
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Desymmetrization of unactivated bis-alkenes via chiral Brønsted acid-catalysed hydroamination†
Zhang-Long
Yu‡
a,
Yong-Feng
Cheng‡
 a,
Na-Chuan
Jiang
a,
Jian
Wang
a,
Li-Wen
Fan
a,
Yue
Yuan
a,
Zhong-Liang
Li
b,
Qiang-Shuai
Gu
*bc and
Xin-Yuan
Liu
*a
a,
Na-Chuan
Jiang
a,
Jian
Wang
a,
Li-Wen
Fan
a,
Yue
Yuan
a,
Zhong-Liang
Li
b,
Qiang-Shuai
Gu
*bc and
Xin-Yuan
Liu
*a
aShenzhen Grubbs Institute, Department of Chemistry, Guangdong Provincial Key Laboratory of Catalysis, Southern University of Science and Technology, Shenzhen 518055, China. E-mail: liuxy3@sustech.edu.cn
bAcademy for Advanced Interdisciplinary Studies, Southern University of Science and Technology, Shenzhen 518055, China. E-mail: guqs@sustech.edu.cn
cShenzhen Key Laboratory of Small Molecule Drug Discovery and Synthesis, Department of Chemistry, Southern University of Science and Technology, Shenzhen 518055, China
First published on 20th May 2020
Abstract
Although great success has been achieved in catalytic asymmetric hydroamination of unactivated alkenes using transition metal catalysis and organocatalysis, the development of catalytic desymmetrising hydroamination of such alkenes remains a tough challenge in terms of attaining a high level of stereocontrol over both remote sites and reaction centers at the same time. To address this problem, here we report a highly efficient and practical desymmetrising hydroamination of unactivated alkenes catalysed by chiral Brønsted acids with both high diastereoselectivity and enantioselectivity. This method features a remarkably broad alkene scope, ranging from mono-substituted and gem-/1,2-disubstituted to the challenging tri- and tetra-substituted alkenes, to provide access to a variety of diversely functionalized chiral pyrrolidines bearing two congested tertiary or quaternary stereocenters with excellent efficiency under mild and user-friendly synthetic conditions. The key to success is indirect activation of unactivated alkenes by chiral Brønsted acids via a concerted hydroamination mechanism.
Introduction
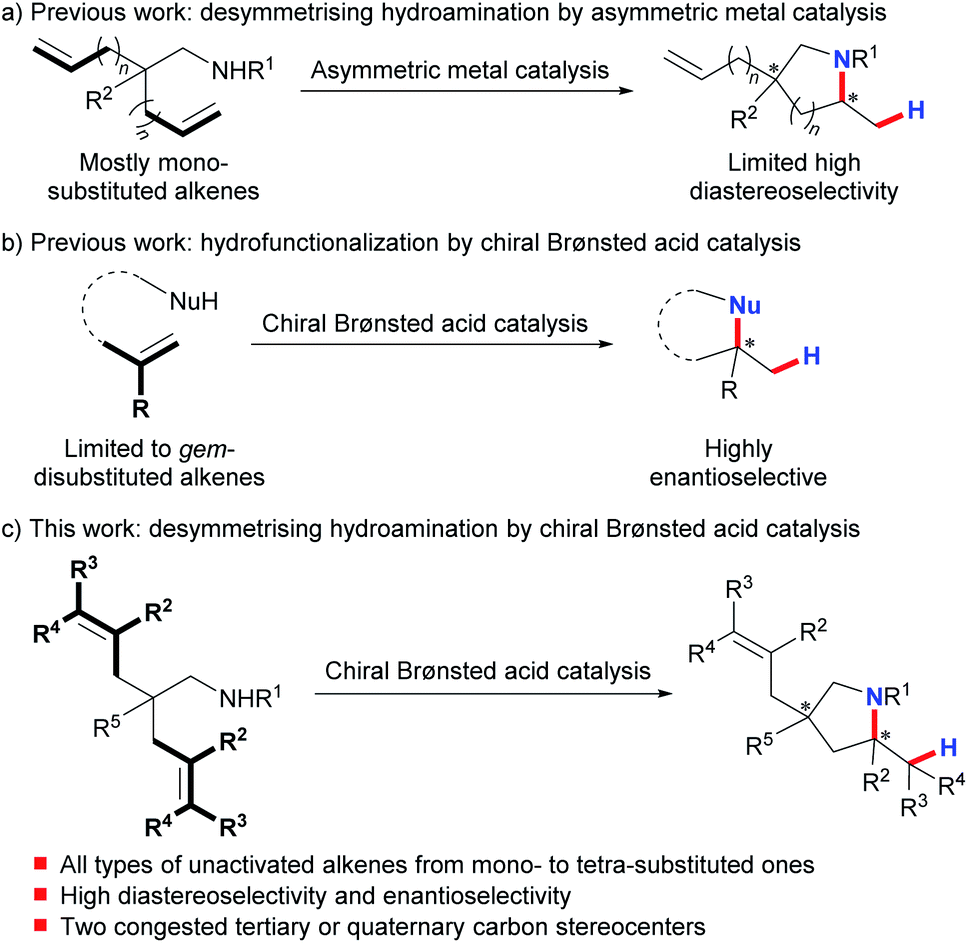
The desymmetrization of prochiral or meso compounds represents a powerful tool for the preparation of complex chiral organic molecules with congested tertiary and quaternary stereocenters, which are ubiquitous in many natural products and biologically relevant molecules.1 This field has been extensively investigated using transition metal catalysis2 and organocatalysis.3 On the other hand, much effort has been devoted over the past decades to the development of asymmetric hydroamination of alkenes as an efficient way to prepare enantioenriched amines.4 In sharp contrast, desymmetrising hydroamination has been reported under metal catalysis with poorly controlled diastereoselectivity.5–7 And only recently have Sadow et al. successfully achieved highly stereoselective desymmetrising hydroamination of two mono-substituted alkene substrates using their developed chiral zirconium complex (Scheme 1a).8 Although of limited functional group compatibility due to the presence of the highly air- and moisture-sensitive early-transition-metal catalyst, this work represents the state-of-the-art in this field. Nonetheless, the limited number of highly stereoselective examples (≥10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr, ≥90% ee) as well as the confined alkene scope highlights the challenge in attaining a high level of stereocontrol over both remote sites and reaction centers at the same time. Clearly, a conceptually different approach is very desirable to achieve a highly efficient and practical desymmetrising hydroamination with a broad alkene scope under mild synthetic conditions.
:1 dr, ≥90% ee) as well as the confined alkene scope highlights the challenge in attaining a high level of stereocontrol over both remote sites and reaction centers at the same time. Clearly, a conceptually different approach is very desirable to achieve a highly efficient and practical desymmetrising hydroamination with a broad alkene scope under mild synthetic conditions.
 | ||
| Scheme 1 Desymmetrising hydroamination by asymmetric metal catalysis and chiral Brønsted acid catalysis. | ||
Chiral Brønsted acids have recently evolved into fundamentally significant tools for asymmetric catalysis.9 Nevertheless, activation of unactivated alkenes by chiral Brønsted acids for nucleophilic attack10 has been impeded by their inherently low basicity. And until recently, List's,10d Terada's,10e and our groups10c have independently disclosed highly enantioselective hydrofunctionalization of minimally functionalized geminally disubstituted alkenes (Scheme 1b). This limited alkene scope emphasizes the remaining difficulty and also untapped space in applying chiral Brønsted acids for asymmetric hydrofunctionalization of unactivated alkenes. To this end, we herein describe the successful realization of desymmetrising hydroamination of unactivated alkenes via indirect activation of alkenes by chiral Brønsted acids through a concerted pathway under mild conditions (Scheme 1c).11 Advantageously, this process tolerates not only common mono-substituted and gem-/1,2-disubstituted unactivated alkenes but also the challenging tri- and tetra-substituted ones. Thus, it provides a straightforward access to diversely functionalized chiral pyrrolidines bearing two congested tertiary or quaternary stereocenters with both high diastereoselectivity and enantioselectivity. These enantioenriched pyrrolidines constitute the core structures of many biologically active natural products and pharmaceutical chemicals.12
Results and discussion
Desymmetrising hydroamination of unactivated alkenes
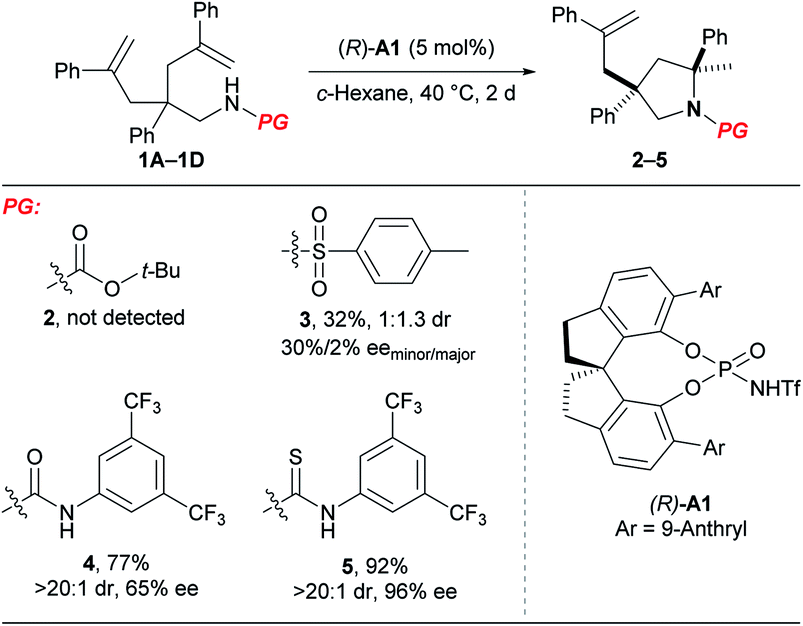
At the outset, we examined different N-protecting groups for their potential to efficiently participate in the desymmetrising hydroamination of geminally disubstituted alkenes in the presence of a chiral N-triflyl phosphoramide (R)-A1 (Table 1).13 The tert-butoxycarbonyl-protected amine 1A failed to undergo the desired hydroamination reaction, while the tosyl-protected one 1B afforded the corresponding hydroamination product 3 with promising enantioselectivity albeit of low yield and diastereoselectivity. Fortunately, the use of an N-arylaminocarbonyl protecting group in 1C resulted in remarkable improvements in both the yield and the diastereoselectivity, although the enantioselectivity was only moderate (65% ee). These encouraging results have prompted us to further investigate the thiocarbonyl analog 1D, which delivered superior stereoselectivity as well as reactivity. The higher acidity of a thiourea compared with a urea may account for the improvements over reactivity and stereoselectivity by invoking stronger hydrogen bonding interactions with the chiral Brønsted acid.| a Reactions were run on a 0.1 mmol scale at 40 °C; isolated yields were shown; dr and ee values were determined by 1H NMR and HPLC analysis, respectively. |
|---|
|
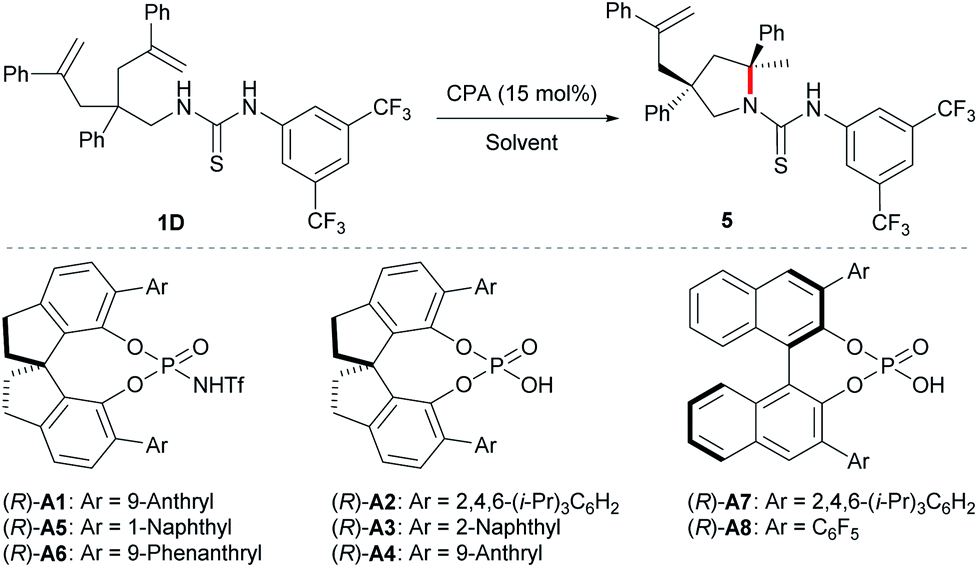
With the optimal protecting group for amine in hand, we then carried out further condition optimization by screening other Brønsted acids, solvents, catalyst loadings, and reaction temperatures (Table 2). The results indicated that: (i) the reactivity and enantioselectivity were greatly affected primarily by chiral Brønsted acid catalysts while the diastereoselectivity remained excellent all the time (entries 1–8); (ii) more acidic chiral N-triflyl phosphoramides13 generally provided better enantiocontrol as well as reactivity compared with chiral phosphoric acids (entries 1, 5, and 6 vs. entries 2–4, 7, and 8); (iii) solvent significantly affected the reaction rate (entries 1 and 9–13) and c-hexane provided the highest yield within the shortest reaction time (entry 13). Although the reaction worked at room temperature, slight heating guaranteed high reactivity with a catalyst loading as low as 5 mol% (entries 13–16). Therefore, the optimal reaction conditions are as follows: 5 mol% (R)-A1 in cyclohexane at 40 °C.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | Time (h) | Yield (%) | Dr | Ee (%) |
| a Reactions were run on a 0.025 mmol scale at room temperature; yield was determined by 1H NMR of the crude reaction residue; ee was determined by HPLC analysis on a chiral stationary phase. b 10 mol% (R)-A1. c 5 mol% (R)-A1. d The reaction was run at 40 °C. | ||||||
| 1 | (R)-A1 | CCl4 | 24 | 91 | >20:1 |
97 |
| 2 | (R)-A2 | CCl4 | 72 | 12 | >20:1 |
0 |
| 3 | (R)-A3 | CCl4 | 48 | 64 | >20:1 |
43 |
| 4 | (R)-A4 | CCl4 | 72 | 68 | >20:1 |
27 |
| 5 | (R)-A5 | CCl4 | 72 | 70 | >20:1 |
90 |
| 6 | (R)-A6 | CCl4 | 72 | 78 | >20:1 |
93 |
| 7 | (R)-A7 | CCl4 | 72 | 75 | >20:1 |
70 |
| 8 | (R)-A8 | CCl4 | 48 | 82 | >20:1 |
86 |
| 9 | (R)-A1 | CH2Cl2 | 72 | 85 | >20:1 |
99 |
| 10 | (R)-A1 | CH3CN | 72 | 88 | >20:1 |
99 |
| 11 | (R)-A1 | EtOAc | 17 | 88 | >20:1 |
99 |
| 12 | (R)-A1 | THF | 72 | 75 | >20:1 |
99 |
| 13 | (R)-A1 | c-Hexane | 4 | 92 | >20:1 |
99 |
| 14 | (R)-A1b | c-Hexane | 24 | 92 | >20:1 |
97 |
| 15 | (R)-A1c | c-Hexane | 96 | 82 | >20:1 |
96 |
| 16d | (R)-A1c | c-Hexane | 24 | 92 | >20:1 |
96 |
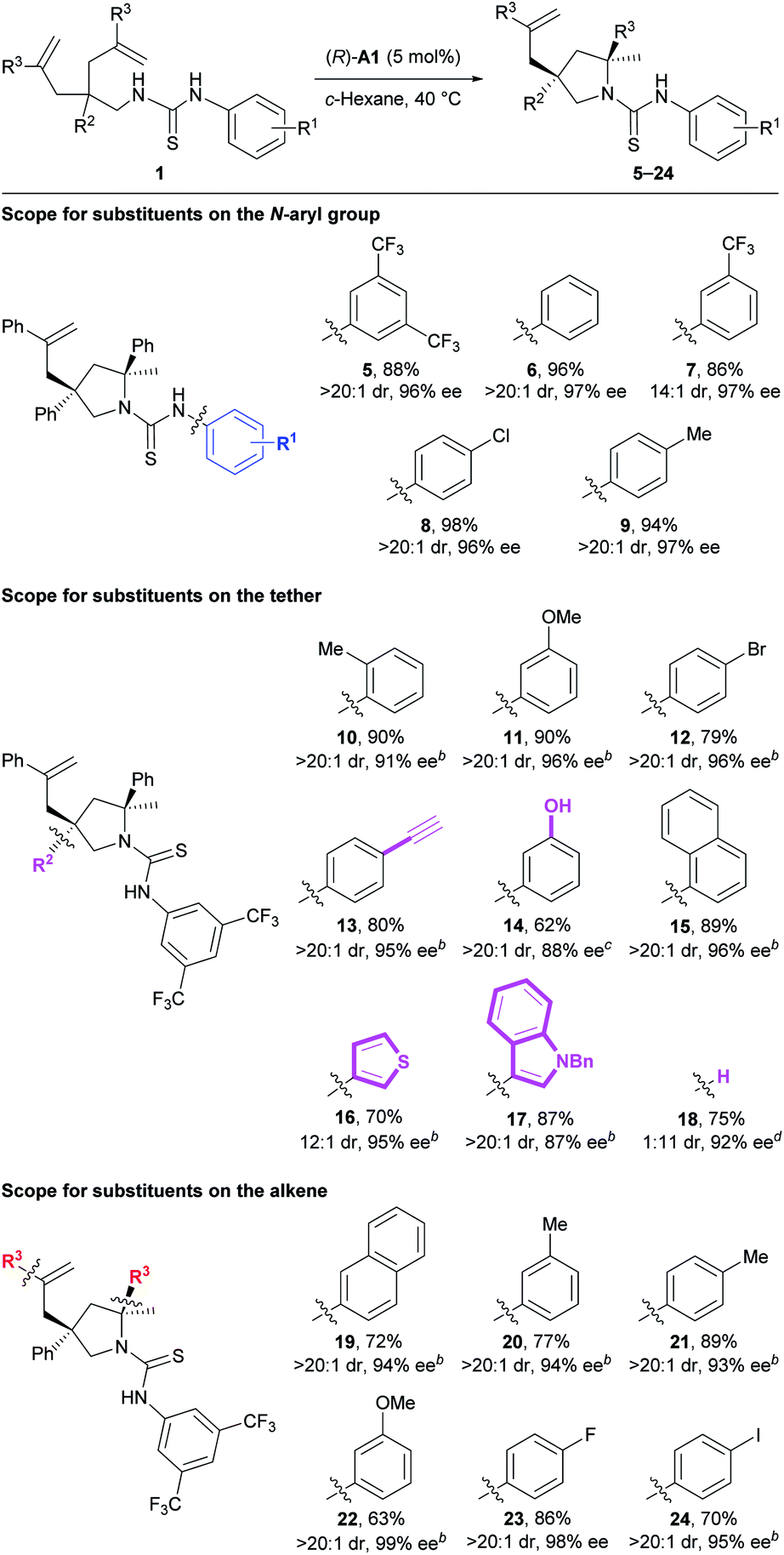
Next, we investigated the substrate scope of the current method (Table 3). Firstly, the N-aryl groups possessing electron-withdrawing (CF3 and Cl) substituents, a slightly electron-donating (Me) substituent, or no substituents on either the para- or meta-positions all provided high reaction efficiency and stereoselectivity (5–9). Secondly, for the aryl ring on the tether, various substituents of different electronic properties at the ortho-, meta-, or para-positions were all well tolerated, giving the expected products 10–12 as apparently single diastereoisomers in 79–90% yields with 91–96% ee. In addition, a reactive terminal triple bond appended on this aryl ring readily survived our reaction conditions to afford the corresponding product 13. Further, a polar unprotected phenol group on the tether was compatible with the reaction conditions to deliver product 14. Besides these monocyclic aryl rings, a bicyclic naphthalene ring was also suitable for this reaction to provide product 15. Of particular note is that two substrates bearing relatively labile electron-rich heteroaryl rings on the tether both underwent the current reaction smoothly to deliver the desired products 16 and 17 with excellent results. Nonetheless, no reaction occurred for substrates bearing polar 3-pyridinyl or unprotected 3-indolyl groups on the tether (for structures, see Fig. S1†), possibly due to the disruption of necessary substrate–catalyst hydrogen bonding interactions. Interestingly, the deleterious effect caused by removing the aryl group (R2) on the tether (33% yield, 1.2:1 dr, 42% and 13% ee) was readily overcome mainly by facile switching the chiral Brønsted acid to (R)-A2. Accordingly, product 18 bearing one tertiary stereocenter and one quaternary stereocenter was forged in 75% yield with excellent stereoselectivity (11:1 dr, 92% ee) under modified conditions. Thirdly, for the aryl ring on the alkene moiety, both electron-rich and -deficient phenyl rings as well as a naphthalene ring all were well accommodated in this transformation, smoothly delivering the corresponding products 19–24. Overall, the good compatibility with aryl halides, terminal alkynes, and electron-rich heterocycles, as mentioned above, as well as the remaining unreacted alkenes leaves a huge synthetic space for further derivatization.
|
a Reactions were run on a 0.10 mmol scale at 40 °C; isolated yields were shown; dr and ee values were determined by 1H NMR and HPLC analysis, respectively.
b The reaction was run at 60 °C.
c The reaction was run using (R)-A1 (15 mol%) in CCl4 at 60 °C on a 0.050 mmol scale.
d The reaction was run using (R)-A2 (Table 1, 15 mol%) in 1,2-dichlorobenzene at 60 °C for 24 h; In the major diastereomer, the unreacted alkene side chain is trans toward the phenyl ring on the pyrrolidine ring of 18; results under standard conditions shown above: 33%, 1:1.2 dr, 13% and 42% ee.
|
|---|
|
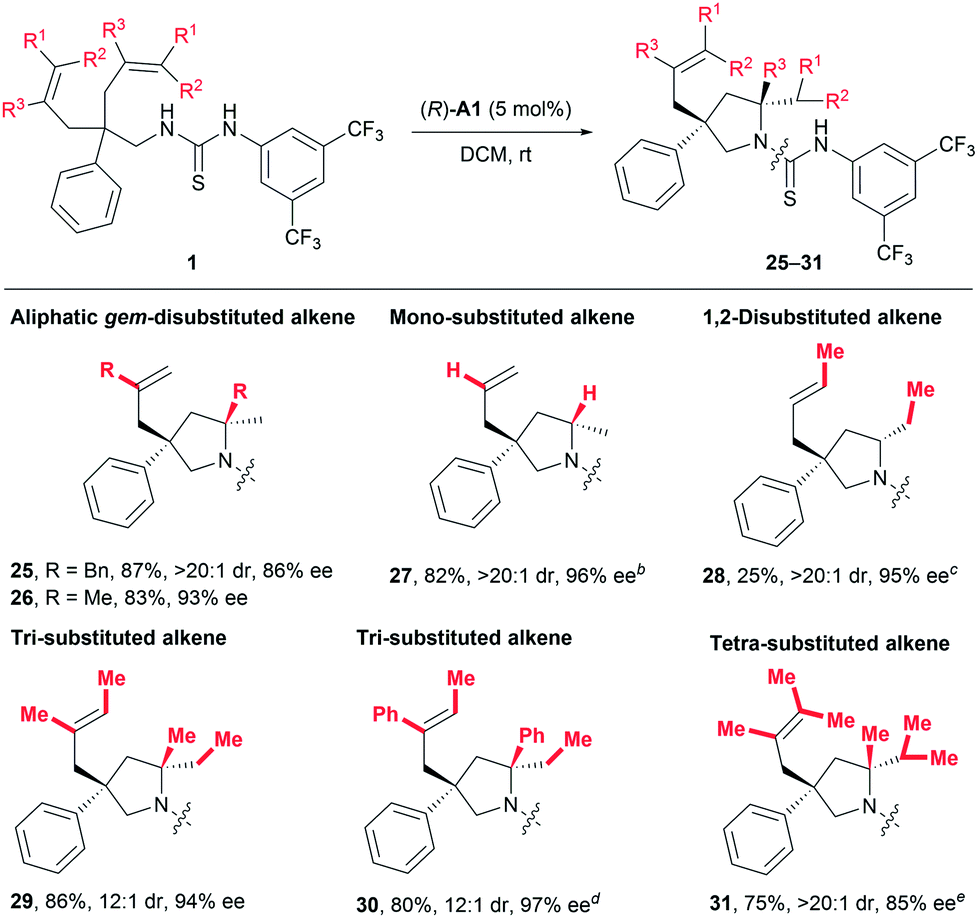
As discussed in the introduction, the application of common chiral Brønsted acids for activating unactivated alkenes towards nucleophilic attack via direct protonation has been prohibited by the fact that such alkenes are not basic enough. Given the above-mentioned robust catalytic activity for styrene-type alkenes, we were naturally eager to challenge our catalytic system by exploring other types of alkenes (Table 4). To our delight, substrates featuring aliphatic geminally disubstituted alkenes were also applicable for this reaction, and the desired products 25 and 26 were obtained with excellent yields and stereoselectivity. More encouragingly, a substrate bearing even-challenging monosubstituted alkenyl groups also underwent the current reaction smoothly, delivering the desired product 27 in 82% yield with 96% ee and larger than 20:1 dr. Encouraged by these results, we continued to examine less activated 1,2-disubstituted alkenes and obtained the desired product with excellent stereoselectivity (28, >20:1 dr, 95% ee), albeit in 25% yield, using 30 mol% catalyst (R)-A1. Most importantly, one major advantage of the current process over metal-catalysed ones would lie on efficient desymmetrising hydroamination of sterically demanding multisubstituted alkenes. In this regard, both tri- and tetra-substituted alkenes bearing electronically distinct alkyl and phenyl substituents all readily gave rise to products 29–31 in 75–86% yields with excellent enantio- and diastereoselectivity (Table 4). Of particular note is that such sterically demanding multisubstituted alkenes are generally problematic for stereoselective hydroamination, which has only been rarely investigated with poor enantiocontrol.14 On the whole, all these results clearly indicate a broad substrate scope and good functional group tolerance, highlighting the generality of this transformation.
| a Reactions were run on a 0.10 mmol scale at room temperature; isolated yields are shown; dr and ee values were determined by 1H NMR and HPLC analysis, respectively. b The reaction was run in c-hexane and (R)-A1 (15 mol%) was used. c The reaction was run in c-hexane at 60 °C and (R)-A1 (30 mol%) was used. d (R)-A1 (15 mol%) was used. e (R)-A1 (20 mol%) was used. |
|---|
|
Transformation and application
To illustrate the synthetic applicability of this transformation, we performed a gram-scale preparation of 5 (Scheme 2), which showed no changes in both reaction efficiency and stereoselectivity. In addition, we have readily removed the N-arylaminothiocarbonyl group in product 5 by straightforward treatment with tert-butylamine at elevated temperature, furnishing free secondary amine 32 in 85% yield (Scheme 2). More importantly, this group also provided additional synthetic potential for further derivatization. For example, 5 underwent a hydroarylative cyclization under the catalysis of InBr3 to yield spirocyclic pyrrolidines 33 as a 1.1:1 mixture of diastereomers in 81% overall yield. Next, we converted the remaining thiocarbonyl in one diastereomer of 33 into carbonyl using Bi(NO3)3·5H2O, and further forged a new fused ring by subsequent smooth oxidation with [bis(trifluoroacetoxy)iodo]benzene (PIFA) (Scheme 2). Remarkably, the resulting polycyclic compound 34 and its precursor 33 possess the core structural elements of many biologically active natural alkaloids like gracilamine15 and hinckdentine A16 (Scheme 2). An important aspect of the transformations discussed above is that no significant enantiopurity erosion has ever occurred, setting a solid foundation for future applications in asymmetric synthesis of enantioenriched azaheterocycles.
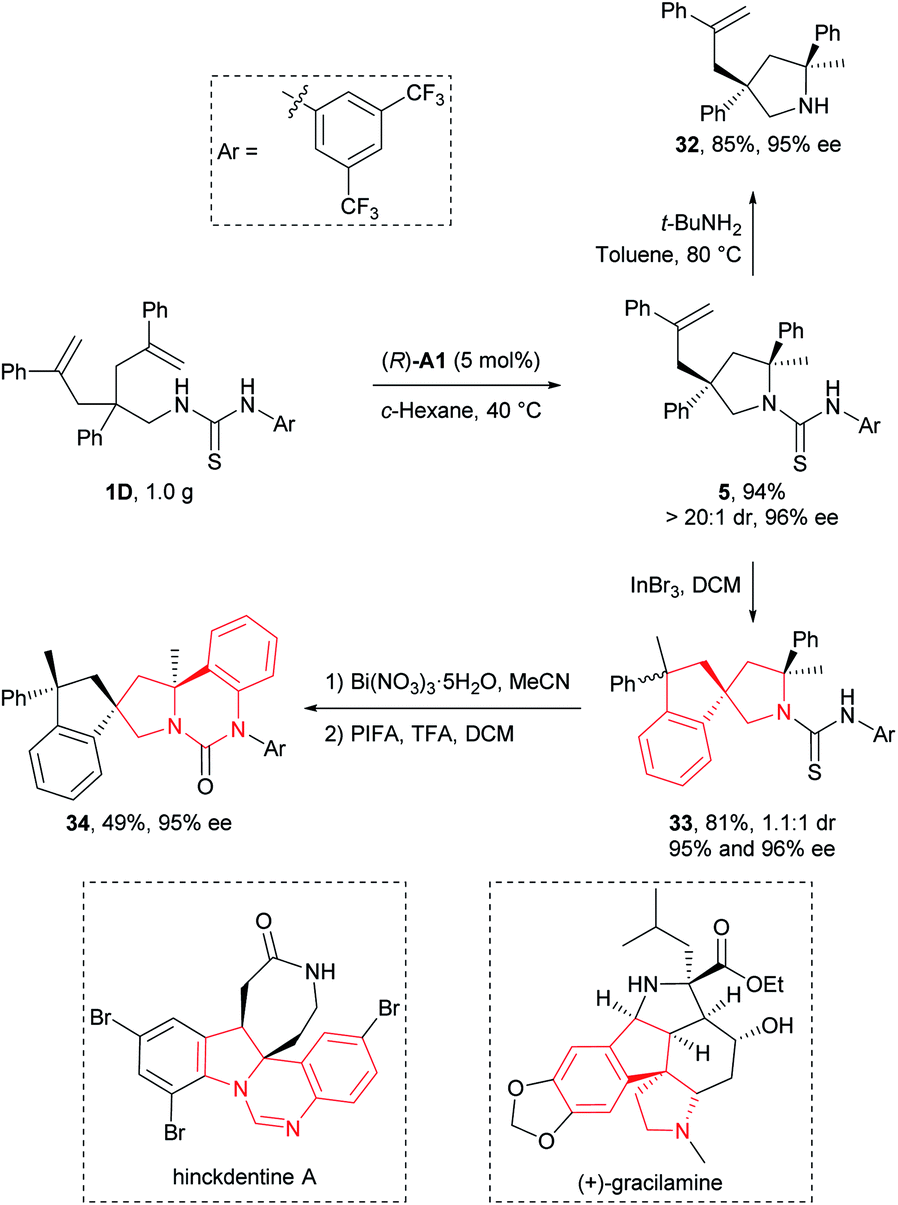 | ||
| Scheme 2 Representative product transformation and application. | ||
Mechanistic investigations
To get insights into the reaction mechanism, we have conducted some control experiments (Scheme 3). When the N–H bond in the N-arylaminothiocarbonyl group was masked with a methyl group, the reaction failed to occur under the standard reaction conditions (Scheme 3a). This is consistent with a speculative multiple hydrogen bonding formation between phosphoric acid and the thiourea moiety, which may have prepared the substrate in an organized conformation for subsequent hydroamination. Furthermore, deuterated 1F (Scheme 3b), prepared by overnight stirring with D2O, afforded d-30 as an almost single diastereomer in regard to C6 under the otherwise standard conditions. This result strongly supports a concerted hydroamination pathway. On the basis of the above mechanistic investigations and previous studies,5–7 we propose that the desymmetrising hydroamination reaction proceeds through a concerted mechanism. Thus, the chiral phosphoramide indirectly activates alkenes through a hydrogen bonding relay with the N–H in the N-arylaminothiocarbonyl protecting group while its relatively basic oxygen deprotonates the protected amine by forming hydrogen bonding with the remaining N–H (Scheme 3c). This extensive hydrogen-bonding formation may have significantly lowered the original high barrier for direct protonation of unactivated alkenes other than the geminally disubstituted ones by a common chiral Brønsted acid. On the basis of the proposed mechanism and given the absolute configuration of 16 determined by X-ray crystallographic analysis (Scheme 3d and Fig. S2;† CCDC 1853110), we have accordingly set up a possible optimal transition state (Scheme 3c) that has led to the formation of most of our hydroamination products according to the Simón and Goodman's model.17 In this transition state, the si-face attack of the geminally disubstituted alkenes by protected amines was favored due to the steric hindrance between the Ar1 groups on the alkenes and the bulky 3,3′-substituents on the chiral phosphoramide catalyst during the re-face approaching. The trans configuration of the sterically demanding Ar1 and Ar2 groups alleviated their otherwise severe pseudo 1,3-diaxial interaction. As a proof of this model, the unreacted alkene side chain is trans to the Ar1 group in 18, as determined by 2D NMR studies, where the steric interaction between the relative bulky side chain and the Ar1 group became predominant.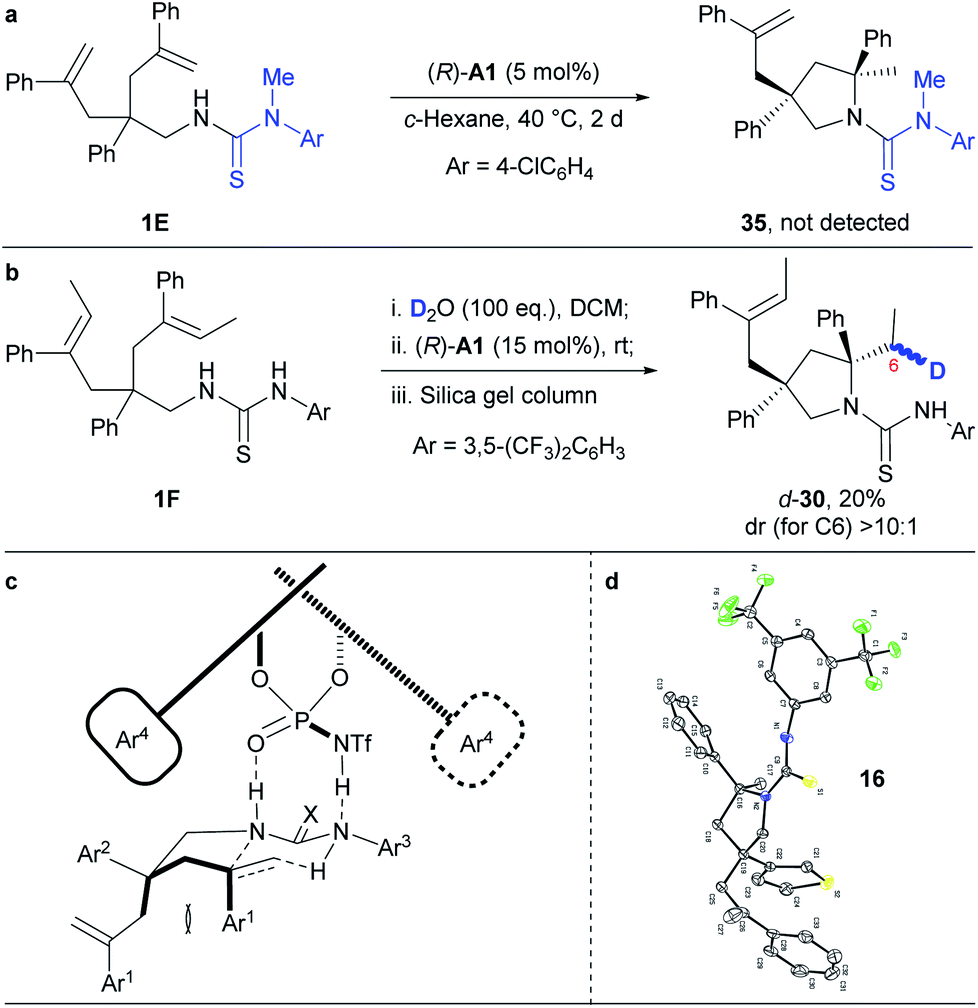 | ||
| Scheme 3 Mechanistic study. | ||
Conclusion
In summary, chiral Brønsted acids have been successfully employed to realize highly diastereoselective and enantioselective desymmetrising hydroamination of unactivated alkenes. In contrast to traditional metal-catalysed desymmetrising hydroamination of unactivated alkenes, this protocol readily accommodates mono-substituted, gem-disubstituted, 1,2-disubstituted, tri-substituted, and tetra-substituted unactivated alkenes with good functional group tolerance. Thus, this mild reaction provides rapid access to a diverse range of enantioenriched pyrrolidines with multiple congested carbon stereocenters. Further studies including detailed investigation of the origin of stereocontrol and the development of desymmetrization of other types of alkenes with chiral Brønsted acids are currently underway.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the National Natural Science Foundation of China (No. 21722203, 21831002, and 21801116), Guangdong Basic and Applied Basic Research Foundation (No. 2019A1515110822), Guangdong Provincial Key Laboratory of Catalysis (No. 2020B121201002), Guangdong Innovative Program (No. 2019BT02Y335), Shenzhen Special Funds (No. JCYJ20170412152435366), Shenzhen Nobel Prize Scientists Laboratory Project (No. C17783101), Shenzhen Key Laboratory of Small Molecule Drug Discovery and Synthesis (No. ZDSYS20190902093215877), and SUSTech Special Fund for the Construction of High-Level Universities (No. G02216303) is greatly appreciated.Notes and references
- For selected recent reviews, see: (a) K. S. Petersen, Tetrahedron Lett., 2015, 56, 6523–6535 CrossRef CAS PubMed; (b) A. Borissov, T. Q. Davies, S. R. Ellis, T. A. Fleming, M. S. W. Richardson and D. J. Dixon, Chem. Soc. Rev., 2016, 45, 5474–5540 RSC; (c) Z. Wang, W.-X. Hong and J. Sun, Curr. Org. Chem., 2016, 20, 1851–1861 CrossRef CAS; (d) X.-P. Zeng, Z.-Y. Cao, Y.-H. Wang, F. Zhou and J. Zhou, Chem. Rev., 2016, 116, 7330–7396 CrossRef CAS PubMed; (e) T. G. Saint-Denis, R.-Y. Zhu, G. Chen, Q.-F. Wu and J.-Q. Yu, Science, 2018, 359, eaao4798 CrossRef PubMed; (f) M. Wang, M. Feng, B. Tang and X. Jiang, Tetrahedron Lett., 2014, 55, 7147–7155 CrossRef CAS.
- For selected reports on transition metal-catalysed enantioselective desymmetrization reactions, see: (a) S. J. Malcolmson, S. J. Meek, E. S. Sattely, R. R. Schrock and A. H. Hoveyda, Nature, 2008, 456, 933–937 CrossRef CAS PubMed; (b) D. H. T. Phan, K. G. M. Kou and V. M. Dong, J. Am. Chem. Soc., 2010, 132, 16354–16355 CrossRef CAS PubMed; (c) J. Y. Lee, Y. S. You and S. H. Kang, J. Am. Chem. Soc., 2011, 133, 1772–1774 CrossRef CAS PubMed; (d) F. Zhou, C. Tan, J. Tang, Y.-Y. Zhang, W.-M. Gao, H.-H. Wu, Y.-H. Yu and J. Zhou, J. Am. Chem. Soc., 2013, 135, 10994–10997 CrossRef CAS PubMed; (e) B. M. Partridge, J. Solana González and H. W. Lam, Angew. Chem., Int. Ed., 2014, 53, 6523–6527 CrossRef CAS PubMed; (f) W. Zi and F. D. Toste, Angew. Chem., Int. Ed., 2015, 54, 14447–14451 CrossRef CAS PubMed; (g) W. Yang, Y. Liu, S. Zhang and Q. Cai, Angew. Chem., Int. Ed., 2015, 54, 8805–8808 CrossRef CAS PubMed; (h) Q.-F. Wu, P.-X. Shen, J. He, X.-B. Wang, F. Zhang, Q. Shao, R.-Y. Zhu, C. Mapelli, J. X. Qiao, M. A. Poss and J.-Q. Yu, Science, 2017, 355, 499–503 CrossRef CAS PubMed; (i) J. Hartung and R. H. Grubbs, J. Am. Chem. Soc., 2013, 135, 10183–10185 CrossRef CAS PubMed; (j) T. Saget and N. Cramer, Angew. Chem., Int. Ed., 2013, 52, 7865–7868 CrossRef CAS PubMed; (k) G. Chen, W. Gong, Z. Zhuang, M. S. Andrä, Y.-Q. Chen, X. Hong, Y.-F. Yang, T. Liu, K. N. Houk and J.-Q. Yu, Science, 2016, 353, 1023–1027 CrossRef CAS PubMed; (l) J. Fu, Z. Ren, J. Bacsa, D. G. Musaev and H. M. L. Davies, Nature, 2018, 564, 395–399 CrossRef CAS PubMed.
- For selected reports on chiral Brønsted acid-catalysed enantioselective desymmetrization reactions, see: (a) K. Mori, T. Katoh, T. Suzuki, T. Noji, M. Yamanaka and T. Akiyama, Angew. Chem., Int. Ed., 2009, 48, 9652–9654 CrossRef CAS PubMed; (b) Q. Gu, Z.-Q. Rong, C. Zheng and S.-L. You, J. Am. Chem. Soc., 2010, 132, 4056–4057 CrossRef CAS PubMed; (c) K. Mori, Y. Ichikawa, M. Kobayashi, Y. Shibata, M. Yamanaka and T. Akiyama, J. Am. Chem. Soc., 2013, 135, 3964–3970 CrossRef CAS PubMed; (d) Z. Chen and J. Sun, Angew. Chem., Int. Ed., 2013, 52, 13593–13596 CrossRef CAS PubMed; (e) J.-B. Gualtierotti, D. Pasche, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2014, 53, 9926–9930 CrossRef CAS PubMed; (f) S.-S. Meng, Y. Liang, K.-S. Cao, L. Zou, X.-B. Lin, H. Yang, K. N. Houk and W.-H. Zheng, J. Am. Chem. Soc., 2014, 136, 12249–12252 CrossRef CAS PubMed; (g) S. Prévost, N. Dupré, M. Leutzsch, Q. Wang, V. Wakchaure and B. List, Angew. Chem., Int. Ed., 2014, 53, 8770–8773 CrossRef PubMed; (h) J.-Y. Du, C. Zeng, X.-J. Han, H. Qu, X.-H. Zhao, X.-T. An and C.-A. Fan, J. Am. Chem. Soc., 2015, 137, 4267–4273 CrossRef CAS PubMed; (i) Z. Wang, F. K. Sheong, H. H. Y. Sung, I. D. Williams, Z. Lin and J. Sun, J. Am. Chem. Soc., 2015, 137, 5895–5898 CrossRef CAS PubMed; (j) J. Y. See, H. Yang, Y. Zhao, M. W. Wong, Z. Ke and Y.-Y. Yeung, ACS Catal., 2018, 8, 850–858 CrossRef CAS; (k) S.-S. Meng, P. Yu, Y.-Z. Yu, Y. Liang, K. N. Houk and W.-H. Zheng, J. Am. Chem. Soc., 2020, 142, 8506–8513 CrossRef CAS PubMed For selected reports on other organocatalyst-catalysed enantioselective desymmetrization reactions, see: (l) F. Liu, X. Bugaut, M. Schedler, R. Fröhlich and F. Glorius, Angew. Chem., Int. Ed., 2011, 50, 12626–12630 CrossRef CAS PubMed; (m) Z. Ke, C. K. Tan, F. Chen and Y.-Y. Yeung, J. Am. Chem. Soc., 2014, 136, 5627–5630 CrossRef CAS PubMed.
- For selected recent reviews, see: (a) T. E. Müller, K. C. Hultzsch, M. Yus, F. Foubelo and M. Tada, Chem. Rev., 2008, 108, 3795–3892 CrossRef PubMed; (b) S. R. Chemler, Org. Biomol. Chem., 2009, 7, 3009–3019 RSC; (c) J. Hannedouche and E. Schulz, Chem.–Eur. J., 2013, 19, 4972–4985 CrossRef CAS PubMed; (d) E. Bernoud, C. Lepori, M. Mellah, E. Schulz and J. Hannedouche, Catal. Sci. Technol., 2015, 5, 2017–2037 RSC; (e) L. Huang, M. Arndt, K. Gooßen, H. Heydt and L. J. Gooßen, Chem. Rev., 2015, 115, 2596–2697 CrossRef CAS PubMed; (f) C. Michon, M.-A. Abadie, F. Medina and F. Agbossou-Niedercorn, J. Organomet. Chem., 2017, 847, 13–27 CrossRef CAS; (g) J. Chen and Z. Lu, Org. Chem. Front., 2018, 5, 260–272 RSC; (h) J. Hannedouche and E. Schulz, Organometallics, 2018, 37, 4313–4326 CrossRef CAS; (i) M. T. Pirnot, Y.-M. Wang and S. L. Buchwald, Angew. Chem., Int. Ed., 2016, 55, 48–57 CrossRef CAS PubMed.
- For late-transition-metal-catalysed enantioselective desymmetrising hydroamination, see: P. Gao, G. Sipos, D. Foster and R. Dorta, ACS Catal., 2017, 7, 6060–6064 CrossRef CAS.
- For main-group-metal-catalysed enantioselective desymmetrising hydroamination, see: (a) X. Zhang, T. J. Emge and K. C. Hultzsch, Angew. Chem., Int. Ed., 2012, 51, 394–398 CrossRef CAS PubMed; (b) P. H. Martínez, K. C. Hultzsch and F. Hampel, Chem. Commun., 2006, 2221–2223 RSC.
- For early-transition-metal-catalysed enantioselective desymmetrising hydroamination, see: (a) D. V. Gribkov, K. C. Hultzsch and F. Hampel, Chem.–Eur. J., 2003, 9, 4796–4810 CrossRef CAS PubMed; (b) D. V. Gribkov, F. Hampel and K. C. Hultzsch, Eur. J. Inorg. Chem., 2004, 2004, 4091–4101 CrossRef; (c) D. V. Gribkov, K. C. Hultzsch and F. Hampel, J. Am. Chem. Soc., 2006, 128, 3748–3759 CrossRef CAS PubMed; (d) D. V. Vitanova, F. Hampel and K. C. Hultzsch, J. Organomet. Chem., 2007, 692, 4690–4701 CrossRef CAS; (e) M. C. Wood, D. C. Leitch, C. S. Yeung, J. A. Kozak and L. L. Schafer, Angew. Chem., Int. Ed., 2007, 46, 354–358 CrossRef CAS PubMed; (f) A. L. Reznichenko and K. C. Hultzsch, Organometallics, 2010, 29, 24–27 CrossRef CAS; (g) R. O. Ayinla, T. Gibson and L. L. Schafer, J. Organomet. Chem., 2011, 696, 50–60 CrossRef CAS; (h) K. Manna, M. L. Kruse and A. D. Sadow, ACS Catal., 2011, 1, 1637–1642 CrossRef CAS; (i) K. Manna, S. Xu and A. D. Sadow, Angew. Chem., Int. Ed., 2011, 50, 1865–1868 CrossRef CAS PubMed; (j) A. Otero, A. Lara-Sánchez, C. Nájera, J. Fernández-Baeza, I. Márquez-Segovia, J. A. Castro-Osma, J. Martínez, L. F. Sánchez-Barba and A. M. Rodríguez, Organometallics, 2012, 31, 2244–2255 CrossRef CAS; (k) K. Manna, W. C. Everett, G. Schoendorff, A. Ellern, T. L. Windus and A. D. Sadow, J. Am. Chem. Soc., 2013, 135, 7235–7250 CrossRef CAS PubMed; (l) L. Hussein, N. Purkait, M. Biyikal, E. Tausch, P. W. Roesky and S. Blechert, Chem. Commun., 2014, 50, 3862–3864 RSC; (m) X. Zhou, B. Wei, X.-L. Sun, Y. Tang and Z. Xie, Chem. Commun., 2015, 51, 5751–5753 RSC.
- K. Manna, N. Eedugurala and A. D. Sadow, J. Am. Chem. Soc., 2015, 137, 425–435 CrossRef CAS PubMed.
- For selected reviews on chiral Brønsted acids, see: (a) T. Akiyama, Chem. Rev., 2007, 107, 5744–5758 CrossRef CAS PubMed; (b) G. Adair, S. Mukherjee and B. List, Aldrichimica Acta, 2008, 41, 31–39 CAS; (c) M. Terada, Chem. Commun., 2008, 4097–4112 RSC; (d) S.-L. You, Q. Cai and M. Zeng, Chem. Soc. Rev., 2009, 38, 2190–2201 RSC; (e) M. Terada, Synthesis, 2010, 2010, 1929–1982 CrossRef; (f) J. Yu, F. Shi and L.-Z. Gong, Acc. Chem. Res., 2011, 44, 1156–1171 CrossRef CAS PubMed; (g) D. Parmar, E. Sugiono, S. Raja and M. Rueping, Chem. Rev., 2014, 114, 9047–9153 CrossRef CAS PubMed; (h) Z. Wang, Z. Chen and J. Sun, Org. Biomol. Chem., 2014, 12, 6028–6032 RSC; (i) T. Akiyama and K. Mori, Chem. Rev., 2015, 115, 9277–9306 CrossRef CAS PubMed; (j) Y.-B. Wang and B. Tan, Acc. Chem. Res., 2018, 51, 534–547 CrossRef CAS PubMed.
- (a) L. Ackermann and A. Althammer, Synlett, 2008, 2008, 995–998 CrossRef; (b) N. D. Shapiro, V. Rauniyar, G. L. Hamilton, J. Wu and F. D. Toste, Nature, 2011, 470, 245–249 CrossRef CAS PubMed; (c) J.-S. Lin, P. Yu, L. Huang, P. Zhang, B. Tan and X.-Y. Liu, Angew. Chem., Int. Ed., 2015, 54, 7847–7851 CrossRef CAS PubMed; (d) N. Tsuji, J. L. Kennemur, T. Buyck, S. Lee, S. Prévost, P. S. J. Kaib, D. Bykov, C. Farès and B. List, Science, 2018, 359, 1501–1505 CrossRef CAS PubMed; (e) J. Kikuchi and M. Terada, Angew. Chem., Int. Ed., 2019, 58, 8458–8462 CrossRef CAS PubMed.
- There are no reports on organocatalytic enantioselective desymmetrising hydroamination reactions. For organocatalytic enantioselective hydroamination reactions, see ref. 10. Also see: (a) M. J. MacDonald, D. J. Schipper, P. J. Ng, J. Moran and A. M. Beauchemin, J. Am. Chem. Soc., 2011, 133, 20100–20103 CrossRef CAS PubMed; (b) M. J. MacDonald, C. R. Hesp, D. J. Schipper, M. Pesant and A. M. Beauchemin, Chem.–Eur. J., 2013, 19, 2597–2601 CrossRef CAS PubMed; (c) A. R. Brown, C. Uyeda, C. A. Brotherton and E. N. Jacobsen, J. Am. Chem. Soc., 2013, 135, 6747–6749 CrossRef CAS PubMed; (d) J.-S. Lin, T.-T. Li, G.-Y. Jiao, Q.-S. Gu, J.-T. Cheng, L. Lv and X.-Y. Liu, Angew. Chem., Int. Ed., 2019, 58, 7092–7096 CrossRef CAS PubMed.
- (a) A. Roy, F. G. Roberts, P. R. Wilderman, K. Zhou, R. J. Peters and R. M. Coates, J. Am. Chem. Soc., 2007, 129, 12453–12460 CrossRef CAS PubMed; (b) L. S. Fernandez, M. S. Buchanan, A. R. Carroll, Y. J. Feng, R. J. Quinn and V. M. Avery, Org. Lett., 2009, 11, 329–332 CrossRef CAS PubMed; (c) X.-D. Zuo, S.-M. Guo, R. Yang, J.-H. Xie and Q.-L. Zhou, Org. Lett., 2017, 19, 5240–5243 CrossRef CAS PubMed.
- For the first report on a chiral BINOL-based N-triflyl phosphoramide catalyst, see: D. Nakashima and H. Yamamoto, J. Am. Chem. Soc., 2006, 128, 9626–9627 CrossRef CAS PubMed.
- Y. Chapurina, H. Ibrahim, R. Guillot, E. Kolodziej, J. Collin, A. Trifonov, E. Schulz and J. Hannedouche, J. Org. Chem., 2011, 76, 10163–10172 CrossRef CAS PubMed.
- (a) S. Tian, W. Zi and D. Ma, Angew. Chem., Int. Ed., 2012, 51, 10141–10144 CrossRef CAS PubMed; (b) Y. Shi, B. Yang, S. Cai and S. Gao, Angew. Chem., Int. Ed., 2014, 53, 9539–9543 CrossRef CAS PubMed; (c) M. Odagi, Y. Yamamoto and K. Nagasawa, Angew. Chem., Int. Ed., 2018, 57, 2229–2232 CrossRef CAS PubMed.
- K. Higuchi, Y. Sato, M. Tsuchimochi, K. Sugiura, M. Hatori and T. Kawasaki, Org. Lett., 2009, 11, 197–199 CrossRef CAS PubMed.
- L. Simón and J. M. Goodman, J. Org. Chem., 2011, 76, 1775–1788 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1853110. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc00001a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |