
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalytic enantioselective arylative cyclizations of alkynyl 1,3-diketones by 1,4-rhodium(I) migration†
Alistair
Groves
ab,
Jinwei
Sun
abc,
Hal R. I.
Parke
ab,
Michael
Callingham
ab,
Stephen P.
Argent‡
b,
Laurence J.
Taylor‡
b and
Hon Wai
Lam
 *ab
*ab
aThe GlaxoSmithKline Carbon Neutral Laboratories for Sustainable Chemistry, University of Nottingham, Jubilee Campus, Triumph Road, Nottingham, NG7 2TU, UK. E-mail: hon.lam@nottingham.ac.uk
bSchool of Chemistry, University of Nottingham, University Park, Nottingham, NG7 2RD, UK
cJiangsu Collaborative Innovation Center of Atmospheric Environment and Equipment Technology, Jiangsu Key Laboratory of Atmospheric Environment Monitoring and Pollution Control, School of Chemistry and Materials Science, Nanjing University of Information Science and Technology, Nanjing, Jiangsu 210044, China
First published on 6th February 2020
Abstract
The enantioselective synthesis of densely functionalized polycarbocycles by the rhodium(I)-catalyzed reaction of arylboronic acids with 1,3-diketones is described. The key step in these desymmetrizing domino addition–cyclization reactions is an alkenyl-to-aryl 1,4-Rh(I) migration, which enables arylboronic acids to function effectively as 1,2-dimetalloarene surrogates.
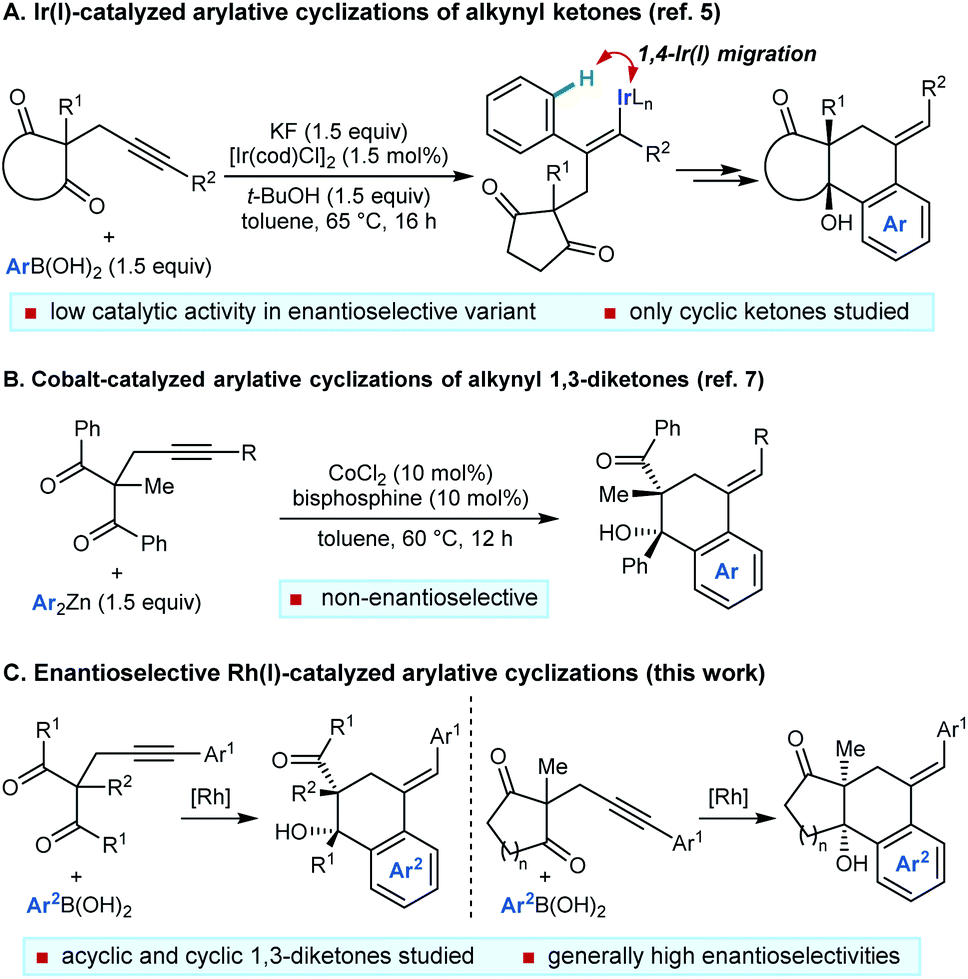
The functionalization of remote C–H bonds offers a powerful method to develop new synthetic methods and achieve transformations that would otherwise be highly challenging.1 Within this field, 1,4-migration of rhodium(I) between two carbon centers2–4 has proven to be highly effective for the catalytic functionalization of remote C–H bonds, which has been used to impressive effect in a range of valuable synthetic methods.4
We have described catalytic arylative cyclizations from the reaction of alkynyl ketones with arylboronic acids, which produce densely functionalized polycarbocycles through a key step involving an alkenyl-to-aryl 1,4-metal migration (Scheme 1A).5 This desymmetrization reaction forms two new carbon–carbon bonds with complete diastereocontrol over two new stereocenters and a trisubstituted alkene. In the non-enantioselective variant of this process, rhodium(I) catalysis was only moderately successful because of the formation of significant quantities of side-products, and the highest yields were obtained using iridium(I) catalysis.5,6 Although preliminary attempts towards an enantioselective variant using chiral bisphosphine–iridium complexes successfully gave products in high enantioselectivities, only modest catalytic activities were observed.5 Furthermore, only cyclic ketones were employed in that study.5 Yan and Yoshikai have reported related cobalt-catalyzed arylative cyclizations of acyclic 1,3-diketones with diarylzinc reagents; however, enantioselective reactions were not described (Scheme 1B).7 Therefore, to increase synthetic utility, there remains a need to discover more effective chiral catalysts that address these limitations by promoting high-yielding and highly enantioselective arylative cyclizations of a wider range substrates,8 including acyclic 1,3-diketones. Here, we report that a chiral bisphosphine–rhodium complex promotes the diastereo- and enantioselective reaction of arylboronic acids with alkynyl 1,3-diketones, for which both acyclic and cyclic 1,3-diketones are effective substrates.
 | ||
| Scheme 1 Catalytic arylative cyclizations involving 1,4-metal migration. | ||
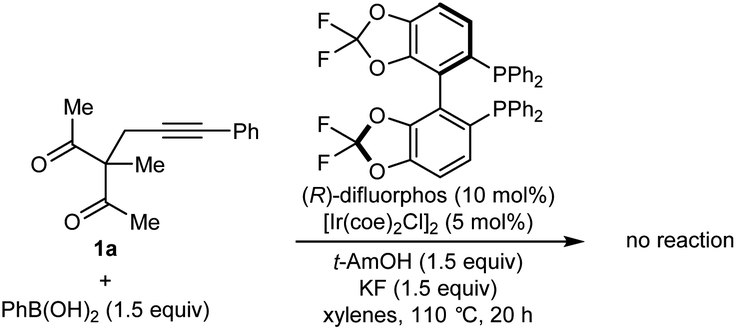
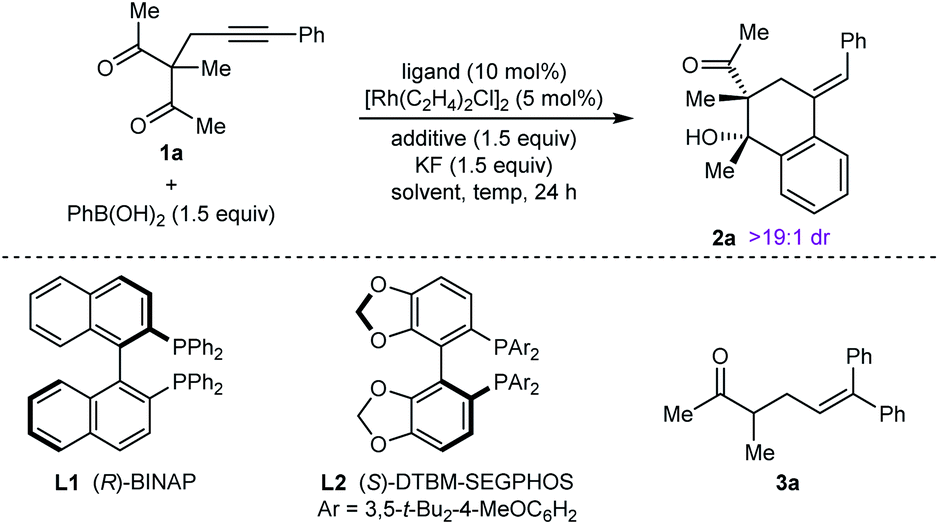
Our experiments began with the arylative cyclization of alkynyl 1,3-diketone 1a with PhB(OH)2 (eqn (1) and Table 1). Application of conditions identical to those described in our
 | (1) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at 70 °C for 24 h gave arylative cyclization product ent-2a in 54% yield (as determined by 1H NMR analysis using 1,4-dimethoxybenzene as an internal standard) as a single diastereomer (>19:1 dr) in 80% ee (Table 1, entry 1).9 Higher enantioselectivity was obtained using (S)-DTBM-SEGPHOS (L2), which gave 2a in 52% NMR yield and 91% ee (entry 2). Changing the protic additive from H2O to t-AmOH (1.5 equiv.) further increased the enantioselectivity (entry 3). The yield of 2a was increased further by raising the temperature to 80 °C (entry 4) and using 2.0 equivalents of PhB(OH)2 (entry 5). Conducting the reaction on a larger scale using 0.30 mmol of 1a gave 2a in 78% yield and 98% ee (entry 6). This experiment also gave a side-product 3a in 5% yield.10 It should be noted that the use of PhB(OH)2 free from triphenylboroxine is very important for good results, as otherwise lower enantioselectivities are observed.11 Finally, repeating the conditions of entry 5 but using [Ir(coe)2Cl]2 in place of [Rh(C2H4)Cl2] led to no reaction, and only unreacted starting material was recovered (entry 7).
:1) at 70 °C for 24 h gave arylative cyclization product ent-2a in 54% yield (as determined by 1H NMR analysis using 1,4-dimethoxybenzene as an internal standard) as a single diastereomer (>19:1 dr) in 80% ee (Table 1, entry 1).9 Higher enantioselectivity was obtained using (S)-DTBM-SEGPHOS (L2), which gave 2a in 52% NMR yield and 91% ee (entry 2). Changing the protic additive from H2O to t-AmOH (1.5 equiv.) further increased the enantioselectivity (entry 3). The yield of 2a was increased further by raising the temperature to 80 °C (entry 4) and using 2.0 equivalents of PhB(OH)2 (entry 5). Conducting the reaction on a larger scale using 0.30 mmol of 1a gave 2a in 78% yield and 98% ee (entry 6). This experiment also gave a side-product 3a in 5% yield.10 It should be noted that the use of PhB(OH)2 free from triphenylboroxine is very important for good results, as otherwise lower enantioselectivities are observed.11 Finally, repeating the conditions of entry 5 but using [Ir(coe)2Cl]2 in place of [Rh(C2H4)Cl2] led to no reaction, and only unreacted starting material was recovered (entry 7).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Ligand | Additive | Solvent | Temp. (°C) | Yieldb (%) | eec (%) |
| a Reactions were conducted with 0.05 mmol of 1a in 1 mL of solvent. b Determined by 1H NMR analysis using 1,4-dimethoxybenzene as an internal standard. c Determined by HPLC analysis on a chiral stationary phase. d The major enantiomer was ent-2a. e Using 2.0 equivalents of PhB(OH)2. f Using 0.30 mmol of 1a in THF (6 mL). g Value in parentheses refers to the yield of side-product 3a, which was also isolated from this experiment. h Using [Ir(coe)Cl2]2 in place of [Rh(C2H4)2Cl2]. i n.r. = no reaction. | ||||||
| 1 | L1 | — | THF:H2O (9:1) |
70 | 54 | −80d |
| 2 | L2 | — | THF:H2O (9:1) |
70 | 52 | 91 |
| 3 | L2 | t-AmOH | THF | 70 | 54 | 95 |
| 4 | L2 | t-AmOH | THF | 80 | 67 | 96 |
| 5e | L2 | t-AmOH | THF | 80 | 70 | 96 |
| 6e,f | L2 | t-AmOH | THF | 80 | 78 (5)g | 98 |
| 7e,h | L2 | t-AmOH | THF | 80 | n.r.i | — |
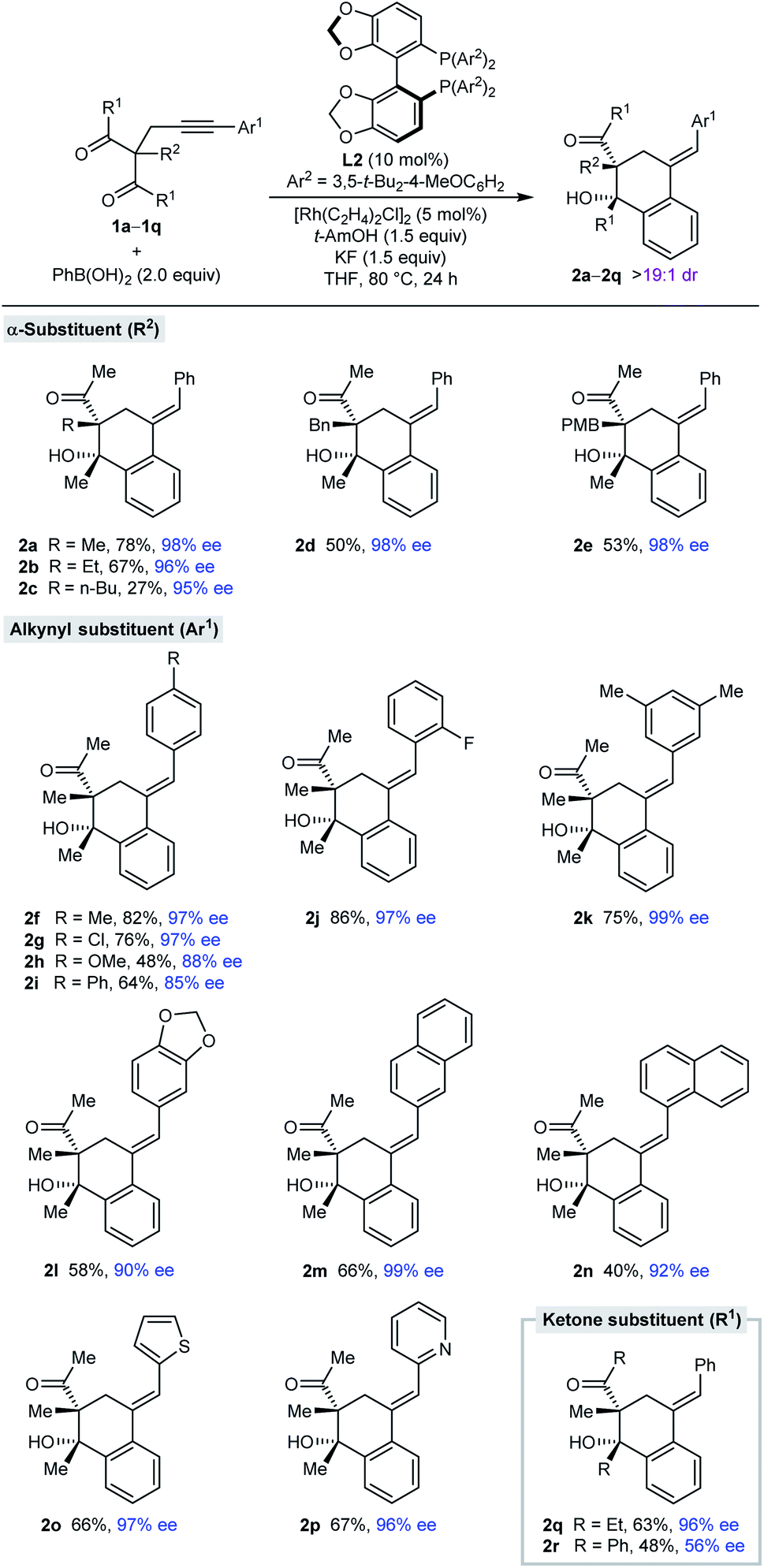
With effective conditions identified (Table 1, entry 6), the scope of this process with respect to the alkynyl acyclic 1,3-diketone 1 was investigated in reactions with PhB(OH)2 (Table 2). Arylative cyclization products 2a–2r were obtained as single observable diastereomers (>19:1 dr as determined by 1H NMR analysis of the crude reaction mixtures) in 27–82% yield and 56–99% ee. Side-products analogous to 3a (see Table 1) were generally detected but not isolated. Changing the α-substituent R2 between the two ketones from methyl (2a) to ethyl (2b), n-butyl (2c), benzyl (2d), or 4-methoxybenzyl (2e) is tolerated. The low yield of 2c results from a low conversion as significant unreacted starting material was observed. The process is also compatible with a range (hetero)aryl groups Ar1 at the alkynyl position, such as 4-substituted phenyl (2f–2i), 2-fluorophenyl (2j), 3,5-dimethylphenyl (2k), 3,4-(methylenedioxy)phenyl (2l), 2-naphthyl (2m), 1-naphthyl (2n), 2-thienyl (2o), and 2-pyridyl (2p) groups. Finally, the ketone substituents can also be varied from methyl to ethyl (2q) or phenyl groups (2r), although the enantioselectivity dropped substantially in the latter case.
| a Reactions were conducted with 0.30 mmol of 1 in THF (6 mL). Yields are of isolated products. Enantiomeric excesses were determined by HPLC analysis on a chiral stationary phase. PMB = para-methoxybenzyl. |
|---|
|
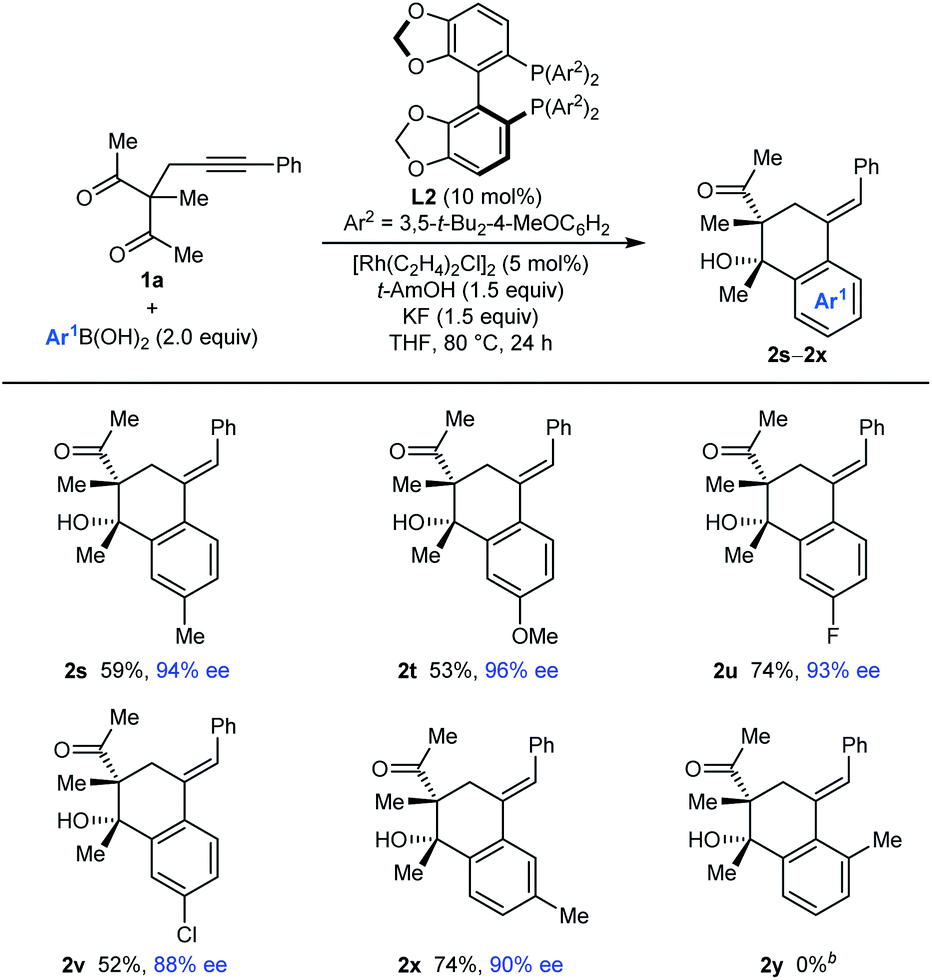
The process is not limited to the use of PhB(OH)2, as shown by the arylative cyclizations of 1a with different arylboronic acids to give 2s–2x in 88–96% ee (Table 3). Various 4-substituted phenylboronic acids containing methyl (2s), methoxy (2t), fluoro (2u), or chloro groups (2v) reacted successfully. When 3-methylphenylboronic acid was employed, 1,4-Rh(I) migration occurred to the least sterically hindered site, para to the methyl group (2x). However, 2-methylphenylboronic acid did not provide any of the arylative cyclization product 2y, and returned mainly unreacted starting material along with what appeared to be small quantities of alkyne hydroarylation products.
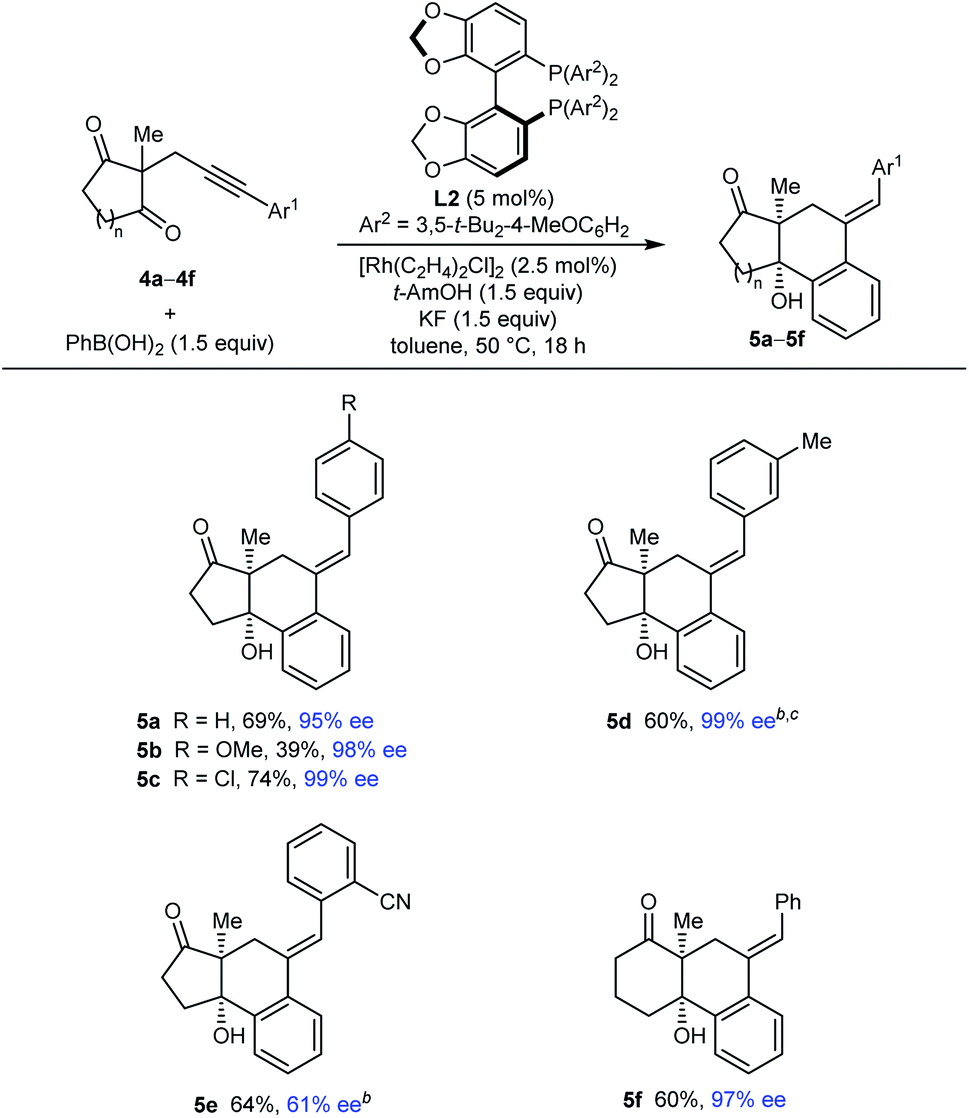
Our attention then turned to the reaction of alkynyl cyclic 1,3-diketones 4, substrates employed in our prior study using iridium catalysis (Tables 4 and 5).5 With toluene as the solvent, these more reactive substrates generally allowed the use of a decreased catalyst loading of 5 mol% and a lower temperature of 50 °C. Furthermore, in most cases, acceptable results were obtained using only 1.5 equivalents of the arylboronic acid. Various substrates 4a–4f underwent arylative cyclization with PhB(OH)2 to give products 5a–5f in 39–74% yield and 61–99% ee (Table 4). Small quantities of side-products resulting from arylrhodation of the alkyne with the regioselectivity opposite to that seen in the formation of products 5 were also observed but generally not isolated (see ESI† for details). As with the acylic 1,3-diketones (Table 2), a range of aryl substituents at the alkyne are tolerated, including phenyl (5a and 5f), 4-methoxyphenyl (5b), 4-chlorophenyl (5c), and 3-methylphenyl (5d). The lower yield of 5b results from the formation of products of alkyne hydroarylation without cyclization. The cyclization of a 2-cyanophenyl-containing substrate 4e proceeded smoothly using a 10 mol% catalyst loading but the product 5e was formed in a modest 61% ee. A six-membered cyclic 1,3-diketone also underwent arylative cyclization with PhB(OH)2 to give 5f in 60% and 97% ee.
| a Reactions were conducted with 0.30 mmol of 4 in toluene (3 mL). Yields are of isolated products. Enantiomeric excesses were determined by HPLC analysis on a chiral stationary phase. b Using 5 mol% of [Rh(C2H4)2Cl]2 and 10 mol% of L2. c Using 2.0 equivalents of the arylboronic acid. d Using 2.4 equivalents of the arylboronic acid. e Unreacted starting material was returned. |
|---|
|
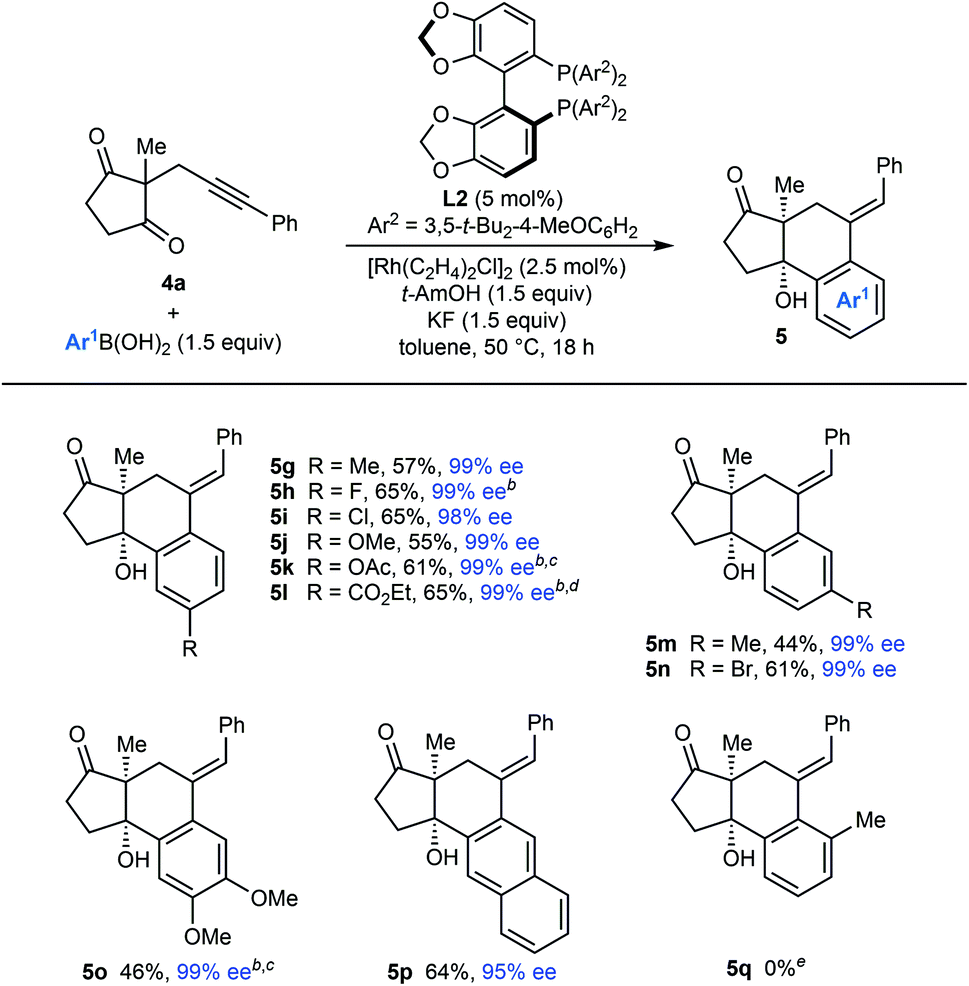
The scope of the arylative cyclization of alkynyl cyclic 1,3-diketones with respect to the arylboronic acid was then explored in reactions with substrate 4a (Table 5). These reactions proceeded in 44–69% yield and gave products 5g–5p in 95–99% ee. The process tolerates diverse 4-substituted phenylboronic acids containing methyl (5g), halide (5h and 5i), methoxy (5j), acetoxy (5k), or carboethoxy groups (5l). 3-Substituted phenylboronic acids (5m and 5n), 3,4-dimethoxyphenylboronic acid (5o), and 2-naphthylboronic acid (5p) also react effectively. Again, where 1,4-Rh(I) migration could occur to two different positions, migration to the sterically less-hindered side was observed (5m–5p). An attempt to form 5q with 2-methylphenylboronic acid was unsuccessful, and returned only unreacted starting material.
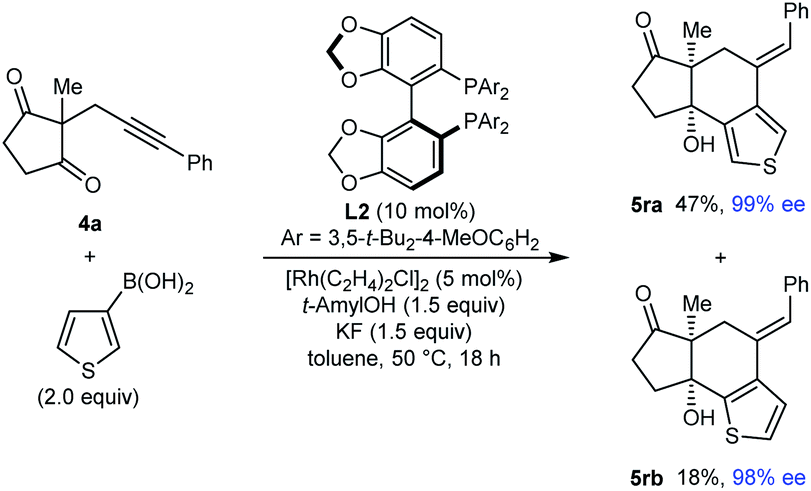
Interestingly, the reaction of substrate 4a with 3-thienylboronic acid gave two products 5ra and 5rb resulting from 1,4-Rh(I) migration to different positions of the thienyl ring before cyclization (eqn (2)).
 | (2) |
Finally, this method is not restricted to the use of 1,3-diketone-containing substrates; substrate 6 containing a single methyl ketone also underwent arylative cyclization to give 7 in 94% yield and 85% ee (eqn (3)).
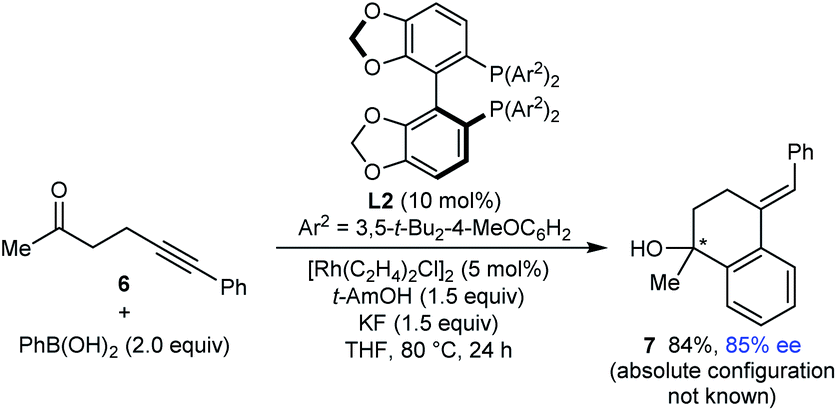 | (3) |
Scheme 2 illustrates a possible catalytic cycle for these reactions, using 1a and PhB(OH)2 as example substrates. First, upon mixing [Rh(C2H4)Cl]2, L2, KF, and t-AmOH, a chiral complex 8 consisting of one bisphosphine bound to one rhodium atom is formed, which could have a chloride, fluoride, or tert-amyl counterion. Transmetalation of 8 with PhB(OH)2 gives an arylrhodium species 9, which can then undergo migratory insertion with the alkyne of 1a to give alkenylrhodium intermediate 10. Alkenyl-to-aryl 1,4-rhodium(I) migration of 10 then provides arylrhodium species 11. The relative configuration of products 2 can be explained by a tentative stereochemical model where cyclization proceeds through a conformation similar to 12, in which: (i) rhodium(I) has a square pyramidal coordination geometry; (ii) the ketone undergoing nucleophilic attack is coordinated to rhodium such that the carbonyl group is aligned with the arylrhodium bond to enable subsequent migratory insertion; and (iii) the second ketone is coordinated to rhodium in an axial position. The relative configuration of products 5 (Tables 4 and 5) is more straightforward to rationalize; because of geometric constraints, nucleophilic addition of the arylrhodium group must occur to the same face of the cyclic 1,3-diketone as that from which the tether connecting the two reacting components projects (as in 14 to give representative product 5a, for example). However, as to exactly how the chiral ligand controls the absolute configuration of the products is not clear at the present time.
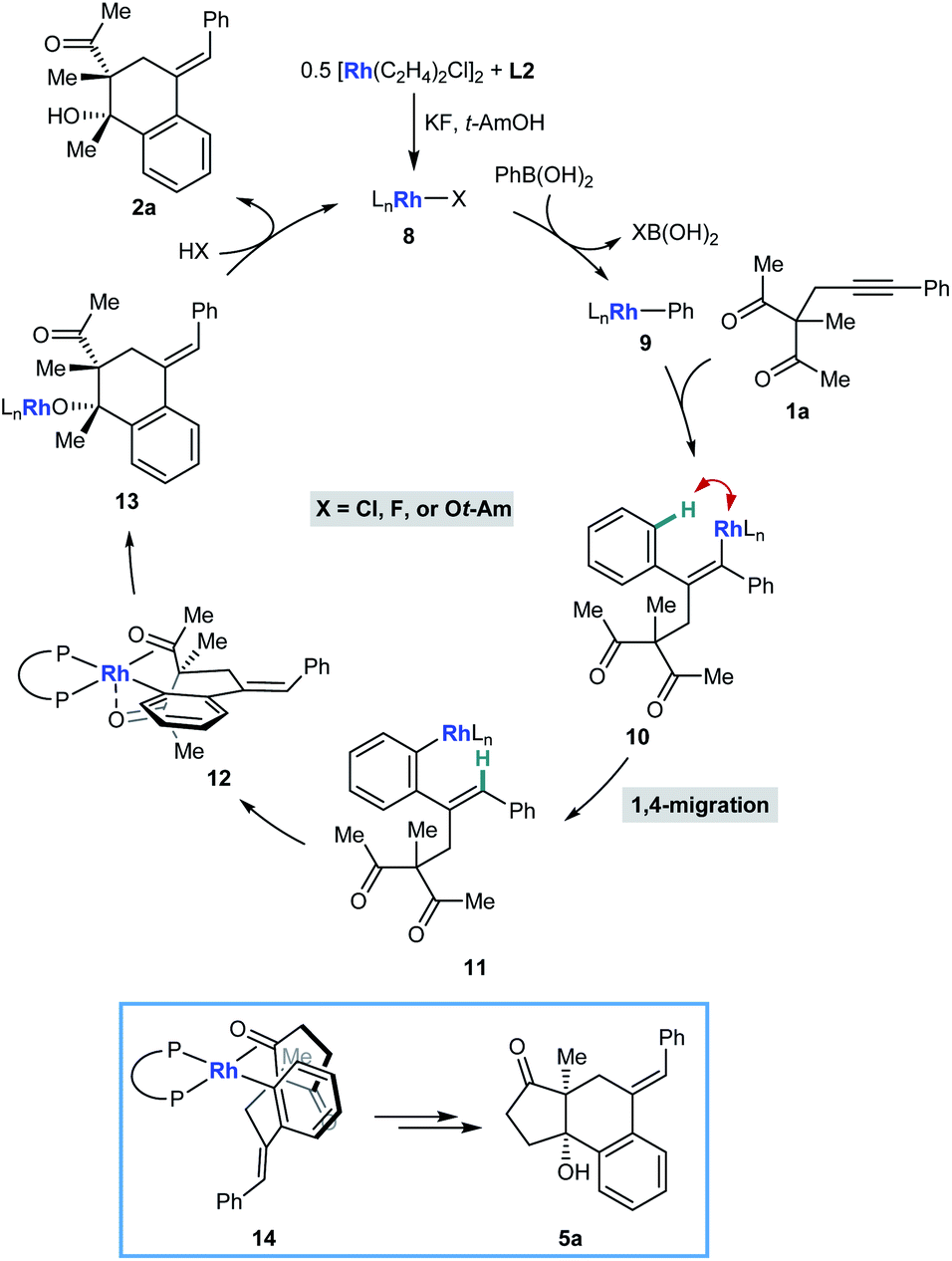 | ||
| Scheme 2 Possible catalytic cycle and rationalization of diastereochemical outcomes. | ||
In conclusion, we have reported rhodium(I)-catalyzed arylative cyclizations of alkynyl 1,3-diketones with arylboronic acids, which involve an alkenyl-to-aryl 1,4-Rh(I) migration as a key step. By using a chiral rhodium(I) complex based upon (S)-DTBM-SEGPHOS, the formation of side-products observed previously5 with [Rh(cod)Cl]2 is significantly reduced, and catalytic activity is greatly increased compared with chiral iridium complexes.5 These desymmetrization reactions provide densely functionalized polycarbocycles with high diastereo- and enantioselectivities, and notably, both acyclic and cyclic 1,3-ketones are effective substrates.12
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Engineering and Physical Sciences Research Council [grant number EP/M50810X/1]; the Leverhulme Trust [grant RPG-2016-341]; the Natural Science Foundation of Jiangsu Province [grant number BK20161307]; the Startup Foundation for Introducing Talent of NUIST (grant number 2016r044); the University of Nottingham; and GlaxoSmithKline.Notes and references
- For selected reviews of remote functionalization reactions, For selected reviews: (a) R. Breslow, Acc. Chem. Res., 1980, 13, 170–177 CrossRef CAS; (b) H. Schwarz, Acc. Chem. Res., 1989, 22, 282–287 CrossRef CAS; (c) H. Jiang, L. Albrecht and K. A. Jorgensen, Chem. Sci., 2013, 4, 2287–2300 RSC; (d) I. Franzoni and C. Mazet, Org. Biomol. Chem., 2014, 12, 233–241 RSC; (e) M. Vilches-Herrera, L. Domke and A. Börner, ACS Catal., 2014, 4, 1706–1724 CrossRef CAS; (f) G. Qiu and J. Wu, Org. Chem. Front., 2015, 2, 169–178 RSC; (g) A. Vasseur, J. Bruffaerts and I. Marek, Nat. Chem., 2016, 8, 209–219 CrossRef CAS; (h) J. Bruffaerts, D. Pierrot and I. Marek, Org. Biomol. Chem., 2016, 14, 10325–10330 RSC; (i) H. Sommer, F. Juliá-Hernández, R. Martin and I. Marek, ACS Cent. Sci., 2018, 4, 153–165 CrossRef CAS.
- Seminal examples: (a) K. Oguma, M. Miura, T. Satoh and M. Nomura, J. Am. Chem. Soc., 2000, 122, 10464–10465 CrossRef CAS; (b) T. Hayashi, K. Inoue, N. Taniguchi and M. Ogasawara, J. Am. Chem. Soc., 2001, 123, 9918–9919 CrossRef CAS PubMed.
- For an early review, see: S. Ma and Z. Gu, Angew. Chem., Int. Ed., 2005, 44, 7512–7517 CrossRef CAS.
- Selected, recent examples of 1,4-rhodium(I) migrations: (a) R. Shintani, S. Isobe, M. Takeda and T. Hayashi, Angew. Chem., Int. Ed., 2010, 49, 3795–3798 CrossRef CAS; (b) K. Sasaki, T. Nishimura, R. Shintani, E. A. B. Kantchev and T. Hayashi, Chem. Sci., 2012, 3, 1278–1283 RSC; (c) J. Zhang, J.-F. Liu, A. Ugrinov, A. F. X. Pillai, Z.-M. Sun and P. Zhao, J. Am. Chem. Soc., 2013, 135, 17270–17273 CrossRef CAS PubMed; (d) R. Shintani, R. Iino and K. Nozaki, J. Am. Chem. Soc., 2014, 136, 7849–7852 CrossRef CAS PubMed; (e) H. B. Hepburn and H. W. Lam, Angew. Chem., Int. Ed., 2014, 53, 11605–11610 CrossRef CAS PubMed; (f) T. Johnson, K.-L. Choo and M. Lautens, Chem. Eur.–J., 2014, 20, 14194–14197 CrossRef CAS PubMed; (g) A. Masarwa, M. Weber and R. Sarpong, J. Am. Chem. Soc., 2015, 137, 6327–6334 CrossRef CAS PubMed; (h) A. Claraz, F. Serpier and S. Darses, ACS Catal., 2017, 7, 3410–3413 CrossRef CAS; (i) B. M. Partridge, M. Callingham, W. Lewis and H. W. Lam, Angew. Chem., Int. Ed., 2017, 56, 7227–7232 CrossRef CAS PubMed; (j) M. Callingham, B. M. Partridge, W. Lewis and H. W. Lam, Angew. Chem., Int. Ed., 2017, 56, 16352–16356 CrossRef CAS PubMed; (k) J. L. Ming and T. Hayashi, Org. Lett., 2018, 20, 6188–6192 CrossRef CAS PubMed; (l) J. L. Ming, Q. Shi and T. Hayashi, Chem. Sci., 2018, 9, 7700–7704 RSC; (m) S.-S. Zhang, T.-J. Hu, M.-Y. Li, Y.-K. Song, X.-D. Yang, C.-G. Feng and G.-Q. Lin, Angew. Chem., Int. Ed., 2019, 58, 3387–3391 CrossRef CAS PubMed; (n) A. Selmani, F. Serpier and S. Darses, J. Org. Chem., 2019, 84, 4566–4574 CrossRef CAS PubMed; (o) L. O'Brien, S. N. Karad, W. Lewis and H. W. Lam, Chem. Commun., 2019, 55, 11366–11369 RSC; (p) A. Selmani and S. Darses, Org. Lett., 2019, 21, 8122–8126 CrossRef CAS PubMed.
- B. M. Partridge, J. Solana González and H. W. Lam, Angew. Chem., Int. Ed., 2014, 53, 6523–6527 CrossRef CAS PubMed.
- For other examples of 1,4-iridium(I) migration, see ref. 4j and; R. E. Ruscoe, M. Callingham, J. A. Baker, S. E. Korkis and H. W. Lam, Chem. Commun., 2019, 55, 838–841. RSC.
- J. Yan and N. Yoshikai, ACS Catal., 2016, 6, 3738–3742 CrossRef CAS.
- For selected examples of other types of enantioselective arylative cyclizations of alkynyl electrophiles, see ref. 4h, n–p and: (a) R. Shintani, K. Okamoto, Y. Otomaru, K. Ueyama and T. Hayashi, J. Am. Chem. Soc., 2005, 127, 54–55 CrossRef CAS PubMed; (b) T. Miura, M. Shimada and M. Murakami, J. Am. Chem. Soc., 2005, 127, 1094–1095 CrossRef CAS PubMed; (c) T. Miura, T. Sasaki, H. Nakazawa and M. Murakami, J. Am. Chem. Soc., 2005, 127, 1390–1391 CrossRef CAS PubMed; (d) R. Shintani, A. Tsurusaki, K. Okamoto and T. Hayashi, Angew. Chem., Int. Ed., 2005, 44, 3909 CrossRef CAS PubMed; (e) J. Song, Q. Shen, F. Xu and X. Lu, Org. Lett., 2007, 9, 2947–2950 CrossRef CAS PubMed; (f) X. Han and X. Lu, Org. Lett., 2010, 12, 108–111 CrossRef CAS PubMed; (g) Z.-T. He, B. Tian, Y. Fukui, X. Tong, P. Tian and G.-Q. Lin, Angew. Chem., Int. Ed., 2013, 52, 5314–5318 CrossRef CAS PubMed; (h) J. Keilitz, S. G. Newman and M. Lautens, Org. Lett., 2013, 15, 1148–1151 CrossRef CAS PubMed; (i) Y. Li and M.-H. Xu, Org. Lett., 2014, 16, 2712–2715 CrossRef CAS PubMed; (j) T. Johnson, K.-L. Choo and M. Lautens, Chem. Eur.–J., 2014, 20, 14194–14197 CrossRef CAS PubMed; (k) F. Serpier, B. Flamme, J.-L. Brayer, B. Folléas and S. Darses, Org. Lett., 2015, 17, 1720–1723 CrossRef CAS PubMed; (l) C. Clarke, C. A. Incerti-Pradillos and H. W. Lam, J. Am. Chem. Soc., 2016, 138, 8068–8071 CrossRef CAS PubMed; (m) C. Yap, G. M. J. Lenagh-Snow, S. N. Karad, W. Lewis, L. J. Diorazio and H. W. Lam, Angew. Chem., Int. Ed., 2017, 56, 8216–8220 CrossRef CAS PubMed; (n) S. N. Karad, H. Panchal, C. Clarke, W. Lewis and H. W. Lam, Angew. Chem., Int. Ed., 2018, 57, 9122–9125 CrossRef CAS.
- The relative and absolute configurations of 2j and 5g were determined by X-ray crystallography, and those of the remaining products were assigned by analogy. CCDC 1959877 and 1959878 contain the ESI crystallographic data for this paper.† Furthermore, the spectroscopic data of 2r are consistent with those reported previously (ref. 7), which suggests the relative configurations of products 2 are identical to those reported in ref. 7.
- Ketone 3a appears to be the result of arylrhodation of the alkyne of 1a with the regioselectivity opposite to that seen in the formation of arylative cyclization product 2a, combined with a retro-Claisen condensation. However, the order of these steps is not currently known.
- See the ESI† for purification of arylboronic acids.
- The research data associated with this publication can be found at: http://dx.doi.org/10.17639/nott.7036.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, full spectroscopic data for new compounds, and crystallographic data for 2j and 5g. CCDC 1959877 and 1959878. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc06309a |
| ‡ To whom enquires regarding X-ray crystallography should be addressed. |
| This journal is © The Royal Society of Chemistry 2020 |