
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The simplest Diels–Alder reactions are not endo-selective†
William J.
Lording
a,
Thomas
Fallon
a,
Michael S.
Sherburn
 *a and
Michael N.
Paddon-Row
*b
*a and
Michael N.
Paddon-Row
*b
aResearch School of Chemistry, Australian National University, Canberra, ACT 2601, Australia. E-mail: michael.sherburn@anu.edu.au
bSchool of Chemistry, University of New South Wales, NSW 2052, Australia
First published on 6th October 2020
Abstract
There is a widespread perception that the high level of endo selectivity witnessed in many Diels–Alder reactions is an intrinsic feature of the transformation. In contrast to expectations based upon this existing belief, the first experimental Diels–Alder reactions of a novel, deuterium-labeled 1,3-butadiene with commonly used mono-substituted alkenic dienophiles (acrolein, methyl vinyl ketone, acrylic acid, methyl acrylate, acrylamide and acrylonitrile) reveal kinetic endo![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :exo ratios close to 1:1. Maleonitrile, butenolide, α-methylene γ-butyrolactone, and N-methylmaleimide behave differently, as does methyl vinyl ketone under Lewis acid catalysis. CBS-QB3 calculations incorporating solvent and temperature parameters give endo:exo product ratios that are in near quantitative agreement with these and earlier experimental findings. This work challenges the preconception of innate endo-selectivity by providing the first experimental evidence that the simplest Diels–Alder reactions are not endo-selective. Trends in behaviour are traced to steric and electronic effects in Diels–Alder transition structures, giving new insights into these fundamental processes.
:exo ratios close to 1:1. Maleonitrile, butenolide, α-methylene γ-butyrolactone, and N-methylmaleimide behave differently, as does methyl vinyl ketone under Lewis acid catalysis. CBS-QB3 calculations incorporating solvent and temperature parameters give endo:exo product ratios that are in near quantitative agreement with these and earlier experimental findings. This work challenges the preconception of innate endo-selectivity by providing the first experimental evidence that the simplest Diels–Alder reactions are not endo-selective. Trends in behaviour are traced to steric and electronic effects in Diels–Alder transition structures, giving new insights into these fundamental processes.
Introduction
The Diels–Alder (DA) reaction1 remains one of the most important reactions in chemical synthesis.2 The most well-known pericyclic reaction unites a diene and a dienophile through a concerted, thermally allowed 4 + 2 cycloaddition, which generates a new six membered ring, two new σ-bonds and up to four contiguous stereocenters.3 The transformation has found wide application in chemical synthesis by virtue of its tolerance towards substitution and the inclusion of diverse functionality within the diene and dienophile.4 The reaction played a central role in the development of theories of organic reactivity, including the conservation of orbital symmetry5 and frontier molecular orbital (FMO) theory.6 The longevity of the DA reaction is unparalleled: it is as significant today as it was 50 years ago. Indeed, synthetic chemistry would be unrecognizable without it.7A fundamental attribute of DA reactions between 1,3-butadienes and substituted olefinic dienophiles is the potential for the formation of endo and exo diastereomeric products. These diastereomeric products result from two distinct transition state structures (TSs) in which a specific dienophile substituent is either closer to (endo) or more distant from (exo) C2 and C3 of the diene (Scheme 1).
 | ||
| Scheme 1 Endo/exo transition structures (TSs) and products in Diels–Alder (DA) reactions. A requirement for distinct endo and exo-stereoisomers is two different groups a reacting site in both diene and dienophile. | ||
Certain structural requirements must be met in the diene and dienophile for the generation of endo and exo diastereomers. Specifically, the substituents on at least one of the two ends of the diene (i.e. C1 and/or C4) and one of the two dienophile carbons must be different. If the dienophile does not fulfil this requirement (e.g. ethylene) then endo- and exo-TSs are not possible. Conversely, if only the diene does not satisfy this condition (as in 1,3-butadiene) then non-equivalent endo and exo-TSs are generated, but they deliver the same cycloadduct.
Early experimental studies on endo/exo stereoselectivity in Diels–Alder reactions by Alder and Stein led to the empirical rule of the “maximum accumulation of unsaturation”.8 Often cited as the “Alder endo rule”, the endo mode of addition is favored by dienophiles bearing unsaturated groups in conjugation with the dienophile's reacting double bond (i.e.Scheme 1,  = COR, CN, etc.). Various theoretical proposals have been advanced to explain the endo selectivity of DA reactions. Secondary orbital interactions (SOIs) are the most widely accepted cause,9 and the two most common types are those proposed by Woodward and Hoffmann (WH SOI)10 and by Salem and Houk (SH SOI).11 The former involves overlap of diene C2 with the carbonyl carbon of the dienophile substituent, and the latter involves overlap of diene C3 with the oxygen of the dienophile carbonyl substituent. Some cycloadditions, for example dimerizations of cyclopentadiene (CPD) and 1,3-butadiene (BD) exhibit bispericyclic TSs, whereupon the SH-type SOI becomes indistinguishable from one of the two σ-bonds being formed.12 The origin of endo/exo selectivity in DA reactions and the existence of SOIs has been debated,13 with other types of interactions being invoked to explain endo-selective DA reactions, amongst them solvent effects,14 electrostatic forces,15 and pre-reaction van der Waals forces.16
= COR, CN, etc.). Various theoretical proposals have been advanced to explain the endo selectivity of DA reactions. Secondary orbital interactions (SOIs) are the most widely accepted cause,9 and the two most common types are those proposed by Woodward and Hoffmann (WH SOI)10 and by Salem and Houk (SH SOI).11 The former involves overlap of diene C2 with the carbonyl carbon of the dienophile substituent, and the latter involves overlap of diene C3 with the oxygen of the dienophile carbonyl substituent. Some cycloadditions, for example dimerizations of cyclopentadiene (CPD) and 1,3-butadiene (BD) exhibit bispericyclic TSs, whereupon the SH-type SOI becomes indistinguishable from one of the two σ-bonds being formed.12 The origin of endo/exo selectivity in DA reactions and the existence of SOIs has been debated,13 with other types of interactions being invoked to explain endo-selective DA reactions, amongst them solvent effects,14 electrostatic forces,15 and pre-reaction van der Waals forces.16
The empirical Alder endo rule is successful in predicting the strong endo-selectivity of kinetically controlled, thermal DA reactions involving rigid and highly activated cyclic dienophiles such as maleic anhydride and benzoquinone. Its extension to thermal reactions of acyclic dienophiles is less clear cut. Furthermore, there are many counterexamples to the endo rule (i.e. reactions exhibiting an exo preference) and this number continues to grow.17–20Exo-selectivity is often attributed to diene and dienophile substitution patterns that generate destabilizing steric strain in endo-TSs,21 although certain catalysts are also effective in promoting exo-selective DA reactions.22
In spite of the large number of mechanistic studies on the Diels–Alder reaction,3 only three experimental studies have been carried out on the endo/exo-selectivity of Diels–Alder reactions involving the parent, archetypal 1,3-butadiene (BD), as summarized in Scheme 2. Thus, the Diels–Alder dimerization of (Z,Z)-1,4-dideutero-1,3-butadiene (Z,Z)-d2-1 (eqn (1)) was found to be very mildly (endo:exo = 56:44)23endo-selective, whereas the Diels–Alder reaction of (Z,Z)-d2-1 with maleic anhydride (eqn (2)) was more strongly endo-selective (endo:exo = 85:15)24 and the DA reaction between E/Z-deutero-1,3-butadiene (E/Z)-d1-1 and cyclopropene (eqn (3)) was very strongly endo-selective (endo:exo >99:1).25 These outcomes were attributed to controlling SOIs.13b,26
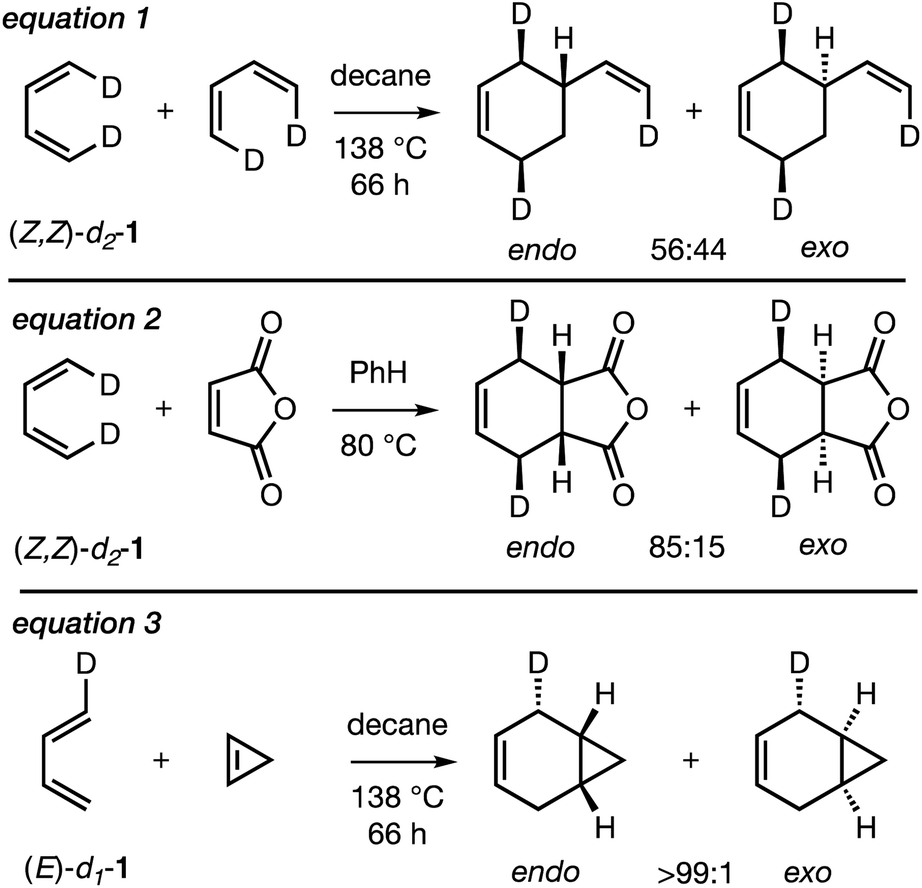 | ||
| Scheme 2 Reported experimental studies on the endo/exo-selectivity of Diels–Alder reactions involving deuterium-labeled 1,3-butadienes (Z,Z)-d2-1 and (E)-d1-1. Both E- and Z-isomers of 1-deutero-1,3-butadiene (eqn (3)) were used but only one stereoisomer is shown for clarity. | ||
The observations of a large difference in the degree of endo selectivity between BD, acting as dienophile (Scheme 2, eqn (1)) and the more reactive maleic anhydride and cyclopropene dienophiles (Scheme 2, eqn (2) and (3), respectively) have attracted recent computational investigations employing distortion–interaction or activation-strain methods,27 as well as energy decomposition analysis techniques.28 In the case of the DA reaction between BD and MA, Fernández and Bickelhaupt attributed high endo selectivity to unfavourable steric interactions in the exo-TS pathway.29 In the case of the cyclopropene BD reaction, Houk and co-workers attribute endo selectivity to several factors including favorable CH⋯π SOIs in the endo-TS.30 The important question of whether endo-selective DA reactions of BD are more generally preferred remains open, since DA reactions of BD with a range of alkene dienophiles bearing substituents covering a broad spectrum of electron withdrawing properties have not yet been reported.
Several computational studies bearing on this issue have appeared, dealing largely with acrolein and acrylonitrile dienophiles. Density functional theory (DFT) and ab initio MO calculations predict moderate to strong endo selectivity for the BD + acrolein Diels–Alder reaction, in the gas phase31 and in solution,32 the degree of endo selectivity being predicted to increase markedly in the Lewis acid catalyzed reaction,33–35 a finding that is consistent with simple frontier MO arguments36 and those relating to diminished Pauli repulsion between the diene and dienophile π-systems.37
Computational results for the Diels–Alder reaction between BD and acrylonitriles are not clear cut. Gas phase Hartree–Fock calculations predict modest exo selectivity for the reactions of BD and CPD with acrylonitrile and maleonitrile.38,39 The predicted exo pathway for the CPD reactions is at variance with the experimentally observed endo mode for this diene with acrylonitrile and maleonitrile.40 However, inclusion of non-specific solvent effects, in the form of self-consistent reaction field theory, reversed the preferred mode to endo for the reaction of both BD and CPD with the two acrylonitriles.39 It was concluded that solvent polarity, not SOIs, is responsible for the endo selectivity in these reactions,39 although the endo selectivity for the reactions with BD remained an experimentally untested prediction. As part of a DFT (B3LYP) study of intramolecular Diels–Alder reactions, we investigated substituent effects on endo/exo selectivities of Diels–Alder reactions between BD and monosubstituted ethylenic dienophiles (CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–Z; Z = CN, CO2Me, CO2H, NO2, CHO, COMe).41 It was found that endo selectivity is predicted for methyl vinyl ketone and acrolein. However, this finding, like those from earlier studies that used the Hartree–Fock procedure,38,39 is unreliable because B3LYP seriously underestimates dispersion energies, thereby skewing the selectivity towards the exo reaction channel.
CH–Z; Z = CN, CO2Me, CO2H, NO2, CHO, COMe).41 It was found that endo selectivity is predicted for methyl vinyl ketone and acrolein. However, this finding, like those from earlier studies that used the Hartree–Fock procedure,38,39 is unreliable because B3LYP seriously underestimates dispersion energies, thereby skewing the selectivity towards the exo reaction channel.
In summary, there exists an important gap in our knowledge—both experimental and computational—concerning the endo/exo selectivity in Diels–Alder reactions involving the most fundamental diene of all, 1,3-butadiene. We have addressed this lacuna and, in this paper, we present the results of our experimental determination of the stereochemical outcomes from the reaction of (1E,3E)-1,4-dideutero-1,3-butadiene 1 with a wide range of dienophiles (Fig. 1). Also presented are the results of DA reactions between the same dienophiles with CPD,42 and a high-level quantum chemical study of these reactions.
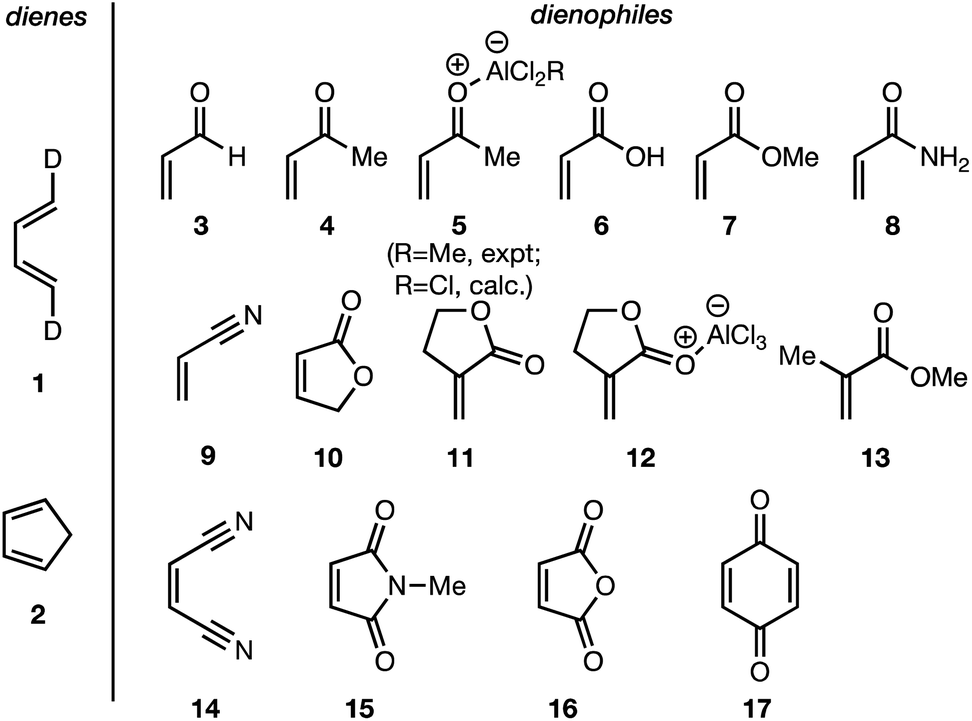 | ||
| Fig. 1 Diels–Alder reactions between the depicted dienes and dienophiles under investigation in this joint experimental–computational study. | ||
Results and discussion
Synthesis of (1E,3E)-1,4-dideutero-1,3-butadiene 1
As discussed above (Scheme 2), previous studies23–25 were carried out with (1E)- and (1Z)-1-deutero-1,3-butadiene, (E/Z)-d1-1, and (1Z,3Z)-1,4-dideutero-1,3-butadiene, (Z,Z)-d2-1. (1E)- and (1Z)-1-deutero-1,3-butadienes are unsuitable for our purposes, since they would lead to mixtures of regioisomeric products with mono-substituted dienophiles. We elected not to repeat the published synthesis of (1Z,3Z)-1,4-dideutero-1,3-butadiene43 due to the involvement of intricate separations and low yields. Ultimately, we targeted the previously unreported (1E,3E)-1,4-dideutero-1,3-butadiene, 1. The requirements for this synthesis would be challenging, since the study mandated access to multigram quantities of this volatile (bp = −4 °C) hydrocarbon in high purity. Our successful two step synthesis of (1E,3E)-1,4-dideutero-1,3-butadiene 1 is shown in Scheme 3. | ||
| Scheme 3 Synthesis of (1E,3E)-1,4-dideutero-1,3-butadiene. | ||
Optimization of the reported44 Pt(IV)-catalyzed iodinative dimerization of acetylene allowed convenient access to (1E,3E)-1,4-diiodo-1,3-butadiene 1 in high stereochemical purity on multigram scale. Metal–halogen exchange of di-iodide 20 using Oshima's trialkylmagnesate reagent45 followed by deutero-demetalation with MeOD furnished the target (1E,3E)-1,4-dideutero-1,3-butadiene 1 in a highly stereoretentive manner (>95% 1E,3E- and >90% d2). Following purification, this compound was kept as a benzene or CH2Cl2 solution for ease of storage and handling.
Diels–Alder reactions
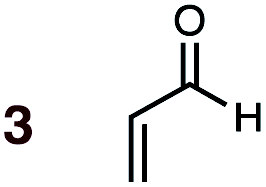
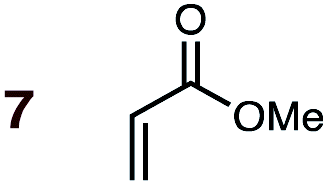
Uncatalyzed cycloaddition reactions between the new, labeled 1,3-butadiene 1 and the dienophiles acrolein 3, methyl vinyl ketone 4, acrylic acid 6, methyl acrylate 7, acrylamide 8, acrylonitrile 9, maleonitrile 14, butenolide 10, α-methylene γ-butyrolactone 11 and N-methylmaleimide 16 were carried out in benzene solution, and the results are summarized in Table 1. Experimental endo:exo ratios were determined by quantitative 800 MHz 1H NMR spectroscopy.46,47 The majority of reactions were carried out with a 1:1 molar ratio of starting diene and dienophile at 1 M concentrations in sealed tubes at 145 °C, the exceptions being the more reactive dienophiles maleonitrile 14 (100 °C) and N-methylmaleimide 15 (20 °C).
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Dienophile | Diene | Adducts | Temp. (°C) | Time | Isolated yield (%) |
Endo:exo (exp.) |
Endo:exo (calc.)a |
(s-cis endo:s-trans endo):(s-cis exo:s-trans exo) (calc.) |
HOMOdiene–LUMOdienophile (eV) |
| a Relative ΔG‡, CBS-QB3, benzene phase. Calculated at the experimental temperature where available. b Experiment conducted with MeAlCl2. c Experimental result from Buono and coworkers.18 d Experimental results from Stephenson and coworkers.24 e Experimental result from Mal and Ray.57 f Experimental result from Stephenson et al. and Klärner et al.23 g Experimental result from Baldwin and Reddy.25 h Experimental result from Wiberg and Bartley.58 | |||||||||
|
1 | 3BD-n, 3BD-x | 145 | 20 h | 37 | 64:36 |
86:14 |
(0.0:5.6):(4.7:9.0) |
4.4 |
| 2 | 3CPD-n, 3CPD-x | 80 | 70 min | 69 | 73:27 |
81:19 |
(0.0:3.4):(3.9:5.9) |
4.0 | |

|
1 | 4BD-n, 4BD-x | 145 | 24 h | 64 | 65:35 |
64:36 |
(0.0:7.8):(1.4:11.1) |
4.7 |
| 2 | 4CPD-n, 4CPD-x | 80 | 20 min | 83 | 79:21 |
86:14 |
(0.0:8.3):(4.5:16.4) |
4.3 | |

|
1 | 5BD-n, 5BD-x | −78 to rt | 20 h | 83 | >95:5b |
99:1 |
(0.0:17.0):(13.0:27.1) |
2.4 |
| 2 | 5CPD-n, 5CPD-x | — | — | — | — | — | — | — | |

|
1 | 6BD-n, 6BD-x | 145 | 40 h | 90 | 60:40 |
60:40 |
(0.0:4.8):(1.1:5.2) |
4.8 |
| 2 | 6CPD-n, 6CPD-x | 80 | 25 min | 94 | 71:29 |
87:13 |
(0.0:5.7):(4.7:10.2) |
4.4 | |
|
1 | 7BD-n, 7BD-x | 145 | 90 h | 83 | 50:50 |
67:33 |
(0.0:4.7):(1.9:5.7) |
5.0 |
| 2 | 7CPD-n, 7CPD-x | 80 | 6.5 h | 86 | 77:23 |
76:24 |
(0.0:4.2):(2.6:8.9) |
4.6 | |
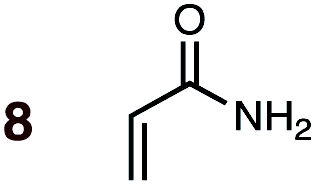
|
1 | 8BD-n, 8BD-x | 145 | 120 h | 9 | 46:54 |
35:65 |
(1.8:7.2):(0.0:11.1) |
5.3 |
| 2 | 8CPD-n, 8CPD-x | 80 | 25 min | 15 | 64:36 |
76:24 |
(0.0:7.4):(2.8:14.8) |
4.9 | |
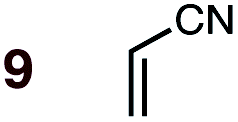
|
1 | 9BD-n, 9BD-x | 145 | 120 h | 76 | 37:63 |
44:56 |
0.6:0.0 |
4.6 |
| 2 | 9CPD-n, 9CPD-x | 80 | 16 h | 87 | 55:45 |
60:40 |
0.0:1.0 |
4.2 | |

|
1 | 10BD-n, 10BD-x | 145 | 120 h | 27 | 73:27 |
82:18 |
0.0:3.9 |
4.9 |
| 2 | 10CPD-n, 10CPD-x | 80 | 68 h | 40 | 80:20 |
90:10 |
0.0:5.3 |
4.5 | |
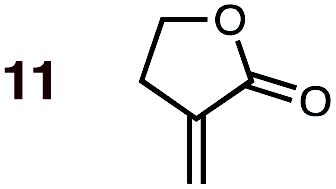
|
1 | 11BD-n, 11BD-x | 145 | 20 h | 87 | 39:61 |
27:73 |
2.5:0.0 |
4.9 |
| 2 | 11CPD-n, 11CPD-x | 80 | 4 h | 72 | 12:88 |
10:90 |
5.3:0.0 |
4.5 | |
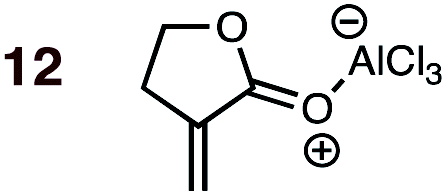
|
1 | 12BD-n, 12BD-x | — | — | — | — | 89:11 |
0.0:5.1 |
3.0 |
| 2 | 12CPD-n, 12CPD-x | −15 °C | — | 72 | 6:94c |
11:89 |
5.2:0.0 |
— | |
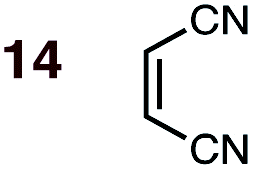
|
1 | 14BD-n, 14BD-x | 100 | 24 h | 75 | 70:30 |
66:43 |
0.0:1.7 |
3.2 |
| 2 | 14CPD-n, 14CPD-x | 20 | 2 h | 81 | 73:27 |
59:41 |
0.0:1.0 |
2.8 | |

|
1 | 15BD-n, 15BD-x | 20 | 24 h | 92 | >99:1 |
99:1 |
0.0:10.9 |
3.5 |
| 2 | 15CPD-n, 15CPD-x | 20 | 15 min | 98 | >99:1 |
>99:1 |
0.0:14.9 |
3.1 | |

|
1 | 16BD-n, 16BD-x | 80 | — | — | 85:15d |
98:2 |
0.0:9.4 |
3.0 |
| 2 | 16CPD-n, 16CPD-x | 25 | — | — | 99.5:0.5d |
97:3 |
0.0:8.6 |
2.6 | |
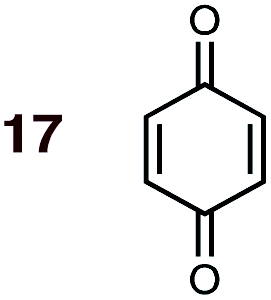
|
1 | 17BD-n, 17BD-x | — | — | — | — | 98:2 |
0.0:9.4 |
2.6 |
| 2 | 17CPD-n, 17CPD-x | 20 | 1 h | 80 | >99:1e |
>99:1 |
0.0:12.7 |
2.2 | |

|
1 | 18BD-n, 18BD-x | 138 | 55 h | 66 | 56:44f |
— | — | — |
| 2 | 18CPD-n, 18CPD-x | — | — | — | — | — | — | — | |
|
1 | 19BD-n, 19BD-x | 0 | 2 h | — | >99:1g |
— | — | — |
| 2 | 19CPD-n, 19CPD-x | 0 | — | 97 | >99:1h |
— | — | — | |
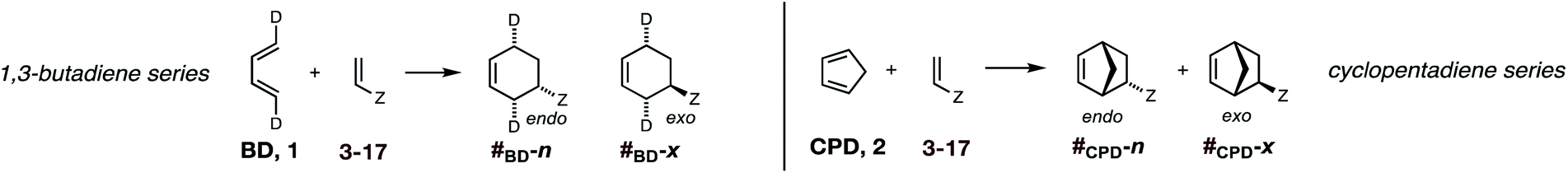
The DA reactions of CPD with the same ten dienophiles were carried out in the same manner, except at a lower temperature. Thus, a 1:1 molar ratio of CPD and dienophile (1 M concentration solutions of both diene and dienophile in benzene) were heated at 80 °C in the majority of cases, with the DA reactions of the more reactive dienophiles maleonitrile 14 and N-methylmaleimide 15 being conducted at 20 °C and the DA reactions of the three least reactive dienophiles employing 3 molar equivalents of CPD.
The majority of DA reactions proceeded cleanly and smoothly, the exceptions being those involving the dienophile acrylamide 8, which was very low yielding due to its poor DA dienophile reactivity and competing polymerization, presumably through Michael addition pathways. Butenolide was also poorly reactive, giving rise to low yielding reactions with both BD and CPD. At the other end of the reactivity scale, acrolein 3 was the most reactive of the mono-substituted dienophiles.
A significant increase in reactivity is seen with dienophiles carrying two activating groups: compare, for example, the reaction temperatures and times of acrylonitrile 9 and maleonitrile 14 with a specific diene, either BD or CPD. As can be seen from inspection of Table 1, uncatalyzed DA reactions in benzene solution are significantly more facile with CPD than with BD. A MeAlCl2-catalyzed (1.1 mol equiv.) reaction between BD and methyl vinyl ketone (cf.5, Fig. 1) was performed at −78 °C with slow warming to ambient temperature over 20 h. This reaction delivered an 83% yield of only the endo product, within the limits of detection (>95:5).
For representative DA reactions involving BD and CPD, different endo:exo ratios of selected products were exposed to the reaction conditions under which they were formed. An unchanged ratio was returned in each case, confirming the kinetic control of these reactions.47 The reaction between labeled 1,3-butadiene 1 and acrylonitrile 9 was also performed at 350 °C in the gas phase, which led to a 50:50 mixture of endo:exo isomers. This result is consistent with a thermodynamically-controlled process.47
A list of additional DA diene–dienophile combinations that were studied computationally are listed in the following Computational methodology section. A small group of previously published experimental and computational results are also included in Table 1.
Computational methodology
Optimized geometries of reactants and transition structures (TSs) and their energies were calculated using the composite ab initio CBS-QB3 method, which is a member of the complete basis set methods developed by Petersson et al.48,49 The CBS-QB3 method uses a B3LYP/6-31G† optimized geometry and frequencies together with CCSD(T), MP4SDQ, and MP2 single-point calculations and a CBS extrapolation to produce accurate energies. The CBS-QB3 method successfully calculates reliable energetics of pericyclic reactions, including DA reactions.50–53 The calculations were carried out for both gas phase reactions and in benzene solvent using the polarizable continuum model (PCM).54,55 Standard states used for calculating free energies were 1 atmosphere of pressure and 298.15 K for gas phase reactions and 1 M for solution phase reactions. Gas phase and solution phase reactions were modeled and the latter data are in better agreement with experimental findings. Solution phase reactions were modeled at both 298.15 K and at the experimental reaction temperature: while both were in good agreement with experimental values, the latter were closer.To check the reliability of the optimized geometries, energies and endo:exo ratios, geometry optimizations were also performed on representative examples using the B3LYP-D3 method with single-point energy refinements then computed at the CCSD(T) level. The TS geometries are extremely close for the two methods, with root-mean-square deviations of only 0.017–0.038 Å across the 8 TSs located. The calculated single point energies and endo/exo ratios are also very similar. See the ESI for details.† Calculations were carried out using the Gaussian 03 or Gaussian 16 packages.56
DA reactions of both BD 1 and CPD 2 with the following fifteen dienophiles were studied: acrolein 3, methyl vinyl ketone (MVK) 4, acrylic acid 6, methyl acrylate 7, acrylamide 8, acrylonitrile 9, maleonitrile 14, butenolide 10, α-methylene γ-butyrolactone 11, N-methylmaleimide (NMM) 15, methyl vinyl ketone·AlCl35, α-methylene γ-butyrolactone·AlCl312, maleic anhydride (MA) 16, benzoquinone (BQ) 17, methyl methacrylate 13. The first ten dienophiles on this list were also examined experimentally. The computed DA reaction of methyl vinyl ketone·AlCl3 serves as a simulacrum for the experiment performed with MeAlCl2 as catalyst. The other DA reactions were calculated to benchmark with literature results, or to provide predictions.
For acyclic dienophiles with conjugating carbonyl groups, specifically 3, 4, 6, 7 and 8, s-cis and s-trans conformations of the dienophilic enone group are possible for both endo- and exo-modes of cycloaddition. In these instances, the calculated endo:exo ratios factor in the relative Boltzmann contributions of each of the four TSs. As can be seen from the data in Table 1, TSs with s-cis conformations are generally preferred.
Additional information is presented in Tables S1 and S2 in the ESI† for DA reactions with BD and CPD, respectively. This includes CBS-QB3 relative H‡298 K and G‡298 K energies (kJ mol−1) in both the gas phase and benzene phase, along with calculated endo:exo product distributions for the thirty DA reactions described above (fifteen each for BD and CPD). B3LYP/6-31G* calculated HOMOdiene–LUMOdienophile energy gaps, as well as atomic polar tensor (APT) charge transfer (CT) from diene to dienophile in TS, and TS dipole moments, in gas phase and benzene phase DA TSs for the thirty DA reactions described above (fifteen each for BD and CPD) are also presented.
The following seven observations based upon the data in Table 1 are noteworthy:
(1) The CBS-QB3 method gives calculated endo:exo product ratios that are generally in very good agreement with experimental data. The agreement between calculated selectivities and these new experimental outcomes validates the theoretical framework of these studies. Benzene phase calculations generally give ratios that are closer to the experimental values than gas phase calculations, although noteworthy (ca. 10–15%) differences in endo:exo ratios between gas and benzene phases are seen only in the cases of acrylamide 8, acrylonitrile 9 and maleonitrile 14. A higher endo-selectivity is predicted in solution in every case (Tables S1 and S3†). Calculated endo:exo product ratios from ΔΔG‡ values corrected to the experimental temperature are also generally closer than are those calculated at 298 K. The widest disparity between experiment and calculation, in energetic terms, is that involving the previously reported experimental result between MA 16 and BD in PhH at 80 °C (ref. 24) (Scheme 2, eqn (3)), with the experimental endo:exo value lower (85:15) than our calculated value (98:2). In light of (a) the closer correlation between calculated and experimental values in other cases, and (b) the much higher endo-selectivity observed with the closely-related dienophiles NMM 15 and BQ 17 (see point 2), we suspect that this previously published experimental value is erroneous.59
(2) Dienophiles carrying two electron withdrawing groups are more strongly endo-selective than are those with one. The three cyclic dienophiles NMM 15, MA 16 and BQ 17 give very high endo-selectivity with both BD and CPD, while the acyclic dienophile maleonitrile 14, with two cis-disposed and powerfully electron-withdrawing cyano-groups, is the least selective of these doubly-activated dienophiles (endo:exo = 70:30 with BD; endo:exo = 73:27 with CPD).
(3) The six mono-substituted dienophiles acrolein 3, methyl vinyl ketone 4, acrylic acid 6, methyl acrylate 7, acrylamide 8, acrylonitrile 9 do not display strong selectivity with BD, giving endo:exo ratios in the range 65:35 to 37:63. We note, however, that endo-selectivity generally diminishes with decreasing electron-withdrawing power, and acrolein 3, methyl vinyl ketone 4, acrylic acid 6, methyl acrylate 7, and acrylamide 8 show an correlation between higher endo-selectivity and smaller HOMOdiene–LUMOdienophile gap (Fig. 2). Dienophiles with two electron-withdrawing groups (point 2) have the smallest HOMOdiene–LUMOdienophile gap, and the highest endo-selectivity. Acrylonitrile 9 and maleonitrile 14, with relatively small HOMOdiene–LUMOdienophile gaps, give anomalously low amounts of endo-products.
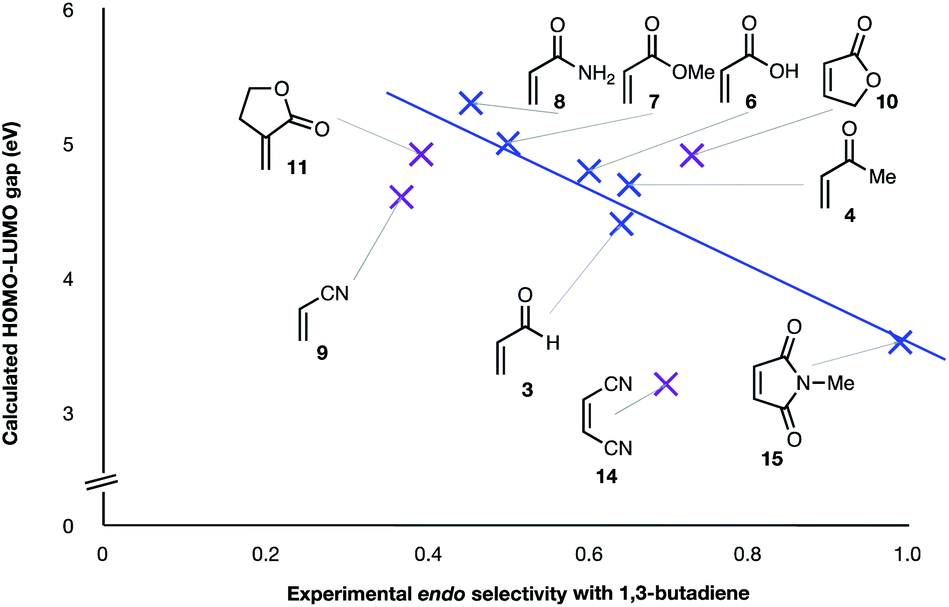 | ||
| Fig. 2 Experimental endo selectivities of ten common dienophiles with 1,3-butadiene and cyclopentadiene (x axis) vs. HOMOdiene–LUMOdienophile gap. Weaker correlations are colored purple. | ||
(4) Both acrylonitrile 9 (endo:exo = 37:63) and α-methylene γ-butyrolactone 11 (endo:exo = 39:61) show an exo preference in their DA reactions with BD (Fig. 2). The former is consistent with earlier gas phase calculations on the DA reaction between BD + acrylonitrile,38,39 but not with those which included electrostatic solvent effects.39 The preferred exo selectivity of α-methylene γ-butyrolactone 11 has been noted previously in its reactions with the bis-TMS ether of (2E,4E)-hexa-2,4-diene-1,6-diol20 and with CPD.18 Parenthetically, the exo-selectivity of the 11 + CPD DA reaction remains high under catalysis with AlCl3 (i.e.12 + CPD), both in calculation and experiment, whereas the 11 + BD reaction is calculated to undergo a switch to strong endo-selectivity under AlCl3 catalysis (i.e.12 + BD).
(5) Whereas the Diels–Alder reaction of methyl acrylate 7 with BD is non-stereoselective (endo:exo = 50:50), the corresponding reaction of butenolide 10 is endo favored (endo:exo = 73:27) (Table 1). This stronger endo preference of butenolide 10 over methyl acrylate 7 is evident in the case of CPD as diene, although the latter dienophile also now displays some degree of endo selectivity (butenolide 10: endo:exo = 80:20; methyl acrylate 7: endo:exo = 77:23).
(6) With the exception of α-methylene γ-butyrolactone 11, the endo:exo ratio increases upon change of diene from BD to CPD. In the case of the α-methylene γ-butyrolactone 11, the percentage endo product falls dramatically upon this change, from 39% to 12% (Fig. 3).
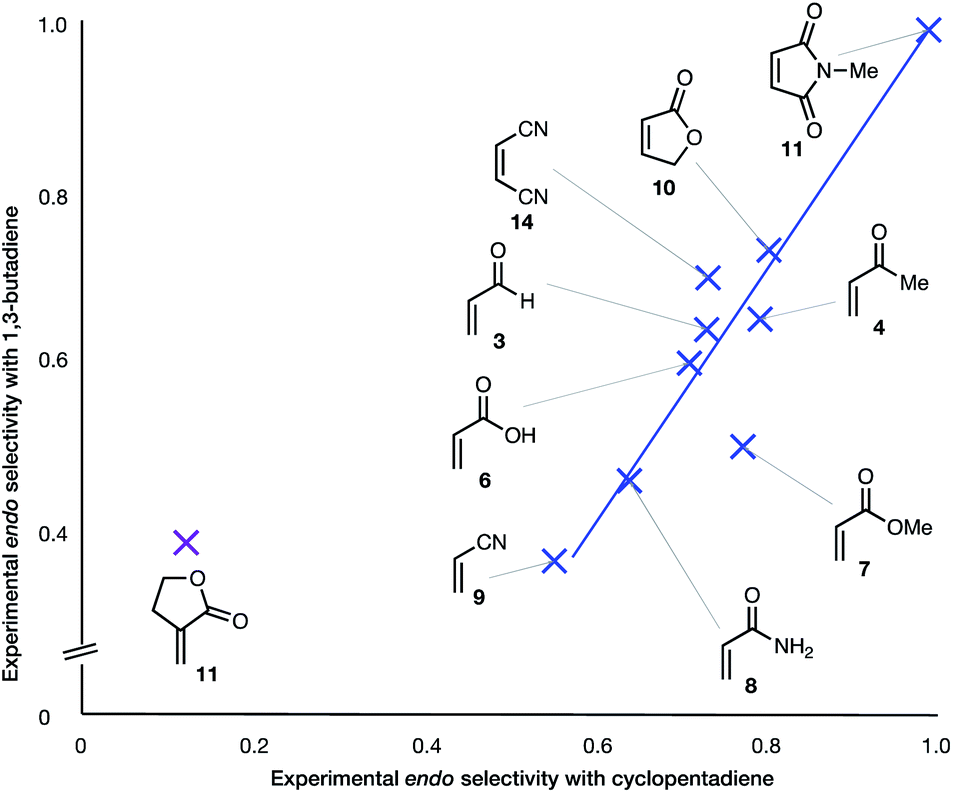 | ||
| Fig. 3 Experimental endo selectivities of ten common dienophiles with 1,3-butadiene (y axis) and cyclopentadiene (x axis), showing an approximate correlation in all but one case. | ||
(7) A significant enhancement in endo preference upon Lewis acid activation is seen in the MeAlCl2-promoted DA reaction between labeled BD and methyl vinyl ketone (cf.5 + BD). When performed uncatalyzed at 145 °C, this reaction (4 + BD) gives a 65:35 endo:exo ratio, whereas the endo isomer is essentially the sole product detected in the Lewis acid catalyzed reaction at 20 °C (Table 1).
At the start of our discussion of these observations, we note that, in the reactions of monosubstituted dienophiles with BD and CPD, the ΔΔG‡ values between endo and exo pathways are less than 3.5 kJ mol−1 (Table 1, entries 1–6). This small energy difference makes the deconvolution of the various contributions to stereocontrol impossible. Our calculations of diene → dienophile charge transfer and dipole moments of TS (see Tables S5 and S6†) neither provided insights into the origins of the selectivity trends described above, nor explanatory information pertaining to the outlying results.
The two most general correlations found in the experimental and computational data are highlighted in Fig. 2 and 3. Fig. 3 shows that, with one exception, the endo/exo-selectivity of DA reactions between dienophiles and both BD and CPD follow similar trends, albeit with slightly higher endo-preferences for CPD. A second general correlation (albeit a rough one) is between the magnitude of the endo-stereoselectivity and the size of the HOMOdiene–LUMOdienophile energy gap (Fig. 2). We previously noted this trend in a broad scope DFT (B3LYP) study (see Introduction).41 This observation of enhanced endo-stereoselectivity with a smaller HOMOdiene–LUMOdienophile energy gap is suggestive of SOIs, although this correlation does not constitute evidence of causation.
The results most worthy of brief discussion are exceptions to these general trends, specifically: (a) the anomalously high proportion of exo-adducts from BD and CPD DA reactions involving maleonitrile 9 and acrylonitrile 14; (b) the enhanced endo-selectivity of butenolide 10 over methyl acrylate 7; and (c) the exo-selectivity of uncatalyzed DA reactions of α-methylene γ-butyrolactone 11, and the divergent stereoselectivities of this dienophile in catalyzed DA reactions (i.e.12) with BD and CPD.
On the anomalous behavior of maleonitrile 9 and acrylonitrile 14
The anomalously low endo-selectivities of DA reactions of acrylonitrile 9 and maleonitrile 14 with BD and CPD can be accounted for by a lack of Salem–Houk (SH) SOIs in DA endo-TSs involving nitriles. The endo-TSs of the reactions of acrylonitrile 9 with BD and CPD, along with those of the dominant s-cis conformation of acrolein 3, are depicted in Fig. 4. | ||
| Fig. 4 CBS-QB3 endo-TSs of uncatalyzed DA reaction of acrolein 3 and acrylonitrile 9 with BD and CPD, highlighting distances between nuclei that may participate in SH-type SOIs. Top row: perspective view; bottom row: view down the forming C–C bonds, from the diene side of the TS. | ||
A comparison of these TSs shows that, whereas the internuclear distances between the acrolein O and diene C2 in DA TSs involving both BD and CPD are 3.42 and 3.23 Å, respectively, in the corresponding acrylonitrile 9 TSs, the N to diene C2 distances are significantly longer, at 4.44 and 4.27 Å, hence unlikely to benefit from stabilizing SH SOIs. A similar situation occurs in the endo-TSs for the DA reactions of BD and CPD with maleonitrile 14. As an aside, the slightly shorter distances seen in CPD DA reactions are due to the shorter C1⋯C4 distance in the 1,3-butadiene moiety of CPD (2.33 Å) relative to BD (2.90 Å): the greater splaying in BD is clearly visible from the lower set of structures depicted in Fig. 4. We also note that internuclear distances (i.e. carbonyl C/nitrile C to diene C3) for WH SOIs are similar for both systems (3.04–3.25 Å), which is again most apparent from inspection of the lower set of structures depicted in Fig. 4.
On the enhanced endo-selectivity of butenolide 10 over methyl acrylate 7
The enhanced endo selectivity of DA reactions between BD/CPD + butenolide 10vs. BD/CPD + methyl acrylate 7 is probably the result of destabilizing steric interactions in exo-TSs involving butenolide 10 (Fig. 5).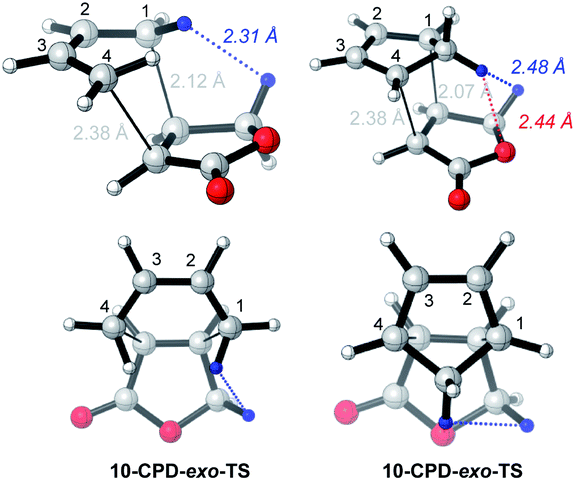 | ||
| Fig. 5 CBS-QB3 exo-TSs of uncatalyzed DA reactions of butanolide 10 with BD and CPD, highlighting close contacts. Top row: perspective view; bottom row: view down the forming C–C bonds, from the diene side of the TS. | ||
Thus, a close contact is identifiable between a proton on the diene and a butenolide dienophile methylene proton which points toward the diene. In the case of BD, an inside methylene proton is close to a butenolide methylene proton (2.31 Å), whereas in the case of CPD, it is the CPD methylene proton directed toward the dienophile that clashes with the same butenolide methylene proton (2.48 Å), in addition to the butenolide ring oxygen (2.44 Å). No such destabilizing steric interaction operates in the exo-TSs involving methyl acrylate for two reasons: (a) methyl acrylate lacks an allylic methylene group; and (b) the preferred TSs involving methyl acrylate have s-cis CC–CO conformations, hence the methoxy group cannot clash with the diene. Parenthetically, a Z-crotonate ester is a cognate of butenolide, since it carries an allylic methyl group and prefers an s-trans CC–CO TS conformation. It is noteworthy that sec-butyl Z-crotonate was more endo selective (by 0.5 kJ mol−1) than methyl acrylate in its reaction with CPD.19 We propose that similar destabilizing steric interactions are operating in this related system.
On the anomalous behavior of α-methylene γ-butyrolactone
The exception to the trend of enhanced endo-selectivity with CPD vs. BD is with the dienophile α-methylene γ-butyrolactone 11, which instead exhibits enhanced exo-selectivity with CPD. Both experimental (published work18) and calculated (this work) Lewis acid catalyzed versions of these reactions provide additional, intriguing results (Table 1). Whereas AlCl3-catalyzed DA reactions of CPD + 12 give the same exo-stereoselectivity as the thermal reaction CPD + 11 (endo:exo = ca. 10:90), our calculations predict a complete reversal in stereoselectivity for the BD + 11 and BD + 12 reactions (thermal: endo:exo = 10:90; catalyzed: endo:exo = 88:12).
The origin of these interesting results with α-methylene γ-butyrolactone 11 (and its AlCl3 complex 12) may be traced to geometrical factors in the TSs of the thermal and Lewis acid-catalyzed DA reactions of this dienophile with BD and CPD. Focusing firstly at TSs of the uncatalyzed reactions (Fig. 6), we can see that all TSs have similar length shorter (1.99–2.03 Å) and longer (2.51–2.61 Å) developing bonds, and similar bond forming asynchronicities, Δras (range = 0.56–0.61 Å).
 | ||
| Fig. 6 CBS-QB3 endo- and exo-TSs of the uncatalyzed DA reaction of α-methylene γ-butyrolactone 11 with BD and CPD, highlighting key destabilizing close contacts, and the direction and magnitude of twist-mode asynchronicity. Top row: perspective view; bottom row: view down the forming C–C bonds, from the diene side of the TS. | ||
As expected, the shorter developing bond is to the β-position of the dienophile enone group. In each TS, a close contact between (a) a dienophile methylene proton pointing toward the diene, and (b) a proton on the diene, is identifiable. In the case of BD, the most significant steric clash (2.31 Å) is between an inside BD methylene proton and a dienophile 11 allylic methylene proton in the endo-TS. An even closer contact (2.21 Å) occurs in the endo-TS of the CPD + 11 TS, which involves the CPD methylene proton directed toward the dienophile. Hence, we can explain the exo-selectivity of uncatalyzed DA reactions of α-methylene γ-butyrolactone 11 with both BD and CPD by identifying that the endo-TSs are disfavored on steric grounds, and that the higher exo-selectivity in the CPD case is due to the steric clash being more severe. The ESI† contains further calculations and additional discussion on the influence of methylene lactone ring size on DA selectivity.
The four TSs of the AlCl3-catalyzed DA reactions of α-methylene γ-butyrolactone 11 (i.e.12) with BD and CPD are depicted in Fig. 7. Again, similar length shorter (1.97–2.00 Å) and longer (2.85–2.92 Å) developing bonds are seen throughout the four TSs, along with similar bond forming asynchronicities, Δras (range = 0.85–0.95 Å). A comparison of the catalyzed with the uncatalyzed DA TSs shows that the longer developing bond is significantly extended in the catalyzed reaction. The same close contacts are present in the four catalyzed reaction TSs as in the uncatalyzed ones. In the endo-TS of the catalyzed CPD + 12 reaction, the distance (2.19 Å) remains similarly close to that seen in the uncatalyzed CPD + 11 reaction (2.21 Å). Furthermore, in the corresponding exo-TS of the same catalyzed and uncatalyzed reactions, the distances are also similar (2.42 and 2.46 Å). Therefore, the similar exo-selectivity for both catalyzed and uncatalyzed reactions involving CPD are understandable on the basis of similar steric effects operating in each pair of TSs. In contrast, in the case of catalyzed BD + 12endo-TS, the distance between the inside BD methylene proton and the dienophile allylic methylene proton is extended (2.41 Å) relative to the uncatalyzed BD + 11endo-TS (2.31 Å). We propose that this extension in the BD endo-TS alleviates destabilizing steric strain and, perhaps with the assistance of SOIs, is the cause of the switch to endo-selectivity under Lewis acid catalysis. The ESI† contains further discussion on the implications of the twist mode asynchronicity differences between the DA TS shown in Fig. 6 and 7.
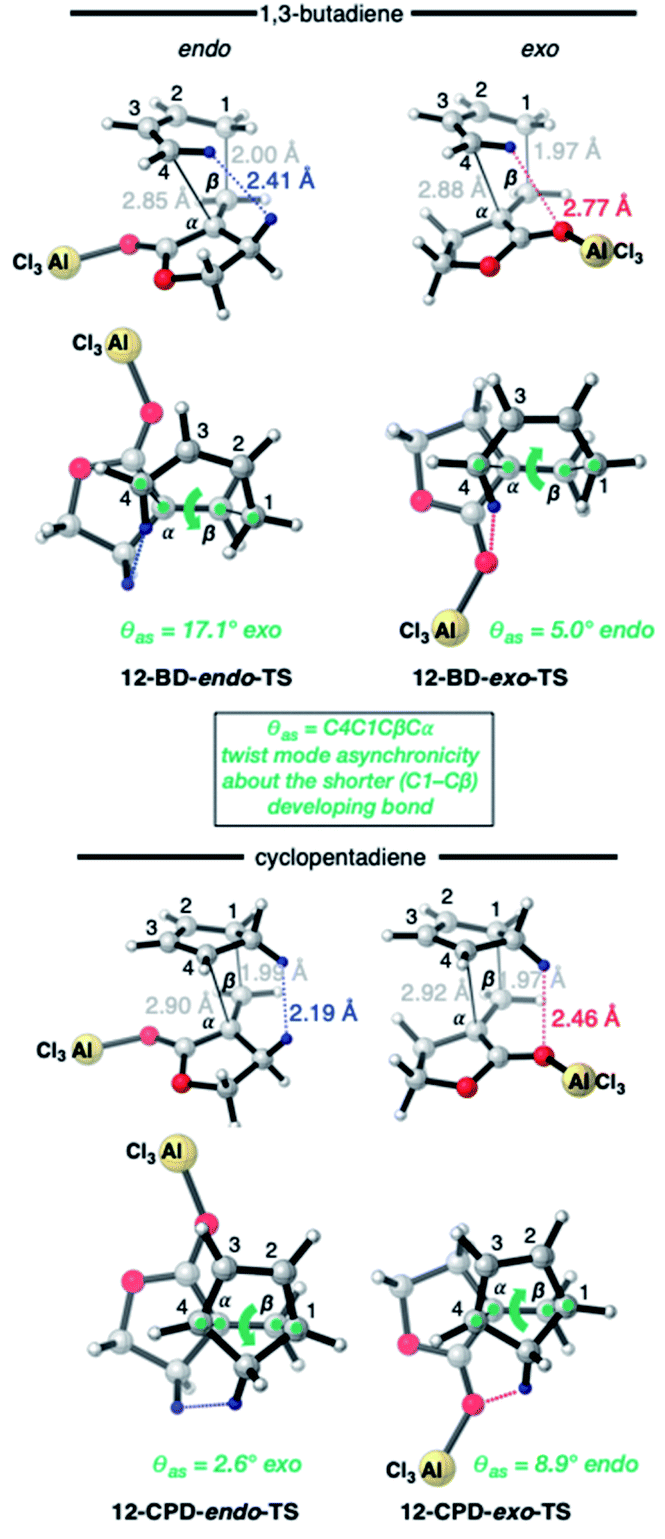 | ||
| Fig. 7 CBS-QB3 endo- and exo-TSs of the AlCl3-catalyzed DA reaction of α-methylene γ-butyrolactone (i.e.12) with BD and CPD, highlighting key destabilizing close contacts, and the direction and magnitude of twist-mode asynchronicity. Top row: perspective view; bottom row: view down the forming C–C bonds, from the diene side of the TS. | ||
Conclusions
In summary, we have conducted the first experimental–computational investigation into the endo:exo selectivity of Diels–Alder reactions between the simplest diene and ten commonly-used dienophiles. The reactions of cyclopentadiene, one of the most commonly-used dienes, with the same ten dienophiles were also performed. This work was facilitated by the first preparative synthesis of (1E,3E)-1,4-dideutero-1,3-butadiene in high stereochemical purity, a compound and synthesis that will find application in other investigations.
The most surprising finding from this study is that the most commonly used mono-substituted alkenic dienophiles (acrolein, methyl vinyl ketone, acrylic acid, methyl acrylate, acrylamide and acrylonitrile) are not endo-selective in thermal Diels–Alder reactions with 1,3-butadiene. Generally, for a given dienophile, endo:exo selectivities for cyclopentadiene are ca. 5–20% higher than with 1,3-butadiene.
The CBS-QB3 method gives calculated endo:exo product ratios that are in very good agreement with experimental findings, hence validating the theoretical framework of this study. These models have broader value to those interested in a deeper understanding of the most important synthetic reaction, and its application in synthesis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Australian Research Council (DP160104322). MNP-R acknowledges that the computational component of this research was undertaken with the assistance of resources provided at the NCI National Facility through the National Computational Merit Allocation Scheme supported by the Australian Government. We thank Dr Tony Willis (ANU) for performing single crystal X-ray analyses of 14CPD-n and 14CPD-x. Computed structures were visualized using CYLview 1.0b: C. Y. Legault, Université de Sherbrooke, 2009, http://www.cylview.org.Notes and references
- O. Diels and K. Alder, Justus Liebigs Ann. Chem., 1928, 460, 98–122 CrossRef CAS.
- A research topic search of SciFinder Scholar conducted in May 2020 produced around 50000 references containing the concept “Diels–Alder”, with >28000 references since the year 2000. Equivalent searches for “metathesis” gave 44000, “Suzuki” gave 34000, “aldol” gave 32000, “Wittig” gave 27000, “Friedel–Crafts” gave 27000, “organocatalysis” gave 19000, “Heck” gave 15000, “Claisen” gave 14000, “electrosynthesis” gave 8000 and “photoredox” gave 7000 references.
- J. Sauer and R. Sustmann, Angew. Chem., Int. Ed., 1980, 19, 779–807 CrossRef.
- F. Fringuelli and A. Taticchi, The Diels–Alder Reaction: Selected Practical Methods, John Wiley & Sons, Chichester, 2002 Search PubMed.
- (a) R. B. Woodward and R. Hoffmann, Angew. Chem., Int. Ed., 1969, 8, 781–853 CrossRef CAS; (b) R. B. Woodward and R. Hoffmann, The Conservation of Orbital Symmetry, Verlag Chemie, Weinheim, 1970 Search PubMed.
- (a) K. Fukui, Acc. Chem. Res., 1971, 4, 57–64 CrossRef CAS; (b) K. Fukui, Science, 1982, 218, 747–754 CrossRef CAS.
- (a) E. J. Corey, Angew. Chem., Int. Ed., 2002, 41, 1650–1667 CrossRef CAS; (b) K. C. Nicolaou, S. A. Snyder, T. Montagnon and G. Vassilikogiannakis, Angew. Chem., Int. Ed., 2002, 41, 1668–1698 CrossRef CAS; (c) K. A. Jorgensen, in Cycloaddition Reactions in Organic Synthesis, ed. S. Kobayashi and K. A. Jorgensen, Wiley VCH, Weinheim, 2002, pp. 151–183 Search PubMed.
- (a) K. Alder, G. Stein, F. v. Buddenbrock, W. Eckardt, W. Frercks and S. Schneider, Justus Liebigs Ann. Chem., 1934, 514, 1–33 CrossRef CAS; (b) K. Alder and G. Stein, Angew. Chem., 1937, 50, 510–519 CrossRef CAS.
- I. Fleming, Frontier Orbital and Organic Chemical Reactions, Wiley, London, 1976 Search PubMed.
- R. B. Woodward and R. Hoffmann, J. Am. Chem. Soc., 1965, 87, 4388–4389 CrossRef.
- (a) L. Salem, J. Am. Chem. Soc., 1968, 90, 553–566 CrossRef CAS; (b) K. N. Houk, Tetrahedron Lett., 1970, 2621–2624 CrossRef CAS.
- (a) P. Caramella, P. Quadrelli and L. Toma, J. Am. Chem. Soc., 2002, 124, 1130–1131 CrossRef CAS; (b) P. Quadrelli, S. Romano, L. Toma and P. Caramella, Tetrahedron Lett., 2002, 43, 8785–8789 CrossRef CAS.
- (a) J. I. Garcia, J. A. Mayoral and L. Salvatella, Acc. Chem. Res., 2000, 33, 658–664 CrossRef CAS; (b) C. S. Wannere, A. Paul, R. Herges, K. N. Houk, H. F. Schaeffer III and P. von Rague Schleyer, J. Comput. Chem., 2007, 28, 344–361 CrossRef CAS.
- J. A. Berson, Z. Hamlet and W. A. Mueller, J. Am. Chem. Soc., 1962, 84, 297–304 CrossRef CAS.
- R. B. Woodward and H. Baer, J. Am. Chem. Soc., 1944, 66, 645–649 CrossRef CAS.
- (a) A. Wassermann, J. Chem. Soc., 1935, 828–839 RSC; (b) A. Wassermann, J. Chem. Soc., 1935, 1511–1514 RSC; (c) A. Wassermann, J. Chem. Soc., 1936, 432–436 RSC; (d) D. Suarez and J. A. Sordo, Chem. Commun., 1998, 385–386 RSC.
- Representative examples of substrate-controlled exo-selective Diels–Alder reactions: (a) J. Wang, Z. Liu, J. Li, Z. Song, C. Hu and Z. Su, J. Org. Chem., 2019, 84, 3940–3952 CrossRef CAS; (b) G. Ho, C. Huang, E. Y. Li, S. Hsu, T. Wu, M. M. L. Zulueta, K. B. Wu and S. Hung, Sci. Rep., 2016, 6, 35147 CrossRef; (c) Z. Liu, X. Lin, N. Yang, Z. Su, C. Hu, P. Xiao, Y. He and Z. Song, J. Am. Chem. Soc., 2016, 138, 1877–1883 CrossRef CAS; (d) S.-J. Min, G. O. Jones, K. N. Houk and S. J. Danishefsky, J. Am. Chem. Soc., 2007, 129, 10078–10079 CrossRef CAS; (e) J. Qi and W. R. Roush, Org. Lett., 2006, 8, 2795–2798 CrossRef CAS; (f) M. Ge, B. M. Stoltz and E. J. Corey, Org. Lett., 2000, 2, 1927–1929 CrossRef CAS; (g) T. Yoon, S. J. Danishefsky and S. de Gala, Angew. Chem., Int. Ed. Engl., 1994, 33, 853–855 CrossRef; (h) W. R. Roush and B. B. Brown, J. Org. Chem., 1992, 57, 3380–3387 CrossRef CAS; (i) W. R. Roush, A. P. Essenfeld, J. S. Warmus and B. B. Brown, Tetrahedron Lett., 1989, 30, 7305–7308 CrossRef CAS.
- F. Fotiadu, F. Michel and G. Buono, Tetrahedron Lett., 1990, 31, 4863–4866 CrossRef CAS.
- J. Furukawa, Y. Kobuke and T. Fueno, J. Am. Chem. Soc., 1970, 92, 6548–6553 CrossRef CAS.
- K. Takeda, I. Imaoka and E. Yoshii, Tetrahedron, 1994, 50, 10839–10848 CrossRef CAS.
- Y.-h. Lam, P. H.-Y. Cheong, J. M. Blasco Mata, S. J. Stanway, V. Gouverneur and K. N. Houk, J. Am. Chem. Soc., 2009, 131, 1947–1957 CrossRef CAS.
- Catalyst-controlled exo-selective Diels–Alder reactions: (a) K. Maruoka, H. Imoto and H. Yamamoto, J. Am. Chem. Soc., 1994, 116, 12115–12116 CrossRef CAS; (b) Z.-J. Jia, Q. Zhou, Q.-Q. Zhou, P.-Q. Chen and Y.-C. Chen, Angew. Chem., Int. Ed., 2011, 50, 8638–8641 CrossRef CAS; (c) J.-H. Zhou, B. Jiang, F.-F. Meng, Y.-H. Xu and T.-P. Loh, Org. Lett., 2015, 17, 4432–4435 CrossRef CAS; (d) D. Yepes, P. Pérez, P. Jaque and I. Fernández, Org. Chem. Front., 2017, 4, 1390–1399 RSC; (e) M. Bakos, Z. Dobi, D. Fegyverneki, Á. Gyömöre, I. Fernández and T. Soós, ACS Sustainable Chem. Eng., 2018, 6, 10869–10875 CrossRef CAS.
- (a) L. M. Stephenson, R. V. Gemmer and S. P. Current, J. Am. Chem. Soc., 1975, 97, 5909–5910 CrossRef CAS; (b) F. G. Klärner, B. Krawczyk, V. Ruster and U. Deiters, J. Am. Chem. Soc., 1994, 116, 7646–7657 CrossRef.
- L. M. Stephenson, D. E. Smith and S. P. Current, J. Org. Chem., 1982, 47, 4170–4171 CrossRef CAS.
- J. E. Baldwin and V. P. Reddy, J. Org. Chem., 1989, 54, 5264–5267 CrossRef CAS.
- (a) Y. Apeloig and E. Matzner, J. Am. Chem. Soc., 1995, 117, 5375–5376 CrossRef CAS; (b) A. Arrieta, F. P. Cossío and B. Lecea, J. Org. Chem., 2001, 66, 6178–6180 CrossRef CAS.
- (a) F. M. Bickelhaupt and K. N. Houk, Angew. Chem., Int. Ed., 2017, 56, 10070–10086 CrossRef CAS; (b) L. P. Wolters and F. M. Bickelhaupt, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2015, 5, 324–343 CAS; (c) D. H. Ess and K. N. Houk, J. Am. Chem. Soc., 2007, 129, 10646–10647 CrossRef CAS.
- (a) M. von Hopffgarten and G. Frenking, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 43–62 CAS; (b) F. M. Bickelhaupt and E. J. Baerends, Rev. Comput. Chem., 2007, 1–86 Search PubMed.
- I. Fernández and F. M. Bickelhaupt, J. Comput. Chem., 2014, 35, 371–376 CrossRef.
- B. J. Levandowski and K. N. Houk, J. Am. Chem. Soc., 2016, 138, 16731–16736 CrossRef CAS.
- R. J. Loncharich, F. K. Brown and K. N. Houk, J. Org. Chem., 1989, 54, 1129–1134 CrossRef CAS.
- S. Kong and J. D. Evanseck, J. Am. Chem. Soc., 2000, 122, 10418–10427 CrossRef CAS.
- D. M. Birney and K. N. Houk, J. Am. Chem. Soc., 1990, 112, 4127–4133 CrossRef CAS.
- J. I. García, J. A. Mayoral and L. Salvatella, J. Am. Chem. Soc., 1996, 118, 11680–11681 CrossRef.
- J. I. García, V. Martinez-Merino, J. A. Mayoral and L. Salvatella, J. Am. Chem. Soc., 1998, 120, 2415–2420 CrossRef.
- K. N. Houk and R. W. Strozier, J. Am. Chem. Soc., 1973, 95, 4094–4096 CrossRef CAS.
- P. Vermeeren, T. A. Hamlin, I. Fernández and F. M. Bickelhaupt, Angew. Chem., Int. Ed., 2020, 59, 6201–6206 CrossRef CAS.
- K. N. Houk, R. J. Loncharich, J. F. Blake and W. L. Jorgensen, J. Am. Chem. Soc., 1989, 111, 9172–9176 CrossRef CAS.
- T. Karcher, W. Sicking, J. Sauer and R. Sustmann, Tetrahedron Lett., 1992, 33, 8027–8030 CrossRef CAS.
- J. Sauer, H. Wiest and A. Mielert, Chem. Ber., 1964, 97, 3183–3207 CrossRef CAS.
- M. N. Paddon-Row, D. Moran, G. A. Jones and M. S. Sherburn, J. Org. Chem., 2005, 70, 10841–10853 CrossRef CAS.
- Several of the cycloadditions of CPD described herein have been previously studied but different solvents and concentrations have been employed and, in some cases, different product ratios have been reported for the same DA reaction. The purpose of the present investigation was to normalize these previous findings by systematically conducting reactions under the same conditions and in the relatively non-polar solvent benzene.
- (a) P. D. Bartlett and G. E. H. Wallbillich, J. Am. Chem. Soc., 1969, 91, 409–414 CrossRef CAS; (b) L. M. Stephenson, R. V. Gemmer and S. P. Current, J. Org. Chem., 1977, 42, 212–214 CrossRef CAS.
- (a) S. A. Mitchenko, V. P. Ananikov, I. P. Beletskaya and Y. A. Ustynyuk, Mendeleev Commun., 1997, 7, 130–131 CrossRef; (b) V. P. Ananikov, S. A. Mitchenko and I. P. Beletskaya, Russ. J. Org. Chem., 2002, 38, 636–650 CrossRef CAS; (c) V. P. Ananikov, A. S. Kashin, O. V. Hazipov, I. P. Beletskaya and Z. A. Starikova, Synlett, 2011, 22, 2021–2024 CrossRef.
- K. Kitagawa, A. Inoue, H. Shinokubo and K. Oshima, Angew. Chem., Int. Ed., 2000, 39, 2481–2483 CrossRef CAS.
- Measured using quantitative 1H NMR spectroscopy at 800 MHz. The methods were developed using non-deuterated samples and varied benzene-d6/DMSO-d6 solvent mixtures in order to attain baseline separation between critical signals.
- See the ESI for details.†.
- (a) J. A. Montgomery Jr, M. J. Frisch, J. W. Ochterski and G. A. Petersson, J. Chem. Phys., 1999, 110, 2822–2827 CrossRef; (b) J. A. Montgomery Jr, M. J. Frisch, J. W. Ochterski and G. A. Petersson, J. Chem. Phys., 2000, 112, 6532–6542 CrossRef.
- (a) M. R. Nyden and G. A. Petersson, J. Chem. Phys., 1981, 75, 1843–1862 CrossRef CAS; (b) G. A. Petersson and M. A. Al-Laham, J. Chem. Phys., 1991, 94, 6081–6090 CrossRef CAS; (c) G. A. Petersson, T. G. Tensfeldt and J. A. Montgomery Jr, J. Chem. Phys., 1991, 94, 6091–6101 CrossRef CAS; (d) G. A. Petersson, K. Malick, W. G. Wilson, J. W. Ochterski, J. A. Montgomery Jr and M. J. Frisch, J. Chem. Phys., 1998, 109, 10570–10579 CrossRef CAS.
- V. Guner, K. S. Khuong, A. G. Leach, P. S. Lee, M. D. Bartberger and K. N. Houk, J. Phys. Chem. A, 2003, 107, 11445–11459 CrossRef CAS.
- Y. Lan, L. Zou, Y. Cao and K. N. Houk, J. Phys. Chem. A, 2011, 115, 13906–13920 CrossRef CAS.
- S. N. Pieniazek and K. N. Houk, Angew. Chem., 2006, 118, 1470–1473 ( Angew. Chem., Int. Ed. , 2006 , 45 , 1442–1445 ) CrossRef.
- M. N. Paddon-Row, A. I. Longshaw, A. C. Willis and M. S. Sherburn, Chem.–Asian J., 2009, 4, 126–134 CrossRef CAS.
- S. Miertuš, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117–129 CrossRef.
- J. Tomasi, B. Mennucci and E. Cancès, J. Mol. Struct.: THEOCHEM, 1999, 464, 211–226 CrossRef CAS.
- (a) M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery Jr, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03, Revision E.01, Gaussian, Inc., Pittsburgh PA, 2003 Search PubMed; (b) M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- D. Mal and S. Ray, Eur. J. Org. Chem., 2008, 3014–3020 CrossRef CAS.
- K. B. Wiberg and W. J. Bartley, J. Am. Chem. Soc., 1960, 82, 6375–6380 CrossRef CAS.
- The previously reported ratio24 was not measured directly but instead was calculated after epoxidation of the DA adduct then exposure to an NMR chemical shift reagent. Whereas the authors provide a general description of how this sequence was performed, no yields, experimental detail or spectra are provided in the communication and no follow-up study could be found.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, compound characterisation, Cartesian coordinates of computed structures, additional TS analyses. CCDC 2005761 and 2005762. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc04553e |
| This journal is © The Royal Society of Chemistry 2020 |