, a proxigram was calculated as a function of the distance to a 5 at% Ir iso-composition surface. The proxigram starts at 1.3 nm distance below the iso-composition surface (Fig. S5a†), which marks the location of the physical surface identified by the occurrence of adsorbed carbon (see Fig. S3b†). Based on the calculated Ir:Ru ratio along this profile (Fig. S5b†), the specimen can be divided into four compositionally distinct zones. Starting from the bottom of the sample, the interface metal–oxide can be seen at a distance of about 28 nm from the iso-composition surface. Above this interface, a thick compositionally uniform zone is observed, ranging from 11.7 to 28.0 nm distance from the Ir iso-composition surface. An average (Ir + Ru):O ratio in this region of 0.47 indicates a nearly stoichiometric (Ir,Ru)IVO2 composition of the oxide (Fig. S5b†). The oxide in this depth interval has an average composition of Ir0.92Ru0.08O2, coinciding with the initial alloy composition in terms of the Ir-to-Ru ratio. The top surface region can further be divided into two compositionally distinct zones. The lower zone, ranging from 4.4–11.7 nm below the iso-composition surface, exhibits a significantly lower Ir:Ru ratio of 6.89, resulting in a composition of Ir0.87Ru0.13O2 in this region. The very top surface region, 1.3–4.3 nm below the iso-composition surface, is again marginally richer in Ir with an average Ir:Ru ratio of 8.01, with an average composition of Ir0.89Ru0.11O2.
Again, the proxigram analysis provides an average picture of the composition of the sample, however, it may not capture localized chemical non-uniformities related to microstructural features within the material. To this end, we again turn to a more detailed analysis by making use of the high spatial resolution of APT, and we compare intragranular regions with grain boundary regions.
The coloured boxes in Fig. 4a and b indicate the analysed regions in Fig. 4c–i. Fig. 4c and d are top-down views on the 25 nm thick slice through the reconstructed dataset, perpendicular to the z-axis. This perspective allows three different grains (I–III) to be distinguished. The ion maps illustrate that the interfaces between those grains are mainly decorated with Ru and RuOx species. Fig. 4e reveals the composition profile across the grain boundary between grains I and II. Please note that the statistical measurement error in localized compositional profiles like these is generally significantly larger than in proxigrams, due to the limited size of the probed volume (i.e. limited number of atoms). While the oxygen content remains essentially the same on both sides of the grain boundary, one can see a clear Ru enrichment in the grain boundary of up to 9 at% (in matrix: 1 at%). At the same time, the Ir content drops to a minimum of 21 at%, with an average of 30 at% away from the grain boundary. The oxygen content of both grains is approximately 68 at%, consistent with the oxygen content of the stoichiometric oxide region derived from the proxigram. The averaged compositions are hence Ir0.96Ru0.04O2 within the grain, and up to approximately Ir0.70Ru0.30O2 in the grain boundary, coinciding with the local compositions of the non-oxidized sample in terms of the Ir-to-Ru ratio.
Furthermore, the boundary/interface between grain II and III also exhibits an increased average Ru content of up to 9 at%, while Ir is depleted with a minimum of 22 at% (Ir0.71Ru0.29O2), similar to the grain boundary in profile 1.
A close-up of the region marked with the dashed square in Fig. 4b, is presented in Fig. 4g. This allows for a closer investigation of the chemical composition along the grain boundary, depicted as Ru iso-point-density surface (1.1 at% per nm3). Looking edge-on onto the grain boundary between the centre and bottom grain in Fig. 4c, one can again observe the higher concentration of oxygen ions on the left side of this boundary.
To better understand how the composition varies with depth, two compositional profiles along the z-direction are presented in Fig. 4h and i. Their positions within the volume, and the profile directions are illustrated as arrows in Fig. 4c and d. A closer inspection of the oxide composition from the bottom of the grain towards the surface, shown in Fig. 4h, reveals a relatively uniform Ir-to-Ru ratio within the entire grain. Again, towards the surface the oxygen content increases with a simultaneous increase of the statistical error in this region, reaching a maximum oxygen content of 78 at% close to the surface. The average composition of this oxide grain, however, can be summarized as Ir0.94Ru0.06O2. This point will be discussed further in ESI Fig. S3.† Contrary to the bulk grain, the composition of the formed oxide in the grain boundary changes significantly from the bottom towards the surface, as depicted in Fig. 4i. As observed previously, the overall Ru content is higher at the interface than in a bulk oxide grain. While the oxygen content remains constant along the boundary with approximately 66 at%, the relative amounts of Ir and Ru change significantly. Within the first 20 nm below the surface, the oxide composition changes from approximately Ir0.75Ru0.25O2 at 18 nm depth, to Ir0.40Ru0.60O2 at 6 nm depth and eventually to Ir0.70Ru0.30O2 at the top-surface.
4. Discussion
4.1. Validation of proposed experimental protocol
The major objective of our protocol is to combine techniques that provide (I) a surface and chemical-state sensitivity to investigate the catalytically relevant top surface of a material, including the oxidation states, and (II) a high spatial resolution to capture spatially confined nanoscale compositional variations.The chemical maps in Fig. 2a and b did not reveal any lateral heterogeneities across the investigated flat top surface. This apparent uniformity of the surface is mainly due to the small grain size of the sputtered thin films which ranges from 5–20 nm. This small grain size consequently results in chemical inhomogeneities on a nanometer scale during oxidation, e.g. solute surface segregation along grain boundaries. As a comparison, Fig. 4c shows that the final cross-sectional area of the analyzed APT volume which is slightly smaller than the spot size of the photon beam during spectromicroscopy (about 120 nm). Localized nano-scale chemical heterogeneities evidenced by APT cannot be identified with SPEM, or other comparable XPS techniques, as the size of those domains falls below its spatial resolution.
The determined surface metal oxidation states of the as-deposited and thermally oxidized samples are consistent with the average surface compositions, as determined by APT, particularly in terms of the stoichiometry of the oxides. Furthermore, the O 1s binding energy measured on the oxide is consistent with an Ir–Ru mixed oxide with a Ru content < 25 at%,32 which we confirm with atom probe. This consistency underlines the strength of the approach of combining APT and SPEM.
In addition, the APT data presented in Fig. 3 reveal a more detailed picture of the non-uniform distribution of Ru within the microstructure of the sputter-deposited thin film alloy. The grain boundary environment appears to be an energetically favorable location for Ru to segregate during the deposition. The amount of Ru segregation can reach up to 45 at% locally, which is beyond the theoretically predicated thermodynamic solubility of Ru in Ir at room temperature (i.e. within the two-phase region).47 Hence, the grain boundaries can be locally partially to fully saturated with Ru. Yet, additional X-ray or electron diffraction data would be required to prove the occurrence of phase separation, and the local formation of a Ru-rich Ru–Ir hcp alloy.
The on average rather Ir-rich surface composition observed in the as-prepared sample reflects the preferential surface configuration of this alloy under vacuum conditions, which can be rationalised by the lower surface energy of Ir as compared to Ru.48,49 In the presence of a gaseous adsorbate, it is known that the surface compositions of alloys can substantially change, or be inverted,35,50–52 as discussed below.
As this sample was subjected to several hours of thermal oxidation at 600 °C in air, there are a number of parallel processes that may take place. As schematically illustrated in Fig. 5a, (1) oxygen adsorbs on the surface of the sample and preferentially penetrates the material through the grain boundaries, which eventually leads to the nucleation and consecutive growth of an oxide. As the oxygen supply is maintained at elevated temperature, the oxide growth front progresses deeper into the material (2). Calculated APT oxygen depth profiles demonstrate that the oxidation front reaches deeper into the material along the grain boundary than within the adjacent bulk oxide grains (Fig. S6†). The oxidation process likely proceeds through grain boundary diffusion of atomic oxygen supplied from the gaseous O2 phase. Once an Ir–Ru mixed oxide is formed in the grain boundary, Ru is stabilized in this location as the RuO2 is entirely miscible in IrO2,53 as opposed to the metallic Ir–Ru system. The presence of an oxygen gas atmosphere drives adsorbate-induced Ru segregation (3) by the strength of the Ru–O bond relative to the Ir–O bond,54 and the potential formation of volatile RuO3 species. However, no pronounced Ru surface enrichment can be observed in the thermally oxidized sample. Even in the regions where diffusion is expected to be the fastest, namely in the triple junction region in Fig. 4c, no pronounced Ru surface enrichment is observed. Moreover, as diffusion is generally enhanced at elevated temperatures, lateral diffusion of dissolved bulk Ru to the grain boundary is conceivable (4). However, we have no experimental evidence for this diffusion from the composition profiles, i.e. no evidence of a diffusion-type of composition profile towards the surface nor Ru depletion adjacent to the boundary. We therefore conclude that either diffusion of Ru in Ir is slow under the experimental conditions (i.e. low diffusivity), or Ru is not thermodynamically driven to segregate to boundaries in the case where diffusion is fast (i.e. high diffusivity).
 | ||
| Fig. 5 (a) Schematic illustration of potential processes occurring along a grain boundary during thermal oxidation: (1) oxygen adsorption on the surface and penetration of the material (absorption) through grain boundaries. (2) Nucleation of an oxide and progression of the oxidation front into the depth of the material. Oxidation front progresses faster along grain boundary, hence reaches deeper, as further shown in Fig. S6.† (3) Surface segregation of Ru driven by the higher free enthalpy of formation of the Ru–O bond, relative to the Ir–O bond. (4) Grain boundary segregation of bulk Ru. (b) Schematic overview of compositionally different regions in thermal oxide sample as a function of depth. UTS = upper top surface; LTS = lower top surface. Left column shows average compositions extracted from a proxigram analysis. The right two columns show the average compositions that were extracted from more localized analyses along an oxide grain and a grain boundary (GB) region, respectively. The arrows on the right indicate the depth at which the composition was measured. Note the strong local deviations between the average (proxigram) compositions and the compositions within the GB. These local deviations from an average composition stress the need for spatially resolved methods to investigate such materials. | ||
In summary, we argue that the oxidation kinetics and the progression of the oxidation front into the material is much faster than the possible diffusional Ru (surface) segregation under the experimental conditions. The oxidation front progressively moves into the material, driving the nucleation and consequent growth of the mixed oxide through the grain boundaries which act as preferential diffusion pathway for oxygen.
XPS, as a method that is well established in catalysis and surface science research, is able to assess the electronic structure and chemistry of surfaces. The spatial resolution in the presented case provides the average chemical state of the surface probed in an atom probe specimen; valuable information that APT is not able to assess. Unlike comparable photoelectron-based microscopy techniques such as classical X-ray photoemission electron microscopy (XPEEM), SPEM imposes less constraints on the measureable sample geometry and surface roughness. Despite XPEEM can provide a higher spatial resolution than SPEM (5.4–18 nm)55,56 it requires atomically flat surfaces to achieve that, which limits application of this method to ideal surfaces. Further, SPEM can be operated under near-ambient pressure conditions, while XPEEM is commonly restricted to UHV conditions.57,58 A major drawback of SPEM is the previously discussed high photon density which bears the risk of damaging sensitive materials (see Fig. S1†). Scanning Auger electron microscopy may be an alternative/complement to SPEM, allowing for the investigation of the lateral distribution of surface oxidation states with a similar surface sensitivity (∼2 nm).59
APT, on the other hand, is a technique among few that can assess the chemical composition of the specimen with a very high spatial resolution. Similar high elemental sensitivities can be reached by e.g. high resolution secondary ion mass spectrometry (nanoSIMS), however, the spatial resolution is limited to approximately 50 nm and to 2D analyses.60 Any three dimensional analysis in a TEM, e.g. tomography that can provide atomically precise reconstructions,61 requires a material that can withstand the highly energetic electron beam. In this context, amorphous Ir-oxides have proven to be rather unstable during TEM analysis though.62
The results from SPEM and APT in the presented case have yielded consistent results. This shows that despite the different probing length scales of the two techniques, they assess the same chemical information. This underlines the validity of the proposed combination of experiments.
4.2. Relevance for OER catalysis
Previous studies on Ir–Ru mixed oxides have shown their superior electrocatalytic activity in OER compared to IrO2.32–34 However, due to the low corrosion resistance of Ru under OER conditions in acidic media, Ru is preferentially dissolved from the bulk. Kasian et al. reported that, due to the loss of Ru in the near-surface region, the activity of the mixed oxide approaches the value corresponding to IrO2.32 The goal of the optimization of these catalysts is to find a sweet spot between high activity and longevity.The currently accepted theory is that IrO2 forms a stable but less active top surface layer on mixed oxides during OER, slowing down the further dissolution of Ru.32,63 This structure can either be achieved by surface segregation of Ir during OER, or by leaching out Ru from the top surface, leaving behind an Ir oxide skeleton. The exact mechanism is difficult to experimentally assess and remains a matter of dispute in the literature. Danilovic et al.33 suggested to use surface segregation as a tool to tailor the surface configuration of metallic Ir–Ru alloys prior to thermal oxidation. Annealing of the initial alloy in UHV leads to the surface segregation of Ir. The IrO2 layer that forms upon consecutive thermal oxidation reduces the Ru dissolution during OER, compared to a non-annealed control sample, however at the expense of a lower OER activity. In our experiments we have not observed any major surface segregation of any of the elements as a result of the applied oxidation conditions.
The current experimental data suggests that the oxide stoichiometry is changing laterally and with depth. This change results in laterally varying reactivity and stability of the catalyst on the surface, but also with depth as the material is corrodes. Material that was originally part of the subsurface can become exposed to the electrolyte and the harsh reaction conditions. Regions with higher Ru content will generally exhibit a higher reactivity and lower stability. The original reactivity in the grain boundary regions, for instance, may further increase if the top surface with a composition of Ir0.70Ru0.30O2 partially dissolves, and more Ru-rich sub-surface regions become exposed, having compositions near Ir0.40Ru0.60O2. We anticipate that the corrosion rate is limited by the supply of Ru from the near-surface region which is expected to be slow at room temperature. Dissolved Ru leaves behind an Ir oxide skeleton which may now decelerate further dissolution of Ru. Yet, the availability of Ru in the near-surface region likely controls the thickness of the leached zone. It remains to be investigated how the mechanical integrity is maintained as the leached zone becomes thicker, and what catalytic properties the possibly less crystalline Ir oxide skeleton exhibits as the reaction progresses.
Changing the surface configuration by the manipulation of the treatment/synthesis parameters prior to OER, we consider to be a promising means to engineer the immediate and long-term performance of a mixed-oxide catalyst. For instance, different thin film deposition conditions (e.g. at elevated temperatures), or heat treatments of the alloys prior to oxidation, offer possibilities to tune the microstructure and local chemistry of the material. Moreover, the temperature and/or oxygen partial pressure in the reaction atmosphere during the thermal oxidation process, may alter the diffusion and oxidation kinetics. An enhanced temperature at lower oxygen partial pressure could, for example, favor the diffusion of Ru within the microstructure of the catalyst before an oxide is formed. Likewise, the kinetics of the oxide growth govern the final oxide grain orientation which is developed at the sample surface. Our data also suggests the formation of differently oxidized grains within one sample, however, to date little is known about the grain orientation dependence of Ir–Ru alloys on the OER activity or corrosion resistance. Coordinatively under-saturated surface sites are generally considered to play a major role in the dissociation and adsorption of water molecules during OER.64–66 Most studies were performed on either oriented oxide samples or single crystals, and the authors collectively reported a dependence of the metal surface oxidation on the crystallographic orientation of the surface, although some results are contradicting.64–67 Nevertheless, regarding the design of new catalysts, one should consider tuning the crystallographic orientation as an additional engineering lever towards better and more sustainable catalyst properties.
The proposed experimental approach is well suited to provide insights into the discussed phenomena in the near-surface region of a material resulting from surface patterning prior to OER. The spatial resolution of these methods offers the opportunity to target specific regions of interest on the surface, and relate the local observations to the underlying microstructure of the material. This combination enables mechanistic insights into e.g. the oxidation process and facilitates the optimization of materials synthesis strategies.
4.3. Outlook
So far, the proposed experimental protocol has proven to deliver complementary information on the chemical changes on a catalyst's surface and its underlying microstructure. The sample design is amenable to withstand the conditions during the experiments.Moving forward, the protocol can now be extended to push the current limitations of the involved methodologies. In the present experiments, the small grain size of the sputter deposited thin films have certainly entailed certain challenges in terms of resolving different chemical surface domains with SPEM. Our APT results have revealed major chemical heterogeneities along microstructural defects (particularly grain boundaries), which unfortunately remained undetected by SPEM. The assessment of the chemical state within the domains on the surface is crucial to better understand the laterally varying adsorbate binding energetics. Hence, we will aim to use model alloys with coarser grains in future experiments that allow the resolution of individual grain boundaries at the surface. Larger grains will facilitate the investigation of the impact of the orientation of individual grains and the misorientation between grains on the formation of oxides or their reaction behavior in OER, by e.g. correlative pre-reaction SEM-based electron backscattered diffraction measurements. The insights gained on the distribution of distinct chemical domains on the surface and their relation to the microstructure of the material can be used to improve the catalyst design and pre-treatment. At last, we expose the engineered catalysts to OER reaction conditions to validate the expected properties gained from the nanoscale materials insights by correlating them to their electrochemical properties.
The sample design is not limited to a thermal oxidation treatment prior to SPEM and APT. The silicon substrate is electrically conductive and well suited to be used as working electrode in an electrochemical cell. This structure offers the opportunity to apply the experimental protocol to electrocatalytic interfaces before and after electrochemical treatment. Our preliminary attempts to perform this kind of experiment have suffered from severe carbon surface contamination after electrochemical treatments. This contamination has prevented us from collecting reliable Ru 3d core level spectra due to the overlap with the C 1s binding energy. Improvements in the experimental protocol are required, namely (1) the prompt preparation of electrochemical oxides before SPEM measurement and/or (2) that the samples are not exposed to air in-between measurements. This can be realized by performing the electrochemical experiments in a glove-box before the samples are moved to UHV/inert gas sample transport containers. Keeping the sample in a protected atmosphere or in high vacuum along the entire experimental chain may reduce the amount of adsorbed carbon species on the surface significantly. Alternatively, the entire electrochemical treatment could be performed in situ which, however, imposes further experimental challenges.
The flexibility of the proposed sample design and workflow offers the opportunity to apply it to other heterogeneous catalysts. The identification and characterization of localized surface features that determine the catalytic properties require a spatial resolution offered by the combination of the proposed methods. Particularly alloy catalyst for gas phase or electrocatalysis present a vast space of research opportunities to answer fundamental questions.
5. Conclusions
In this work we presented a correlative microscopy approach to investigate catalytic surfaces with a high surface sensitivity and at the near-atomic scale. The developed experimental strategy involves scanning photoemission electron microscopy (SPEM) and atom probe tomography (APT) applied on the identical location on a sample surface. This involves a sample design developed to suit electrochemical or thermal oxidation treatments (or similar), and all involved consecutive experimental steps. We show exemplary on an Ir–Ru thermal mixed oxide that SPEM provides surface oxidation states that are consistent with determined atomic-scale composition obtained from APT. Furthermore, we showed intrinsic chemical heterogeneities in sputter-deposited Ir–Ru thin films. Ruthenium segregates to the grain boundaries during the deposition, resulting in a large difference between the nominal and intragranular composition vs. the local composition at the defect, and also fluctuating Ir-to-Ru ratios along such defect structures within the material. We stress that such nanoscale non-uniformities can easily be missed when applying methods with a lower spatial resolution. By comparison to the untreated sample we conclude, that the observed varying oxide compositions along the grain boundary is governed by the intrinsic heterogeneous Ru distribution. Temperature induced diffusion during the thermal oxidation only had a minute effect on the redistribution of Ru. The oxidation kinetics was much faster under the experimental conditions with the oxidation front moving through the material at a high pace, freezing the inherent local Ir-to-Ru ratios by the formation of a mixed oxide. We point out the importance of understanding the microstructure and chemical inhomogeneities of sputter-deposited films for use in electrocatalytic applications, since these may impact the catalyst's performance in OER. We stress with our experiments the need to understand fundamental processes during every step of catalyst preparation and operation at the nanoscale to (1) be able to establish solid structure–function relationships and (2) exploit certain processes as engineering means to tune a catalyst's properties towards a better performance and sustainability.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors wish to thank Elettra Sincrotrone for the allocation of beamtime at the ESCAmicroscopy beamline. The research leading to these results has received funding from the European Community's Horizon 2020 Framework Programme under grant agreement no. 730872. K. S. acknowledges IMPRS-SurMat for funding. U. Tezins and A. Sturm are acknowledged for their technical support at the FIB/APT facilities at MPIE. O. K. acknowledges support from the Helmholtz-Zentrum Berlin GmbH and from the Helmholtz Association Initiative and Networking Fund.References
- A. S. Aricò, S. Siracusano, N. Briguglio, V. Baglio, A. Di Blasi and V. Antonucci, J. Appl. Electrochem., 2013, 43, 107–118 CrossRef.
- V. R. Stamenkovic, D. Strmcnik, P. P. Lopes and N. M. Markovic, Nat. Mater., 2016, 16, 57 CrossRef PubMed.
- G. Monforte, S. Siracusano, V. Baglio and A. S. Aricò, Meeting Abstracts, MA2015-02, 2015, p. 1431 Search PubMed.
- S. Siracusano, N. Hodnik, P. Jovanovic, F. Ruiz-Zepeda, M. Šala, V. Baglio and A. S. Aricò, Nano Energy, 2017, 40, 618–632 CrossRef CAS.
- S. Cherevko, S. Geiger, O. Kasian, N. Kulyk, J.-P. Grote, A. Savan, B. R. Shrestha, S. Merzlikin, B. Breitbach, A. Ludwig and K. J. J. Mayrhofer, Catal. Today, 2016, 262, 170–180 CrossRef CAS.
- S. K. Makineni, A. Kumar, M. Lenz, P. Kontis, T. Meiners, C. Zenk, S. Zaefferer, G. Eggeler, S. Neumeier, E. Spiecker, D. Raabe and B. Gault, Acta Mater., 2018, 155, 362–371 CrossRef CAS.
- S. K. Makineni, M. Lenz, S. Neumeier, E. Spiecker, D. Raabe and B. Gault, Scr. Mater., 2018, 157, 62–66 CrossRef CAS.
- M. Kuzmina, M. Herbig, D. Ponge, S. Sandlöbes and D. Raabe, Science, 2015, 349, 1080–1083 CrossRef CAS PubMed.
- B. Gault, F. de Geuser, L. Bourgeois, B. M. Gabble, S. P. Ringer and B. C. Muddle, Ultramicroscopy, 2011, 111, 683–689 CrossRef CAS PubMed.
- S. Kumar Makineni, S. Sugathan, S. Meher, R. Banerjee, S. Bhattacharya, S. Kumar and K. Chattopadhyay, Sci. Rep., 2017, 7, 11154 CrossRef PubMed.
- R. K. W. Marceau, G. Sha, R. Ferragut, A. Dupasquier and S. P. Ringer, Acta Mater., 2010, 58, 4923–4939 CrossRef CAS.
- M. Herbig, Scr. Mater., 2018, 148, 98–105 CrossRef CAS.
- M. Herbig, D. Raabe, Y. J. Li, P. Choi, S. Zaefferer and S. Goto, Phys. Rev. Lett., 2014, 112, 126103 CrossRef CAS PubMed.
- A. Kwiatkowski da Silva, G. Leyson, M. Kuzmina, D. Ponge, M. Herbig, S. Sandlöbes, B. Gault, J. Neugebauer and D. Raabe, Acta Mater., 2017, 124, 305–315 CrossRef CAS.
- M. J. Yao, E. Welsch, D. Ponge, S. M. H. Haghighat, S. Sandlöbes, P. Choi, M. Herbig, I. Bleskov, T. Hickel, M. Lipinska-Chwalek, P. Shanthraj, C. Scheu, S. Zaefferer, B. Gault and D. Raabe, Acta Mater., 2017, 140, 258–273 CrossRef CAS.
- B. Gault, A. J. Breen, Y. Chang, J. He, E. A. Jägle, P. Kontis, P. Kürnsteiner, A. Kwiatkowski da Silva, S. K. Makineni, I. Mouton, Z. Peng, D. Ponge, T. Schwarz, L. T. Stephenson, A. Szczepaniak, H. Zhao and D. Raabe, J. Mater. Res., 2018, 33, 4018–4030 CrossRef CAS.
- P. Kontis, Z. Li, D. M. Collins, J. Cormier, D. Raabe and B. Gault, Scr. Mater., 2018, 145, 76–80 CrossRef CAS.
- T. Schwarz, G. Stechmann, B. Gault, O. Cojocaru-Mirédin, R. Wuerz and D. Raabe, Prog. Photovoltaics, 2018, 26, 196–204 CAS.
- K. Babinsky, R. De Kloe, H. Clemens and S. Primig, Ultramicroscopy, 2014, 144, 9–18 CrossRef CAS PubMed.
- K. P. Rice, Y. Chen, T. J. Prosa and D. J. Larson, Microsc. Microanal., 2016, 22, 583–588 CrossRef CAS PubMed.
- T. Li, P. A. J. Bagot, E. A. Marquis, S. C. E. Tsang and G. D. W. Smith, J. Phys. Chem. C, 2012, 116, 4760–4766 CrossRef CAS.
- T. Li, P. A. J. Bagot, E. A. Marquis, S. C. E. Tsang and G. D. W. Smith, J. Phys. Chem. C, 2012, 116, 17633–17640 CrossRef CAS.
- T. Li, P. A. J. Bagot, E. A. Marquis, S. C. Edman Tsang and G. D. W. Smith, Ultramicroscopy, 2013, 132, 205–211 CrossRef CAS PubMed.
- P. A. J. Bagot, K. Kruska, D. Haley, X. Carrier, E. Marceau, M. P. Moody and G. D. W. Smith, J. Phys. Chem. C, 2014, 118, 26130–26138 CrossRef CAS.
- T. V. de Bocarmé, M. Moors, N. Kruse, I. S. Atanasov, M. Hou, A. Cerezo and G. D. W. Smith, Ultramicroscopy, 2009, 109, 619–624 CrossRef PubMed.
- P. Felfer, P. Benndorf, A. Masters, T. Maschmeyer and J. M. Cairney, Angew. Chem., Int. Ed., 2014, 53, 11190–11193 CrossRef CAS PubMed.
- K. Tedsree, T. Li, S. Jones, C. W. A. Chan, K. M. K. Yu, P. A. J. Bagot, E. A. Marquis, G. D. W. Smith and S. C. E. Tsang, Nat. Nanotechnol., 2011, 6, 302 CrossRef CAS PubMed.
- T. Li, P. A. J. Bagot, E. Christian, B. R. C. Theobald, J. D. B. Sharman, D. Ozkaya, M. P. Moody, S. C. E. Tsang and G. D. W. Smith, ACS Catal., 2014, 4, 695–702 CrossRef CAS.
- S.-H. Kim, P. W. Kang, O. O. Park, J.-B. Seol, J.-P. Ahn, J. Y. Lee and P.-P. Choi, Ultramicroscopy, 2018, 190, 30–38 CrossRef CAS PubMed.
- S.-H. Kim, J. Y. Lee, J.-P. Ahn and P.-P. Choi, Microsc. Microanal., 2019, 1–9, DOI:10.1017/S1431927618015556.
- T. Li, O. Kasian, S. Cherevko, S. Zhang, S. Geiger, C. Scheu, P. Felfer, D. Raabe, B. Gault and K. J. J. Mayrhofer, Nat. Catal., 2018, 1, 300–305 CrossRef.
- O. Kasian, S. Geiger, P. Stock, G. Polymeros, B. Breitbach, A. Savan, A. Ludwig, S. Cherevko and K. J. J. Mayrhofer, J. Electrochem. Soc., 2016, 163, F3099–F3104 CrossRef CAS.
- N. Danilovic, R. Subbaraman, K. C. Chang, S. H. Chang, Y. Kang, J. Snyder, A. P. Paulikas, D. Strmcnik, Y. T. Kim, D. Myers, V. R. Stamenkovic and N. M. Markovic, Angew. Chem., Int. Ed., 2014, 53, 14016–14021 CrossRef CAS PubMed.
- V. A. Saveleva, L. Wang, W. Luo, S. Zafeiratos, C. Ulhaq-Bouillet, A. S. Gago, K. A. Friedrich and E. R. Savinova, J. Phys. Chem. Lett., 2016, 7, 3240–3245 CrossRef CAS PubMed.
- M. T. Greiner, J. Cao, T. E. Jones, S. Beeg, K. Skorupska, E. A. Carbonio, H. Sezen, M. Amati, L. Gregoratti, M.-G. Willinger, A. Knop-Gericke and R. Schlögl, ACS Catal., 2018, 8, 2286–2295 CrossRef CAS.
- M. Amati, B. Aleman, B. Bozzini, L. Gregoratti, H. Sezen and M. Kiskinova, Surf. Sci., 2016, 652, 20–25 CrossRef CAS.
- P. Zeller, M. Amati, H. Sezen, M. Scardamaglia, C. Struzzi, C. Bittencourt, G. Lantz, M. Hajlaoui, E. Papalazarou, M. Marino, M. Fanetti, S. Ambrosini, S. Rubini and L. Gregoratti, Phys. Status Solidi A, 2018, 215, 1800308 CrossRef.
- L. Gregoratti, A. Barinov, E. Benfatto, G. Cautero, C. Fava, P. Lacovig, D. Lonza, M. Kiskinova, R. Tommasini, S. Mähl and W. Heichler, Rev. Sci. Instrum., 2004, 75, 64–68 CrossRef CAS.
- K. Thompson, D. Lawrence, D. J. Larson, J. D. Olson, T. F. Kelly and B. Gorman, Ultramicroscopy, 2007, 107, 131–139 CrossRef CAS PubMed.
- V. Pfeifer, T. E. Jones, J. J. Velasco Vélez, C. Massué, R. Arrigo, D. Teschner, F. Girgsdies, M. Scherzer, M. T. Greiner, J. Allan, M. Hashagen, G. Weinberg, S. Piccinin, M. Hävecker, A. Knop-Gericke and R. Schlögl, Surf. Interface Anal., 2016, 48, 261–273 CrossRef CAS.
- V. OrPfeifer, T. E. Jones, J. J. Velasco Velez, C. Massue, M. T. Greiner, R. Arrigo, D. Teschner, F. Girgsdies, M. Scherzer, J. Allan, M. Hashagen, G. Weinberg, S. Piccinin, M. Havecker, A. Knop-Gericke and R. Schlogl, Phys. Chem. Chem. Phys., 2016, 18, 2292–2296 RSC.
- K. S. Kim and N. Winograd, J. Catal., 1974, 35, 66–72 CrossRef CAS.
- R. Kötz, H. J. Lewerenz and S. Stucki, J. Electrochem. Soc., 1983, 130, 825–829 CrossRef.
- R. Kötz, H. Neff and S. Stucki, J. Electrochem. Soc., 1984, 131, 72–77 CrossRef.
- O. C. Hellman, J. A. Vandenbroucke, J. Rüsing, D. Isheim and D. N. Seidman, Microsc. Microanal., 2000, 6, 437–444 CrossRef CAS PubMed.
- B. Gault, M. P. Moody, J. M. Cairney and S. P. Ringer, Mater. Today, 2012, 15, 378–386 CrossRef CAS.
- H. Okamoto, J. Phase Equilib., 1992, 13, 565–567 CrossRef CAS.
- S. Zafeiratos, S. Piccinin and D. Teschner, Catal. Sci. Technol., 2012, 2, 1787 RSC.
- L. Vitos, A. V. Ruban, H. L. Skriver and J. Kollár, Surf. Sci., 1998, 411, 186–202 CrossRef CAS.
- S. Piccinin, C. Stampfl and M. Scheffler, Surf. Sci., 2009, 603, 1467–1475 CrossRef CAS.
- B. C. Han, A. Van der Ven, G. Ceder and B.-J. Hwang, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72(20), 205409 CrossRef.
- J. A. Herron and M. Mavrikakis, Catal. Commun., 2014, 52, 65–71 CrossRef CAS.
- E. N. Balko and C. R. Davidson, J. Inorg. Nucl. Chem., 1980, 42, 1778–1781 CrossRef CAS.
- K. W. Frese, Surf. Sci., 1987, 182, 85–97 CrossRef CAS.
- R. Koenenkamp, Aberration correction in photoemission microscopy and applications in photonics and plasmonics, United States, 2017 Search PubMed.
- T. Schmidt, A. Sala, H. Marchetto, E. Umbach and H. J. Freund, Ultramicroscopy, 2013, 126, 23–32 CrossRef CAS PubMed.
- H. Sezen, B. Alemán, M. Amati, M. Dalmiglio and L. Gregoratti, ChemCatChem, 2015, 7, 3665–3673 CrossRef CAS.
- H. Sezen, M. Al-Hada, M. Amati and L. Gregoratti, Surf. Interface Anal., 2018, 50, 921–926 CrossRef CAS.
- E. Martinez, P. Yadav, M. Bouttemy, O. Renault, Ł. Borowik, F. Bertin, A. Etcheberry and A. Chabli, J. Electron Spectrosc. Relat. Phenom., 2013, 191, 86–91 CrossRef CAS.
- K. L. Moore, E. Lombi, F.-J. Zhao and C. R. M. Grovenor, Anal. Bioanal. Chem., 2012, 402, 3263–3273 CrossRef CAS PubMed.
- Y. Yang, C.-C. Chen, M. C. Scott, C. Ophus, R. Xu, A. Pryor, L. Wu, F. Sun, W. Theis, J. Zhou, M. Eisenbach, P. R. C. Kent, R. F. Sabirianov, H. Zeng, P. Ercius and J. Miao, Nature, 2017, 542, 75–79 CrossRef CAS PubMed.
- E. Willinger, C. Massué, R. Schlögl and M. G. Willinger, J. Am. Chem. Soc., 2017, 139, 12093–12101 CrossRef CAS PubMed.
- V. A. Saveleva, L. Wang, D. Teschner, T. Jones, A. S. Gago, K. A. Friedrich, S. Zafeiratos, R. Schlogl and E. R. Savinova, J. Phys. Chem. Lett., 2018, 9, 3154–3160 CrossRef CAS PubMed.
- C. Roy, R. R. Rao, K. A. Stoerzinger, J. Hwang, J. Rossmeisl, I. Chorkendorff, Y. Shao-Horn and I. E. L. Stephens, ACS Energy Lett., 2018, 3, 2045–2051 CrossRef CAS.
- R. R. Rao, M. J. Kolb, J. Hwang, A. F. Pedersen, A. Mehta, H. You, K. A. Stoerzinger, Z. Feng, H. Zhou, H. Bluhm, L. Giordano, I. E. L. Stephens and Y. Shao-Horn, J. Phys. Chem. C, 2018, 122, 17802–17811 CrossRef CAS.
- E. Özer, C. Spöri, T. Reier and P. Strasser, ChemCatChem, 2017, 9, 597–603 CrossRef.
- M. Scohy, S. Abbou, V. Martin, B. Gilles, E. Sibert, L. Dubau and F. Maillard, ACS Catal., 2019, 9(11), 9859–9869 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ta10818a |
| This journal is © The Royal Society of Chemistry 2020 |