
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEngineering a metal–organic framework derived Mn–N4–CxSy atomic interface for highly efficient oxygen reduction reaction†
Huishan
Shang
a,
Zhuoli
Jiang
a,
Danni
Zhou
a,
Jiajing
Pei
b,
Yu
Wang
c,
Juncai
Dong
d,
Xusheng
Zheng
e,
Jiatao
Zhang
 a and
Wenxing
Chen
*a
a and
Wenxing
Chen
*a
aBeijing Key Laboratory of Construction Tailorable Advanced Functional Materials and Green Applications, School of Materials Science and Engineering, Beijing Institute of Technology, Beijing 100081, China. E-mail: wxchen@bit.edu.cn
bState Key Lab of Organic-Inorganic Composites, Beijing University of Chemical Technology, Beijing 100029, China
cShanghai Synchrotron Radiation Facility, Shanghai Institute of Applied Physics, Chinese Academy of Science, Shanghai 201800, China
dBeijing Synchrotron Radiation Facility (BSRF), Institute of High Energy Physics, Chinese Academy of Sciences, Beijing 100049, China
eNational Synchrotron Radiation Laboratory (NSRL), University of Science and Technology of China, Hefei 230029, China
First published on 20th May 2020
Abstract
Atomic interface engineering is an effective pathway to regulate the performance of single metal atom catalysts for electrochemical reactions in energy applications. Herein, we construct a sulfur modified Mn–N–C single atom catalyst through a metal–organic framework derived atomic interface strategy, which exhibits outstanding ORR activity with a half-wave potential of 0.916 V vs. RHE in alkaline media. Moreover, operando X-ray absorption spectroscopy analysis indicates that the isolated bond-length extending the low-valence Mn–N4–CxSy moiety serves as an active site during the ORR process. These findings suggest a promising method for the advancement of single atom catalysis.
Introduction
Development of energy conversion devices, such as fuel cells and metal–air batteries, is an important approach to resolving the energy and environmental issues,1 where the oxygen reduction reaction (ORR) plays a key role in the implementation of effective electrochemical processes.2 Although platinum-based ORR catalysts have proved to be highly active, the prohibitive cost, rarity in earth reserves and low stability greatly hinder their application.3 Recently, earth-abundant metal materials have emerged as promising alternatives to replace Pt, but generally their performances are far from satisfactory.4 In spite of that, tremendous efforts have been devoted to find novel electrocatalysts; the rational use of existing catalytic components to realize the maximum activity still needs to be strengthened.Metal–carbon hybrids represent an attractive type of ORR catalysts due to their tunable electronic structure and high electrical conductivity.5–12 Particularly, a nitrogen-doped carbon matrix has been discovered to introduce anchoring sites for immobilizing isolated active metallic centers to form metal–nitrogen–carbon (M–N–C) atomic interfaces, which may boost the ORR reactivity.13–23 However, recent research also revealed that the N atoms in M–N–C possessed strong electronegativity, which would result in the increase of the adsorption energy of ORR intermediates (such as OOH*, O* and OH*) and consequently decreased the kinetic activity.24–26 Reasonable regulation of the local atomic configuration at the M–N–C atomic interface can optimize the adsorption strength of the ORR intermediates and subsequently lower the potential barriers.27–29 Resembling nitrogen, sulfur is a p-block element, but has lower electronegativity. Hence, the introduction of alien S is expected to modify the electronic structures of M–N–C by adjusting the electron-withdrawing/donating properties, thus realizing the enhancement of the ORR activity.30–34
In this work, we report the construction of a Mn single atom catalyst anchored on a sulfur and nitrogen modified carbon support (denoted as MnSAs/S–NC). Impressively, the achieved MnSAs/S–NC exhibited excellent activity for the ORR under alkaline conditions, with a high half-wave potential (E1/2) of 0.916 V vs. RHE, which was much better than that of Pt/C. Comprehensive X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) analyses revealed that the electronic and atomic synergistic mechanism between the Mn single atom and the S, N co-modified carbon support resulted in excellent ORR activity. Through operando experiments, we found that bond-extending low valence Mn–N4–CxSy species at the atomic interface of MnSAs/S–NC served as active sites during the oxygen reduction process.
As illustrated in Fig. 1a, the synthesis of MnSAs/S–NC consisted of two steps. Firstly, the manganese precursor was encapsulated in zeolitic imidazolate frameworks (ZIF-8) through a self-assembly process, which was denoted as Mn-ZIF-8. Then the mixture of the prepared Mn-ZIF-8 and S pieces was pyrolysed under an Ar atmosphere and subsequently leached using sulfuric acid to produce the S, N co-doped carbon matrix, which contained Mn–N–C active sites. Three comparison samples were also synthesized through a similar method but with some modifications, including MnSAs/NC (S free), S–NC (S, N co-modified carbon) and NC (N-modified carbon).
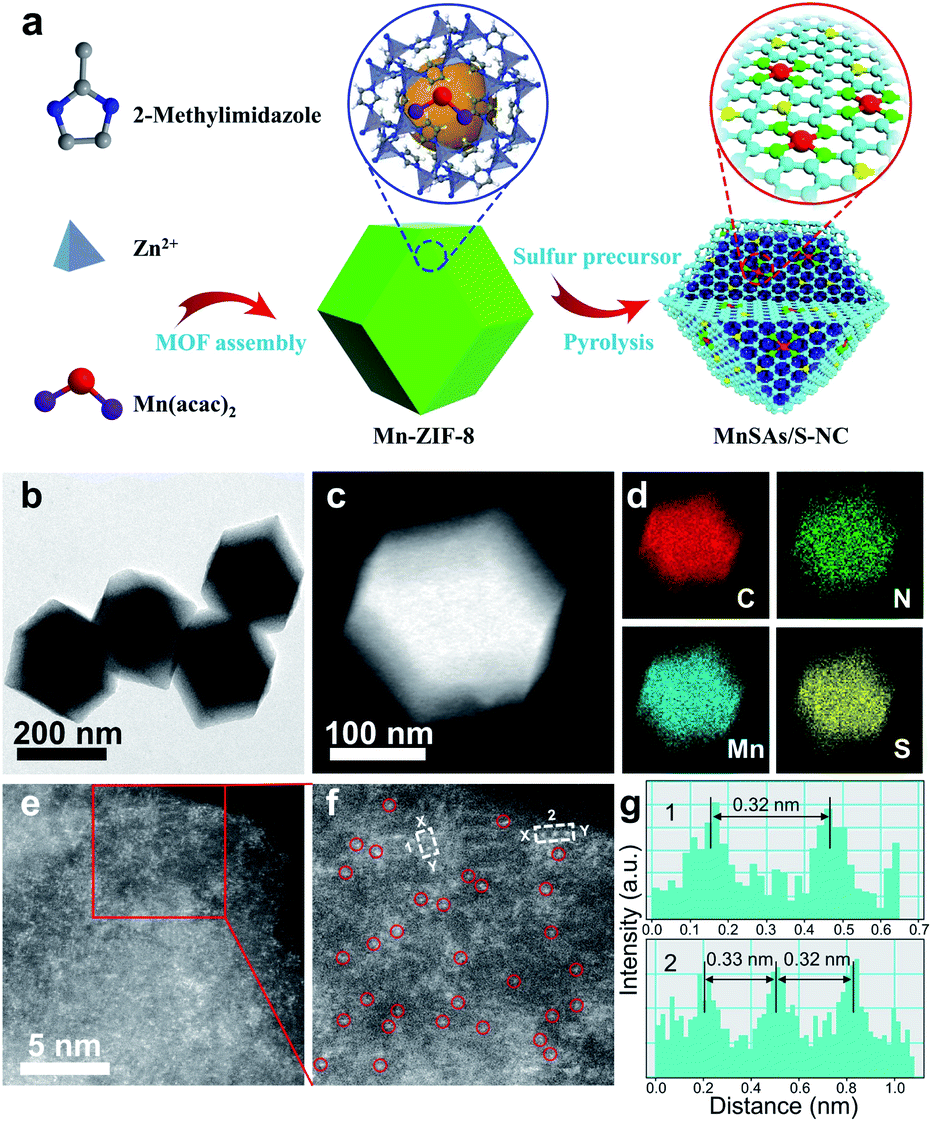 | ||
| Fig. 1 (a) Schematic illustration for the preparation of MnSAs/S–NC. (b) TEM, (c) STEM and (d) EDS images of MnSAs/S–NC. (e) HAADF-STEM image and (f) the magnified image of MnSAs/S–NC. (g) The corresponding intensity profiles along the line X–Y in (f). | ||
The X-ray diffraction (XRD) pattern of MnSAs/S–NC displayed only two broad shoulder peaks at ∼26° and ∼44°, which could be ascribed to the (002) and (010) planes of graphite carbon, no other diffraction peaks related to the Mn-based compounds were found (Fig. S1 and S2†). The morphology of the MnSAs/S–NC was observed by transmission electron microscopy (TEM), and it was found that no nanoparticles of Mn species were detected (Fig. 1b, c and S3†). Energy-dispersive X-ray spectroscopy (EDS) indicated that C, N, Mn and S were distributed uniformly over the entire architecture (Fig. 1d). The Mn loading in the MnSAs/S–NC was as high as 1.34 wt%, according to the inductively coupled plasma optical emission spectrometry (ICP-OES) analysis. The atomic dispersion of Mn species on the carbon substrate was confirmed directly by STEM, equipped with a probe spherical aberration corrector (Fig. 1e, f and S4†). In Fig. 1f, the Mn single atoms were clearly observed as bright dots marked with red circles for better observation in the high-magnification HAADF-STEM image. Moreover, the intensity profiles along X–Y in Fig. 1f, indicated that the Mn atoms are separated by at least 0.32 nm, significantly exceeding the Mn-effective atomic radius (Fig. 1g), which further demonstrated that the Mn species was atomically dispersed in the MnSAs/S–NC. Moreover, the STEM, EDS and HAADF-STEM characterization experiments of MnSAs/NC are also exhibited in Fig. S5 and S6.†
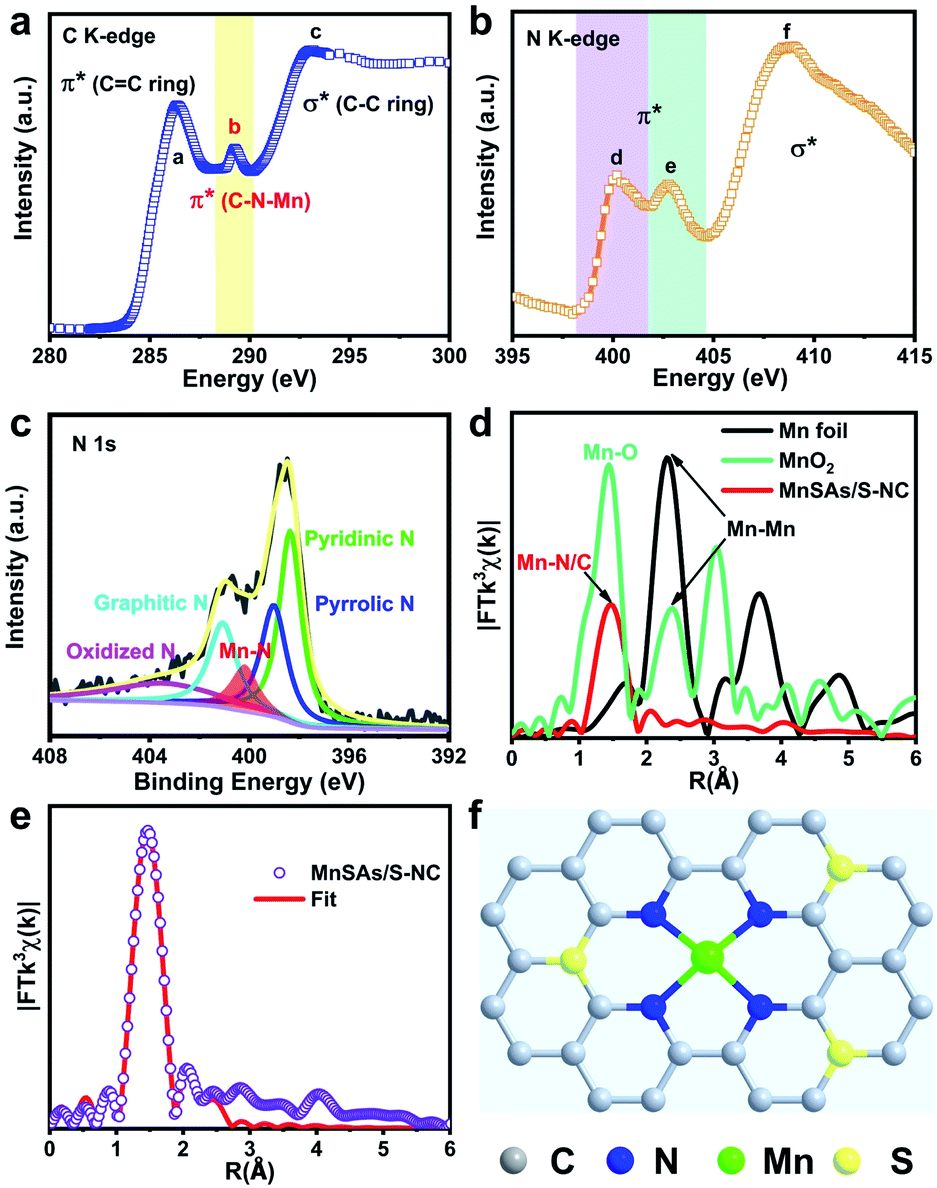
In order to investigate the atomic and electronic interaction between Mn, S, N and C in the MnSAs/S–NC, soft X-ray absorption near-edge spectroscopy (XANES) measurements were performed, which was element-resolved and interface-configuration-sensitive.35 As shown in Fig. 2a, the C K-edge spectra were dominated by three strong resonances with peak centers of 286.4 eV (peak a), 289.1 eV (peak b), and 293.1 eV (peak c). These resonances could be ascribed to the dipole transition of the C 1s core electron into the antibonding orbitals of 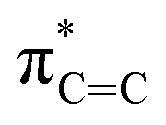 ,
, 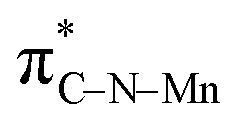 , and
, and 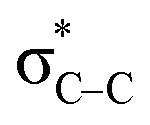 , respectively. This result suggested the existence of the Mn–N–C bonding at the atomic interface. In the N K-edge XANES spectra of MnSAs/S–NC (Fig. 2b), three obvious peaks were observed at 400.2 (peak d), 402.7 (peak e) and 408.6 eV (peak f), arising from pyridinic N, pyrrolic ring N and graphitic N. X-ray photoelectron spectroscopy (XPS) measurements were carried out to further probe the compositions and chemical state of the elements in MnSAs/S–NC. The core-level scan XPS spectra of N 1s, as illustrated in Fig. 2c, comprised five main peaks that resulted from pyridinic N (398.4 eV), pyrrolic N (399.2 eV), graphitic N (401.1 eV), oxide N (403.1 eV) and Mn–N (400.2 eV). In the high-resolution XPS spectra of C 1s shown in Fig. S7a,† the MnSAs/S–NC exhibited three main peaks located at 284.7 eV, 285.8 eV and 289.4 eV that could be attributed to C
, respectively. This result suggested the existence of the Mn–N–C bonding at the atomic interface. In the N K-edge XANES spectra of MnSAs/S–NC (Fig. 2b), three obvious peaks were observed at 400.2 (peak d), 402.7 (peak e) and 408.6 eV (peak f), arising from pyridinic N, pyrrolic ring N and graphitic N. X-ray photoelectron spectroscopy (XPS) measurements were carried out to further probe the compositions and chemical state of the elements in MnSAs/S–NC. The core-level scan XPS spectra of N 1s, as illustrated in Fig. 2c, comprised five main peaks that resulted from pyridinic N (398.4 eV), pyrrolic N (399.2 eV), graphitic N (401.1 eV), oxide N (403.1 eV) and Mn–N (400.2 eV). In the high-resolution XPS spectra of C 1s shown in Fig. S7a,† the MnSAs/S–NC exhibited three main peaks located at 284.7 eV, 285.8 eV and 289.4 eV that could be attributed to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, CN and C–C. The S 2p spectrum (Fig. S7b†) gave peaks at 165.2 eV and 164.0 eV, originating from S 2p1/2 and S 2p3/2 of the C–S–C structure in the metal–organic framework derived carbon frameworks.
C, CN and C–C. The S 2p spectrum (Fig. S7b†) gave peaks at 165.2 eV and 164.0 eV, originating from S 2p1/2 and S 2p3/2 of the C–S–C structure in the metal–organic framework derived carbon frameworks.
 | ||
| Fig. 2 (a) C K-edge, (b) N K-edge XANES spectra and (c) the N 1s XPS spectra of MnSAs/S–NC. (d) FT k3-weighted Mn K-edge EXAFS spectra of MnSAs/S–NC and the references (Mn foil and MnO2). (e) FT-EXAFS fitting curves of MnSAs/S–NC at the Mn K-edge. (f) Schematic atomic interface model of MnSAs/S–NC. | ||
Hard XAFS measurements is a powerful technique to probe the local environment at the atomic level.36 As shown in Fig. 2d, the Fourier transform (FT) of the EXAFS curve of the MnSAs/S–NC catalyst exhibited only one dominant peak at 1.47 Å, which was assigned to the back scattering of Mn–N coordination. Meanwhile, the peaks derived from the Mn–Mn peaks at 2.31 and 2.38 Å in the FT curves of Mn foil and MnO2 were not observed. This precluded the aggregation of Mn-related particles or clusters, and further demonstrated the isolated feature of Mn species in MnSAs/S–NC. Quantitative EXAFS fitting analyses were conducted at the Mn K-edge to extract the structural parameters. The fitting results are exhibited in Fig. 2e, S8 and Table S1.† The best-fitting results for MnSAs/S–NC clearly showed that the Mn atom was connected by four N atoms at the first coordination shell, with a bond length of 1.98 Å, suggesting the formation of Mn–N4 configuration at the atomic interface. Combining the above results, we constructed a probable atomic structure model for MnSAs/S–NC, as shown in Fig. 2f, which was denoted as Mn–N4–CxSy. In order to further investigate the local structure of the Mn single atom, the XANES calculation was carried out, as shown in Fig. S9,† which exhibited that the calculated spectrum was in good agreement with the experimental one, based on the atomic model in Fig. 2f. This suggested that pyridinic-N derived Mn–N–C species in the MnSAs/S–NC might favored a superior ORR catalytic performance.
The ORR test was evaluated in a three-electrode system using O2-saturated 0.1 M KOH as the electrolyte (Fig. S10†). Firstly, Fig. S11† displays the CV curves of MnSAs/S–NC under N2- and O2-saturated solutions, indicating efficient ORR catalysis on the MnSAs/S–NC. Fig. 3a exhibits the linear sweep voltammetry (LSV) curves of MnSAs/S–NC and the counterparts, which were obtained at a rotation speed of 1600 rpm. The half-wave potential (E1/2) of MnSAs/S–NC was 0.916 V, which was obviously more positive compared to MnSAs/NC (0.891 V), and even 65 mV positive than that of Pt/C (0.851 V), a widely used benchmark. Specifically, the MnSAs/S–NC sample exhibited a kinetic current density (Jk) of 8.9 mA cm−2 at 0.90 V, which was 4.9 times higher than that of Pt/C (1.8 mA cm−2) (Fig. 3b). For comparison, NC and S–NC demonstrated rather low Jk (0.08 mA cm−2 and 0.49 mA cm−2) and E1/2 (0.68 V and 0.80 V, respectively), indicating that the Mn–N sites might play important roles in the ORR instead of the N–C or S–N–C sites. Moreover, the ORR activity for MnSAs/S–NC surpassed the other listed non-precious single atom catalysts (Fig. 3c and Table S2†), indicating that the MnSAs/S–NC was among the best electrocatalysts for the ORR in an alkaline medium.
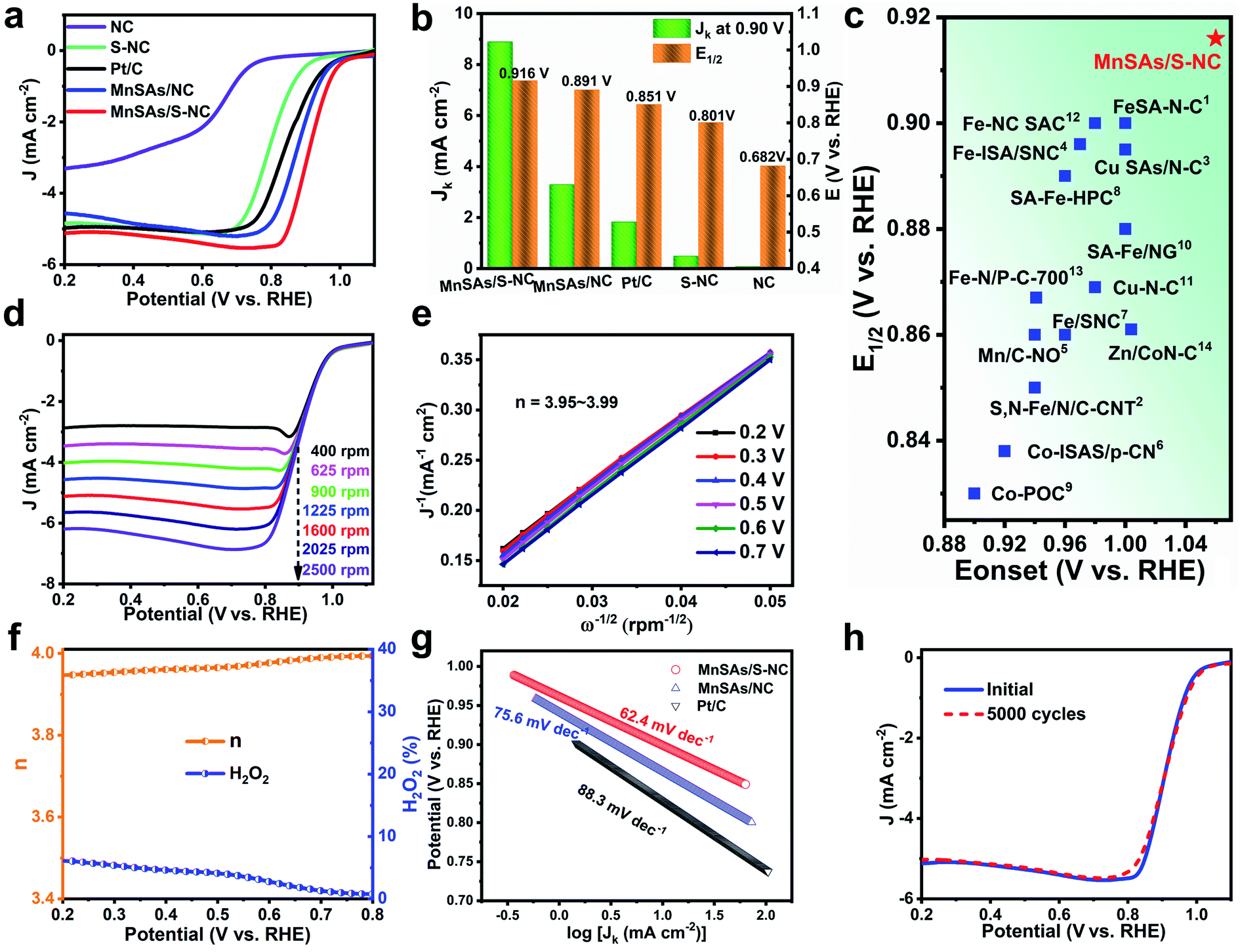 | ||
| Fig. 3 (a) Polarization curves for MnSAs/S–NC, MnSAs/NC, S–NC, NC and commercial Pt/C. (b) Comparison of Jk at 0.90 V and E1/2 for MnSAs/S–NC and the reference catalysts. (c) Comparison of Eonset and E1/2 values for our catalysts with some recently reported ORR catalysts listed in Table S2.† (d) The ORR polarization curves for MnSAs/S–NC at different rotating rates and the corresponding K–L plots (e). (f) Electron-transfer number (n) and H2O2 yield of MnSAs/S–NC from 0.2 to 0.8 V (vs. RHE). (g) Tafel slopes of MnSAs/S–NC, MnSAs/NC and Pt/C for the ORR. (h) ORR polarization curve before and after 5000 cycles. | ||
The kinetics activity of MnSAs/S–NC was evaluated by recording LSV curves at various rotating speeds (400–2500 rpm). As shown in Fig. 3d, current density increased proportionally with the rotation rates because of the shortened O2 diffusion distance. The resulting linear Koutecky–Levich (K–L) curves indicated similar slopes at different redox potentials, as well as analogous first-order reaction kinetics relative to the dissolved O2 concentration. The calculated electron transfer numbers (n) over the MnSAs/S–NC in the potentials ranging from 0.2 to 0.7 V were determined to be 3.95–3.99 (Fig. 3e), which were very close to the theoretical value of Pt/C (4.0) (Fig. S12 and S13†), implying that the ORR process catalyzed by MnSAs/S–NC ideally executed a high-efficiency 4e− pathway. In addition, rotating ring disk electrode tests were conducted to investigate the electron-transfer mechanism (Fig. 3f). From 0.2 to 0.8 V, the electron transfer number of MnSAs/S–NC was in the range of 3.95–3.99 and H2O2 yield remained below 7%, further evidencing a direct four-electron reduction pathway. The Tafel slope of MnSAs/S–NC was calculated to be 62.4 mV dec−1, lower than that of MnSAs/NC (75.6 mV dec−1) and Pt/C (88.3 mV dec−1) (Fig. 3g), further demonstrating the more desirable ORR kinetics for MnSAs/S–NC. In addition, the methanol crossover effect was detected through MnSAs/S–NC electrodes cycling in a methanol-containing solution. The CV curves of the MnSAs/S–NC catalyst showed almost no change when 0.5 M CH3OH was added into the electrolyte (Fig. S14†). These results demonstrated the high tolerance to methanol of the MnSAs/S–NC catalyst.37 Apart from catalytic activity, durability is also important for practical utilization. The durability of the MnSAs/S–NC catalyst was estimated by potential cycling from 0.2 to 1.1 V at a scan rate of 50 mV s−1 for 5000 cycles. After cycling tests, there was very little change in E1/2, suggesting its excellent stability (Fig. 3h). Moreover, the durability of MnSAs/NC for the ORR was also given as reference (Fig. S15†). The chronoamperometry at 0.90 V vs. RHE for MnSAs/S–NC exhibited little activity decline after the 60 h test (Fig. S16†). Fig. S17 and S18† reveal that Mn species were still atomically anchored in the carbon matrix. Additionally, the ORR performance of MnSAs/S–NC in acidic solution was also measured (Fig. S19 and Table S3†).
To reveal the nature of the Mn active site under ORR conditions, the changes in local atomic environments of MnSAs/S–NC were studied by operando XAFS measurements.38,39 The tests were carried out in a homemade organic glass electrochemical cell. The XAFS data were recorded at the open-circuit voltage (OCV) and the representative potential of E1/2 (0.916 V vs. RHE). Fig. 4a presents the XANES spectra at the Mn K-edge of MnSAs/S–NC collected under different conditions, along with the data of Mn foil, MnO, Mn2O3 and MnO2 as standards. From the dry sample to OCV, the position of absorption edge was just slightly shifted. When 0.916 V vs. RHE was applied, an obvious shift towards lower energy occurred, implying a distinct decrease in the Mn oxidation state in MnSAs/S–NC during the ORR. The fitted average oxidation states from XANES curves are shown in Fig. 4b and S20.† It was found that the mean valence state of Mn decreased from 2.80 to 2.51 for the catalyst between OCV and 0.916 V.
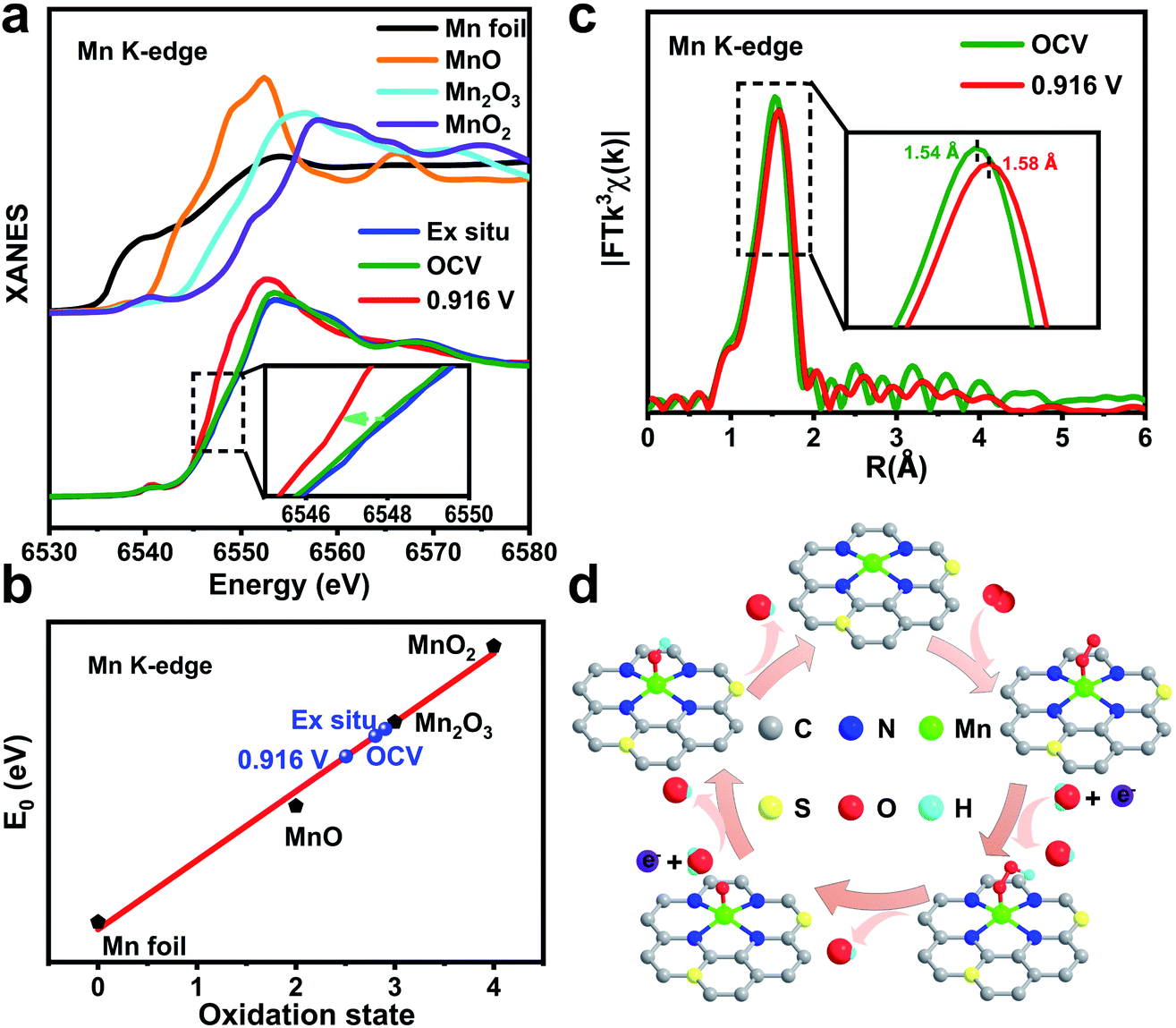 | ||
| Fig. 4 (a) Operando XANES spectra recorded at the Mn K-edge of MnSAs/S–NC from the OCV condition to 0.916 V during the electrocatalytic ORR. (b) The fitted average oxidation states of Mn from XANES spectra. (c) Mn K-edge EXAFS spectra of MnSAs/S–NC at various potentials during the ORR. (d) The proposed ORR mechanism for the MnSAs/S–NC. C: grey; N: blue; S: yellow; Mn: green; H: cyan; O: red. | ||
Furthermore, the local environment evolution of the single Mn sites in MnSAs/S–NC was detected by operando EXAFS spectroscopy. Fig. 4c shows the corresponding k3-weighted FT-EXAFS spectra at different applied potentials. The FT curves still exhibited one main peak, assigned to Mn–N coordination. However, the Mn–N peaks displayed a slightly high-R shift from 1.54 Å to 1.58 Å, implying some changes in the coordination environment of the Mn sites, which was reflected by the extension in the Mn–N bond. Quantitatively, the least-squares EXAFS curve-fitting analysis for the first coordination shell of the Mn centre was carried out by considering two scattering paths of Mn–N and Mn–O (Fig. S21, S22 and Table S4†). According to the fitted structural parameters, the mean bond length for Mn–N under the catalytic conditions increased from 1.99 Å (OCV) to 2.02 Å (0.916 V vs. RHE), which was possibly induced by chemical adsorption of the oxygen species. The most reasonable coordination arrangement was considered as the Mn–N4–CxSy moiety connected with ORR intermediates (Fig. 4d), which acted as the real active site for the ORR. We performed additional operando XAS measurements to evaluate the reversibility of Mn K-edge XAFS spectra of MnSAs/S–NC (Fig. S23–S25 and Table S5†). The atomically dispersed Mn site was susceptible to the valence induced by chemisorbed oxygen species under catalytic conditions. The low-valence Mn species possess a high reactivity towards the adsorption of oxygen. As a comparison, the operando XAS experiments of MnSAs/NC were also performed (Fig. S26–S30 and Table S6†). Overall, the formation of the bond-length extending low-valence HOO–Mn–N4–CxSy, O–Mn–N4–CxSy and HO–Mn–N4–CxSy sites during the ORR process may bring about the boosted ORR activity.
Conclusions
In summary, we report a metal–organic framework derived sulfur modified Mn–N–C single atom ORR catalyst through atomic interface engineering. The active site was confirmed by structure-sensitive XAFS measurements. Through XPS, soft XANES and hard XAFS measurements, the atomic interface structure model of Mn–N4–CxSy was proposed. Benefiting from the desirable atomic structure, MnSAs/S–NC showed excellent ORR performance in alkaline media. Operando XAFS reveal that the improved ORR activity of MnSAs/S–NC derived from the formation of the bond-length extending low-valence oxygen linked Mn–N4–CxSy sites during the ORR process. Our findings provide the possibility for engineering the local environment of catalytic active sites and pave the way for developing advanced oxygen-involved electrode catalysts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 51631001, 21801015, 51872030, 21643003, 51702016, 51501010, and 21703219), the Beijing Institute of Technology Research Fund Program for Young Scholars (3090012221909) and the Fundamental Research Funds for the Central Universities. This paper is supported by the opening project of the State Key Laboratory of Explosion Science and Technology (Beijing Institute of Technology). The opening project number is ZDKT18-01. The authors thank the BL14W1 in the Shanghai Synchrotron Radiation Facility (SSRF), and BL10B and BL12B in the National Synchrotron Radiation Laboratory (NSRL) for help with characterization. The authors thank the characterized work supported by the Analysis & Testing Center of Beijing Institute of Technology. The authors acknowledge the critical testing work supported by the Beijing Zhongkebaice Technology Service Co., Ltd.Notes and references
- Y. Li and H. Dai, Chem. Soc. Rev., 2014, 43, 5257–5275 RSC.
- M. Shao, Q. Chang, J. Dodelet and R. Chenitz, Chem. Rev., 2016, 116, 3594–3657 CrossRef CAS PubMed.
- A. Kulkarni, S. Siahrostami, A. Patel and J. K. Nørskov, Chem. Rev., 2018, 118, 2302–2312 CrossRef CAS PubMed.
- W. Xia, A. Mahmood, Z. Liang, R. Zou and S. Guo, Angew. Chem., Int. Ed., 2016, 55, 2650–2676 CrossRef CAS PubMed.
- C. Zhu, H. Li, S. Fu, D. Du and Y. Lin, Chem. Soc. Rev., 2016, 45, 517–531 RSC.
- Y. Liang, Y. Li, H. Wang, J. Zhou, J. Wang, T. Regier and H. Dai, Nat. Mater., 2011, 10, 780–786 CrossRef CAS PubMed.
- B. Y. Xia, Y. Yan, N. Li, H. B. Wu, X. W. Lou and X. Wang, Nat. Energy, 2016, 1, 15006 CrossRef CAS.
- J. Wang, W. Liu, G. Luo, Z. Li, C. Zhao, H. Zhang, M. Zhu, Q. Xu, X. Wang, C. Zhao, Y. Qu, Z. Yang, T. Yao, Y. Li, Y. Lin, Y. Wu and Y. Li, Energy Environ. Sci., 2018, 11, 3375–3379 RSC.
- P. Su, H. Xiao, J. Zhao, Y. Yao, Z. Shao, C. Li and Q. Yang, Chem. Sci., 2013, 4, 2941–2946 RSC.
- W. Gu, L. Hu, W. Hong, X. Jia, J. Li and E. Wang, Chem. Sci., 2016, 7, 4167–4173 RSC.
- X. Li, H. Rong, J. Zhang, D. Wang and Y. Li, Nano Res., 2020 DOI:10.1007/s12274-020-2755-3.
- Z. Zhuang, Q. Kang, D. Wang and Y. Li, Nano Res., 2020 DOI:10.1007/s12274-020-2827-4.
- C. Zhu, S. Fu, Q. Shi, D. Du and Y. Lin, Angew. Chem., Int. Ed., 2017, 56, 13944–13960 CrossRef CAS PubMed.
- R. Jiang, L. Li, T. Sheng, G. Hu, Y. Chen and L. Wang, J. Am. Chem. Soc., 2018, 140, 11594–11598 CrossRef CAS PubMed.
- C. Cheng, S. Li, Y. Xia, L. Ma, C. Nie, C. Roth, A. Thomas and R. Haag, Adv. Mater., 2018, 30, 1802669 CrossRef PubMed.
- Y. Yang, K. Mao, S. Gao, H. Huang, G. Xia, Z. Lin, P. Jiang, C. Wang, H. Wang and Q. Chen, Adv. Mater., 2018, 30, 1801732 CrossRef PubMed.
- J. Z. Li, M. J. Chen, D. A. Cullen, S. Hwang, M. Y. Wang, B. Y. Li, K. X. Liu, S. Karakalos, M. Lucero, H. G. Zhang, C. Lei, H. Xu, G. E. Sterbinsky, Z. X. Feng, D. Su, K. L. More, G. F. Wang, Z. B. Wang and G. Wu, Nat. Catal., 2018, 1, 935–945 CrossRef CAS.
- S. Wei, Y. Wang, W. Chen, Z. Li, W.-C. Cheong, Q. Zhang, Y. Gong, L. Gu, C. Chen, D. Wang, Q. Peng and Y. Li, Chem. Sci., 2020, 11, 786–790 RSC.
- S. Sun, G. Zhang, N. Gauquelin, N. Chen, J. Zhou, S. Yang and X. Sun, Sci. Rep., 2013, 3, 1775 CrossRef.
- L. Zhang, R. Si, H. Liu, N. Chen, Q. Wang, K. Adair and X. Sun, Nat. Commun., 2019, 10, 4936 CrossRef PubMed.
- N. Cheng, L. Zhang, K. Doyle-Davis and X. Sun, Electrochem. Energy Rev., 2019, 2, 539 CrossRef.
- X. Zhang, A. Chen, M. Zhong, Z. Zhang, X. Zhang, Z. Zhou and X. H. Bu, Electrochem. Energy Rev., 2019, 2, 29–104 CrossRef CAS.
- J. J. Cai, Q. Y. Zhou, B. Liu, X. F. Gong, L. Zhao, X. L. Sui and Z. B. Wang, Nanoscale, 2020, 12, 973–982 RSC.
- H. Xu, D. Cheng, D. Cao and X. Zeng, Nat. Catal., 2018, 1, 339–348 CrossRef CAS.
- Y. Han, Y. Wang, R. Xu, W. Chen, L. Zheng, A. Han, Y. Zhu, J. Zhang, H. Zhang, J. Luo, C. Chen, Q. Peng, D. Wang and Y. Li, Energy Environ. Sci., 2018, 11, 2348–2352 RSC.
- K. Yuan, D. F. Lützenkirchen-Hecht, L. Li, L. Shuai, Y. Li, R. Cao, M. Qiu, X. Zhuang, Y. Chen, M. K. H. Leung and U. Scherf, J. Am. Chem. Soc., 2020, 142, 2404–2414 CrossRef CAS PubMed.
- H. Wu, H. Li, X. Zhao, Q. Liu, J. Wang, J. Xiao, S. Xie, R. Si, F. Yang, S. Miao, X. Guo, G. Wang and X. Bao, Energy Environ. Sci., 2016, 9, 3736–3745 RSC.
- F. Li, G. F. Han, H. J. Noh, S. J. Kim, Y. L. Lu, H. Y. Jeong, Z. P. Fu and J. B. Baek, Energy Environ. Sci., 2018, 11, 2263–2269 RSC.
- P. Yin, T. Yao, Y. Wu, L. Zheng, Y. Lin, W. Liu, H. Ju, J. Zhu, X. Hong, Z. Deng, G. Zhou, S. Wei and Y. Li, Angew. Chem., Int. Ed., 2016, 55, 10800–10805 CrossRef CAS PubMed.
- P. Chen, T. Zhou, L. Xing, K. Xu, Y. Tong, H. Xie, L. Zhang, W. Yan, W. Chu, C. Wu and Y. Xie, Angew. Chem., Int. Ed., 2017, 56, 610–614 CrossRef CAS PubMed.
- H. Shen, E. Gracia-Espino, J. Ma, K. Zang, J. Luo, L. Wang, S. Gao, X. Mamat, G. Hu, T. Wagberg and S. Guo, Angew. Chem., Int. Ed., 2017, 56, 13800–13804 CrossRef CAS PubMed.
- Y. Mun, S. Lee, K. Kim, S. Kim, S. Lee, J. W. Han and J. Lee, J. Am. Chem. Soc., 2019, 141, 6254–6262 CrossRef CAS PubMed.
- Y. Chen, S. Ji, S. Zhao, W. Chen, J. Dong, W. C. Cheong, R. Shen, X. Wen, L. Zheng, A. I. Rykov, S. Cai, H. Tang, Z. Zhuang, C. Chen, Q. Peng, D. Wang and Y. Li, Nat. Commun., 2018, 9, 5422 CrossRef CAS PubMed.
- Q. Li, W. Chen, H. Xiao, Y. Gong, Z. Li, L. Zheng, X. Zheng, W. Yan, W.-C. Cheong, R. Shen, N. Fu, L. Gu, Z. Zhuang, C. Chen, D. Wang, Q. Peng, J. Li and Y. Li, Adv. Mater., 2018, 30, 1800588 CrossRef PubMed.
- P. Chen, N. Zhang, S. Wang, T. Zhou, Y. Tong, C. Ao, W. Yan, L. Zhang, W. Chu, C. Wu and Y. Xie, PNAS, 2019, 116, 6635–6640 CrossRef CAS PubMed.
- J. E. Penner-Hahn, Coord. Chem. Rev., 1999, 190–192, 1101–1123 CrossRef CAS.
- Y. Chen, S. Ji, Y. Wang, J. Dong, W. Chen, Z. Li, R. Shen, L. Zheng, Z. Zhuang, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2017, 56, 6937–6941 CrossRef CAS.
- J. K. Li, S. Ghoshal, W. T. Liang, M.-T. Sougrati, F. Jaouen, B. Halevi, S. McKinney, G. McCool, C. R. Ma, X. X. Yuan, Z.-F. Ma, S. Mukerjee and Q. Y. Jia, Energy Environ. Sci., 2016, 9, 2418–2432 RSC.
- A. Zitolo, N. Ranjbar-Sahraie, T. Mineva, J. Li, Q. Jia, S. Stamatin, G. F. Harrington, S. M. Lyth, P. Krtil, S. Mukerjee, E. Fonda and F. Jaouen, Nat. Commun., 2017, 8, 957 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and supplementary characterization. See DOI: 10.1039/d0sc02343d |
| This journal is © The Royal Society of Chemistry 2020 |