
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSMolESY: an efficient and quantitative alternative to on-instrument macromolecular 1H-NMR signal suppression†
Panteleimon G.
Takis
 *ab,
Beatriz
Jiménez
ab,
Caroline J.
Sands
ab,
Elena
Chekmeneva
ab and
Matthew R.
Lewis
ab
*ab,
Beatriz
Jiménez
ab,
Caroline J.
Sands
ab,
Elena
Chekmeneva
ab and
Matthew R.
Lewis
ab
aSection of Bioanalytical Chemistry, Division of Systems Medicine, Department of Metabolism, Digestion and Reproduction, Imperial College London, South Kensington Campus, London, SW7 2AZ, UK
bNational Phenome Centre, Department of Metabolism, Digestion and Reproduction, Imperial College London, Hammersmith Campus, IRDB Building, London, W12 0NN, UK. E-mail: p.takis@imperial.ac.uk
First published on 27th May 2020
Abstract
One-dimensional (1D) proton-nuclear magnetic resonance (1H-NMR) spectroscopy is an established technique for measuring small molecules in a wide variety of complex biological sample types. It is demonstrably reproducible, easily automatable and consequently ideal for routine and large-scale application. However, samples containing proteins, lipids, polysaccharides and other macromolecules produce broad signals which overlap and convolute those from small molecules. NMR experiment types designed to suppress macromolecular signals during acquisition may be additionally performed, however these approaches add to the overall sample analysis time and cost, especially for large cohort studies, and fail to produce reliably quantitative data. Here, we propose an alternative way of computationally eliminating macromolecular signals, employing the mathematical differentiation of standard 1H-NMR spectra, producing small molecule-enhanced spectra with preserved quantitative capability and increased resolution. Our approach, presented in its simplest form, was implemented in a cheminformatic toolbox and successfully applied to more than 3000 samples of various biological matrices rich or potentially rich with macromolecules, offering an efficient alternative to on-instrument experimentation, facilitating NMR use in routine and large-scale applications.
Introduction
1H-NMR spectroscopy is a well-established technique used throughout clinical, population-scale, pharmaceutical and agricultural product research for qualitative and quantitative analyses of small molecules (SMs) in complex samples.1–3 It is also increasingly used to measure the abundance of larger structures such as lipoprotein species in blood plasma and serum4 and indirectly estimate NMR-invisible components of biofluids.5 All these types of measurements are captured in the single most common experiment in metabolomics and clinical research applications, the 1H-NMR one-dimensional general profile experiment with solvent signal suppression (e.g. one dimensional nuclear Overhauser effect spectroscopy, 1D-NOESY pulse sequence).6 However, broad baseline signals arising from macromolecular content (e.g. proteins and lipids abundant in serum/plasma, urine from subjects with pathological conditions, cerebrospinal fluid, saliva, tissues, cell lysates, pancreatic fluid, and food matrices such as milk, oil etc.)2,7–10 can overlap with and even hide the SM signals in a spectrum, inhibiting the efficiency of their deconvolution for annotation and quantification. More importantly, variability in macromolecule concentrations among samples results in baseline fluctuations which hinder the robust determination of SM contribution to diagnostic phenotypic signatures and/or fingerprinting by multivariate analysis (MVA).These issues may be addressed by physically removing the macromolecules, for example, by ultra-centrifugation with filtering,11,12 but the time and cost required for sample processing, potential for introducing procedural variability, and negative impact to the integrity of the sample itself all undermine the key strengths of NMR as a high-throughput, intrinsically precise and non-destructive technique.1,2 Instead, the more practical and routinely applied approach is to suppress resonances from macromolecular-derived signals on-instrument. This is accomplished by performing an ancillary “spin-echo” experiment such as the Carr–Purcell–Meiboom–Gill (CPMG)13 pulse sequence which filters macromolecular signals via transverse relaxation times (T2), generating a 1D spectrum of slow relaxing signals, mainly belonging to SMs. The approach is sufficiently reproducible although imperfect in its suppression of broad resonances (Fig. 1A) given the time limit for large cohort studies (e.g. metabolomics) and unsuitable for direct absolute quantification, as the signal integral is modulated by the high variability of T2 values for each proton spin system from each SM.14 It is also time consuming, contributing substantially to the acquisition time required by standard profiling workflows (ESI Fig. S1†). The approach is therefore costly, especially at the scale required for the routine analysis of samples from epidemiology cohorts, food industry quality control, and other large-scale applications.
 | ||
| Fig. 1 SMolESY analytical reproducibility and performance in various matrices. (A) 1D-NOESY, CPMG and SMolESY spectra of albumin titration (0–225 mM). CPMG spectra exhibit ineffective suppression of albumin signals (light blue boxed areas), whereas SMolESY achieves their complete attenuation. Moreover, SMolESY maintains SMs' (herein impurities) fingerprint. 1D-NOESY, CPMG and SMolESY spectra of (B) bovine milk and (C) olive oil, focused on the fatty acids-lipids aliphatic groups 1H-NMR region. It is clearly shown that SMolESY supersedes the routine CPMG spectrum (light blue boxed areas), enhancing the resolution by effectively narrowing the broad NMR signals of the aliphatic chains and increasing resolution. In addition, SMolESY affords the direct quantification by integration of several SMs, which are easily detected/assigned compared to both 1D-NOESY and CPMG spectra, where spectral deconvolution is needed. (D) Integrals of five 1D-NOESY 1H-NMR signals from cytidine in the artificial mixture of metabolites in 9 concentrations were correlated with the SMolESY with a linear correlation (R2 > 0.985), passing through the origin (dashed circle), and the statistical one-way ANOVA tests (ESI Table S1†) confirmed all intercepts/slopes coincidence (horizontal/vertical error bars show ±1% integration error). Regardless of signals multiplicity (doublets with different j-coupling, multiplets, triplet), SMolESY shows intra-metabolites analytical reproducibility. (E–H) PCA of a urine dataset produces the same results for both 1D-NOESY and SMolESY, capturing similar cumulative variability, whereas loading plots point at the same variables for groups discrimination. | ||
As a more efficient and higher performance alternative, we have developed a novel computationally derived experiment, “SMolESY” (Small Molecule Enhancement SpectroscopY), which reliably increases resolution and depletes macromolecular signals directly from the 1H 1D-NMR spectrum with no intensity modulation. The approach relies on mathematical differentiation, previously used for improving the spectral resolution of various spectroscopic techniques (e.g. near-infrared, electron-spin resonance, and NMR).15–18 By calculating the first partial derivative of the imaginary data of the NMR spectrum (see paragraph Differentiation of imaginary spectral data – basic theory in the Experimental section and ESI Fig. S2†), SMolESY yields a profile of SMs free from large molecule signal baseline interference and sample-to-sample fluctuation. As the approach does not rely on T2 or j-coupling constant modulation,19 the inherent quantitative quality of the conventional 1H-NMR spectrum is preserved. Furthermore, the resolution of SMs derived signals is enhanced by as much as three-fold,20 enabling the annotation of otherwise overlapping signals and further facilitating their quantification. However, it is also commonly understood that derivatives are prone to instability when applied to signals of very low intensity, and therefore the practical effects of a reduced signal-to-noise ratio (s/n) required evaluation. Herein, we demonstrate that despite the lower s/n, for the case of SMs of biologically relevant complex mixtures, the signal's limit of detection (LOD) is not functionally affected. To our knowledge, the application of our approach (even in its simplest form of differentiation without combined with any traditional or modern signal denoising filters21) to biofluids or complex matrices of large cohort studies, with the view to suppressing signals of macromolecules across entire spectra in a systematic way, has never been reported or tested. Based upon our findings, the SMolESY experiment may be used to functionally replace and additionally improve upon several weaknesses of traditionally used spin-echo experiments, particularly in the NMR-based metabolomics field.
Results and discussion
The approach was applied to 1H-NMR 1D-NOESY spectra from various datasets of varying sample matrix complexity in order to systematically evaluate SMolESY performance.SMolESY performance for macromolecular spectral background attenuation
The first set of 1D-NOESY and CPMG spectra were generated from a series of pure human serum albumin solutions at concentrations designed to span and exceed those found in normal human blood (Fig. 1A) and from two food matrices, namely of bovine milk and olive oil, respectively (Fig. 1B and C). In all cases, the corresponding SMolESY spectra showed complete attenuation of large molecule derived broad signals resulting in zero-baselines across the whole spectral area. In the model albumin solutions SMolESY signals from SMs which belong to impurities embedded in the protein reagent appeared highlighted as they are the only observable resonance on the reprocessed spectra (Fig. 1A). For the milk solution, the commonly applied CPMG experiment in the NMR-based metabolomics pipeline resulted in unsuppressed 1H-NMR broad signals of fatty acid chains (Fig. 1B), whereas SMolESY provided an effective broad signals attenuation. Furthermore, many resonating signals of milk metabolites7 on the edges of broad signals were sufficiently enhanced by SMolESY, so as to be easily assigned and quantified, which was not the case from their corresponding CPMG spectrum. The same effect was observed for the methyl group signal of olive oil saturated fatty acids which is fully deconvolved from other methyl groups, as well as for the triplet of the methyl terminal group of linolenic acid8 which is easily separated by the 13C satellites of other protons (Fig. 1C).Validation of SMolESY signal integrity and intra-metabolite reproducibility: application to free and very low macromolecular content matrices
The second dataset was generated from synthetic mixtures of metabolites in varying concentrations (see section Artificial mixtures preparation in Experimental section) designed to enable assessment of SMolESY fidelity across a comprehensive set of 1H-NMR peak shapes and multiplicities (e.g. triplet, quartet etc.). The relationship between 1D-NOESY and SMolESY peak integrals (Fig. 1D, S3 and Table S1 (ESI)†) was strongly linear with coefficient of determination (R2) values >0.98 and passing through the origin regardless of signal multiplicity. This is demonstrated by evaluation of the uniquely shaped 1H-NMR signals for five different 1H spin systems present in cytidine (Fig. 1D) and further signals from six other SMs (ESI Fig. S3 and Table S1†). In addition, one-way ANOVA tests for the curves of different spin systems from the same metabolite proved that both slopes and intercepts coincide (Table S1†), indicating preservation of the 1D-NOESY qualitative signal response in the SMolESY spectra.SMolESY was then applied on a third dataset consisting of publicly available 1D-NOESY spectra from normal human urine samples22 (see paragraph Plasma – urine spectra employed for the present study in the Experimental section). Urine's complex SM composition in the virtual absence of macromolecules was used to assess SMolESY's preservation of SM signal information. Principal Component Analysis (PCA) on both the 1D-NOESY and SMolESY urine spectral datasets produced score plots with the same pattern of sample groups with similar cumulative captured variability (85% and 82.7% respectively for the two first components) and loading plots with the same pattern of variable weightings (Fig. 1E–H). 3D score plots from the same analyses are described in ESI Fig. S4.† The result demonstrates that the multivariate information sets recovered from each spectral type are equivalent, providing support for SMolESY's use in classical metabolomics (pursuit of diagnostic and prognostic chemical patterns) and “fingerprinting” applications. Beyond the intended validation, the use case itself is of potential value as numerous pathological conditions can significantly increase urinary excretion of macromolecules such as albumin and lipids.23,24 Although metabolically interesting in their own right, the presence of such lipid/protein signals in urine samples can also confound any subsequent SM multivariate analyses and quantitation, since these signals would not be attenuated by NMR experiments routinely applied to urine samples or by pre-processing methods, for example, normalization.25 SMolESY therefore has an ability to salvage otherwise compromised spectra from specimen in sample sets where macromolecules would not be expected or planned for.
Application of SMolESY into large plasma cohorts
For the third dataset, SMolESY spectra were produced from a collection of 3020 1D-NOESY profiles of human plasma samples from two different cohorts (2026 plasma-heparin and 994 plasma-EDTA samples) (see paragraph Plasma – urine spectra employed for the present study in the Experimental section) so as to increase sample content variability and compared to their corresponding CPMG spectra. Pearson correlation (ESI Fig. S1†) between the SMolESY and CPMG spectra showed that 73% of transformed peaks were highly correlated with r > 0.90 (Fig. 2 and S5 (ESI)†). The remaining 27% of peaks correspond to either CPMG peaks convolved with poorly suppressed broad signals (Fig. 2A–I, N–O, S5 and S6 (ESI)†), those lying on the edges of signals from highly abundant metabolites, or those not visible in the CPMG because of significant peak overlap but resolved in the SMolESY spectra (ESI Fig. S6†). Evaluation of the differentiation's effect on spectral s/n in exemplar analytes revealed an average decrease of 30% in SMolESY versus CPMG spectra (see Experimental section “Signal-to-noise ratio (s/n) and peak picking calculations”). However, the net effect of SMolESY on retrievable SM information across the entire spectrum appeared positive, with a 34% increase in the number of detected signals over the CPMG. Together, these results confirm the inter-spectra reproducibility of SMolESY and its outperformance on the CPMG, since the total number of SMs SMolESY NMR visible features is higher than in the CPMG. Only four broad signals (representing less than 1% of the total transformed peaks) were less well represented in the SMolESY spectra than the CPMG (Fig. 2L and S5J (ESI)†). These signals corresponded to the broad linewidth to the half-height (Δv1/2) of the urea NMR peak (CPMG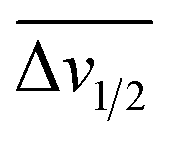 ∼ 40 Hz) and very low abundance unknown metabolites (1.5 < signal-to-noise ratio < 2.2, CPMG 10 Hz) (Fig. 2L and S5J (ESI)†). It is well known that quantitation of any SM containing labile 1H is compromised when water suppression techniques are used, hence the urea signal is excluded from the MVA in the majority of studies.26Ad hoc experiments are therefore necessary in to accurately quantify these types of metabolites.26 Additionally, statistical correlation analyses by statistical total correlation spectroscopy (STOCSY) (Fig. 3)27 were applied to several low to high structural complexity molecules in the SMolESY plasma data, further confirming that correlation structure in the dataset is preserved and additionally demonstrating the potential for enhanced metabolite identification in SMolESY spectra arising from the improvement in signal resolution (Fig. 3). For instance, the signal multiplicities of several 1H spin systems for L-threonine (Fig. 3D) and L-proline (Fig. 3F) were better resolved, leading not only to a better assignment of their signals but also to a substantial deconvolution of other SM signals in several spectral areas confounded by broad NMR signals of plasma lipoproteins.
∼ 40 Hz) and very low abundance unknown metabolites (1.5 < signal-to-noise ratio < 2.2, CPMG 10 Hz) (Fig. 2L and S5J (ESI)†). It is well known that quantitation of any SM containing labile 1H is compromised when water suppression techniques are used, hence the urea signal is excluded from the MVA in the majority of studies.26Ad hoc experiments are therefore necessary in to accurately quantify these types of metabolites.26 Additionally, statistical correlation analyses by statistical total correlation spectroscopy (STOCSY) (Fig. 3)27 were applied to several low to high structural complexity molecules in the SMolESY plasma data, further confirming that correlation structure in the dataset is preserved and additionally demonstrating the potential for enhanced metabolite identification in SMolESY spectra arising from the improvement in signal resolution (Fig. 3). For instance, the signal multiplicities of several 1H spin systems for L-threonine (Fig. 3D) and L-proline (Fig. 3F) were better resolved, leading not only to a better assignment of their signals but also to a substantial deconvolution of other SM signals in several spectral areas confounded by broad NMR signals of plasma lipoproteins.
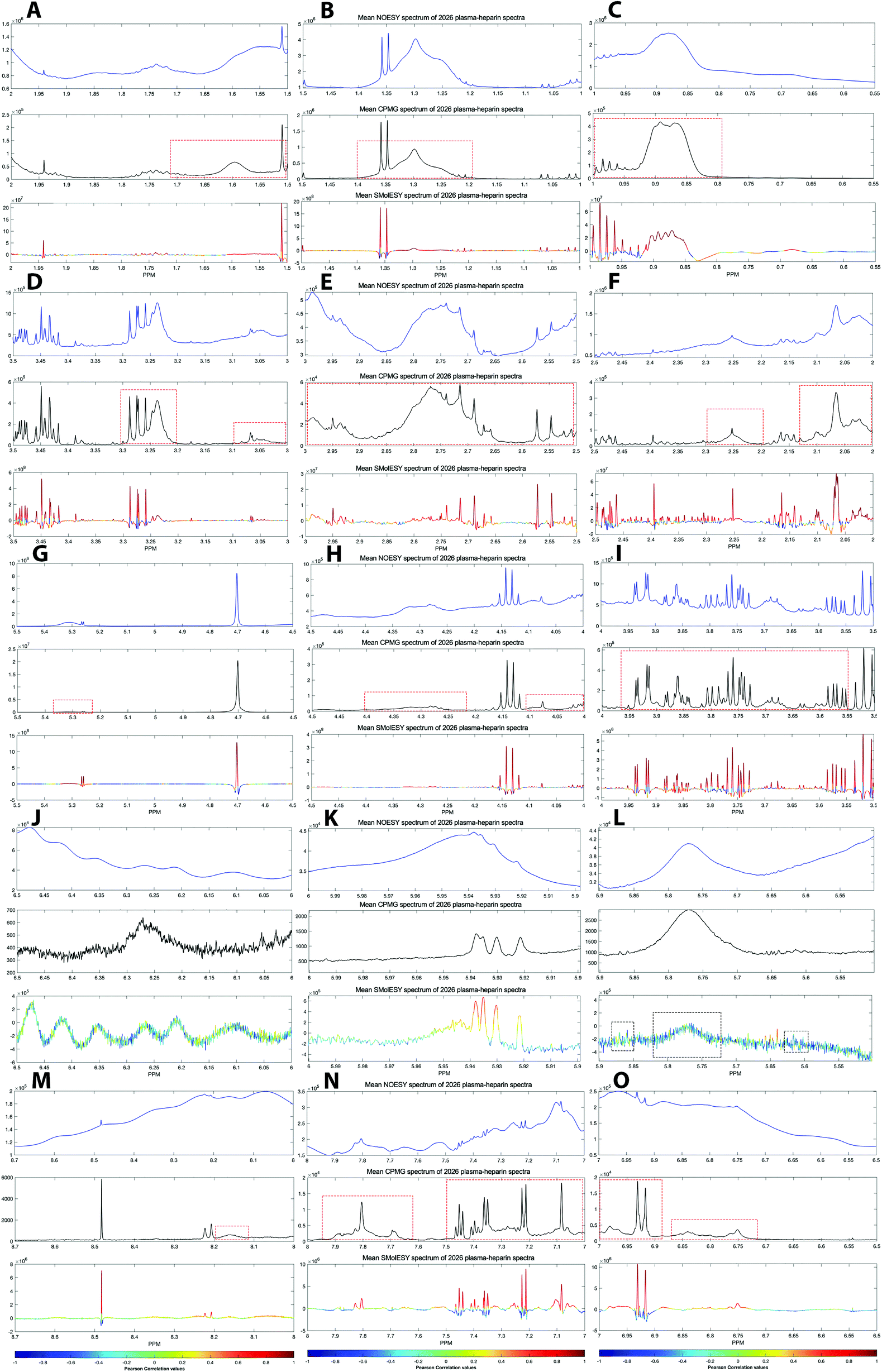 | ||
| Fig. 2 SMolESY performance in more than 2000 plasma-heparin samples. (A–O) Mean spectrum of 2026 plasma-heparin 1D-NOESY (upper panel), CPMG (middle panel) and SMolESY (bottom panel) spectra zoomed at ∼0.5 ppm window from 0.55–8.7 ppm. The mean SMolESY spectrum is colored according to the Pearson coefficients from SMolESY versus CPMG signals correlation in 2026 spectra (ESI Fig. S1†). The majority of highly resolved SMolESY signals are linearly correlated to the CPMG and >99.5% of CPMG features of SMs are maintained, while successfully suppressing the broad signals of macromolecules in contrast to CPMG (examples of unsuppressed CPMG broad signals are highlighted by red dashed boxes). It is noted that broad signal of urea along with 3–4 broad signals of very low abundance (i.e. <1.5 times the CPMG noise) metabolites are recovered by SMolESY but are highly suppressed and exhibit low correlation to the CPMG (black dashed boxes in panel (L)). | ||
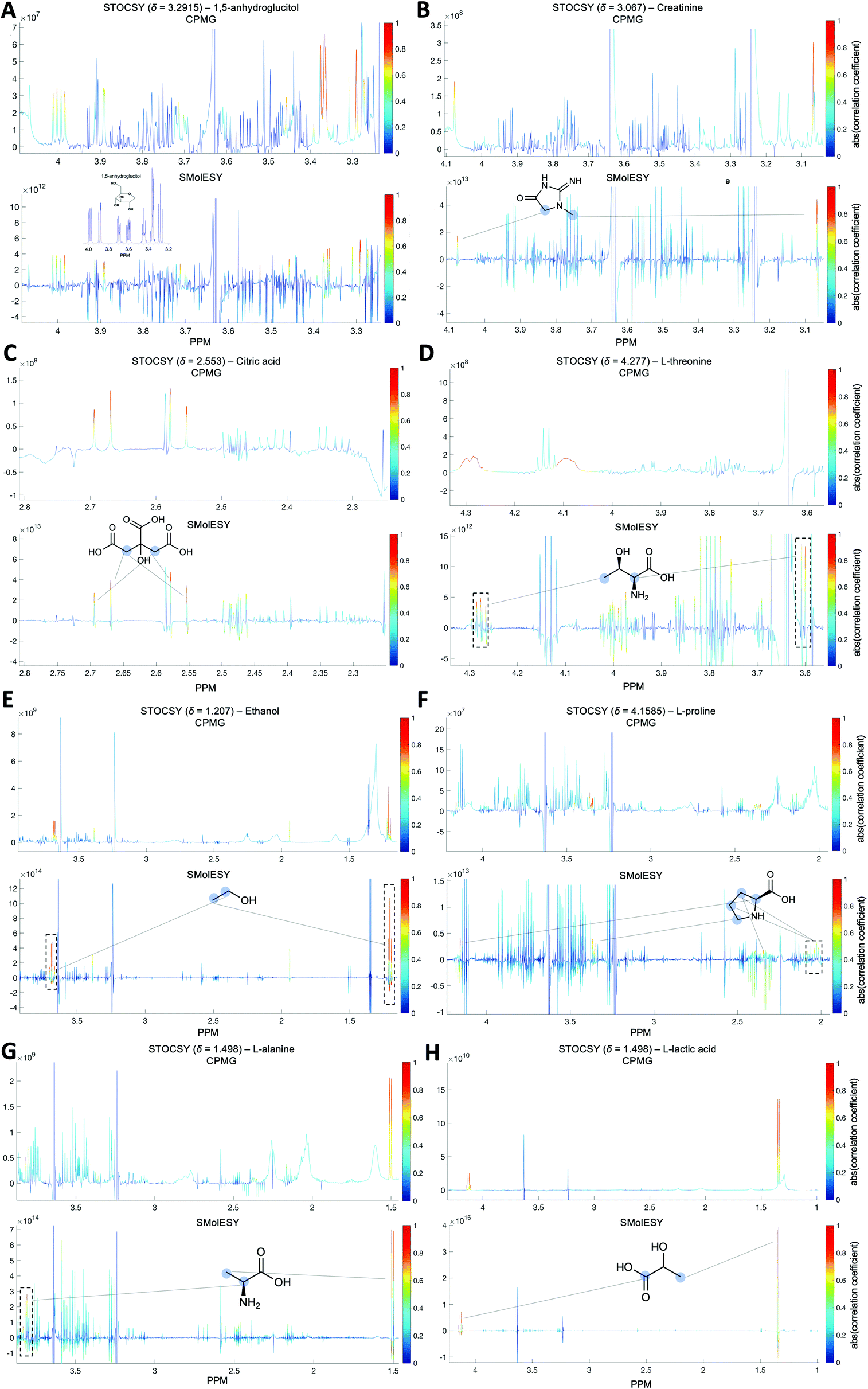 | ||
| Fig. 3 STOCSY analyses between SMolESY and CPMG 994 plasma-EDTA spectra. (A) 1,5-anhydroglucitol, (B) creatinine, (C) citric acid, (D) L-threonine, (E) ethanol, (F) L-proline, (G) L-alanine and (H) L-lactic acid metabolites assignment by STOCSY in the SMolESY(bottom panel) spectra outperforms CPMG (upper panel), indicating higher correlation values among all signals and maintaining metabolites NMR fingerprint. STOCSY in the SMolESY spectra shows correlations between all spin systems of L-threonine and L-proline (black dashed boxes) in contrast to CPMG. Moreover, STOCSY for L-alanine and ethanol exhibits all expected correlations for both metabolites' signals (i.e. one doublet and one quartet for L-alanine, one triplet and one quartet for ethanol), in contrary to the CPMG spectra which fail to map the spin systems multiplicity. Light blue circles indicate the corresponding spin systems of each metabolite. Chemical shift values of “driver” peaks (mentioned in the title of each panel) for the metabolites were taken from the mean spectrum of SMolESY spectra. | ||
Both correlation and STOCSY results confirm the efficacy and fidelity of SMolESY, with more 1D-NOESY SM features maintained (>99%) than those visible by CPMG owing to the resolution enhancement. It is noteworthy that the resolution enhancement of SM peaks due to Δv1/2 narrowing is further improved by the complete removal of broad signals background.
SMolESY employment and implementation to NMR-based metabolomics and analytical studies
Two final key remaining characteristics for the successful implementation of SMolESY to metabolomics and analytical studies are spectral binning and absolute quantification. These were addressed using NMR experiments where 17 common biological metabolites in various known concentrations were spiked-in to a real plasma matrix to provide a SMs profile against a constant macromolecular background (see paragraph Artificial mixtures preparation – spiking experiments in the Experimental section).‡Comparison between spectral bins of SMolESY and CPMG spectral bins indicated a strong linear correlation for all spiked metabolites (R2 > 0.98) (Fig. 4A–C and S7†), even in cases where resonances overlapped with broad macromolecular signals (e.g.Fig. 4B and C). Furthermore, the ease of quantification as well as immediate deconvolution of the SM signals by SMolESY is exemplified in a randomly selected plasma spectrum, where the immediate identification and integration of above 20 metabolites' signals at high resolution and without interference from broad signals or baseline distortions (Fig. 4D–W) is accomplished. The metabolite quantification by straightforward integration of SMolESY features (see paragraph SMolESY signals integration procedure in the Experimental section) was compared to outputs of standard 1D-NOESY peaks' deconvolution and fitting algorithms (Bruker Biospin, http://www.bruker.com, commercially available IVDr quantitation4 and in-house algorithms). SMolESY-based quantification results for the tested spiked metabolites follow a linear correlation with spiked concentrations, as well as with the measured values from deconvolved/fitted 1D-NOESY data (Fig. 5). In addition, the calculated relative root mean square error (RRMSE) values (ESI Fig. S8†) from the quantification of 12 spiked metabolites (Fig. 5), clearly demonstrate that direct SMolESY signal integration has the propensity to provide substantially less error in the absolute quantification than the deconvolution algorithm. For instance, 1D-NOESY signals employed for the quantification of L-isoleucine (Fig. 4E), L-valine (Fig. 4F) and acetone (Fig. 4L) via deconvolution, resonate on top/foothills of very broad signals (i.e. require baseline removal through fitting) and thus, owing to cumulative baseline fitting errors, exhibit higher RRMSE values than when quantified via direct integration of SMolESY signals (ESI Fig. S8†).
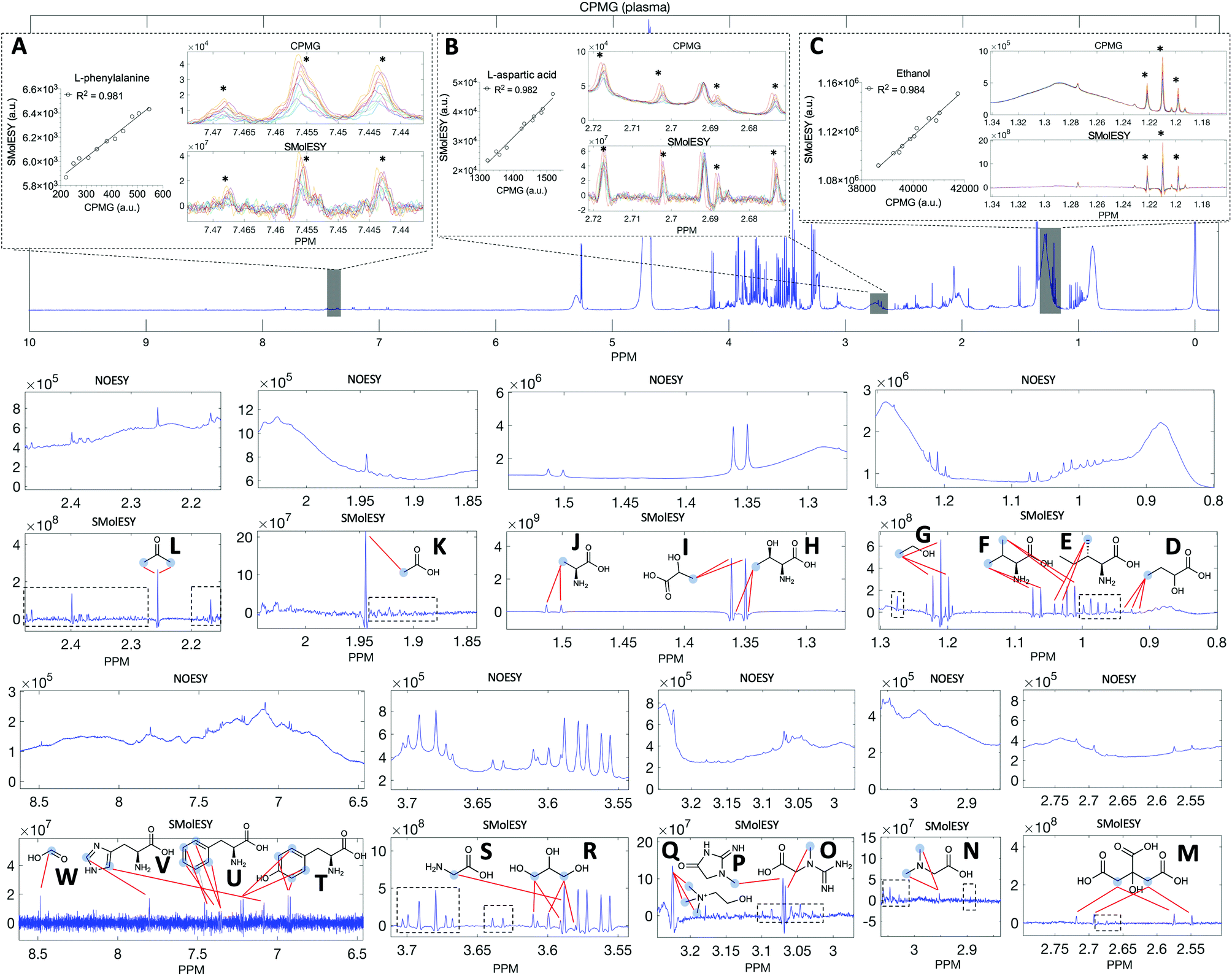 | ||
| Fig. 4 SMolESY for binning and assignment-quantification. Comparison of SMolESY and CPMG spectral bins including signals of (A) L-phenylalanine, (B) L-aspartic acid and (C) ethanol spiked (11 concentrations) in a human plasma sample. Linear regression curves indicate the excellent reproducibility of SMolESY R2 > 0.98 while ourperforming CPMG in broad signal suppression (error bars are omitted due to ∼0 error in bin integration). SMolESY signals (light blue circles – red lines) from >20 plasma metabolites: (D) 2-hydroxybutyric acid, (E) L-isoleucine, (F) L-valine, (G) ethanol, (H) L-threonine, (I) L-lactic acid, (J) L-alanine, (K) acetic acid, (L) acetone, (M) citric acid, (N) N,N-dimethylglycine, (O) creatine, (P) creatinine, (Q) choline, (R) glycerol, (S) glycine, (T) L-tyrosine, (U) L-phenylalanine, (V) L-histidine, (W) formic acid and many more metabolites (black dashed boxes) are completely deconvolved from the macromolecular content in contrary to the commonly employed for quantification 1D-NOESY spectrum and their direct integration could provide metabolites' concentration values. | ||
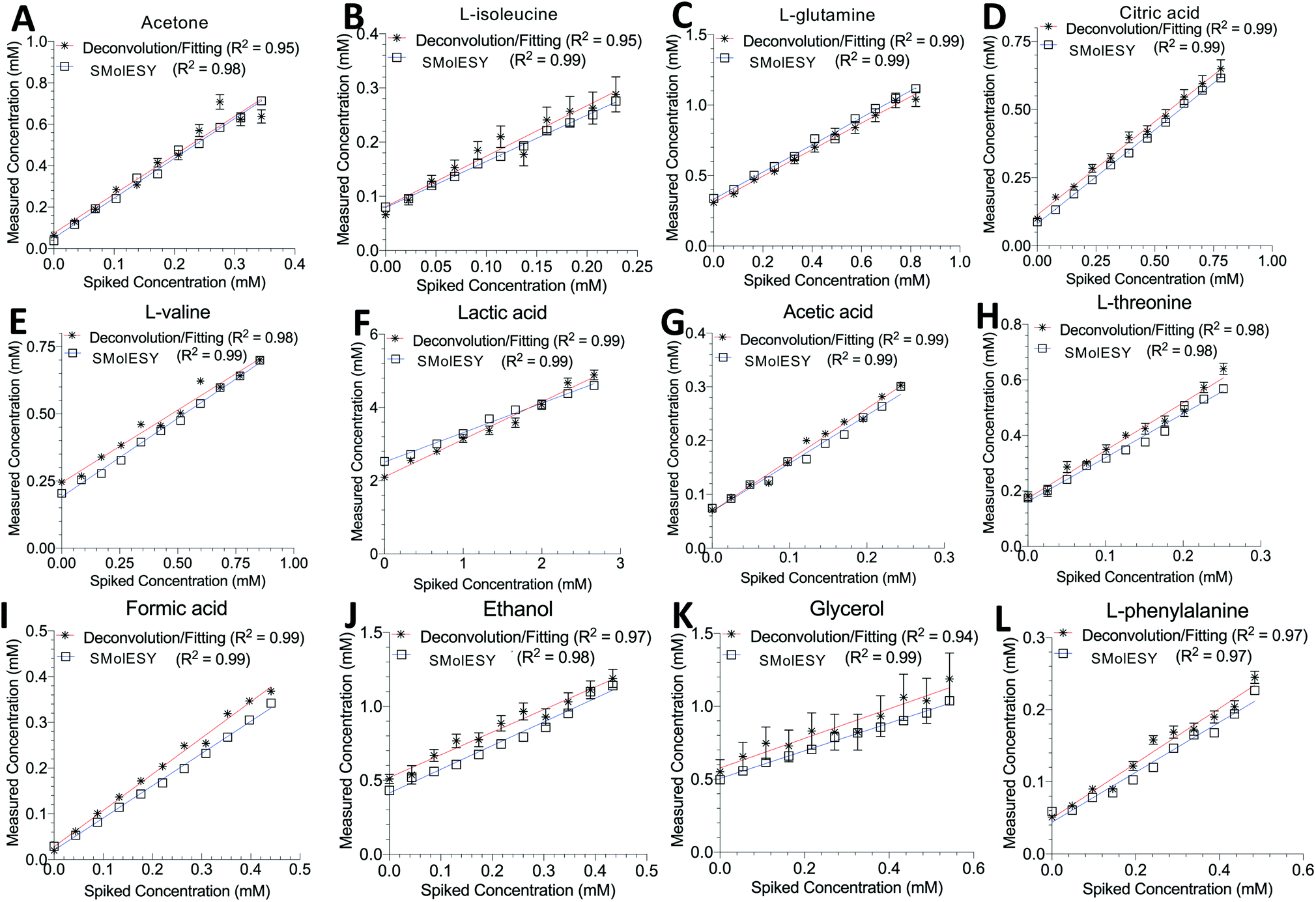 | ||
| Fig. 5 SMolESY for absolute quantification. Absolute quantification was performed for 11 concentrations of several spiked metabolites: (A) acetone, (B) L-isoleucine, (C) L-glutamine, (D) citric acid, (E) L-valine, (F) lactic acid, (G) acetic acid, (H) L-threonine, (I) formic acid, (J) ethanol, (K) glycerol and (L) L-phenylalanine in a plasma matrix by SMolESY (i.e. by direct integration of the transformed signals of each metabolite) and 1D-NOESY by deconvolution/fitting algorithms (herein by the commercially available IVDr platform from Bruker Biospin)4 and plotted against the spiked concentration values. Linear regression analyses clearly show the applicability of SMolESY for absolute quantification (R2 > 0.97), and all calculated concentrations based upon SMolESY data are in reasonable agreement with the deconvolution results. It should be noted that no calibration was applied to the SMolESY integrals so as to account for e.g. T1 relaxation times differences between 1H spin systems from different chemical groups28etc., whereas these refinements are implemented into the IVDr platform of Bruker Biospin. Hence, some slight discrepancies can be observed between SMolESY and the Bi-QUANT-PS™ values due to this refinement. The calculation of absolute concentration values is based upon the ERETIC signal (and its transformation) produced during the acquisition of 1D-NOESY data by Bruker. It is noteworthy that the instant quantification via integration has no computational cost and the deconvolution/fitting algorithms are prone to higher errors (see vertical error bars and calculated relative root mean square error (RRMSE) values ESI Fig. S8†) compared to the integration process (±∼1%) of the already deconvoluted signals in the “clean” baseline of SMolESY spectra. For the IVDr data, plotted error bars are taken from the Δ values produced by the corresponding reports. | ||
To facilitate the implementation of SMolESY in both targeted (direct metabolite signal integration), untargeted (profiling/fingerprinting) and quantitative NMR (qNMR) applications, we created a cheminformatic toolbox, “SMolESY_platform”, for producing and processing SMolESY data from raw NMR spectra (ESI Fig. S9,† see paragraph “SMolESY_platform” toolbox details in the Experimental section). It is freely available for download at: https://github.com/pantakis/SMolESY_platform.§
The compromising effect that common macromolecules (proteins, lipids and polysaccharides) exhibit on individual quantitative SM measurements and on the broader SM profile has yet to be adequately addressed. Consequently, modern standard protocols for biofluid, cell extracts,29 food30 and other rich in macromolecules complex mixtures analysis rely on a sequence of experiments, each of which is individually flawed in application to the most common of biofluids (e.g. blood products). Whereas 1D-NOESY is ineffective at detecting SMs contribution from MVA, CPMG cannot be used for accurate and reliable quantification. Here we demonstrate that the computational transformation of the standard 1D 1H-NMR experiment yields both high fidelity spectral SM profiles and data from which quantitative chemical measurements can be extracted.
Systematic evaluation of SMolESY clearly demonstrates its ability to cleanly suppress macromolecular signals in synthetic test cases (albumin titration), common agricultural products (milk and oil), and human plasma. In all cases, the suppression of macromolecular signals resulted in the enhancement of SM-derived information from the SMolESY's ability to reproduce the SM-derived information captured by the 1D 1H-NMR with high fidelity, ensuring the transformation is not detrimental to SM signals. SMolESY implementation both on a large cohort of more than 3000 individuals' plasma samples and >100 urine samples showed an outstanding reproducibility with virtually no loss of metabolic information. Although the approach does risk decreasing the s/n of very broad signals such as those from highly exchangeable and/or interacting protons of small molecules (e.g. urea), generally such signals are of low fidelity in 1H-NMR analyses unless specific experiments26 or sample preparation procedures31 are employed. This risk can be further mitigated by applying smoothing algorithms such as traditional or advanced approaches for signal denoising from acoustics, radio astronomy etc.15,17,20,21 on the SMolESY data acquired by our toolbox. An example of a denoising filter application is provided in ESI Fig. S10.† However, such approaches are not suitable for automation and must be undertaken with care as they may introduce artifactual NMR signals.
Additionally, the method helps recover signals in crowded regions of the spectrum and areas where macromolecule signals appear. This, combined with the expected enhancement to spectral resolution when calculating signal derivatives, facilitates the chemical assignment of SMs by increasing the analytical specificity. Furthermore, the linear mathematical transformation preserves the quantitative aspects of the data, given the appropriate calibration and reference signal from reference compounds or electronically produced by the PULCON experiments.32 SMolESY can therefore be used directly for absolute quantification without the need for complex and computationally expensive deconvolution algorithms typically applied to 1D experiments and unlike spin-echo pulse sequence experiments altogether.
Importantly, these improvements can also be realized post hoc by retrospective application of SMolESY to existing 1D 1H-NMR raw spectra. This could be of major importance for NMR analysis of sample types with low physiological macromolecular content (e.g. urine) for which spin-echo experiments are not routinely acquired, yet which occasionally or in pathological conditions (e.g. albuminuria) can contain macromolecules. Moreover, SMolESY is also readily applicable to historical datasets increasing its value and making them comparable with new processed datasets. Its application only requires high resolution 1H-NMR data (>65k data points) input which is the established norm within modern high quality metabolomics and analytical studies.22,33
Conclusions
SMolESY is well suited for the enhancement of SMs profiles in NMR spectra derived from complex sample types exhibiting broad and confounding macromolecular signals. In its simplest form as a partial derivative, 1D 1H-NMR spectra are transformed yielding effective suppression of macromolecular signals and enhanced clarity and resolution of small molecule signals. The quantitative capacity of the original data is preserved and, despite variable reductions in the s/n measured across the spectrum, the total chemical information recovered from SMolESY is greater than that from CPMG (demonstrated in human plasma and serum as major biological matrices of interest). Thus, the validation set presented here and applications to various sample types establish SMolESY as a functional in silico replacement for the routine CPMG experiment (or other spin echo variants).The approach may further enable higher throughput sample preparation procedures by precluding the removal of macromolecules from sample types where such preparation is routine practice (e.g. for the NMR study of various food matrices30,34). SMolESY is therefore of major significance in biomedical research, food industry, environmental sciences and indeed any other applications where 1H-NMR is applied to chemically complex samples with abundant macromolecules. The approach is particularly pertinent for large cohort studies where up to 30% acquisition time could be saved compared to the conventional NMR-metabolomics pipeline (ESI Fig. S1†). Since 1H-NMR is emerging as the dominant technique for large scale application to biofluid analysis (e.g. supporting molecular epidemiology and biobanking efforts) and increasingly used for routine quality control assessment of agricultural products, we believe the time and cost savings provided by SMolESY will support the future application of NMR in these contexts.
Experimental
Differentiation of imaginary spectral data – basic theory
Herein, the numerical differentiation (first derivative) of spectral data was calculated by the “gradient” function integrated in MATLAB programming suite (MathWorks, version R2019b). In general, the first derivative of a signal is the rate of change of y (i.e. intensity data) with x (ppm data), dy/dx, which in practice is the slope of the tangent to the signal at each point across the ppm axis.15 It has been shown that the maximum intensity of a signal in the derivative spectrum is inversely proportional to its linewidth to the half-height (Δv1/2), and therefore very broad signals are significantly suppressed while the Δv1/2 of sharp signals are further narrowed35 (see below). Importantly, and in contrast to differentiation of the real data, the 1st derivative of the imaginary data yields positive peak intensities (>0 baseline, see below) owing to the gradient of all signals described by the imaginary data. Derivative spectroscopy could enhance the resolution of a signal, whereas a broad signal could be completely attenuated, which could be easily described in the following equations.Assuming a Fourier transformed Lorentzian signal f(x) across a specific frequency region equals to:35–37
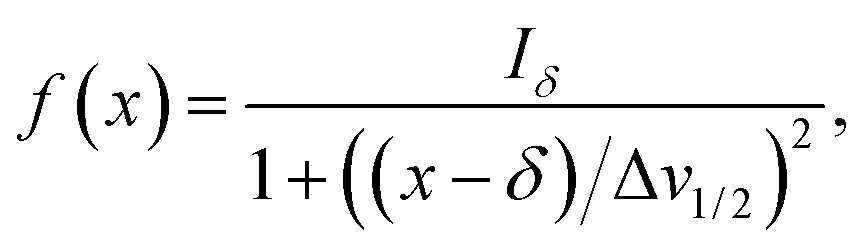 | (1) |
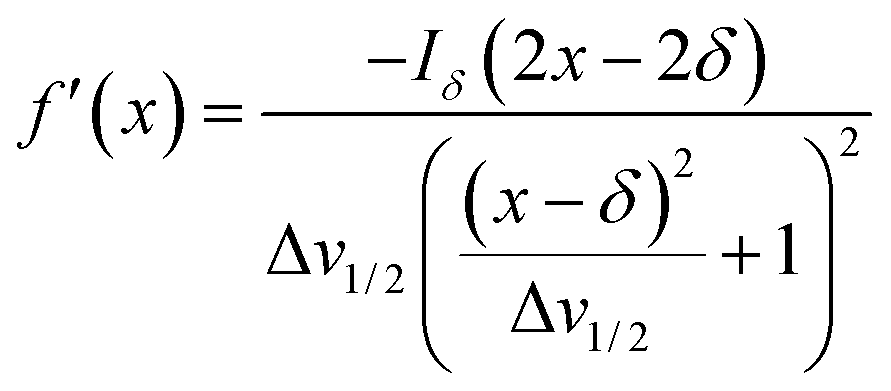 | (2) |
From eqn (2), it can be seen that signals with large Δv1/2 (e.g. broad signals of macromolecules) are highly suppressed to zero (Iδ ∼ 0), whereas sharp signals (i.e. small Δv1/2 values) are sharpened, thus enhancing spectral resolution.
The 1st numerical derivative of the real data from an NMR spectrum (after Fourier transform and phase correction) produces an antisymmetric signal (positive on one side and negative on the other) (ESI Fig. S2A†), whereas the 1st derivative of the imaginary data, due to its gradient (namely positive–negative maxima per signal) (ESI Fig. S2B†), produces a positive transformed signal which exhibits the same δ as the real data without applying any symmetrisation algorithms. The transformed signal from the imaginary spectral data exhibits no chemical shifting compared to the real spectrum (ESI Fig. S2C†) and is immediately employable for any NMR-based metabolomics or analytical study. Furthermore, as differentiation is a linear technique the amplitude of the transformed signal is directly proportional to the original, theoretically retaining its quantitative nature.15 The same signal (i.e. at the positive side of the baseline) could be produced by the 2nd derivative of the real data of the NMR spectrum multiplied by −1 or the 2nd power derivative,35 however, the signal-to-noise ratio is decreased (ESI Fig. S2D†) compared to the 1st derivative.
Reagents
All reagents employed for the artificial mixtures of metabolites, spiking experiments and buffer composition were purchased from Sigma Aldrich.Software
All scripts for the correlation analyses were coded in the MATLAB programming suite (MathWorks, version R2019b). The linear regression analyses, statistical comparisons between slopes and intercepts (i.e. one-way ANOVA tests), as well as their plotting, were performed by Prism 8 (GraphPad Software, Inc, 2019). Multivariate statistics was performed using the MATLAB based PLS_Toolbox (Eigenvector Research, Inc., Manson, WA, USA 98831, version 8.7.1 (2019) software available at http://www.eigenvector.com).Artificial mixtures preparation – spiking experiments
The albumin concentrations in the NMR samples were 0, 7.5, 15.0, 37.5, 75.0, 150.0 and 225.0 mM. The selected metabolites and their concentration for the initial artificial mixture were: cytidine (1 mM), benzoic acid (1 mM), citric acid (0.25 mM), caprylic acid (1 mM), L-isoleucine (0.375 mM), creatinine (0.5 mM), L-glutamic acid (0.5 mM), L-glutamine (0.625 mM), hippuric acid (0.625 mM), L-phenylalanine (0.8 mM), and L-tryptophan (0.375 mM). Artificial mixtures of small MW metabolites contained 50% plasma buffer (see below the plasma buffer composition) and 50% of the aqueous mixture of metabolites in different concentrations. After the initial mixture, eight sequential (and equal) dilutions were performed, resulting in nine different samples. Among these metabolites, we depict (Fig. 1D, S3 and Table S1 (ESI)†) those that exhibit a high variety of signal complexity (i.e. spin systems multiplicity) so as to test the reproducibility of their signals (i.e. integrals) when applying SMolESY.The spiked 17 metabolites in a human plasma sample along with their different concentrations are summarized in the ESI Table S2.† Ten different concentrations of each metabolite were spiked in a new plasma sample, so ∼17 × 10 ≈ 170 plus 17 non-spiked (in total 187) samples were prepared and their corresponding NMR spectra were acquired.
NMR samples preparation and spectra acquisition details
The total number of plasma (>3200) and urine (∼100) NMR samples were prepared following the established standard operating procedures for metabolomics analyses.33 Namely, the plasma NMR samples consisted of 50% plasma buffer [75 mM Na2HPO4; 6.2 mM NaN3; 4.6 mM sodium trimethylsilyl [2,2,3,3-d4]propionate (TMSP) in H2O with 20% (v/v) 2H2O; pH 7.4] and 50% of blood plasma and urine NMR samples consisted of 10% urine buffer [1.5 M KH2PO4 dissolved in 99.9% 2H2O, pH 7.4, 2 mM NaN3 and 5.8 mM 3-(trimethyl-silyl)propionic acid-d4 (TSP)] and 90% of urine. The cow milk sample was prepared following the same protocol used for blood products and additional centrifugation cycle was required in order to remove extra fat content. The olive oil sample was prepared by diluting the sample 10% in deuterated chloroform.Solution 1H NMR spectra of all samples were acquired using a Bruker IVDr 600 MHz spectrometer (Bruker BioSpin) operating at 14.1 T and equipped with a 5 mm PATXI H/C/N with 2H-decoupling probe including a z-axis gradient coil, an automatic tuning-matching (ATM) and an automatic refrigerated sample changer (Sample-Jet). Temperature was regulated to 300 ± 0.1 K and 310 ± 0.1 K for urine and plasma samples, respectively.
For each blood sample, three NMR experiments were acquired in automation: a general profile 1H NMR water presaturation experiment using a one-dimensional pulse sequence where the mixing time of the 1D-NOESY experiment is used to introduce a second presaturation time, a spin echo edited experiment using the Carr–Purcell–Meiboom–Gill (CPMG) pulse sequence which filters out signals from fast T2 relaxing protons from molecules with slow rotational correlation times such as proteins and other macromolecules, and a 2D J-resolved experiment. Each experiment had a total acquisition time of approximately four minutes [32 scans were acquired for the 1D-NOESY (98![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 304 data points, spectral width of 18029 Hz) and the 1D-CPMG (73728 data points, spectral width of 12019 Hz) experiments while two scans and 40 planes were acquired for the 2D J-resolved experiment].
304 data points, spectral width of 18029 Hz) and the 1D-CPMG (73728 data points, spectral width of 12019 Hz) experiments while two scans and 40 planes were acquired for the 2D J-resolved experiment].
For each urine sample two NMR experiments were acquired as previously published in ref. 33. Free induction decays of all 1D-spectra were multiplied by an exponential function equivalent to 0.3 Hz line-broadening before applying Fourier transform. All Fourier transformed spectra were automatically corrected for phase and baseline distortions and referenced to the TSP singlet at 0 ppm.
Plasma – urine spectra employed for the present study
In the present study, we randomly selected ∼3000 plasma 1H-NMR spectra (both 1D-NOESY and CPMG) from the National Phenome Centre repository, previously acquired for various clinical and epidemiology phenotyping studies. Of these, approximately 1000 plasma spectra corresponded to plasma-EDTA samples, and the rest (∼2000) to heparin plasma samples. Both heparin and EDTA collected plasma samples were selected for the SMolESY validation in order to highlight the broad applicability of this approach.Urine 1D-NOESY 1H-NMR spectra were taken from a publicly available study (available at Metabolights, accession number: MTBLS694).
Signal-to-noise ratio (s/n) and peak picking calculations
Signal-to-noise ratios (s/n) of selected 1H NMR signals from the CPMG and SMolESY NMR profiles, namely, from L-alanine, glucose, L-phenylalanine and formic acid were calculated as the ratio of peak intensity at maximum height to the standard deviation of the noise for each of the 3020 CPMG and SMolESY plasma spectra. Noise was calculated as indicated in ref. 19. The selected signals resonate at different spectral areas with variable amount of noise and exhibit different multiplicities. The whole number of “peak-picked” signals was calculated by using the “findpeaks” Matlab function, implementing as threshold the calculated level of noise for the CPMG and SMolESY spectra, respectively.Multivariate analyses (MVA) details
Principal component analysis (PCA) was performed for the 1D-NOESY and the SMolESY 1H-NMR urine datasets, after excluding H2O spectral region (4.7–4.84 ppm) for the spectral width 0.5–10 ppm. Both 1D-NOESY and SMolESY 1H-NMR spectra were calibrated to TSP signal (singlet) at 0 ppm. No binning was applied to the data, all data points for each spectrum were used as variables. For the PCA analysis, NMR data was mean centered and no normalization was applied so as to have intact signals contribution to the PCA analysis.“SMolESY_platform” toolbox details
In order to facilitate the implementation of SMolESY data into NMR metabolomics analyses and NMR-based analytical deconvolution of complex matrices content, we created a graphical user interface toolbox (SMolESY_platform) for the SMolESY data generation, exportation as well as pre-treatment for any metabolomics pipeline (ESI Fig. S9†). Our software enables the loading of the ordinary biofluids 1D-NMR spectra and their transformation to SMolESY along with the visualization of both 1H-NMR and SMolESY spectra for any comparison. Furthermore, it offers the opportunity for the calibration of SMolESY data to a reference peak (for example, to the anomeric doublet resonance of glucose at ∼5.25 ppm for plasma/serum/cerebrospinal fluid/pancreatic juice). In addition, the SMolESY_platform provides: (i) a semi-automated alignment and integration of SMolESY signals for absolute quantification (i.e. targeted approach) and (ii) a variable shaped binning algorithm for untargeted metabolomics studies (i.e. diseases fingerprinting etc.). Both signals and bin-tables (i.e. bucket tables) integration values can be exported for further statistical analyses. Full details of SMolESY_platform functionalities and features can be found at: https://github.com/pantakis/SMolESY_platform.SMolESY signals integration procedure
To calculate the absolute concentration of each metabolite from the SMolESY spectra, we directly integrated the signals, taking the transformed ERETIC signal (i.e. in our case it was the Quantref electronic signal generated by Bruker Biospin) as a reference. In the following figure the example of L-alanine (left panel) and ERETIC (right panel) SMolESY signal integration procedure is illustrated: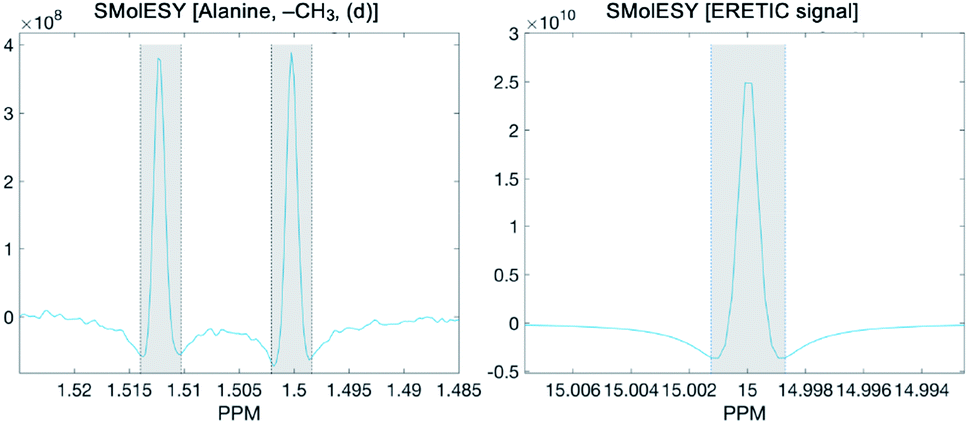
It is noted that an integration function is incorporated in the SMolESY_platform toolbox.
Author contributions
P. G. T. conceived the methodology, wrote the software code and performed statistical analyses of NMR data. B. J. contributed to the methodology development, designed NMR experiments along with P. G. T. and refined the manuscript. C. J. S. communicated ideas for the statistical analyses of spectra comparisons and refined the manuscript. E. C. performed statistical correlation analyses (STOCSY) for the assignment of metabolites in the plasma NMR spectra and refined the manuscript. M. R. L. supervised the study and communicated ideas for the methodology development as well as data handling and composed the final version of the manuscript along with P. G. T.Conflicts of interest
Imperial College London has filed a patent application for an invention disclosed in the paper. The application designates Dr Panteleimon G. Takis and Dr Matthew R. Lewis as inventors.Note added after first publication
This article replaces the version published on 1st June 2020, which contained errors in a duplication of eqn (1) instead of correctly showing eqn (2).Acknowledgements
We would like to acknowledge Nikita Harvey and Dr Samantha L. Lodge for their significant contribution to the preparation of the metabolite mixtures, the NMR samples as well as several NMR spectra acquisition. Infrastructure support was provided by the National Institute for Health Research (NIHR) Imperial Biomedical Research Centre (BRC). This work was supported by the Medical Research Council and National Institute for Health Research [grant number MC_PC_12025] and infrastructure support was provided by the National Institute for Health Research (NIHR) Imperial Biomedical Research Centre (BRC).Notes and references
- P. G. Takis, V. Ghini, L. Tenori, P. Turano and C. Luchinat, TrAC, Trends Anal. Chem., 2019, 120, 115300 CrossRef.
- A. Vignoli, V. Ghini, G. Meoni, C. Licari, P. G. Takis, L. Tenori, P. Turano and C. Luchinat, Angew. Chem., Int. Ed., 2019, 58, 968–994 CrossRef CAS PubMed.
- A.-H. Emwas, R. Roy, T. R. McKay, L. Tenori, E. Saccenti, A. N. G. Gowda, D. Raftery, F. Alahmari, L. Jaremko, M. Jaremko and S. D. Wishart, Metabolites, 2019, 9, 123 CrossRef CAS PubMed.
- B. Jiménez, E. Holmes, C. Heude, R. F. Tolson, N. Harvey, S. L. Lodge, A. J. Chetwynd, C. Cannet, F. Fang, J. T. M. Pearce, M. R. Lewis, M. R. Viant, J. C. Lindon, M. Spraul, H. Schäfer and J. K. Nicholson, Anal. Chem., 2018, 90, 11962–11971 CrossRef PubMed.
- P. G. Takis, H. Schäfer, M. Spraul and C. Luchinat, Nat. Commun., 2017, 8, 1–11 CrossRef CAS PubMed.
- R. T. Mckay, Concepts Magn. Reson., Part A, 2011, 38, 197–220 CrossRef.
- M. S. Klein, N. Buttchereit, S. P. Miemczyk, A.-K. Immervoll, C. Louis, S. Wiedemann, W. Junge, G. Thaller, P. J. Oefner and W. Gronwald, J. Proteome Res., 2012, 11, 1373–1381 CrossRef CAS PubMed.
- L. Mannina, A. P. Sobolev and A. Segre, Spectrosc. Eur., 2003, 15, 6–14 CAS.
- C. Ingallina, A. Cerreto, L. Mannina, S. Circi, S. Vista, D. Capitani, M. Spano, A. P. Sobolev and F. Marini, Metabolites, 2019, 9, 65 CrossRef CAS PubMed.
- N. Cortese, G. Capretti, M. Barbagallo, A. Rigamonti, P. G. Takis, G. Castino, D. Vignali, G. Maggi, F. Gavazzi, C. Ridolfi, G. Nappo, G. Donisi, M. Erreni, R. Avigni, D. Rahal, P. Spaggiari, M. Roncalli, P. Cappello, F. Novelli, P. Monti, A. Zerbi, P. Allavena, A. Mantovani and F. Marchesi, Cancer Immunol. Res., 2020, 8, 493–505 CrossRef PubMed.
- G. A. Nagana Gowda, Y. N. Gowda and D. Raftery, Anal. Chem., 2015, 87, 706–715 CrossRef CAS PubMed.
- S. Ravanbakhsh, P. Liu, T. C. Bjordahl, R. Mandal, J. R. Grant, M. Wilson, R. Eisner, I. Sinelnikov, X. Hu, C. Luchinat, R. Greiner and D. S. Wishart, PLoS One, 2015, 10, e0124219 CrossRef PubMed.
- H. Y. Carr and E. M. Purcell, Phys. Rev., 1954, 94, 630–638 CrossRef CAS.
- R. Barrilero, N. Ramírez, J. C. Vallvé, D. Taverner, R. Fuertes, N. Amigó and X. Correig, J. Proteome Res., 2017, 16, 1847–1856 CrossRef CAS PubMed.
- H. Mark and J. Workman, in Chemometrics in Spectroscopy, ed. H. Mark and J. Workman, Academic Press, Amsterdam, 2007, pp. 339–378 Search PubMed.
- B. J. Harden, S. R. Nichols and D. P. Frueh, J. Am. Chem. Soc., 2014, 136, 13106–13109 CrossRef CAS PubMed.
- C. Cobas, Magn. Reson. Chem., 2018, 56, 1140–1148 CrossRef CAS PubMed.
- C. Cobas, Magn. Reson. Chem., 2020, 58, 512–519 CrossRef CAS PubMed.
- A. Rodriguez-Martinez, J. M. Posma, R. Ayala, N. Harvey, B. Jimenez, A. L. Neves, J. C. Lindon, K. Sonomura, T. A. Sato, F. Matsuda, P. Zalloua, D. Gauguier, J. K. Nicholson and M. E. Dumas, Anal. Chem., 2017, 89, 11405–11412 CrossRef CAS PubMed.
- T. O'Haver, An Introduction to Signal Processing in Chemical Analysis, https://terpconnect.umd.edu/%7Etoh/spectrum/TOC.html Search PubMed.
- Recent Trends in Denoising Tutorial: Publications, https://web.archive.org/web/20090807095156/http://www.stanford.edu/%7Eslansel/tutorial/publications.htm.
- C. J. Sands, A. M. Wolfer, G. D. S. Correia, N. Sadawi, A. Ahmed, B. Jiménez, M. R. Lewis, R. C. Glen, J. K. Nicholson and J. T. M. Pearce, Bioinformatics, 2019, 35, 5359–5360 CrossRef PubMed.
- M. T. James, B. R. Hemmelgarn, N. Wiebe, N. Pannu, B. J. Manns, S. W. Klarenbach and M. Tonelli, Lancet, 2010, 376, 2096–2103 CrossRef.
- S. R. Khan, P. A. Glenton, R. Backov and D. R. Talham, Kidney Int., 2002, 62, 2062–2072 CrossRef CAS PubMed.
- A. H. Emwas, E. Saccenti, X. Gao, R. T. McKay, V. A. P. M. dos Santos, R. Roy and D. S. Wishart, Metabolomics, 2018, 14, 31 CrossRef PubMed.
- L. Liu, H. Mo, S. Wei and D. Raftery, Analyst, 2012, 137, 595–600 RSC.
- O. Cloarec, M.-E. Dumas, A. Craig, R. H. Barton, J. Trygg, J. Hudson, C. Blancher, D. Gauguier, J. C. Lindon, E. Holmes and J. Nicholson, Anal. Chem., 2005, 77, 1282–1289 CrossRef CAS PubMed.
- F. A. A. Mulder, L. Tenori and C. Luchinat, Angew. Chem., Int. Ed., 2019, 58, 15283–15286 CrossRef CAS PubMed.
- N. Aranibar and M. D. Reily, Methods Mol. Biol., 2014, 1104, 223–236 CrossRef CAS PubMed.
- E. Ralli, M. Amargianitaki, E. Manolopoulou, M. Misiak, G. Markakis, S. Tachtalidou, A. Kolesnikova, P. Dais and A. Spyros, Methods Mol. Biol., 2018, 1738, 203–211 CrossRef CAS PubMed.
- P. Charisiadis, V. Exarchou, A. N. Troganis and I. P. Gerothanassis, Chem. Commun., 2010, 46, 3589–3591 RSC.
- G. Wider and L. Dreier, J. Am. Chem. Soc., 2006, 128, 2571–2576 CrossRef CAS PubMed.
- A. C. Dona, B. Jiménez, H. Schäfer, E. Humpfer, M. Spraul, M. R. Lewis, J. T. M. Pearce, E. Holmes, J. C. Lindon and J. K. Nicholson, Anal. Chem., 2014, 86, 9887–9894 CrossRef CAS PubMed.
- A. Spyros, Nucl. Magn. Reson., 2016, 45, 269–307 CAS.
- J. T. M. Pearce, T. J. Athersuch, T. M. D. Ebbels, J. C. Lindon, J. K. Nicholson and H. C. Keun, Anal. Chem., 2008, 80, 7158–7162 CrossRef CAS PubMed.
- E. W. Weisstein, Lorentzian Function, http://mathworld.wolfram.com/LorentzianFunction.html, accessed 1 December 2019 Search PubMed.
- J. Keeler, Understanding NMR Spectroscopy, John Wiley & Sons, Ltd, 2nd edn, 2011 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: The experimental scheme of NMR based metabolomics pipeline for biofluids with macromolecular content (e.g. proteins, lipoproteins, lipids etc.) – SMolESY contribution; examples of enhanced spectral resolution by the imaginary NMR spectral part differentiation; validation of SMolESY intra-metabolites signals reproducibility; extra PCA analysis; SMolESY performance in 994 plasma-EDTA samples; mean spectrum of 994 plasma-EDTA samples spectra focusing on the 3.5–4.0 1H-NMR ppm region; SMolESY application and reproducibility validation to spectra binning; SMolESY errors evaluation for absolute quantification; an overview of the SMolESY_platform graphical user interface (GUI) toolbox; statistical analyses results for SMolESY intra-metabolites signals reproducibility tests; computer code for the calculation of the Pearson correlation values; Example of SMolESY signals denoising. See DOI: 10.1039/d0sc01421d |
| ‡ NMR spectra (raw data) of several spiked metabolites in real plasma matrices could be freely downloaded from the Metabolights database, under the study identifier: MTBLS715 (after the data curation period). |
| § Source code and compiled versions for Windows and MacOS of “SMolESY_platform” are freely available at: https://github.com/pantakis/SMolESY_platform. |
| This journal is © The Royal Society of Chemistry 2020 |