
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Carbonate-promoted C–H carboxylation of electron-rich heteroarenes†
Tyler M.
Porter
and
Matthew W.
Kanan
 *
*
Department of Chemistry, Stanford University, Stanford, California 94305, USA. E-mail: mkanan@stanford.edu
First published on 5th October 2020
Abstract
C–H carboxylation is an attractive transformation for both streamlining synthesis and valorizing CO2. The high bond strength and very low acidity of most C–H bonds, as well as the low reactivity of CO2, present fundamental challenges for this chemistry. Conventional methods for carboxylation of electron-rich heteroarenes require very strong organic bases to effect C–H deprotonation. Here we show that alkali carbonates (M2CO3) dispersed in mesoporous TiO2 supports (M2CO3/TiO2) effect CO32−-promoted C–H carboxylation of thiophene- and indole-based heteroarenes in gas–solid reactions at 200–320 °C. M2CO3/TiO2 materials are strong bases in this temperature regime, which enables deprotonation of very weakly acidic bonds in these substrates to generate reactive carbanions. In addition, we show that M2CO3/TiO2 enables C3 carboxylation of indole substrates via an apparent electrophilic aromatic substitution mechanism. No carboxylations take place when M2CO3/TiO2 is replaced with un-supported M2CO3, demonstrating the critical role of carbonate dispersion and disruption of the M2CO3 lattice. After carboxylation, treatment of the support-bound carboxylate products with dimethyl carbonate affords isolable esters and the M2CO3/TiO2 material can be regenerated upon heating under vacuum. Our results provide the basis for a closed cycle for the esterification of heteroarenes with CO2 and dimethyl carbonate.
Introduction
C–H carboxylation (Scheme 1) is a compelling alternative to conventional syntheses of carboxylic acids that utilize oxidative transformations or more functionalized substrates and has attracted attention as a way to expand the use of CO2 in chemical production.1–3 However, carboxylation faces the challenge of overcoming the low reactivity of C–H bonds and CO2, and it lacks the large intrinsic driving force of other C–H functionalizations such as oxidation or amination. The insertion of CO2 into C–H bonds to form a carboxylic acid is actually endergonic on account of the small ΔH and negative ΔS, while C–H carboxylation is exergonic (depending on base strength) because of the driving force from deprotonation. Increased interest in this transformation over the last several years has led to a number of methods that encompass both acid–base2,4–9 (ionic) and radical mechanisms2,10–14 for C–H activation. Despite these recent advances, most methods for C–H carboxylation under conventional, solution-phase conditions require highly reactive, resource-intensive reagents to activate C–H bonds. As such, the development of alternatives that use benign, regenerable reagents is critical to create opportunities for scalable CO2 utilization.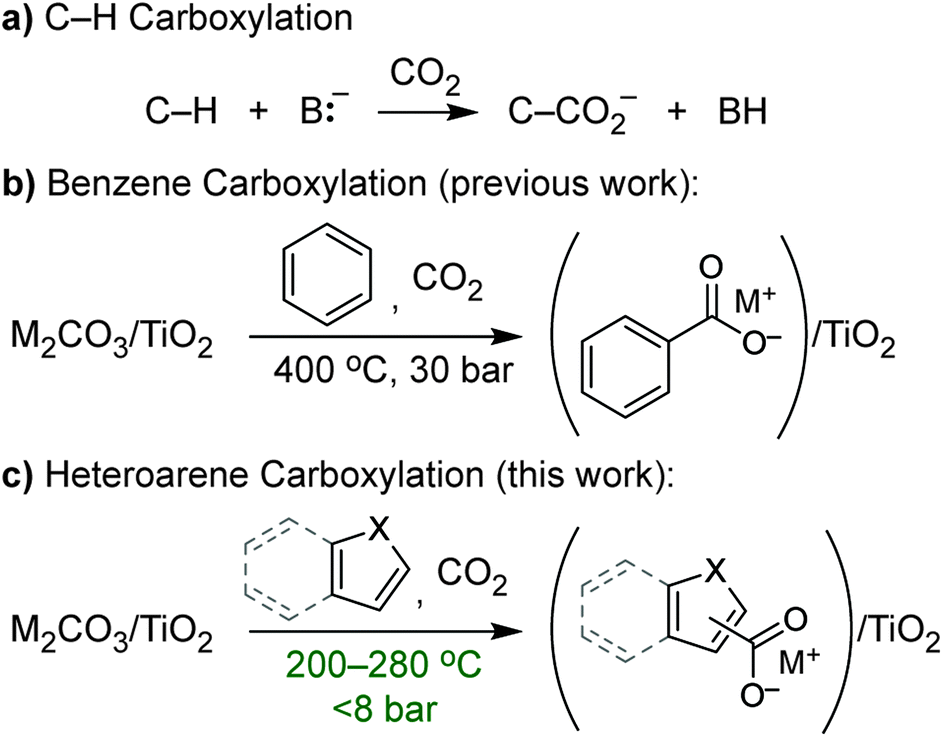 | ||
| Scheme 1 (a) General scheme for base-promoted C–H carboxylation; (b) C–H carboxylation of benzene using dispersed alkali carbonates (M2CO3/TiO2); (c) C–H carboxylation of heteroarenes using M2CO3/TiO2. | ||
The carboxylation of aromatic substrates is of particular interest for the synthesis of a wide variety of both fine and commodity chemicals.2,15–19 Because of the high bond dissociation enthalpy (BDE) of aromatic C–H bonds, acid–base (ionic) activation of the substrate has been the most commonly employed strategy. For some substrates, deprotonation of an X–H bond (X = heteroatom) generates a nucleophilic intermediate that undergoes C–H carboxylation via an electrophilic aromatic substitution (EAS) mechanism. The classic example is the Kolbe–Schmidt reaction used for aspirin synthesis, in which phenol is transformed into salicylate by reaction with hydroxide and CO2.20 While the carboxylation of indoles and pyrroles has been achieved similarly,6,8,21 these reactions have required the use of superstoichiometric LiOtBu to deprotonate the N–H bonds.
Apart from these special cases, carboxylation of (hetero)arene substrates via acid–base chemistry requires direct activation of the C–H bond to generate a reactive carbon-centered nucleophile. Within the last decade, several groups have demonstrated Brønsted-base-promoted carboxylation of (hetero)arenes in organic solvents at near ambient CO2 pressure.4,6–9,21 Hu et al. have shown that relatively acidic heteroarenes (pKa up to 28 in organic solvent) can be carboxylated using Cs2CO3 as the base in refluxing DMF.4,7 The carboxylation of electron-rich heteroarenes beyond this pKa threshold, however, has required much stronger bases. For example, the carboxylation of benzothiophene (pKa of 33 in THF) was not possible under these same conditions.4 Recently, Kondo et al. have demonstrated the carboxylation of a diverse set of (benzo)thiophenes and (benzo)furans by reaction with excess LiOtBu, CsF, and crown ether at 160 °C under a CO2 atmosphere.9 However, these conditions were not able to carboxylate protected indoles, such as 1-methylindole, whose C2 carbon has a pKa near 38. Carboxylation of electron rich heteroarenes functionalized with an amide directing group has been achieved using Ni catalysis with stoichiometric KOtBu and Mn0.22
Arenes have pKas that generally lie beyond what can be measured (pKa > 40). Researchers have developed methods to carboxylate arenes that are functionalized with a directing group by using a Rh or Pd species to catalyze C–H activation.23–25 In addition to the directing group and catalyst, these methods also require a strong base (KOtBu) or Lewis acid activator (AlMe(OMe)2) to engender reactivity. In the absence of a directing group, solution-phase arene C–H carboxylation requires an extremely strong base such as Schlosser's base,26 or stoichiometric aluminum reagents.25,27
Apart from acid–base strategies, a very recent report by König et al. has described a photoredox method to carboxylate of (hetero)arenes under mild conditions in which the substrate is activated by one-electron photoreduction and Cs2CO3 serves as the stoichiometric base.28 This method affords moderate to high yields across a variety of substrates, although it is presently incompatible with some classes of (hetero)arene substrates and uses relatively high loadings of a photocatalyst requiring multi-step synthesis.
We previously showed that simple alkali carbonates (M2CO3) can promote C–H carboxylation of very weakly acidic substrates in solvent-free, alkali salt media at elevated temperature.29–32 This transformation is particularly useful for converting a monocarboxylate substrate into a dicarboxylate product, where the substrate enables the formation of a molten reaction medium.33 More recently, we demonstrated that M2CO3 dispersed into mesoporous TiO2 (M2CO3/TiO2, Fig. 1a) promotes the carboxylation of benzene and other aromatic hydrocarbon C–H bonds in gas–solid reactions (Scheme 1b).34 Dispersion in mesopores disrupts the bulk M2CO3 crystal structure, creating an amorphous material that can attain superbase reactivity, even in the presence of CO2. This carbonate-promoted C–H carboxylation of aromatic hydrocarbons takes place at moderate pressures and temperatures of ∼400 °C. In this study, we begin to assess the generality and selectivity of this strategy using electron-rich heteroarenes, which have somewhat more acidic C–H bonds. We show that gas–solid carbonate-promoted C–H carboxylation occurs at substantially lower temperatures for these substrates and that selective reactions are possible in the presence of multiple C–H bonds (Scheme 1c, Fig. 1b). For thiophene substrates, the selectivity and mechanistic studies support a carboxylation pathway that proceeds via C–H deprotonation by the amorphous CO32−, as seen previously with arenes. For more nucleophilic indole substrates, however, carboxylation proceeds via electrophilic aromatic substitution, which provides a new pathway for CO2 utilization enabled by dispersed carbonate materials.
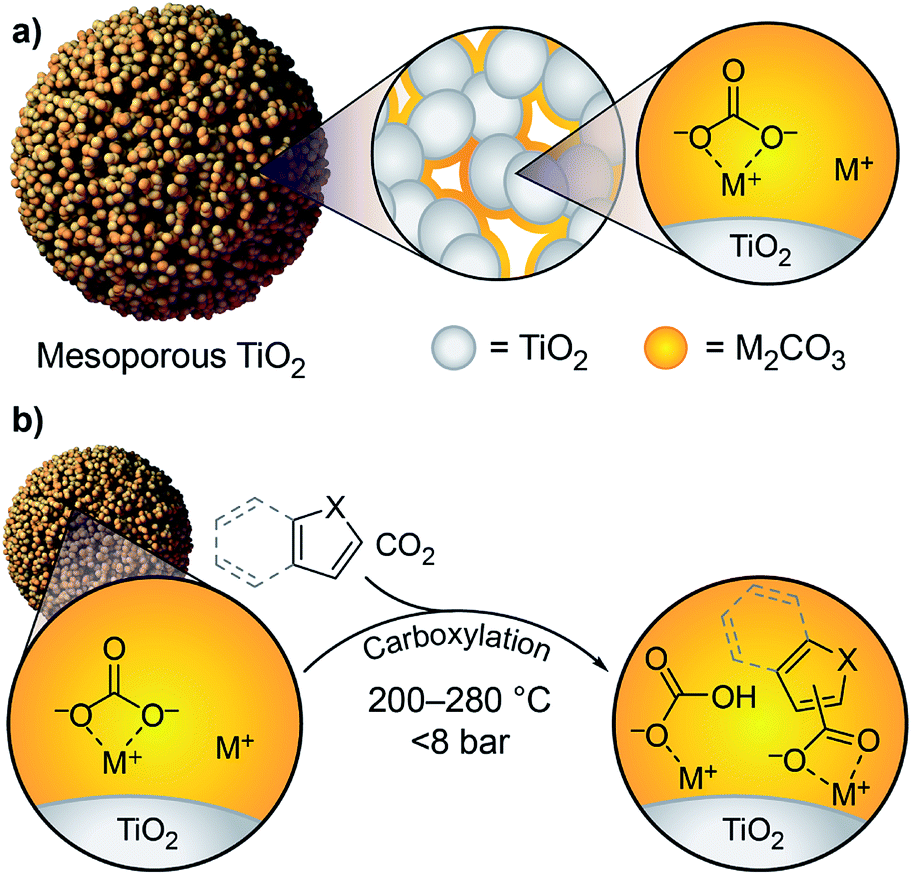 | ||
| Fig. 1 (a) Schematic view of M2CO3/TiO2 showing the M2CO3 dispersed over the mesoporous TiO2 surface; (b) heteroarene C–H carboxylation promoted by M2CO3/TiO2. Note that MHCO3 is shown for simplicity, but under the reaction conditions it is thermally unstable and will convert to 0.5 equiv. M2CO3, H2O, and CO2. | ||
Results and discussion
Acidity calculations
Thiophene- and indole-based heterocycles were selected as C–H carboxylation substrates to probe the effects of C–H acidity and π-nucleophilicity. The C–H acidities were evaluated by using density functional theory (DFT) to calculate the standard enthalpy change for heterolytic bond dissociation in the gas phase (ΔacidH°, also known as the gas phase acidity) (Fig. 2 and S1†).35,36 ΔacidH° provides a way to compare the thermodynamics of deprotonation irrespective of whether the pKa can be measured. Benzene, which reacts with dispersed carbonates at ∼400 °C, has a ΔacidH° of 401 kcal mol−1; its pKa is too large to be measured but has been estimated to be >43.36 The most acidic C–H bonds in each heterocycle were found to be more acidic (lower ΔacidH°) than benzene by 15–23 kcal mol−1, while the separation between the two most acidic C–H bonds in each substrate was 6–11 kcal mol−1. For comparison, the experimental pKa values of benzothiophene (C2), thiophene (C2), and 1-methylindole (C2) are 32, 33, and 38 according to measurements performed in THF.36,37 Additional DFT calculations to determine solution state pKa values showed good agreement to these experimental values (Fig. S2†). | ||
| Fig. 2 (a) Schematic depiction of gas-phase heterolytic C–H bond dissociation. The standard enthalpy of this reaction is the gas phase acidity. (b) Calculated gas phase acidities and experimental pKas of the most acidic C–H bonds in the heteroarene substrates. Benzene is included as a reference point. | ||
Carbonate-promoted C–H carboxylation reactions
C–H carboxylation reactions were performed in a sealed vessel containing M2CO3 dispersed on TiO2 (M2CO3/TiO2, M+ = Cs+, K+, Na+), heterocycle substrate, and CO2 (see ESI† for detailed experimental procedures). In most cases, the substrate was placed within a glass culture tube in the reactor to ensure that only volatilized substrate would be able to react with the M2CO3/TiO2 material (Fig. S3†). The products were isolated by aqueous extraction from the TiO2 support and quantified by 1H NMR (Fig. S4–S9†). In all cases, control experiments using M2CO3 without the TiO2 support showed no reactivity, whereas M2CO3/TiO2 promoted C–H carboxylation in varying degrees depending on the identity of M+. Additional control experiments showed minimal reactivity with the mesoporous TiO2 support alone.We first assessed the temperature dependence of C–H carboxylation under a common set of conditions using 1.5 mmol substrate, a CO2 loading corresponding to 4–5 bar at the reaction temperature, and a reaction time of 3 h (Fig. 3 and S10†). The relatively low substrate loading corresponded to a maximum pressure of ∼2.5 bar at the highest temperature evaluated (320 °C). Thus, the overall pressure of the reactor at temperature was <8 bar for all of the reactions in this temperature screen. For benzothiophene (Fig. 3a), the onset of carboxylation reactivity was observed at 200 °C. Optimal results were seen at 280 °C, where 190 μmol of benzothiophene carboxylation product was obtained per gram of TiO2 (190 μmol g−1 TiO2) with a 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of 2-carboxylate to 3-carboxylate isomers for Cs2CO3/TiO2. Using K2CO3/TiO2, 207 μmol g−1 TiO2 of benzothiophene carboxylate product was obtained with a 25:1 product ratio (Fig. 3a). While both the yield and selectivity declined at higher temperatures, the carboxylation selectivity followed the C–H acidities, consistent with a mechanism gated by C–H deprotonation (see below). In contrast to Cs+ and K+, much lower reactivity was observed with Na2CO3/TiO2, suggesting that this material is a weaker base in gas–solid reactions.
:1 ratio of 2-carboxylate to 3-carboxylate isomers for Cs2CO3/TiO2. Using K2CO3/TiO2, 207 μmol g−1 TiO2 of benzothiophene carboxylate product was obtained with a 25:1 product ratio (Fig. 3a). While both the yield and selectivity declined at higher temperatures, the carboxylation selectivity followed the C–H acidities, consistent with a mechanism gated by C–H deprotonation (see below). In contrast to Cs+ and K+, much lower reactivity was observed with Na2CO3/TiO2, suggesting that this material is a weaker base in gas–solid reactions.
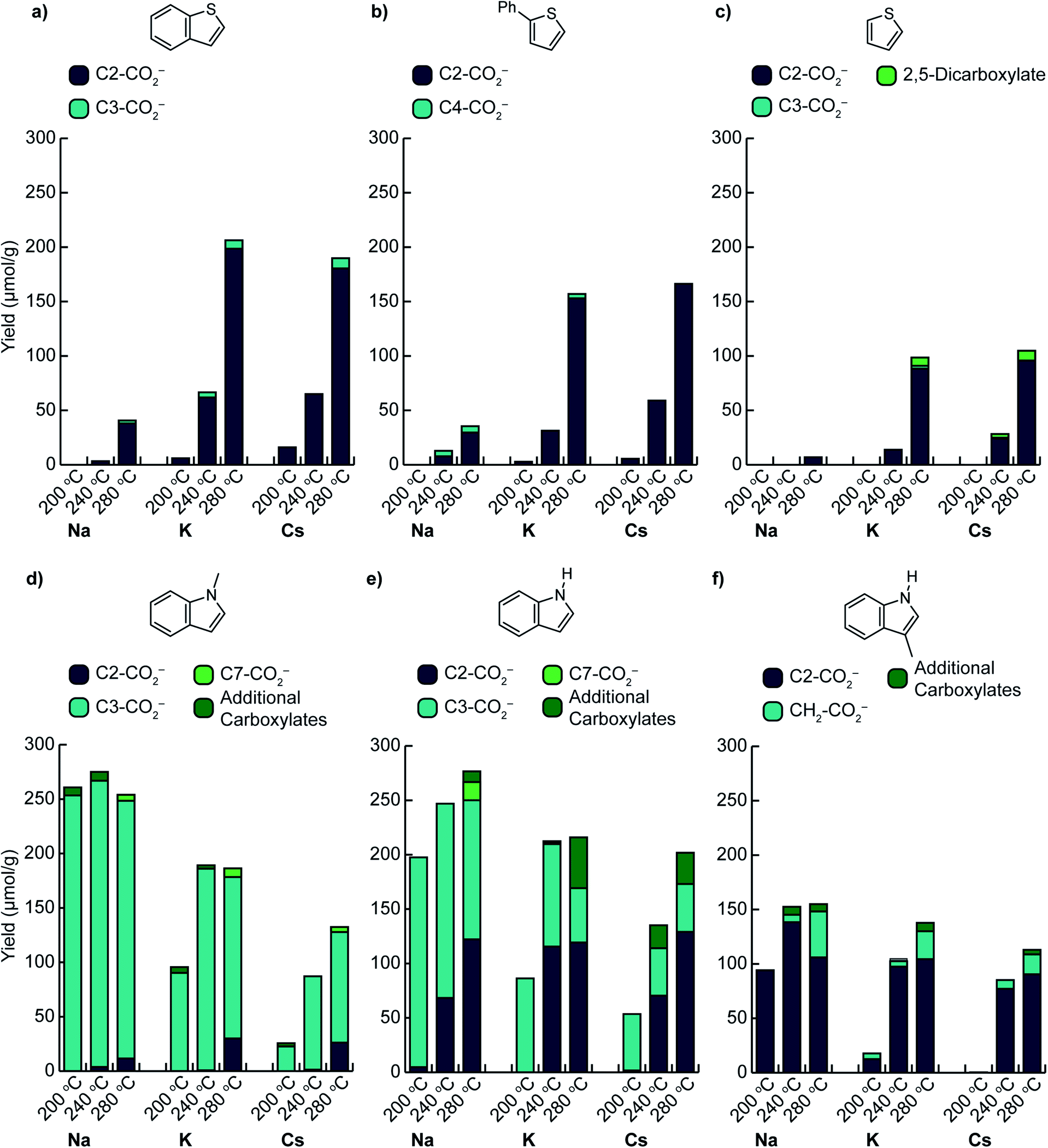 | ||
| Fig. 3 Summary of C–H carboxylation for heteroarenes using M2CO3/TiO2 and CO2 at various temperatures ((a) benzothiophene, (b) 2-phenylthiophene, (c) thiophene, (d) 1-methylindole, (e) indole, (f) 3-methylindole). Reactions were performed using glass culture tubes to separate un-vaporized organics from the M2CO3/TiO2 materials. Conditions: 250 mg M2CO3/TiO2, 1.5 mmol heterocycle, 2.5 bar CO2 at 298 K, 3 h reaction time. | ||
Comparison of benzothiophene carboxylation with our previous results for benzene carboxylation further highlights the effect of C–H acidity on carboxylation. Whereas >200 μmol g−1 TiO2 of carboxylate products were obtained for benzothiophene at 280 °C and <8 bar total pressure, the maximum yields for benzene carboxylation using the same M2CO3/TiO2 materials were ∼100 μmol g−1 TiO2 at 420–440 °C and ∼30 bar total pressure. Thus, reducing the C–H bond acidity (ΔacidH°) by 23 kcal mol−1 enables higher yielding carboxylation reactions under substantially milder conditions (100 °C lower temperature, 1/3 the total pressure). Furthermore, the benzothiophene results also demonstrate that a 7 kcal mol−1 separation in C–H acidity (C2 vs. C3 position) is sufficient for selective C–H carboxylation.
Because of its high boiling point (221 °C), the vapor pressure of benzothiophene is expected to reach its saturation pressure at T ≤ 240 °C under the conditions used for the data in Fig. 3a (see Table S1† for saturation vapor pressures calculated using the Clausius–Clapeyron equation). As a result, the vapor pressure of benzothiophene varies by ∼5× over the 200–320 °C range examined. To deconvolute temperature dependence from substrate pressure dependence, a series of carboxylation reactions were performed at 280 °C for 3 h using different amounts of benzothiophene corresponding to calculated pressures ranging from 0.5 bar to 3.5 bar, which is approximately the saturation pressure at 280 °C. The total benzothiophene carboxylate yield showed a modest variation from 150 μmol g−1 TiO2 to 210 μmol g−1 TiO2 over this range (Fig. S17†). Thus, the temperature dependence of the benzothiophene carboxylation yield in Fig. 3a is primarily a result of the temperature effect on the rate constant.
Phenylthiophene reacted in a very similar manner to benzothiophene. The onset of carboxylation was observed at 200 °C with very high selectivity for the 5-phenylthiophene-2-carboxylate isomer (derived from the most acidic C–H bond) observed up to 280 °C. Comparable yields were observed for Cs2CO3/TiO2 and K2CO3/TiO2, while substantially lower yields were seen for Na2CO3/TiO2 (Fig. 3b). The carboxylate yield varied by ∼50% over a 7-fold variation in phenylthiophene pressure (0.5–3.5 bar) at 320 °C (Fig. S17†). The similarity in the temperature- and pressure-dependent yields for both benzothiophene and phenylthiophene is reflected in their nearly identical ΔacidH° values for their two most acidic C–H bonds, suggesting that the same mechanism is operative for both substrates. Notably, although separating the substrate with a culture tube in the reactor ensures that it can only interact with the M2CO3via the gas phase, the carboxylation reactions with low-volatility substrates like benzothiophene and phenylthiophene proceed in comparable or better yield when the two are combined directly (Fig. S11†).
In contrast to the heterocycle pressure dependence, increasing CO2 pressures were found to significantly inhibit C–H carboxylation for both substrates (Fig. S18†). The CO2 pressure dependence was evaluated for benzothiophene and phenylthiophene at 280 °C and 320 °C, respectively. Interestingly, inspection of the culture tubes for both substrates post-reaction revealed increasing amounts of un-vaporized heterocycle with increasing CO2 partial pressure (Fig. S18†). While the calculated saturation pressures indicate that all of the 1.5 mmol of substrate should be vaporized at these temperatures, this observation suggests that CO2 dissolves in the substrate upon melting and lowers its vapor pressure substantially. An additional contributing factor may be that higher CO2 pressure results in the formation of polycarbonate species (e.g. C2O52−) on the M2CO3/TiO2 material, which are weaker bases than CO32−, thereby reducing the rate of C–H deprotonation.
C–H carboxylation was also possible with thiophene itself. In the temperature screen performed with 1.5 mmol substrate (Fig. 3c), all of the thiophene is expected to be volatilized over the 200–320 °C range because of its relatively low boiling point (84 °C). The corresponding thiophene pressures range from 2–2.5 bar. In contrast to benzothiophene and phenylthiophene, no thiophene carboxylates were observed at 200 °C, which is consistent with the 6 kcal mol−1 higher ΔacidH° for its C(2)–H bond (Fig. 2). The formation of thiophene-2-carboxylate was observed beginning at 240 °C, with optimal results at 280 °C, where 96 μmol g−1 TiO2 was formed along with 9 μmol g−1 TiO2 thiophene-2,5-dicarboxylate when using Cs2CO3/TiO2. Comparable yields were obtained with K2CO3/TiO2, while Na2CO3/TiO2 was much less effective. The observation of thiophene-2,5-dicarboxylate indicates that the initially formed monocarboxylate product undergoes a second C–H carboxylation on the support. In addition to the thiophene carboxylates, ∼25 μmol g−1 TiO2 of propionate was produced across the temperature range of 240–320 °C (Table S12†). This product arises from an unknown decomposition pathway starting from thiophene or a thiophene carboxylate. The yield of thiophene carboxylates was improved by increasing the thiophene pressure to 5 bar, with a comparable proportion of propionate byproduct (Fig. S17†). In contrast to benzothiophene and phenylthiophene, essentially no CO2 pressure dependence was observed for thiophene at 280 °C. Given the much higher volatility of thiophene, CO2 has a negligible effect on its vapor pressure at this temperature.
We next investigated the effects of increasing the nucleophilicity of the heterocycle by switching from thiophene to indole substrates.38 To avoid the complication of an acidic N–H bond, we first evaluated 1-methylindole. The most acidic C–H position of this substrate is C(2)–H, whose ΔacidH° (384 kcal mol−1) is very close to the C(2)–H bond of thiophene (Fig. 2). The most nucleophilic position, however, is C3,39 which has a much less acidic C–H bond (ΔacidH° of C(3)–H is 11 kcal mol−1 higher than C(2)–H). Surprisingly, C–H carboxylation occurred readily at 200 °C with a strong preference for the C3 position (Fig. 3d). Moreover, the yield increased substantially as the alkali cation size was decreased, resulting in the highest yields for reactions using Na2CO3/TiO2. Optimal results were obtained using Na2CO3/TiO2 at 200 °C, with a yield of 250 μmol g−1 TiO2 for the C3-carboxylate (Fig. 3d and S8†). At higher temperatures (T > 240 °C) the C2-carboxylate was observed as an additional minor product. The selective formation of the C3 carboxylate is consistent with an EAS mechanism in which C–C bond formation precedes C–H deprotonation. Further support was found in the kinetic isotope effect for C–H carboxylation and DFT calculations (see below). Previously reported methods have achieved selective C3 carboxylation of 1-methylindole with CO2, but have required the use of stoichiometric organoaluminum reagents.25,40,41 Na2CO3/TiO2 provides a benign and much less resource-intensive alternative.
Selective C3 carboxylation was also observed with indole at 200 °C using M2CO3/TiO2 (Fig. 3e). The M2CO3 dependence followed the same trend as for 1-methylindole, with optimal results obtained using Na2CO3/TiO2. Because the N–H functionality on indole is much more acidic than the C–H bonds (≥35 kcal mol−1 difference in ΔacidH°), it is likely that indole is rapidly deprotonated by M2CO3/TiO2 to form indolide, which can react reversibly with CO2 to form indole-1-carboxylate (N–CO2−). Given the very low acidity of the C–H bond at C3 (ΔacidH° = 397 kcal mol−1, Fig. 2), the selectivity for C3 carboxylation at 200 °C is consistent with an EAS mechanism in which deprotonated indole is the reactive nucleophile.39 Beyond 200 °C, however, the reaction yielded a mixture of C3, C2, and C7 carboxylation products. At 280 °C, C2 carboxylation accounted for 44–64% of the total carboxylation products depending on the choice of M2CO3. Substitution at C2 is commonly seen alongside C3 in solution-phase EAS reactions with indole.42 Both methylindole and indole showed very similar pressure dependences on both heterocycle and CO2 partial pressure (Fig. S17 and S18†).
Finally, 3-methylindole (skatole) was evaluated to assess the effects of blocking carboxylation at C3. Carboxylation was observed at C2 with a similar temperature dependence as seen for C2 carboxylation of indole (Fig. 3f). Using Na2CO3/TiO2, 94 μmol g−1 TiO2 of the C2-carboxylate was obtained at 200 °C. Increasing the temperature to 240 °C boosted the yield to 138 μmol g−1 TiO2, although minor amounts of additional carboxylates were observed at this temperature, including the product of methyl carboxylation. To our knowledge, C2 carboxylation of 3-methylindole with CO2 has not previously been achieved.
Kinetic isotope effects and DFT calculations to probe C–H carboxylation mechanism
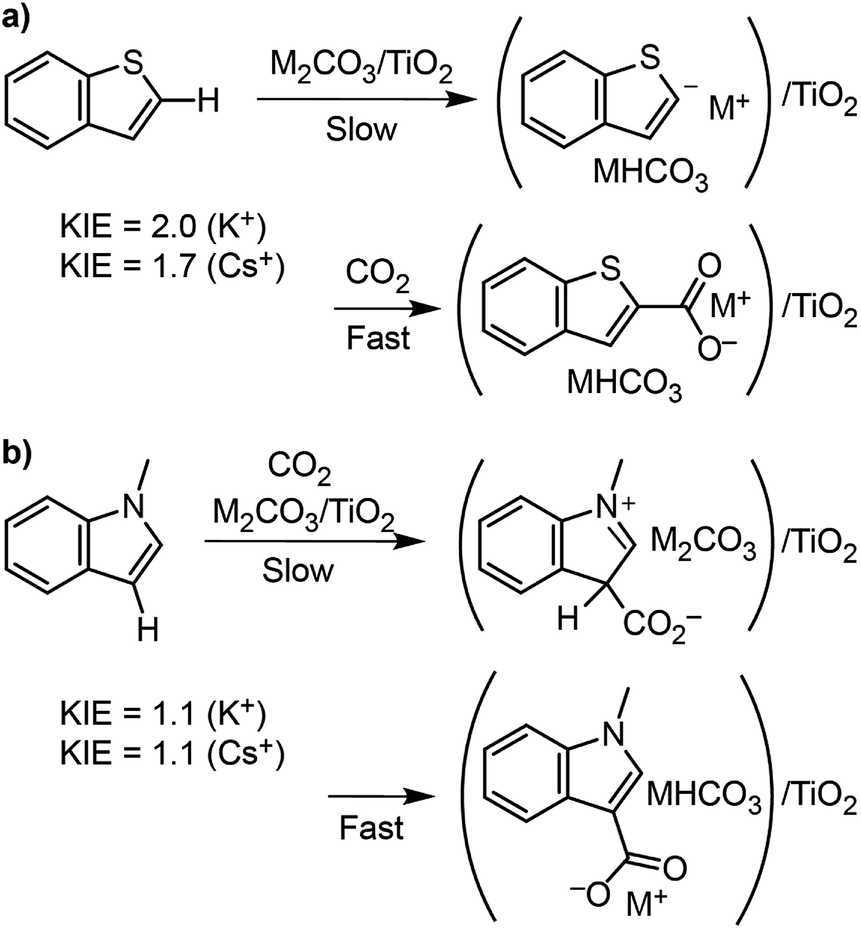
To better understand the differences in thiophene- vs. indole-based heteroarene C–H carboxylation, kinetic isotope effects (KIEs) were measured using intermolecular competition experiments.43 C–H carboxylation reactions were performed for 1-methylindole (200 °C, 1.5 h) and benzothiophene (260 °C, 0.5 h) using various ratios of protiated and deuterated substrate (Fig. 4).34 KIE values of 2.0 and 1.7 were observed for C2 carboxylation of benzothiophene using K2CO3/TiO2 and Cs2CO3/TiO2, respectively. These values are consistent with a mechanism in which C–H deprotonation is slow and the resulting carbanion reacts rapidly with CO2 (Scheme 2) and does not support an EAS mechanism. In addition, previous studies of benzothiophene substitution with strong electrophiles have shown selective substitution at C3, indicating that this is the preferred position for EAS reactivity.39,44,45 The KIE values for benzothiophene are similar to what we have previously observed for benzene C–H carboxylation using the same M2CO3/TiO2 materials,34 as well as solid base-catalyzed reactions that feature rate-determining deprotonation.46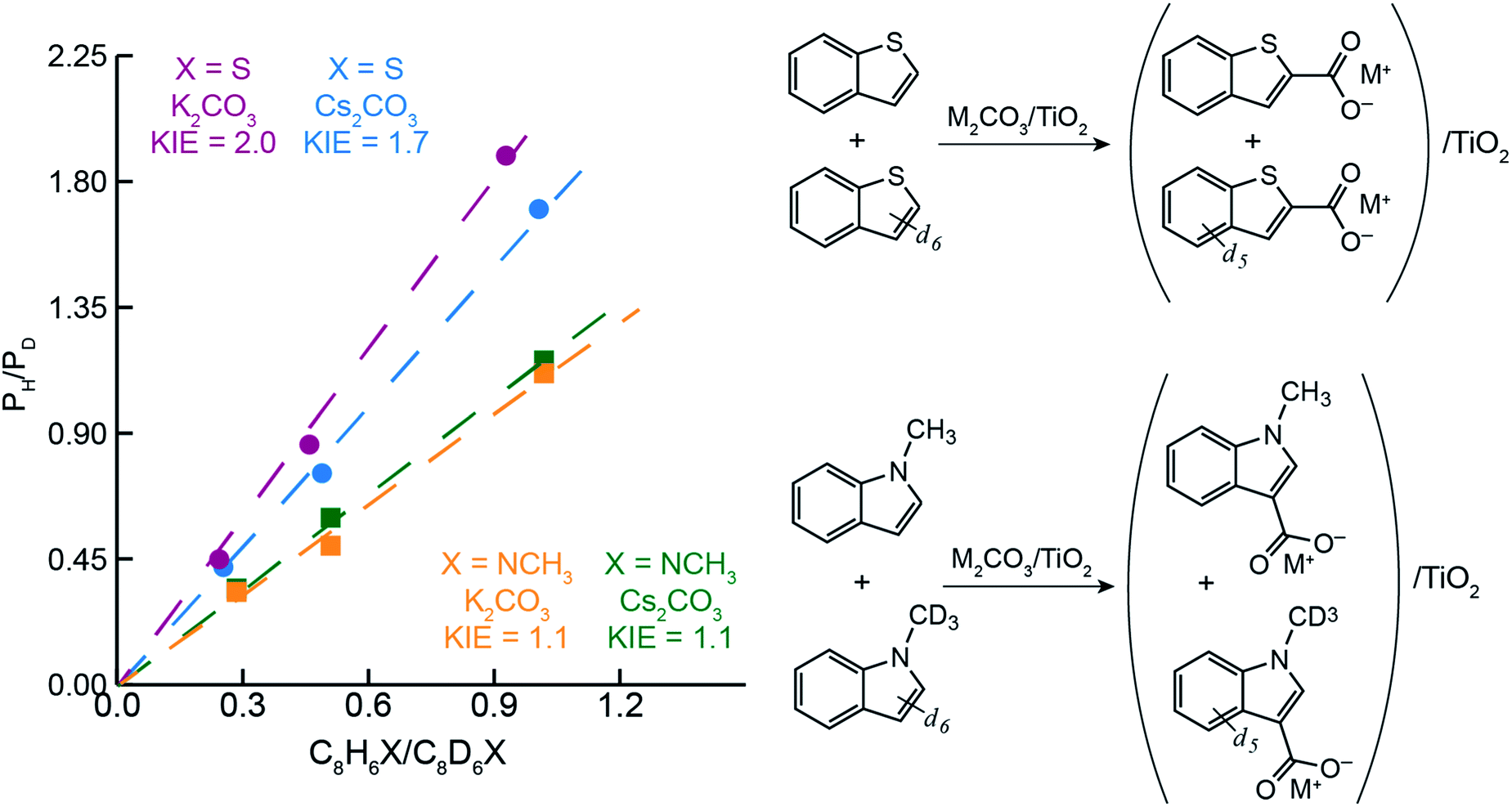 | ||
| Fig. 4 Ratio of protiated to deuterated products obtained from CO32−-promoted C–H carboxylation vs. the ratio of C8H6X to C8D6X (where X = S, NCH3, or NCD3) at 260 °C and 0.5 h for benzothiophene (top-right) and 200 °C and 1.5 h for methylindole (bottom-right). | ||
 | ||
| Scheme 2 Proposed C–H carboxylation mechanisms for (a) benzothiophene and (b) 1-methylindole. Note that MHCO3 is shown for simplicity, but under the reaction conditions it is thermally unstable and will convert to 0.5 equiv. M2CO3, H2O, and CO2. | ||
In contrast to benzothiophene, a KIE value of 1.1 was observed for C3 carboxylation of 1-methylindole, which is within NMR quantification error of 1.0. The disparity in KIE values for these two substrates indicates distinct mechanisms for their C–H carboxylation reactivity. The lack of a KIE for 1-methylindole is consistent with an EAS mechanism at 200 °C in which attack of the π system on CO2 precedes C–H deprotonation (Scheme 2). To our knowledge, an EAS reaction between CO2 and a neutral substrate has not previously been reported. DFT calculations were performed to assess the feasibility of such a pathway with 1-methylindole. Calculations performed using either vacuum or low dielectric solvents (ε < 9) failed to identify a transition state or putative EAS intermediate, suggesting that a gas-phase reaction between 1-methylindole and CO2 is unlikely. With a higher dielectric (ε > 20), however, an EAS transition state was identified that is ∼30 kcal mol−1 higher in energy than the substrates (Fig. S19†). Interestingly, the zwitterionic intermediate resulting from CO2 addition was very close in energy to the transition state, indicating that the reverse reaction is extremely rapid. Together, the KIE and DFT results suggest that the carboxylation of methylindole takes place via an EAS mechanism with substrate that is adsorbed onto the M2CO3/TiO2 material. The amorphous carbonate provides a dielectric to stabilize the transition state for CO2 addition and a proximal base that can immediately deprotonate the putative zwitterionic intermediate. The higher yield for Na2CO3/TiO2 may reflect a stronger adsorption of 1-methylindole because of the higher charge density for Na+. Further studies incorporating atomistic modeling of the amorphous carbonate surface are needed to assess this pathway more thoroughly. Nonetheless, the DFT results indicate that an EAS-like mechanism is possible.
Carboxylate esterification and M2CO3/TiO2 regeneration
In our previous study of arene C–H carboxylation, we showed that arene carboxylates could be isolated as methyl esters with concomitant regeneration of the M2CO3/TiO2 material by subjecting the carboxylation product to flowing CO2 and methanol at elevated temperatures.34 The same procedure was unsuccessful for isolating heteroarene carboxylate esters because their high boiling points (>300 °C) necessitated temperatures that led to decomposition under the reaction conditions. The use of flowing CO2 and dimethyl carbonate enabled isolation of methyl esters, but the yields were <50% (Fig. S20†). Instead, it was found that we could isolate the ester at near quantitative yields by heating the supported heteroarene carboxylate ((RCOOM)/TiO2) in neat dimethyl carbonate at 160 °C within a stainless-steel batch reactor (Fig. S21†). Subsequent heating of the support material under vacuum resulted in regeneration of M2CO3/TiO2. After establishing optimal carboxylation and methylation conditions, we assessed the ability of M2CO3/TiO2 to catalyze a closed heteroarene esterification cycle over multiple iterations (Fig. 5). When a single sample of Cs2CO3/TiO2 was used for 5 cycles, methyl benzothiophene-2-carboxylate was isolated as the only detectable product by NMR (Fig. S16†) from each cycle with an average yield of 150 μmol g−1 TiO2. In each cycle following the methylation step, an aliquot of the support (∼50 mg) was analyzed by aqueous extraction and 1H NMR to detect unreacted, supported carboxylate. In all cases, no supported carboxylates were observed, indicating complete methylation. Over the five cycles, no indication of catalyst degradation was observed (Fig. 5). These results support previous observations of the ability for dispersed carbonates to catalyze a closed esterification cycle,34 and extend the substrate scope to include heteroarenes.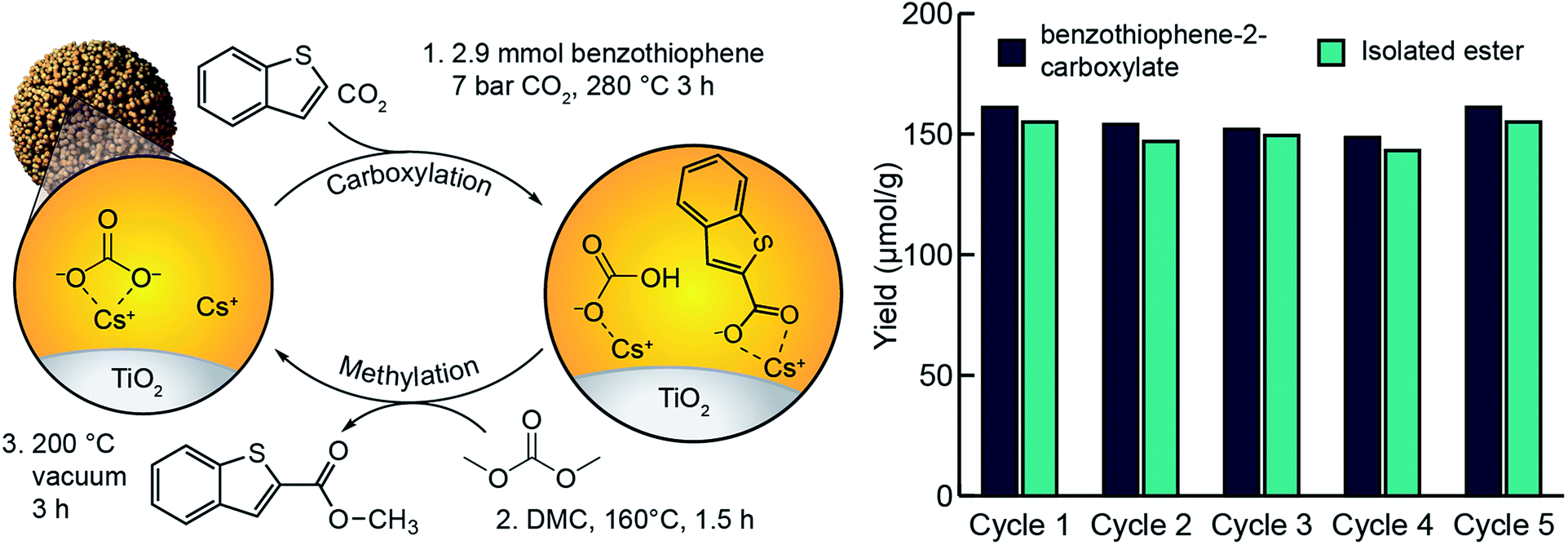 | ||
| Fig. 5 Cycling steps and yields for benzothiophene esterification using Cs2CO3/TiO2. Reaction conditions: carboxylation at 280 °C for 3 h, esterification at 160 °C for 1.5 h, followed by a regeneration cycle at 200 °C for 3 h under reduced pressure. | ||
Conclusion
Conventional solution-phase methods for C–H carboxylation of aromatic substrates with low C–H acidity have relied on the use of highly reactive and resource-intensive organic bases. Our results show that CO32− can serve as a benign, regenerable base for C–H carboxylation via a gas–solid reaction utilizing a dispersed, amorphous carbonate material. Compared to reactions with benzene and other arenes using the same M2CO3/TiO2 materials, the heteroarene carboxylations investigated here reach higher yields (up to 250 μmol g−1 TiO2) under substantially milder conditions (200 °C lower temperature, 1/3 the total pressure). Thiophene-based heterocycles react preferentially at the most acidic C–H bond. The temperature-dependent selectivity and KIE measured for benzothiophene are consistent with a mechanism in which C–H deprotonation is followed by C–C bond formation. In contrast, indole-based heterocycles react preferentially at the most nucleophilic position (C3). DFT calculations and the absence of a significant KIE support an EAS mechanism for the carboxylation of 1-methylindole, which nonetheless requires dispersed carbonate. The combination of CO32−-promoted C–H carboxylation and methylation with dimethyl carbonate provides a two-step cycle to convert aromatic heteroarenes into methyl esters with regeneration of M2CO3/TiO2. Ongoing work seeks to improve the efficiency of this cycle by using alternative supports to increase the loading of reactive carbonate and access reactivity at lower temperatures.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Science under award DE-SC0020394 and the TomKat Center for Sustainable Energy. T. M. P. gratefully acknowledges support from the Camille and Henry Dreyfus Foundation Postdoctoral Program in Environmental Chemistry. The authors would like to thank Dr Stephen Lynch and the Stanford NMR facility for technical assistance with NMR collection and Dr Jan Meisner for insightful discussions regarding the DFT calculations. The authors also thank the Stanford Research Computing Center for providing computational resources and support.References
- I. Tomassi, Direct Carboxylation of C(sp3)–H and C(sp2)–H Bonds with CO2 by Transition-Metal-Catalyzed and Base-Mediated Reactions, Catalysts, 2017, 7, 380 CrossRef.
- J. Luo and I. Larrosa, C–H Carboxylation of Aromatic Compounds through CO2 Fixation, ChemSusChem, 2017, 10(17), 3317–3332 CrossRef CAS.
- J. Hong, M. Li, J. Zhang, B. Sun and F. Mo, C–H Bond Carboxylation with Carbon Dioxide, ChemSusChem, 2019, 12(1), 6–39 CrossRef CAS.
- O. Vechorkin, N. Hirt and X. Hu, Carbon Dioxide as the C1 Source for Direct C–H Functionalization of Aromatic Heterocycles, Org. Lett., 2010, 12(15), 3567–3569 CrossRef CAS.
- B. Pieber, T. Glasnov and C. O. Kappe, Flash carboxylation: fast lithiation–carboxylation sequence at room temperature in continuous flow, RSC Adv., 2014, 4(26), 13430–13433 RSC.
- W.-J. Yoo, T. V. Q. Nguyen, M. G. Capdevila and S. Kobayashi, Lithium tert-Butoxide-Mediated Carboxylation Reactions of Unprotected Indoles and Pyrroles with Carbon Dioxide, Heterocycles, 2015, 90(2), 1196–1204 CrossRef CAS.
- S. Fenner and L. Ackermann, C–H carboxylation of heteroarenes with ambient CO2, Green Chem., 2016, 18(13), 3804–3807 RSC.
- J. Luo, S. Preciado, P. Xie and I. Larrosa, Carboxylation of Phenols with CO2 at Atmospheric Pressure, Chem.–Eur. J., 2016, 22(20), 6798–6802 CrossRef CAS.
- M. Shigeno, K. Hanasaka, K. Sasaki, K. Nozawa-Kumada and Y. Kondo, Direct Carboxylation of Electron-Rich Heteroarenes Promoted by LiO-tBu with CsF and [18]Crown-6, Chem.–Eur. J., 2019, 25(13), 3235–3239 CAS.
- H. Seo, M. H. Katcher and T. F. Jamison, Photoredox activation of carbon dioxide for amino acid synthesis in continuous flow, Nat. Chem., 2017, 9(5), 453–456 CrossRef CAS.
- N. Ishida, Y. Masuda, Y. Imamura, K. Yamazaki and M. Murakami, Carboxylation of Benzylic and Aliphatic C–H Bonds with CO2 Induced by Light/Ketone/Nickel, J. Am. Chem. Soc., 2019, 141(50), 19611–19615 CrossRef CAS.
- Q.-Y. Meng, T. E. Schirmer, A. L. Berger, K. Donabauer and B. König, Photocarboxylation of Benzylic C–H Bonds, J. Am. Chem. Soc., 2019, 141(29), 11393–11397 CrossRef CAS.
- H. Seo, L. V. Nguyen and T. F. Jamison, Using Carbon Dioxide as a Building Block in Continuous Flow Synthesis, Adv. Synth. Catal., 2019, 361(2), 247–264 CrossRef CAS.
- C. S. Yeung, Photoredox Catalysis as a Strategy for CO2 Incorporation: Direct Access to Carboxylic Acids from a Renewable Feedstock, Angew. Chem., Int. Ed., 2019, 58(17), 5492–5502 CrossRef CAS.
- Carbon Dioxide as Chemical Feedstock, ed. M. Aresta, Wiley-VCH, Weinheim, 2010 Search PubMed.
- M. Börjesson, T. Moragas, D. Gallego and R. Martin, Metal-Catalyzed Carboxylation of Organic (Pseudo)halides with CO2, ACS Catal., 2016, 6(10), 6739–6749 CrossRef.
- L. J. Gooßen, N. Rodríguez and K. Gooßen, Carboxylic Acids as Substrates in Homogeneous Catalysis, Angew. Chem., Int. Ed., 2008, 47(17), 3100–3120 CrossRef.
- D. Kumar, N. Maruthi Kumar, K.-H. Chang and K. Shah, Synthesis and anticancer activity of 5-(3-indolyl)-1,3,4-thiadiazoles, Eur. J. Med. Chem., 2010, 45(10), 4664–4668 CrossRef CAS.
- H. Maag, Prodrugs of Carboxylic Acids, in Prodrugs: Challenges and Rewards Part 1, ed. V. J. Stella, R. T. Borchardt, M. J. Hageman, R. Oliyai, H. Maag and J. W. Tilley, Springer New York, New York, NY, 2007, pp. 703–729 Search PubMed.
- O. Boullard, H. Leblanc and B. Besson, Salicylic Acid, Ullmann's Encyclopedia of Industrial Chemistry, 2000 Search PubMed.
- W.-J. Yoo, M. G. Capdevila, X. Du and S. Kobayashi, Base-Mediated Carboxylation of Unprotected Indole Derivatives with Carbon Dioxide, Org. Lett., 2012, 14(20), 5326–5329 CrossRef CAS.
- C. Pei, J. Zong, S. Han, B. Li and B. Wang, Ni-Catalyzed Direct Carboxylation of an Unactivated C–H Bond with CO2, Org. Lett., 2020, 22(17), 6897–6902 CrossRef CAS.
- H. Mizuno, J. Takaya and N. Iwasawa, Rhodium(I)-Catalyzed Direct Carboxylation of Arenes with CO2 via Chelation-Assisted C–H Bond Activation, J. Am. Chem. Soc., 2011, 133(5), 1251–1253 CrossRef CAS.
- L. Fu, S. Li, Z. Cai, Y. Ding, X.-Q. Guo, L.-P. Zhou, D. Yuan, Q.-F. Sun and G. Li, Ligand-enabled site-selectivity in a versatile rhodium(II)-catalysed aryl C–H carboxylation with CO2, Nat. Catal., 2018, 1(6), 469–478 CrossRef CAS.
- T. Suga, H. Mizuno, J. Takaya and N. Iwasawa, Direct carboxylation of simple arenes with CO2 through a rhodium-catalyzed C–H bond activation, Chem. Commun., 2014, 50(92), 14360–14363 RSC.
- M. Schlosser, H. C. Jung and S. Takagishi, Selective mono- or dimetalation of arenes by means of superbasic reagents, Tetrahedron, 1990, 46(16), 5633–5648 CrossRef CAS.
- G. A. Olah, B. Török, J. P. Joschek, I. Bucsi, P. M. Esteves, G. Rasul and G. K. Surya Prakash, Efficient Chemoselective Carboxylation of Aromatics to Arylcarboxylic Acids with a Superelectrophilically Activated Carbon Dioxide—Al2Cl6/Al System, J. Am. Chem. Soc., 2002, 124(38), 11379–11391 CrossRef CAS.
- M. Schmalzbauer, T. D. Svejstrup, F. Fricke, P. Brandt, M. J. Johansson, G. Bergonzini and B. König, Redox-Neutral Photocatalytic C–H Carboxylation of Arenes and Styrenes with CO2, Chem, 2020 DOI:10.1016/j.chempr.2020.08.02.
- A. Banerjee, G. R. Dick, T. Yoshino and M. W. Kanan, Carbon dioxide utilization via carbonate-promoted C–H carboxylation, Nature, 2016, 531(7593), 215–219 CrossRef CAS.
- G. R. Dick, A. D. Frankhouser, A. Banerjee and M. W. Kanan, A scalable carboxylation route to furan-2,5-dicarboxylic acid, Green Chem., 2017, 19(13), 2966–2972 RSC.
- A. Banerjee and M. W. Kanan, Carbonate-Promoted Hydrogenation of Carbon Dioxide to Multicarbon Carboxylates, ACS Cent. Sci., 2018, 4(5), 606–613 CrossRef CAS.
- A. W. Lankenau and M. W. Kanan, Polyamide monomers via carbonate-promoted C–H carboxylation of furfurylamine, Chem. Sci., 2020, 11(1), 248–252 RSC.
- A. D. Frankhouser and M. W. Kanan, Phase Behavior That Enables Solvent-Free Carbonate-Promoted Furoate Carboxylation, J. Phys. Chem. Lett., 2020, 11(18), 7544–7551 CrossRef CAS.
- D. J. Xiao, E. D. Chant, A. D. Frankhouser, Y. Chen, A. Yau, N. M. Washton and M. W. Kanan, A closed cycle for esterifying aromatic hydrocarbons with CO2 and alcohol, Nat. Chem., 2019, 11(10), 940–947 CrossRef CAS.
- E. V. Anslyn and D. A. Dougherty, Modern Physical Organic Chemistry, University Science Books, 2006 Search PubMed.
- K. Shen, Y. Fu, J.-N. Li, L. Liu and Q.-X. Guo, What are the pKa values of C–H bonds in aromatic heterocyclic compounds in DMSO?, Tetrahedron, 2007, 63(7), 1568–1576 CrossRef CAS.
- R. R. Fraser, T. S. Mansour and S. Savard, Acidity measurements in THF. V. Heteroaromatic compounds containing 5-membered rings, Can. J. Chem., 1985, 63(12), 3505–3509 CrossRef CAS.
- H. Mayr, B. Kempf and A. R. Ofial, π-Nucleophilicity in Carbon–Carbon Bond-Forming Reactions, Acc. Chem. Res., 2003, 36(1), 66–77 CrossRef CAS.
- T. Eicher, S. Hauptmann and A. Speicher, Five-Membered Heterocycles: Sections 5.1–5.21, The Chemistry of Heterocycles, 2003, pp. 52–121 Search PubMed.
- K. Nemoto, S. Tanaka, M. Konno, S. Onozawa, M. Chiba, Y. Tanaka, Y. Sasaki, R. Okubo and T. Hattori, Me2AlCl-mediated carboxylation, ethoxycarbonylation, and carbamoylation of indoles, Tetrahedron, 2016, 72(5), 734–745 CrossRef CAS.
- S. Tanaka, K. Watanabe, Y. Tanaka and T. Hattori, EtAlCl2/2,6-Disubstituted Pyridine-Mediated Carboxylation of Alkenes with Carbon Dioxide, Org. Lett., 2016, 18(11), 2576–2579 CrossRef CAS.
- M. Westermaier and H. Mayr, Electrophilic Allylations and Benzylations of Indoles in Neutral Aqueous or Alcoholic Solutions, Org. Lett., 2006, 8(21), 4791–4794 CrossRef CAS.
- E. M. Simmons and J. F. Hartwig, On the Interpretation of Deuterium Kinetic Isotope Effects in C–H Bond Functionalizations by Transition-Metal Complexes, Angew. Chem., Int. Ed., 2012, 51(13), 3066–3072 CrossRef CAS.
- V. Ji Ram, A. Sethi, M. Nath and R. Pratap, Chapter 5 – Five-Membered Heterocycles, in The Chemistry of Heterocycles, ed. V. Ji Ram, A. Sethi, M. Nath and R. Pratap, Elsevier, 2019, pp. 149–478 Search PubMed.
- J. K. Kajorinne, J. C. M. Steers, M. E. Merchant and C. D. MacKinnon, Green halogenation reactions for (hetero)aromatic ring systems in alcohol, water, or no solvent, Can. J. Chem., 2018, 96(12), 1087–1091 CrossRef CAS.
- Y. Ono and H. Hattori, Characterization of Solid Base Catalysts, in Solid Base Catalysis, ed. Y. Ono and H. Hattori, Springer Berlin Heidelberg, Berlin, Heidelberg, 2011, pp. 11–68 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc04548a |
| This journal is © The Royal Society of Chemistry 2020 |