
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMeeting key synthetic challenges in amanitin synthesis with a new cytotoxic analog: 5′-hydroxy-6′-deoxy-amanitin†
Alla
Pryyma
 ,
Kaveh
Matinkhoo
,
Antonio A. W. L.
Wong
and
David M.
Perrin
*
,
Kaveh
Matinkhoo
,
Antonio A. W. L.
Wong
and
David M.
Perrin
*
Department of Chemistry, University of British Columbia, 2036 Main Mall, Vancouver, B.C. V6T 1Z1, Canada. E-mail: dperrin@chem.ubc.ca
First published on 15th October 2020
Abstract
Appreciating the need to access synthetic analogs of amanitin, here we report the synthesis of 5′-hydroxy-6′-deoxy-amanitin, a novel, rationally-designed bioactive analog and constitutional isomer of α-amanitin, that is anticipated to be used as a payload for antibody drug conjugates. In completing this synthesis, we meet the challenge of diastereoselective sulfoxidation by presenting two high-yielding and diastereoselective sulfoxidation approaches to afford the more toxic (R)-sulfoxide.
Introduction
α-Amanitin, the deadliest of the amatoxins produced by the death-cap mushroom Amanita phalloides, is a potent, orally available inhibitor of RNA polymerase II (pol II) (Ki ∼ 10 nM),1 that has been validated as a payload for targeted cancer therapy.2 First described in 1907![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 3 and isolated in 1941,4 α-amanitin is a compact bicyclic octapeptide that has been indispensable for probing RNA pol II-catalysed transcription in eukaryotes. While the toxin makes numerous contacts with RNA pol II, the lack of synthetic access has limited the ability to generate discrete analogs to probe these contacts, most notably the role of the 6′-hydroxyl group on the indole. Of the few naturally occurring amatoxins wherein the 6′-OH is replaced by H, amaninamide and amanin are only 25–50% as toxic as the natural product (Fig. 1) indicating moderate importance of the OH group.5
3 and isolated in 1941,4 α-amanitin is a compact bicyclic octapeptide that has been indispensable for probing RNA pol II-catalysed transcription in eukaryotes. While the toxin makes numerous contacts with RNA pol II, the lack of synthetic access has limited the ability to generate discrete analogs to probe these contacts, most notably the role of the 6′-hydroxyl group on the indole. Of the few naturally occurring amatoxins wherein the 6′-OH is replaced by H, amaninamide and amanin are only 25–50% as toxic as the natural product (Fig. 1) indicating moderate importance of the OH group.5
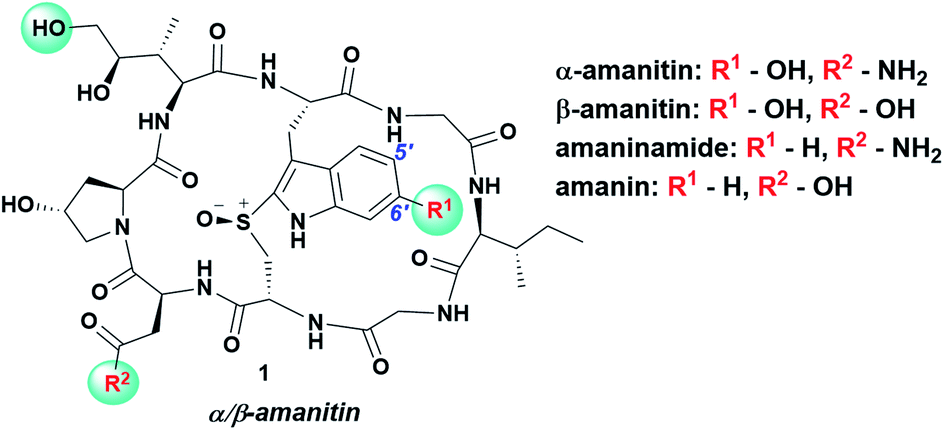 | ||
| Fig. 1 Chemical structure of α/β-amanitin and structure–activity relationships of natural and synthetic amatoxins with indole modifications; a 2–4 fold-increase in Ki as a result of indole modification is relative to the 6′-OH containing counterparts; commonly used conjugation sites are highlighted. | ||
Its putative contribution to affinity is understood from two apparent interactions seen in a recent co-crystal structure of α-amanitin-RNA pol II: (i) the 6′-hydroxyl group forms an H-bond with Ser-782 accounting for the difference in Ki's between mammalian and yeast RNA pol II; (ii) the hydroxyl group increases the electron density of the indole which forms a cation–π interaction with Arg-749.6 Besides contributing to toxicity, the 6′-OH provides a preferred handle for chemical functionalization for grafting to antibodies.
Recently, α-amanitin has been showcased as a highly effective payload in antibody drug conjugates (ADCs).2a Unlike other ADC payloads, α-amanitin targets both dividing and quiescent cells and shows potential for preventing cancer relapse from drug-tolerant cell subpopulations.7 Examples include ADCs for targeting human epidermal growth factor receptor 2 and prostate specific membrane antigen.8 With but a few exceptions,9 nearly all bioconjugates to a cytotoxic amanitin have emerged from naturally-sourced α-amanitin. To date, conjugation handles used for α-amanitin-based bioconjugates include the δ-hydroxyl of (2S,3R,4R)-4,5-dihydroxyisoleucine (DHIle),2a the asparagine side chain,10 and the 6′-hydroxyl of the tryptathionine staple.11 Of these, increasingly, the latter has been favored as a site for bioconjugation to the intact toxin.
As biosynthetic fermentation is currently low-yielding,12 synthesis will likely serve as the main means for accessing large amounts of toxin needed to address structure–activity relationships and provide new handles for bioconjugation. This heightened interest in α-amanitin has inspired three total syntheses13 that address synthetic challenges embedded in three key components: (i) an enantioselective synthesis of DHIle, (ii) creation of an oxidatively challenged 6′-OH-tryptathionine-sulfoxide that demands formal oxidation at positions 2 and 6 of the indole, and (iii) stereoselective oxidation of the thioether to give (R)-sulfoxide, which is approximately 8-fold more toxic than the (S)-sulfoxide.13a
In 2018, Perrin et al. opted for a 6-boronated-L-tryptophan as a masked phenol equivalent that underwent oxidative fluorocyclization followed by Savige–Fontana reaction to form the tryptathionine staple14 with the pendent boronate.13a Oxidative deborylation, peptide elongation, and macrocyclization gave the S-deoxy-amanitin that was readily sulfoxidized with moderate diastereoselectivity (ca. 2:1) favoring the (R)-sulfoxide.
In 2020, Süssmuth et al. described a convergent “5 + 1 + 2” strategy whereby the tryptathionine staple was synthesized first by reacting protected cysteine-sulfenyl chloride with orthogonally protected 6-OBn-L-tryptophan. The tryptathionine was then elaborated to a pentapeptide followed by addition of DHIle and Pro–Asn and macrolactamization.13b A third report advanced a scalable synthesis of 6-OAc-Trp, and utilized Savige–Fontana reaction14 to introduce the 6′-OH-tryptathionine staple.13c While all three reports provide access to 6′-OH-tryptathionine, they noted the severe challenges of selective oxidative thiolation of the electron-rich indole.
In contrast to the immediate interest in accessing the 6′-hydroxyl group, interest in generating the (R)-sulfoxide remains academic. Nevertheless, because Nature exclusively produces the (R)-sulfoxide, logic suggests that the (R)-sulfoxide may provide privileged pharmacological/toxicological properties. As such, diastereoselective access of the (R)-sulfoxide continues to pose a significant synthetic challenge that was met with but moderate selectivity in the first total synthesis and remained unprogressed in the two subsequent ones.
Despite these synthetic efforts, there remains a demand for biologically active and more synthetically accessible α-amanitin analogs that: (i) capture the putative π–cation interaction for enhanced affinity and (ii) provide an indole-situated conjugation handle for ADC development. Towards these ends, herein we report the synthesis of a constitutional isomer of near-native toxicity that introduces a 5′-OH-tryptathionine, which is readily achieved from cheap, commercially available 5-OH-L-tryptophan (5-OH-Trp).
In addition, we identify new methodologies to create the (R)-sulfoxide with high diastereoselectivity (>15:1) along with the (S)-sulfoxide diastereomer and sulfone for comparison. Anticipating success with chemoselective conjugation akin to aforementioned conjugates to the 6′-OH-indole, this work now expands the toolbox of available synthetic amanitin analogs for consideration as payloads for use in translational applications.
Results
In approaching the synthetic challenge of creating a hydroxy-tryptathionine staple, we appreciated the high natural abundance of 5-OH-tryptophan in a number of biosynthetic pathways along with its commercial availability as a single enantiomer. Hence, we hypothesized that 5-OH-L-tryptophan could be introduced in lieu of the synthetically-demanding 6-OH-L-tryptophan. We also reasoned that the 5′-hydroxyl could also engage in an H-bonding interaction with Ser-782. A docking study validated this hypothesis: the 5′-OH-6′-deoxy-amanitin (2) was docked into the RNA pol II using Autodock Vina molecular docking program.15The most energetically favorable pose of 5′-OH-6′-deoxy-amanitin was compared to the crystal structure pose of α-amanitin (Fig. 2). Further assessment using PyMOL Molecular Graphics System confirmed that the aforementioned key interactions between the enzyme and the indole ring are still achievable and the modified indole does not alter the overall confirmation of the fit.16 Importantly, the docking pose suggested that the 5′-hydroxyl group should still form the anticipated H-bond with Ser-782 (Fig. 2, green line). Based on these observations, we reasoned that the incorporation of 5-OH-tryptophan should maintain the toxicity of the new amanitin analog.
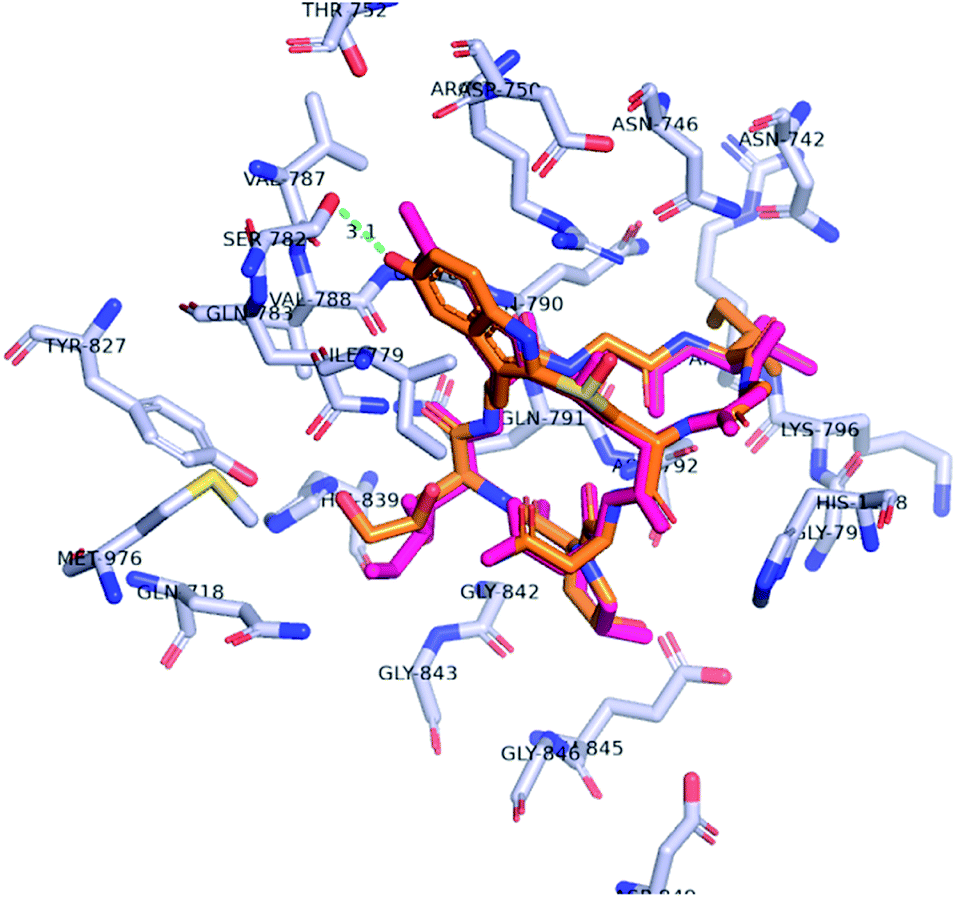 | ||
| Fig. 2 Docking pose of 5′-OH-6′-deoxy-amanitin 2 showing favorable interactions with RNA-pol II; 5′-OH-6′-deoxy-amanitin 2 – orange, α-amanitin 1 – magenta, green dashed line – distance between Ser-782 and indole alcohol. | ||
Encouraged by this result, we undertook a synthesis of 5′-OH-6′-deoxy-amanitin (2) that would rely on two key components: (i) introduction of the fully protected DHIle-NHS ester as previously reported, and (ii) synthesis and characterization of the 3a-fluoro-pyrroloindoline produced from 5-OH-tryptophan that could be subjected to late-stage Savige–Fontana reaction to give the 5′-OH-tryptathionine (Scheme 1). In addition to this synthetic feat, we addressed mid- and late-stage sulfoxidation with high diastereoselectivity to afford the desired (R)-sulfoxide.
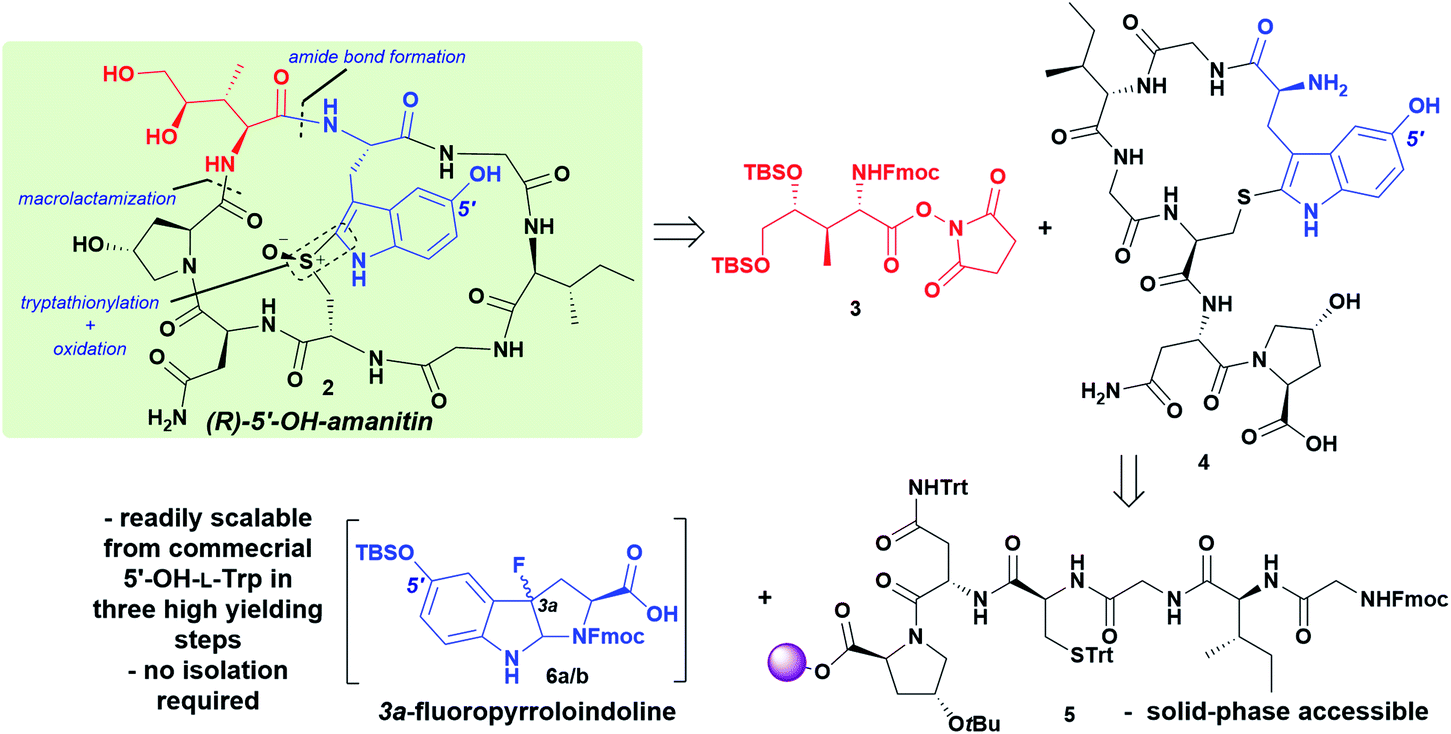 | ||
| Scheme 1 Retrosynthesis of 5′-OH-6′-deoxy-amanitin 2, showing major disconnection sites and key intermediates. DHIle-NHS ester 3 is coupled to the monocyclic 5′-OH-heptapeptide 4 obtained from a 5-OH-tryptophan-derived FPI precursor 6a/b and solid-phase accessible hexapeptide 5. | ||
Following the approach used in the first total synthesis of α-amanitin13a and supported by more recent work on the oxidative fluorocyclization of tryptophan to give fluoropyrroloindoline (FPI) for Savige–Fontana tryptathionylation,17 we started with commercially available 5-OH-tryptophan. Following Fmoc-protection, oxidative fluorocyclization with N-fluoro-2,4,6-trimethylpyridinium triflate (FP-T300) gave a diastereomeric pair of fluoropyrroloindolines (syn-cis/anti-cis). Unfortunately, these decomposed upon isolation, presumably due to the electron-rich indole ring (data not shown). To mitigate this problem, we employed a facile protection of the hydroxyl group as a tert-butyldimethylsilyl ether that greatly improved the stability of the diastereomeric pair of FPI's 6a/b. Thus, TBS-protected 8 was treated with FP-T300 in DCM to yield 6a/b as a diastereomeric mixture in 87% yield (Scheme 2, 6a/bsyn-cis/anti-cis, 1.8:1).
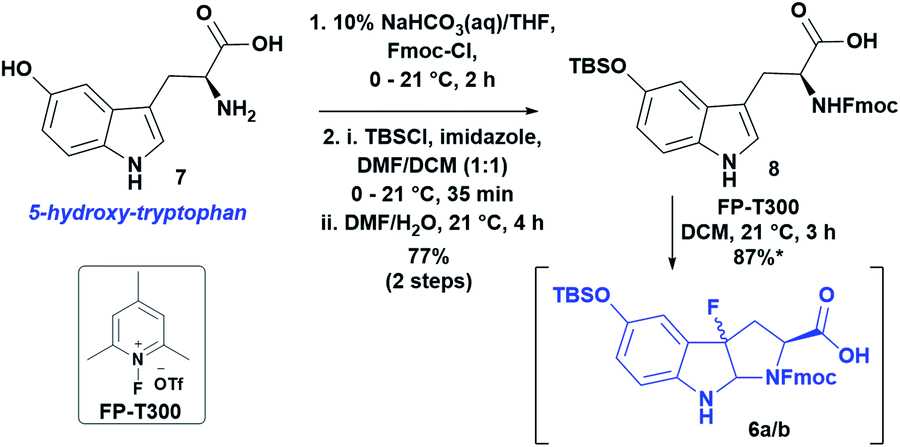 | ||
| Scheme 2 Synthesis of Nα-Fmoc-FPI(5-OTBS)-OH (6a/b) as a pair of diastereomers; *NMR yield. | ||
With fluorocyclization proceeding in high yield, we directly coupled the diastereomeric mixture 6a/b to hexapeptide 5 that had been prepared on the solid-phase using standard Fmoc solid-phase peptide synthesis conditions (Scheme 3). It is easily appreciated that the 1.8:1 diastereomeric distribution of the FPI precursor is immaterial since the stereochemistry is lost during the Savige–Fontana reaction. Fmoc-deprotection, followed by TFA/DCM (5:1) treatment resulted in global deprotection, resin cleavage, and thioetherification (Savige–Fontana reaction) to afford the desired 5′-OH-tryptathionine-containing monocycle 4. The monocyclic heptapeptide 4 was coupled with the fully protected NHS-ester of DHIle (3).13a Subsequent Fmoc-deprotection revealed a free amine, while TBS deprotection decreased steric encumbrance to facilitate macrocyclization that successfully effected 11, the penultimate and first appreciably toxic species that would await sulfoxidation to give the target (R)-5′-OH-6′-deoxy-amanitin (Scheme 3).
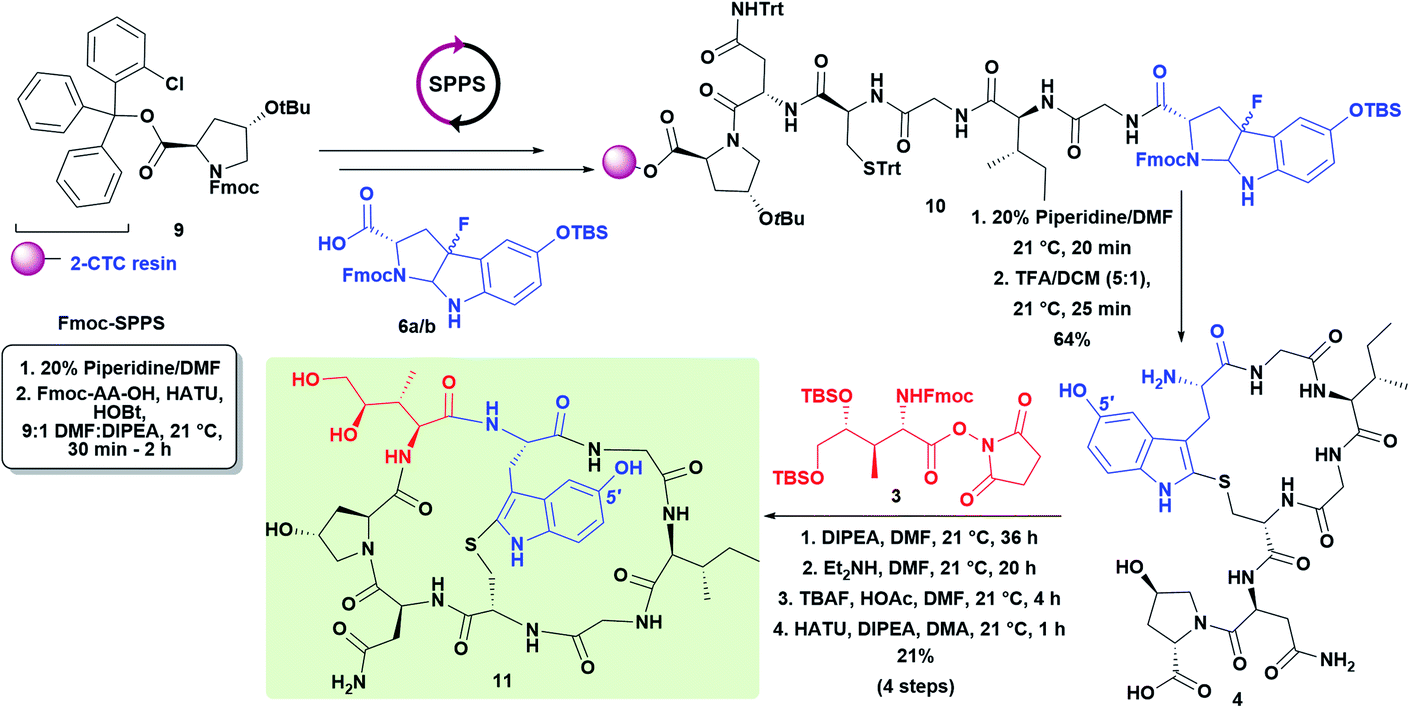 | ||
| Scheme 3 Synthesis of 5′-OH-tryptathionine and incorporation of DHIle to give bicyclic precursor 11. | ||
With a robust synthetic approach to a new thioether analog with equal toxicity compared to S-deoxy-amanitin (vide infra), the final obstacle towards 5′-OH-6′-deoxy-amanitin was the diastereoselective sulfoxidation of the tryptathionine bridge to the (R)-configured sulfoxide. Of note, in the α-amanitin series, the (R)-sulfoxide is the most potent of the toxins, followed by the equipotent sulfone and thioether and then the unnatural (S)-sulfoxide that shows 8-fold reduced potency.18 The relationship between the stereochemical configuration of the sulfoxide and the biological activity has fascinated scientists for decades as numerous efforts fail to provide a structural rationalization for this intriguing difference in toxicity (vide infra, discussion).5b,18,19
Achieving diastereoselective sulfoxidation still poses an unmet synthetic challenge in this context. Previously, it was shown that oxidation of the thioether with H2O2 in acetic acid produces sulfoxides in a 1:2 ratio favoring the (S)-diastereomer.5b,20 In the 2018 report, m-chloroperbenzoic acid (mCPBA)-mediated sulfoxidation successfully yielded the (R)-sulfoxide in modest ∼2:1 excess over (S)-sulfoxide. Inexplicably, selectivity could be tuned by varying the reaction solvent; iPrOH/EtOH and tBuOH/EtOH co-mixtures were effective at providing moderate selectivity for the desired (R)-sulfoxide while polar solvents (e.g. DMF, HOAc, CF3CH2OH) greatly favored the (S)-sulfoxide.13a,c
In order to verify that 11 would undergo similar sulfoxidation as S-deoxy-amanitin, we commenced our investigation by subjecting it to mCPBA treatment in iPrOH/EtOH (2:1) (Fig. 3). The HPLC and mass spectrometry analysis of the reaction revealed full consumption of 11 after one hour and formation of three distinct products in a ratio of 4.4:4.6:1, which were identified by mass spectrometry as two sulfoxides and a sulfone (see Fig. S2 in ESI†). This transformation was accompanied by a predictable change in the UV-Vis absorbance signifying a modification of the chromophore. The first of the two sulfoxides eluting from the HPLC was tentatively assigned as the desired (R)-sulfoxide based on the shorter HPLC retention time observed for α-amanitin with respect to its (S)-sulfoxide diastereomer. It is well-known that the (S)-sulfoxides in a series of amanitin analogs are significantly less potent inhibitors of RNA pol II in vitro, compared to the corresponding (R)-sulfoxides5c (although previous reports did not provide unambiguous structural elucidation and relied only on correlations with chromatographic retention times and toxicity to assign stereochemistry).
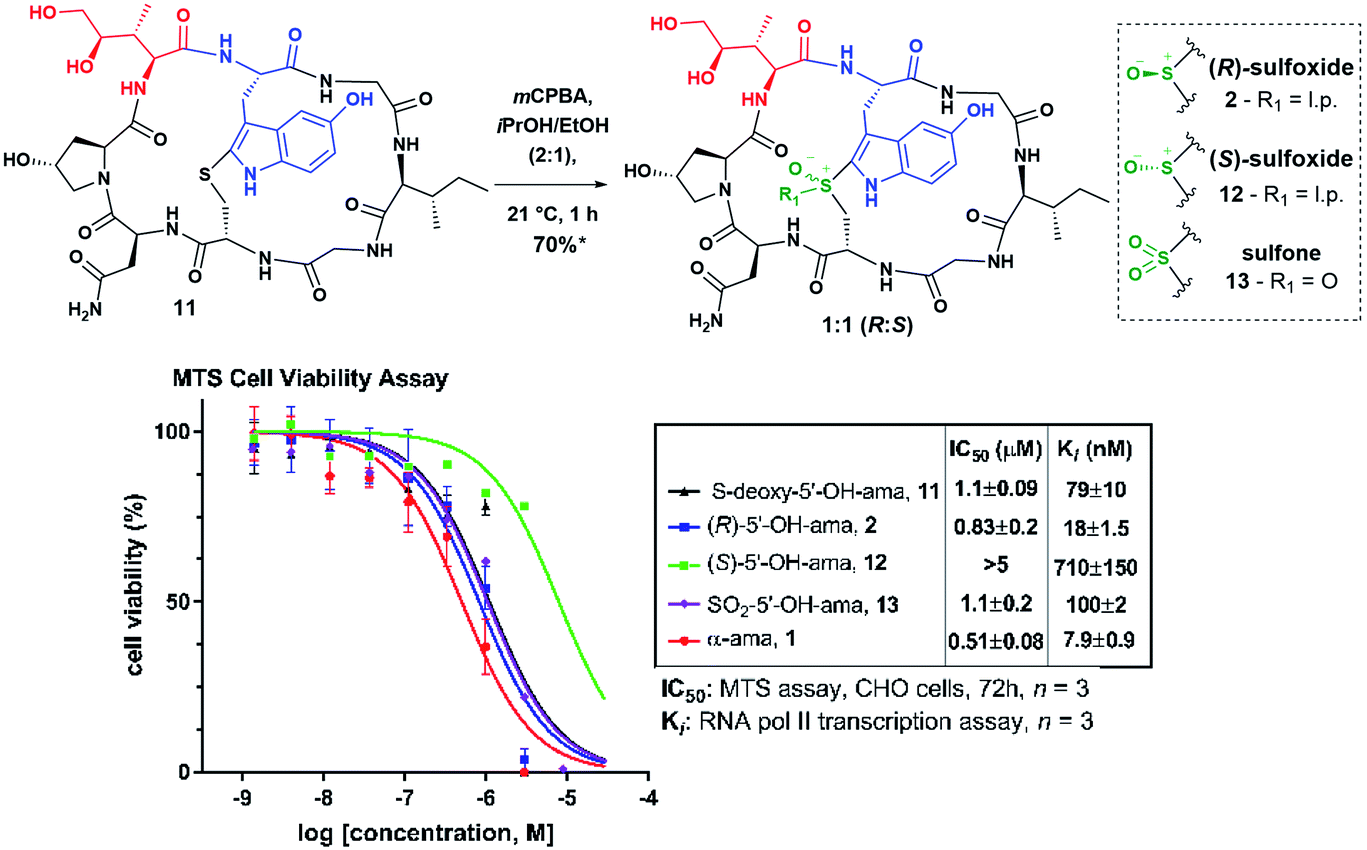 | ||
| Fig. 3 MTS and RNA pol II assay results of oxidized products; *yield in sulfoxide; IC50: CHO cells, MTS, 72 h, n = 3; Ki: RNA pol II transcription assay, n = 3. | ||
Applying the correlation between chromatographic retention times of these compounds and authentic α-amanitin to make a tentative stereochemical assignment, we then assessed the biological activity of the new amanitin analogs 2 and 11–13 using MTS colorimetric cell viability and RNA pol II transcription assays (Fig. 3). To our delight, MTS assay on Chinese Hamster Ovary (CHO) cells revealed that 2 and 11–13 showed toxicities in keeping with the chromatographic elution profile observed for the (R)-, (S)-sulfoxides and the sulfone of α-amanitin (Fig. 3) and leading us to assign a tentative (R)-designation to the sulfoxide born on 2.
Whereas 2 was nearly as cytotoxic as α-amanitin itself, strikingly, the putative (S)-sulfoxide 12 showed significantly reduced cytotoxicity even compared to (S)-sulfoxide of α-amanitin.13a,18,21 While many synthetic amanitin analogs reported in the recent studies have relied solely on IC50 values from cytotoxicity assays as a measure of activity,9,22 such analysis assumes equal cellular uptake. To obtain a direct measure of activity against the intracellular target RNA pol II, we performed an in vitro transcription assay using HeLa-Scribe nuclear extract transcription system and [32P]-α-rGTP. Interestingly, the Ki values for compounds 2 and 11–13 were spread over a far greater range compared to the IC50 values (Fig. 3). Thioether 11 and sulfone 13 showed similar affinity (79 ± 10 and 100 ± 2.2 nM, respectively), while (R)-sulfoxide 2 was approximately five-fold more active (18 ± 1.5 nM) and the (S)-sulfoxide 12 showed greatly reduced inhibitory activity (710 ± 150 nM) compared to (R)-sulfoxide 2.
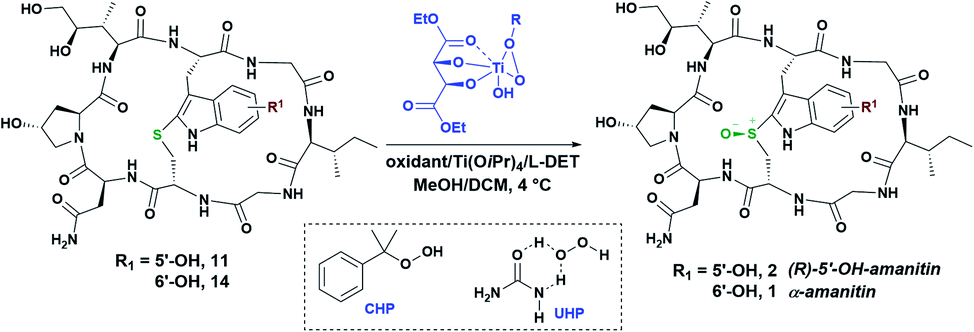
The near-native toxicity of 2 would suggest significant potential for use as an ADC payload and prompted us to develop diastereoselective sulfoxidation methods in anticipation of future translational needs. Hence, we turned our attention to catalytic sulfoxidation as it represents one of the dominant methods for enantioselective sulfoxidation. One of the earliest and most convenient methods for asymmetric sulfoxidation employs titanium tetra-isopropoxide [Ti(OiPr)4] and (+)-diethyl-L-tartrate (L-DET) as a ligand.23 The initial sulfoxidation screening in the presence of Ti(OiPr)4 and L-DET was carried out with two different oxidizing agents, cumene hydroperoxide (CHP) and urea hydroperoxide (UHP), in MeOH:DCM (1:1) at 4 °C (Table 1, entries 1–3). Use of CHP as an oxidant yielded (S)-sulfoxide in excess (Table 1, entry 1) while UHP gave the (R)-sulfoxide in 2.6-fold excess when used in the presence of L-DET:Ti(OiPr)4 (Table 1, entry 2). In the absence of L-DET:Ti(OiPr)4, UHP gave the (S)-sulfoxide in 2.8-fold excess (Table 1, entry 3). When L-DET:Ti(OiPr)4 were used in 4:1 ratio, the diastereoselectivity was improved to 95:5 (R:S) when applied to 100-nmol (Table 1, entry 4) (lower selectivity was observed on a 5-nmol scale). Use of excess L-DET ligand with respect to Ti(OiPr)4 (4:1 ratio) in 1:1 MeOH/DCM at 4 °C over 20 hours (Table 1, entry 4), yielded the desired (R)-sulfoxide in 71% yield and without over-oxidation to the sulfone. Nevertheless, approximately 25% of thioether 11 remained unconsumed after 20 hours. Increasing the amount of UHP in the presence of L-DET:Ti(OiPr)4 (8:1) and using DCM as the major solvent improved both the yield and the diastereoselectivity (5-nmol scale, Table 1, entry 5). To illustrate the utility of this protocol in production of α-amanitin, we subjected S-deoxy-amanitin 14 to these optimized conditions. Gratifyingly, the (R)-sulfoxide was obtained in 84% yield and with high diastereoselectivity (Table 1, entry 6 and Fig. 4). The product identity was confirmed by HPLC co-injection with an authentic sample of α-amanitin, illustrating the first case of a highly diastereoselective access to α-amanitin (see Fig. S7, ESI†).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Conditions | Substrate | (R:S) |
11 or 12 (%) | Yield (%) |
|
a (R:S) distribution was determined by HPLC.
b HPLC sulfoxide yield.
c Isolated yield.
|
|||||
| 1 | CHP/Ti(OiPr)4/L-DET (1:1:2), DCM/MeOH (1:1), 17 h |
11 | 1:1.1 |
39 | 61b |
| 2 | UHP/Ti(OiPr)4/L-DET (1:1:2), DCM/MeOH (1:1), 17 h |
11 | 2.6:1 |
44 | 66b |
| 3 | UHP, DCM/MeOH (1:1), 17 h |
11 | 1:2.8 |
20 | 80b |
| 4 |
UHP/Ti(OiPr)
4
/
L
-DET (1.2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :0.8:3.2), DCM/MeOH (1:1), 20 h :0.8:3.2), DCM/MeOH (1:1), 20 h
|
11 |
19:1
|
25 | 71 |
| 5 | UHP/Ti(OiPr)4/L-DET (2.5:1:8), DCM/MeOH (3:1), 17 h |
11 | 6.4:1 |
4 | 96b |
| 6 |
UHP/Ti(OiPr)
4
/
L
-DET (2.5:1:8), DCM/MeOH (3:1), 17 h
|
14 |
>50:1
|
16 | 84 |
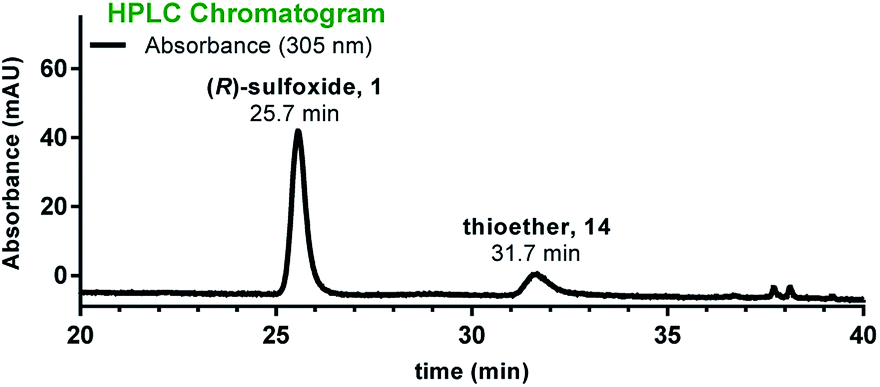 | ||
| Fig. 4 HPLC chromatogram of the sulfoxidation reaction run in the presence of Ti(OiPr)4 and L-DET showing formation of α-amanitin 1 as the major diastereomer along with unreacted thioether 14. | ||
While these results represent the first-ever successful approach to a highly diastereoselective installation of the (R)-sulfoxide in any amanitin analog, we wondered if there could be yet other means of accessing the desired (R)-sulfoxide without resorting to chiral catalysis. Appreciating that the monocyclic heptapeptide intermediate 4 likely adopts a different geometry compared to the bicycle 11, we hypothesized that the sulfoxidation of this intermediate may offer a unique opportunity for inducing the desired (R)-configuration at an earlier stage in the synthesis. We started our initial screening of the sulfoxidation with mCPBA in iPrOH/EtOH (2:1); two products eluted from the HPLC one minute apart. The first product, designated as compound 15, appearing to be a single diastereomer, was found to be a sulfoxide by MS but of unknown stereochemistry, while the second proved to be an inseparable mixture of the second sulfoxide and sulfone 16 (Fig. 5). To reduce the amount of sulfoxide/sulfone by-products, solvent and temperature screening was performed (Table S4,† entries 1–16). The lowest amount of the by-products was obtained with dimethylformamide (DMF) as the solvent (Table S4,† entry 11). The sulfoxidation was further optimized by varying the reaction temperature (Table S4,† entries 14–16). Temperatures below 0 °C were found to be optimal for suppressing the production of sulfoxide/sulfone. Running the reaction in DMF at −20 °C on a 1-μmol scale yielded the first sulfoxide in 5-fold excess over the second sulfoxide and inseparable sulfone (Fig. 5). This protocol resulted in complete consumption of the thioether 4 in less than 5 minutes.
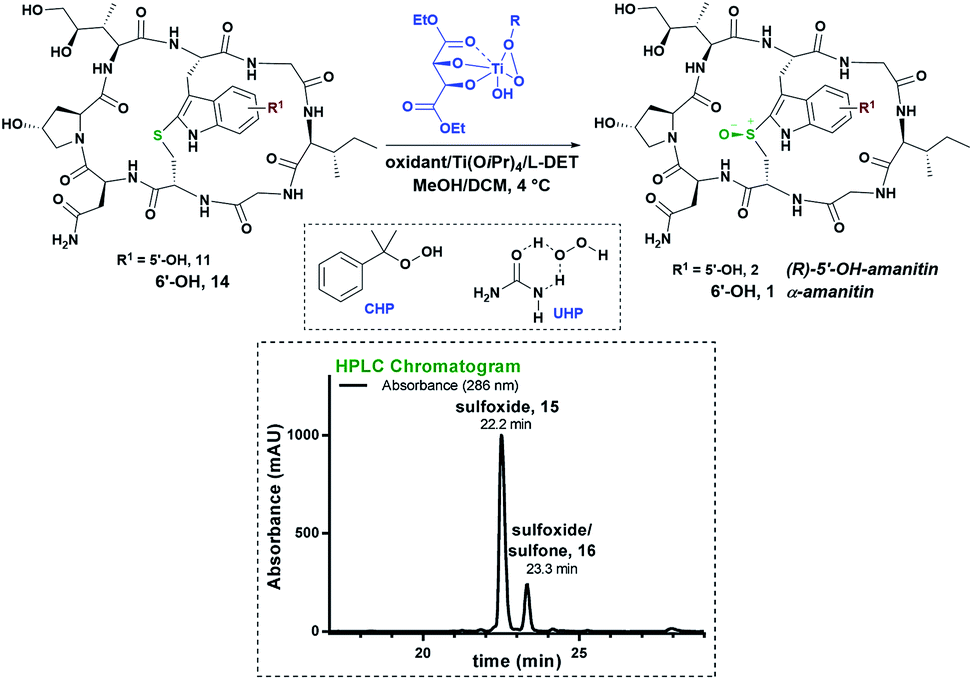 | ||
| Fig. 5 Oxidation of heptapeptide 4 to (R)-sulfoxide and (S)-sulfoxide/sulfone in 5:1 ratio with mCPBA as an oxidant in DMF at −20 °C; HPLC chromatogram of mCPBA oxidation; in reporting this, we show 15 to have undefined stereochemistry at sulphur, which was determined subsequently. | ||
To confirm the stereochemical configuration of the monocyclic sulfoxide, 15 was converted to the final bicyclic 5′-OH-6′-deoxy-amanitin 2 by the following approach (Scheme 4): the heptapeptide sulfoxide 15 was coupled to a suitably protected dihydroxyisoleucine to give the octapeptide, which was then Fmoc- and TBS-deprotected and finally macrolactamized to produce the bicyclic amanitin analog 2 (Scheme 4). The final product was analysed by HPLC, including co-injection with an “authentic sample” of (R)-5′-OH-6′-deoxy-amanitin (2) (Fig. 2), that was obtained through an mCPBA oxidation of the bicyclic precursor 11 following a previously reported procedure for α-amanitin synthesis (Scheme 4A).13a
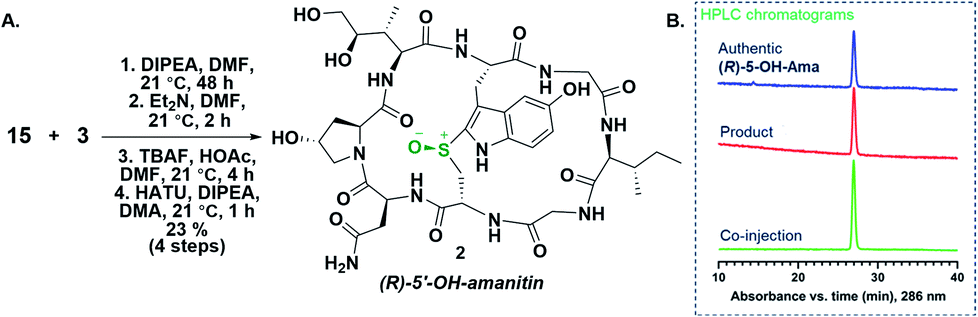 | ||
| Scheme 4 Synthesis of the (R)-5′-OH-6′-deoxy-amanitin 2 to confirm the stereochemistry of the heptapeptide sulfoxidation, incorporation of dihydroxyisoleucine and macrolactamization (A). HPLC chromatogram of the sulfoxide containing 5′-OH-6′-deoxy-amanitin obtained through sulfoxide 15 and co-injection with (R)-5′-OH-6′-deoxy-amanitin 2 produced via a reported mCPBA oxidation route confirming (R)-sulfoxide formation during heptapeptide sulfoxidation (B). | ||
Based on the HPLC co-injection data, we therefore confirmed that the sulfoxide 15 was indeed of the (R)-configuration (Scheme 4B). This method thus represents a highly diastereoselective, mild, facile, and cost-effective approach for introduction of (R)-sulfoxide at an earlier stage in the synthesis and, importantly, before the incorporation of the costly DHIle or other analogs that may be of interest.
To examine whether this approach is generally applicable to other monocyclic precursors, we attempted this mCPBA sulfoxidation on the monocyclic 5′-OH-pentapeptide S4, analogous to the building block employed in the “5 + 1 + 2” strategy developed by Süssmuth and coworkers. Somewhat surprisingly, this led to formation of two sulfoxides in 1:2 ratio. Similarly, when the monocyclic octapeptide (seco-5′-OH-6′-deoxy-octapeptide S3) was subjected to mCPBA in DMF, two sulfoxides were formed with little diastereoselectivity (1:1.7) suggesting that neither the pentapeptide nor the octapeptide adopts a conformation that is preordered to confer highly diastereoselective attack by the oxidant.
In an attempt to rationalize the outstanding selectivity in oxidizing the heptapeptide to obtain the (R)-sulfoxide, we examined the differences in geometries between the bicyclic octapeptide 11 and monocyclic heptapeptide 4. Based on the geometry-optimized structure of the heptapeptide monocycle 4, we hypothesize that the extensive H-bonding interactions observed between Asn-1, Hyp-2, 5′-OH-Trp-4 (Fig. 6A) and the backbone may cause an increase in steric bulk on the si-face of the indole, disfavoring oxidant attack on the pro-S lone pair on the sulfur atom while leaving the pro-R lone pair more solvent-accessible.
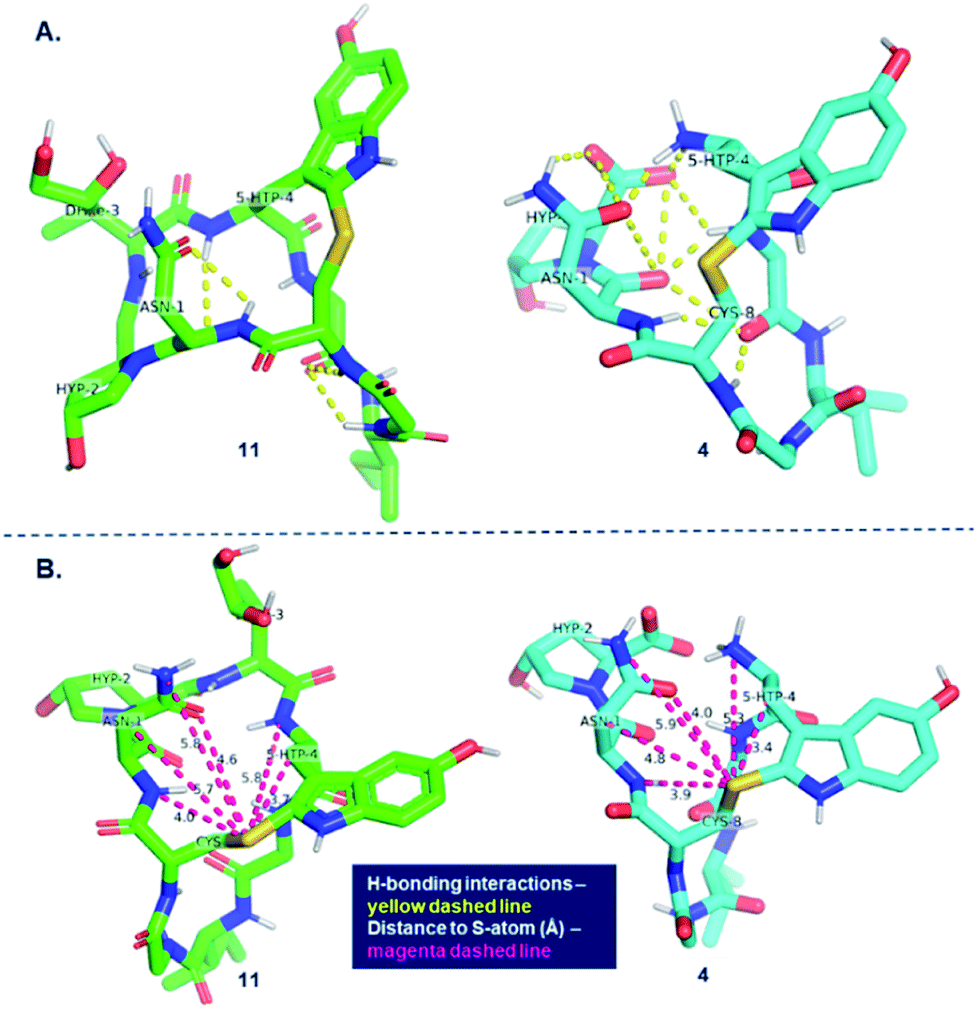 | ||
| Fig. 6 Modelling of bicyclic precursor 11 (in green, left) and monocyclic heptapeptide precursor 4 (in blue, right) shows greater exposure of the pro-R lone pair compared to the pro-S lone pair; H-bonding interactions highlighted in yellow (A); distances from neighboring atoms to S-atom (B). | ||
This can be observed by measuring distances between sulfur and the neighboring atoms in the heptapeptide monocycle 4 and comparing these to bicycle 11 (Fig. 6B). Based on this analysis, five out of six neighboring atoms are in closer proximity to sulfur in the heptapeptide. The Asn-1 side chain is noteworthy since it shows the largest degree of displacement. In contrast, the conformation of amanitin 11 shows that both lone pairs on the sulfur atom share a similarly congested environment thus explaining the generally poor diastereoselectivity.
Finally, to complete this study, we evaluated certain key products by circular dichroism (CD) to prove for key structural elements such as α-helices, β-sheets, and β-turns that are consistently found in non-random portions of proteins. Amatoxins are characterized by a type-II β-turn in ring II (formed by Trp4–Gly5–Ile6–Gly7–Cys8), which is recognized to be crucial for binding, and numerous natural toxins in this class as well as synthetic derivatives have been characterized by CD. Notably, (R)-sulfoxide 2 strongly matched the conformation of α-amanitin; however, the nearly-equipotent thioether 11 gave a spectrum that differed significantly from both 2 and α-amanitin in the 210–240 nm range (Fig. 7). The (S)-sulfoxide 12 and sulfone 13 both differed significantly from 2 and α-amanitin at nearly all wavelengths, and while both showed more similar CD-spectra, these showed vastly different cytotoxicities and Ki values in vitro. A similar trend is observed for the heptapeptides. The implications of these finding are discussed below.
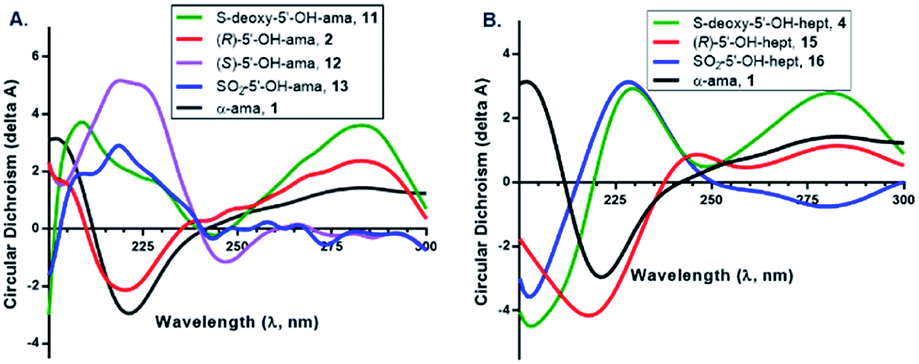 | ||
| Fig. 7 CD spectra of amanitins 2, 11–13 (A) and heptapeptides 4, 15, 16 (B). | ||
Discussion
Herein we describe the synthesis of a new amanitin analog that is a constitutional isomer of the natural product. In light of the importance of the 6′-OH for both enhancing toxicity and providing a conjugation handle, and given the synthetic challenges in accessing the 6-OH-tryptophan or its equivalents, we aimed to design an analog that would: (i) impart greater electron density into the indole system, (ii) provide an H-bond for added recognition, and (iii) preserve a conjugation handle on the indole while bypassing the need to install the synthetically challenging 6′-OH-tryptathionine staple. To direct our efforts, we first examined the interactions between the indole core and the enzyme in the crystal structure along with the biological activity scope of the known indole-modified analogs that have been prepared from the natural product.A review of the vast literature on derivatives of naturally sourced amatoxins suggests that the indole core is relatively tolerant to modifications. Small modifications generally result in negligible reductions in RNA pol II affinity (Fig. 1). For example, 6′-ether derivatives have been synthesized successfully in the past by treating α-amanitin with alkyl halides.24 Similarly, a two-fold reduction in affinity was observed when α-amanitin was methylated or ethylated at position-6 to give 6′-OR-α-amanitin (R = Me, Et); methyl and ethyl ethers were two-fold less active in an RNA pol II inhibition assay. However, longer chain lipophilic derivatives exhibited a much-reduced affinity towards RNA pol II even though their cell toxicity was increased likely due to improved cell penetration capacity.24 Other 7′-modified derivatives of α-amanitin have been synthesized, and this position was generally shown to be quite tolerant to modifications. Amine and arylazo substitutions were introduced at the 7′-position of tryptophan on α-amanitin.25 In 1982, Morris et al. evaluated a series of six secondary-amine modifications at the 7′-position and seven 7′-arylazo-amatoxins for affinity towards RNA pol II and III. All of the tested analogs were active towards RNA pol II, with Ki values up to 8 times that of α-amanitin.26 These reports further underscored the plasticity that the indole core offers and encouraged us to develop a new tryptophan-modified analog while addressing the remaining challenge of diastereoselective sulfoxidation.
As part of our characterization, we provide CD spectra to demonstrate what appears to be conformational differences between the thioether, (R)- and (S)-sulfoxides along with the sulfone. Since the sulfoxides show dramatic differences in the CD spectra, it suggests that they adopt significantly different conformations. Previously, however, full structural elucidation by NMR spectroscopy unexpectedly showed that the (R)- and (S)-sulfoxides had nearly overlapping structures,18 and while in the crystalline state, structures of the (S)-sulfoxide and sulfone were nearly identical.19a To date, molecular dynamics calculations have provided little additional insight into an enduring mystery as to why the (S)-sulfoxide is uniquely less active than the sulfone, the (R)-sulfoxide27 and the thioether. Hence, in terms of this work, a structural rationalization for the differences in CD spectra or cytotoxicity/Ki values, which correlate to those seen with other amanitins, is not readily intuited.
While the advantages of using an (R)-sulfoxide over either the thioether or sulfone as an ADC payload remain to be assessed in preclinical animal models and human clinical studies, it is reasonable to hypothesize that Nature may have chosen to install the (R)-sulfoxide deliberately to enhance systemic toxicity. Here, we have identified a means of achieving highly diastereoselective sulfoxidation on the precursor heptapeptide, which now enables production of a configurationally stable precursor that awaits conjugation with DHIle, the most synthetically challenging monomer used in the synthesis. In addition, the use of a chiral catalyst to introduce the (R)-sulfoxide late-stage now provides access to late-stage diastereoselective sulfoxidation on bicyclic octapeptides.
The use of chiral sulfoxidation catalysts on S-deoxy-amanitin recapitulated similar diastereoselectivity that we report here for the 5′-hydrox-6′-deoxy analog demonstrating utility for resolving this question in the context of providing the natural product as well. Although we did not evaluate this on the heptapeptide precursor of amanitin, we believe that this method should provide similar results and thus likely augurs for success in accessing the natural product in high diastereomeric excess without resoting to chiral catalysts. Finally, we posit that one might use this approach to accomplish sulfoxidation on amanitin analogs that are produced from circularly-permuted macrocycles resembling phakellistatins.28
Conclusions
5′-OH-6′-deoxy-amanitin, a novel and bioactive analog of α-amanitin that takes advantage of the naturally abundant, commercially available 5-OH-tryptophan to incorporate a conjugation handle, was successfully synthesized. The 5-OTBS-fluoropyrroloindoline (5-OTBS-FPI) was accessed in three simple high yielding steps from commercially available 5-OH-L-tryptophan, in contrast to addressing key oxidations on the synthetically challenging 6-OH-tryptophan. MTS cell viability and RNA pol II inhibition assays demonstrated near-native potency. It is readily appreciated that the 5′-OH can serve as a handle for further conjugation to various antibodies through a multitude of currently available strategies.11a,eThe (R)-sulfoxide 2 is significantly more toxic than the (S)-sulfoxide 12 in the context of this new analog. To access this diastereomer, we developed a late-stage sulfoxidation approach utilizing TiIV/L-DET-based catalytic system that is conveniently applied to the bicyclic toxin as the ultimate synthetic step. We suggest that this methodology has a high potential of being translatable to a broad range of bicyclic amanitin analogs due to structural similarities observed among active amatoxins. In addition, a mild, facile, and cost-effective early stage sulfoxidation approach at the monocycle stage was successfully employed. This new methodology capitalizes on what appear to be unique structural components of the heptapeptide macrocycle that must determine the diastereoselectivity of sulfoxidation that uses readily available reagents. Taken together, these sulfoxidation methods represent a great improvement in diastereoselectivity over the previously reported mCPBA-assisted sulfoxidation. Finally, we believe this highly selective method for (R)-sulfoxide formation may be broadly applicable to use on other monocyclic amanitin precursors and presents a significant advancement for future large scale amanitin syntheses.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from the Canadian Institutes for Health Research #220656 and the Canadian Cancer Society Research Initiative Grant #703374 and thank Dr Maria Ezhova for help with NMR acquisition, Dr Elena Polishchuk and Ms Jessie Chen for help with cytotoxicity assays.Notes and references
- T. J. Lindell, F. Weinberg, P. W. Morris, R. G. Roeder and W. J. Rutter, Science, 1970, 170, 447–449 CrossRef CAS.
- (a) G. Moldenhauer, A. V. Salnikov, S. Luttgau, I. Herr, J. Anderl and H. Faulstich, J. Natl. Cancer Inst., 2012, 104, 622–634 CrossRef CAS; (b) K. C. Nicolaou and S. Rigol, Angew. Chem., Int. Ed., 2019, 58, 11206–11241 CrossRef CAS.
- H. Schlesinger and W. W. Ford, J. Biol. Chem., 1907, 3, 279–283 Search PubMed.
- H. Wieland and R. Hallermayer, Justus Liebigs Ann. Chem., 1941, 548, 1–18 CrossRef CAS.
- (a) K. Baumann, K. Munter and H. Faulstich, Biochemistry, 1993, 32, 4043–4050 CrossRef CAS; (b) A. Buku, R. Altmann and T. Wieland, Justus Liebigs Ann. Chem., 1974, 1580–1586 CrossRef CAS; (c) G. Zanotti, C. Mohringer and T. Wieland, Int. J. Pept. Protein Res., 1987, 30, 450–459 CrossRef CAS.
- X. Y. Liu, L. Farnung, C. Wigge and P. Cramer, J. Biol. Chem., 2018, 293, 7189–7194 CrossRef CAS.
- K. Kume, M. Ikeda, S. Miura, K. Ito, K. A. Sato, Y. Ohmori, F. Endo, H. Katagiri, K. Ishida, C. Ito, T. Iwaya and S. S. Nishizuka, Sci. Rep., 2016, 6, 15 CrossRef.
- (a) J. Anderl, C. Muller, B. Heckl-Ostreicher and R. Wehr, Cancer Res., 2011, 71, 1 Search PubMed; (b) T. Hechler, M. Kulke, C. Mueller, A. Pahl and J. Anderl, Cancer Res., 2014, 74, 1 Search PubMed.
- L. Zhao, J. P. May, A. Blanc, D. J. Dietrich, A. Loonchanta, K. Matinkhoo, A. Pryyma and D. M. Perrin, ChemBioChem, 2015, 16, 1420–1425 CrossRef CAS.
- G. Barbantibrodano and L. Fiume, Nature (London), New Biol., 1973, 243, 281–283 CrossRef CAS.
- (a) K. W. Swiderska, A. Szlachcic, L. Opalinski, M. Zakrzewska and J. Otlewski, Int. J. Mol. Sci., 2018, 19, 16 Search PubMed; (b) L. Bodero, P. L. Rivas, B. Korsak, T. Hechler, A. Pahl, C. Muller, D. Arosio, L. Pignataro, C. Gennari and U. Piarulli, Beilstein J. Org. Chem., 2018, 14, 407–415 CrossRef CAS; (c) M. A. Krzyscik, L. Opalinski and J. Otlewski, Mol. Pharm., 2019, 16, 3588–3599 CrossRef CAS; (d) S. Park, S. Y. Kim, J. Cho, D. Jung, D. Seo, J. Lee, S. Lee, S. Yun, H. Lee, O. Park, B. Seo, S. Kim, M. Seol, S. H. Woo and T. K. Park, Bioconjugate Chem., 2019, 30, 1969–1978 CrossRef CAS; (e) S. Park, S. Y. Kim, J. Cho, D. Jung, D. Seo, J. Lee, S. Lee, S. Yun, H. Lee, O. Park, B. Seo, S. H. Woo and T. K. Park, Bioconjugate Chem., 2019, 30, 1957–1968 CrossRef CAS.
- (a) H. Luo, S. Y. Hong, R. M. Sgambelluri, E. Angelos, X. Li and J. D. Walton, Chem. Biol., 2014, 21, 1610–1617 CrossRef CAS; (b) H. Luo, B. DuBois, R. M. Sgambelluri, E. R. Angelos, X. Li, D. Holmes and J. D. Walton, Toxicon, 2015, 103, 60–64 CrossRef CAS.
- (a) K. Matinkhoo, A. Pryyma, M. Todorovic, B. O. Patrick and D. M. Perrin, J. Am. Chem. Soc., 2018, 140, 6513–6517 CrossRef CAS; (b) M. A. J. Siegert, C. H. Knittel and R. D. Süssmuth, Angew. Chem., Int. Ed., 2020, 59, 5500–5504 CrossRef CAS; (c) C. Lutz, W. Simon, S. Werner-Simon, A. Pahl and C. Muller, Angew. Chem., Int. Ed., 2020, 59, 11390–11393 CrossRef CAS.
- W. E. Savige and A. Fontana, Chem. Commun., 1976, 15, 600–601 RSC.
- (a) O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CAS; (b) D. Seeliger and B. L. de Groot, J. Comput.-Aided Mol. Des., 2010, 24, 417–422 CrossRef CAS.
- W. L. DeLano, The PyMOL Molecular Graphics System, DeLano Scientific, San Carlos CA, USA, 2002 Search PubMed.
- A. Pryyma, Y. J. Bu, Y. Wai, B. O. Patrick and D. M. Perrin, Org. Lett., 2019, 21, 8234–8238 CrossRef CAS.
- T. Wieland, C. Gotzendorfer, J. Dabrowski, W. N. Lipscomb and G. Shoham, Biochemistry, 1983, 22, 1264–1271 CrossRef CAS.
- (a) G. Shoham, W. N. Lipscomb and T. Wieland, J. Am. Chem. Soc., 1989, 111, 4791–4809 CrossRef CAS; (b) G. Shoham, D. C. Rees, W. N. Lipscomb, G. Zanotti and T. Wieland, J. Am. Chem. Soc., 1984, 106, 4606–4615 CrossRef CAS.
- G. Zanotti, C. Birr and T. Wieland, Int. J. Pept. Protein Res., 1981, 18, 162–168 CrossRef CAS.
- (a) A. Buku, T. Wieland, H. Bodenmuller and H. Faulstich, Experientia, 1980, 36, 33–34 CrossRef CAS; (b) T. Wieland, C. Gotzendorfer, G. Zanotti and A. C. Vaisius, Eur. J. Biochem., 1981, 117, 161–164 CrossRef CAS.
- J. P. May, P. Fournier, B. O. Patrick and D. M. Perrin, Chem.–Eur. J., 2008, 14, 3410–3417 CrossRef CAS.
- (a) F. Difuria, G. Modena and R. Seraglia, Synthesis, 1984, 325–326 CrossRef CAS; (b) J. L. Han, V. A. Soloshonok, K. D. Klika, J. Drabowicz and A. Wzorek, Chem. Soc. Rev., 2018, 47, 1307–1350 RSC; (c) P. Pitchen and H. B. Kagan, Tetrahedron Lett., 1984, 25, 1049–1052 CrossRef CAS.
- H. Faulstich, H. Trischmann, T. Wieland and E. Wulf, Biochemistry, 1981, 20, 6498–6504 CrossRef CAS.
- (a) E. Falckpedersen, P. W. Morris and D. L. Venton, Int. J. Pept. Protein Res., 1983, 21, 431–439 CrossRef CAS; (b) P. W. Morris, D. L. Venton and K. M. Kelley, Biochemistry, 1978, 17, 690–698 CrossRef CAS.
- E. Falckpedersen, W. Neuman and P. W. Morris, Biochemistry, 1982, 21, 5164–5170 CrossRef CAS.
- A. B. Pomilio, M. E. Battista and A. A. Vitale, J. Mol. Struct.: THEOCHEM, 2001, 536, 243–262 CrossRef CAS.
- J. P. May and D. M. Perrin, Chem.–Eur. J., 2008, 14, 3404–3409 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc04150e |
| This journal is © The Royal Society of Chemistry 2020 |