
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVersatility and adaptative behaviour of the P^N chelating ligand MeDalphos within gold(I) π complexes†
Miquel
Navarro
 a,
Alberto
Toledo
a,
Sonia
Mallet-Ladeira
b,
E. Daiann
Sosa Carrizo
c,
Karinne
Miqueu
c and
Didier
Bourissou
*a
a,
Alberto
Toledo
a,
Sonia
Mallet-Ladeira
b,
E. Daiann
Sosa Carrizo
c,
Karinne
Miqueu
c and
Didier
Bourissou
*a
aCNRS/Université Paul Sabatier, Laboratoire Hétérochimie Fondamentale et Appliquée (LHFA, UMR 5069), 118 Route de Narbonne, 31062 Toulouse Cedex 09, France. E-mail: dbouriss@chimie.ups-tlse.fr
bInstitut de Chimie de Toulouse (FR 2599), 118 Route de Narbonne, 31062 Toulouse Cedex 09, France
cCNRS/Université de Pau et des Pays de l'Adour, Institut des Sciences Analytiques et de Physico-Chimie pour L'Environnement et les Matériaux (IPREM UMR 5254), Hélioparc, 2 Avenue du Président Angot, 64053 Pau Cedex 09, France
First published on 4th February 2020
Abstract
The hemilabile P^N ligand MeDalphos enables access to a wide range of stable gold(I) π-complexes with unbiased alkenes and alkynes, as well as electron-rich alkenes and for the first time electron-poor ones. All complexes have been characterized by multi-nuclear NMR spectroscopy and whenever possible, by X-ray diffraction analyses. They all adopt a rare tricoordinate environment around gold(I), with chelation of the P^N ligand and side-on coordination of the alkene, including the electron-rich one, 3,4-dihydro-2H-pyrane. The strength of the N → Au coordination varies significantly in the series. It is the way the P^N ligand accommodates the electronic demand at gold, depending on the alkene. Comparatively, when the chelating P^P ligand (ortho-carboranyl)(PPh2)2 is used, gold(I) π-complexes are only isolable with unbiased alkenes. The bonding situation within the gold(I) P^N π-complexes has been thoroughly analyzed by DFT calculations supplemented by Charge Decomposition Analyses (CDA), Natural Bond Orbital (NBO) and Atoms-in-Molecules (AIM) analyses. Noticeable variations in the donation/back-donation ratio, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C weakening, alkene to gold charge transfer and magnitude of the N → Au coordination were observed. Detailed examination of the descriptors for the Au/alkene interaction and the N → Au coordination actually revealed intimate correlation between the two, with linear response of the MeDalphos ligand to the alkene electronics. The P^N ligand displays non-innocent and adaptative character. The isolated P^N gold(I) π-complexes are reactive and catalytically relevant, as substantiated by the chemo and regio-selective alkylation of indoles.
C weakening, alkene to gold charge transfer and magnitude of the N → Au coordination were observed. Detailed examination of the descriptors for the Au/alkene interaction and the N → Au coordination actually revealed intimate correlation between the two, with linear response of the MeDalphos ligand to the alkene electronics. The P^N ligand displays non-innocent and adaptative character. The isolated P^N gold(I) π-complexes are reactive and catalytically relevant, as substantiated by the chemo and regio-selective alkylation of indoles.
Introduction
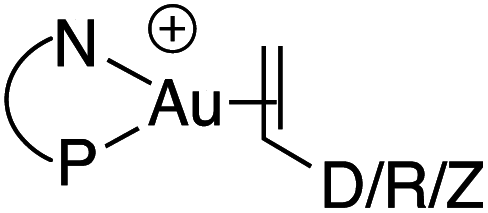
For a long time, gold was considered to be “the noblest of all metals”1 and too chemically inert to be used in catalysis. However, since the discovery of the ability of gold to activate π-bonds towards nucleophilic addition, homogenous gold catalysis has emerged as a very powerful tool in organic synthesis.2 A large number of useful transformations such as the addition of oxygen-, nitrogen- and carbon-nucleophiles to CC multiple bonds, rearrangements and cycloaddition reactions have been developed.3 All these catalytic reactions usually involve the initial π-coordination of a CC multiple bond to gold. Consequently, the isolation of gold π-complexes has received considerable attention.4–6 The prepared compounds serve as models for the transient species and their study provide valuable insight into the factors governing the catalytic transformations. Over the last 15 years, a number of cationic dicoordinate gold(I) π-complexes have been isolated and characterized with N-heterocyclic carbenes (NHCs) or phosphines as ancillary ligands, and electron-unbiased alkenes such as styrene and hex-1-ene (Fig. 1).4,5 A few complexes have also been reported with electron-rich alkenes, such as enamines and enol ethers, for which coordination tends to slip from side-on to terminal.7 In addition, N-based chelating ligands have been occasionally found to form stable tricoordinate gold(I) π-complexes.8 Nonetheless, the number and variety of known π-alkene gold complexes are still limited. Tricoordinate species remain very scarce9 and to the best of our knowledge, gold π-complexes featuring electron-poor alkenes are hitherto known. | ||
| Fig. 1 Alkene gold(I) complexes (D = electron-donating, Z = electron-withdrawing substituents). | ||
With the aim to advance further our knowledge on these key reactive intermediates in gold chemistry, we aimed in this work to address the following question: is it possible for a unique ligand to accommodate the π-coordination of a wide range of alkenes at gold, i.e. unbiased as well as electron-rich and electron-poor alkenes? To this end, we envisioned to take advantage of the higher modularity of bifunctional ligands to find a proper balance in stability/reactivity. Indeed, the alkenes should bind tightly enough to gold to form stable complexes, but the coordination should not result in too strong activation of the π-system to induce oligomerization by polyaddition.
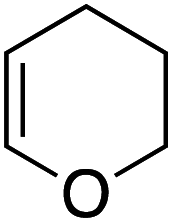
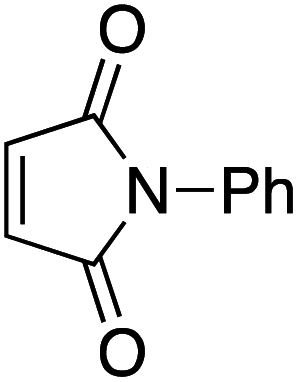
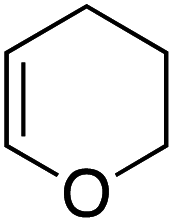
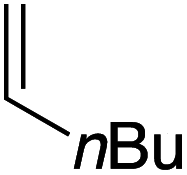
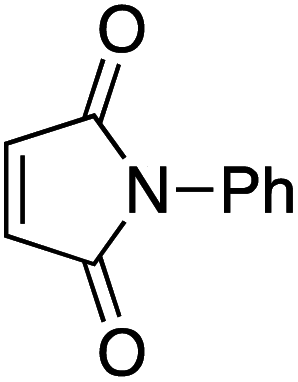
Our recent isolation of tridentate gold(I) complexes of styrene with P^P chelating and P^N hemilabile ligands10 prompted us to screen a range of other π-systems. Gratifyingly, stable gold(I) π-complexes were found to be readily accessible with different unbiased alkenes and alkynes such as hex-1-ene, ethylene and hex-3-yne, as well as 3,4-dihydro-2H-pyrane (DHP) as electron-rich alkene, and methylacrylate (MA) and N-phenylmaleimide (NPM) as electron-poor alkenes. These results are reported hereafter, along with detailed characterization and analysis of the bonding situation by experimental and computational means. This study unveils the faculty of the P^N ligand to respond and accommodate the electron demand at gold by weakening/strengthening the N → Au interaction.
Results and discussion
Synthesis and characterization of (P^N)Au(I) π-complexes
Treatment of MeDalphos gold(I) chloride with AgSbF6 in the presence of an excess (3 equiv.) of alkene or alkyne proved to be a general entry to gold(I) π-complexes, as illustrated by the preparation of the series of complexes 1–7 (Scheme 1).11 The reactions were conveniently monitored by 31P NMR spectroscopy. Filtration of the reaction mixtures through short pads of Celite followed by washing with Et2O afforded the pure gold(I) π-complexes 1–6 in good yields (50–89%). Only complex 7 proved too unstable to be isolated. Since decomposition occurred upon filtration, it was characterized in the presence of excess of N-phenylmaleimide without further purification. Complexes 1–6 are air stable solids. Their 1H and 13C NMR spectra display a new set of well-defined signals for the alkene/alkyne (distinct from those of the free π-system). No chemical exchange was observed between the coordinated and free alkene/alkyne within the NMR time-scale. No sign of alkene/alkyne dissociation was detected either under vacuum. | ||
| Scheme 1 Synthesis of gold(I) π-complexes 1–7. | ||
Coordination to gold(I) induces a marked upfield shift (with exception of complex 5 that shows a downfield shift) of the 1H and 13C NMR signals with respect to the free alkene/alkyne (Table 1 and S2†).11 These shift differences reach up to 2.13 ppm in 1H NMR and 65 ppm in 13C NMR spectroscopy, suggesting substantial gold → π* back-donation (see below). Another conspicuous feature of all π-complexes is the sizeable downfield shift of the N(CH3)21H NMR signal, as compared to the (MeDalphos) AuCl precursor. Interestingly, the magnitude of the 1H NMR downfield shift of the N(CH3)2 signal seems to correlate with the electronic properties of the π-system: Δδ increases from 0.15 ppm for the DHP complex 5 (electron-rich alkene) to 0.82 ppm for the NPM complex 7 (electron-poor alkene) while complexes 1–4 with unbiased alkenes/alkyne exhibit intermediate values (Δδ ∼ 0.5 ppm). Similar trends are observed in 13C NMR [the N(CH3)2 signals shift from 49.4 for DHP to 55.3 ppm for NPM] as well as 31P NMR spectroscopy (the signal shifts from 56.5 ppm for DHP to 65.3 ppm for NPM). These observations suggest that the P^N ligand adjusts its coordination to gold depending on the electronics of the π-system, and that NMR spectroscopy is a sensitive reporter for this adaptative behavior.
|
|
|
|
|
|
|
|
|
|---|---|---|---|---|---|---|---|
| a All data recorded in CD2Cl2. Data for the styrene complex 1 from ref. 10. | |||||||
| δ 1H | Halkene | 7.76 | 6.03 | 5.66 | 4.10 | 4.61 | 4.68 |
| 4.78 | 4.33 | 3.94 | 4.35 | 5.67 | |||
| 4.17 | 3.76 | 4.23 | |||||
| HNMe2 | 2.74 | 3.09 | 3.01 | 3.14 | 3.25 | 3.44 | |
| 2.71 | 2.82 | 2.98 | 3.07 | 3.33 | |||
| δ 13C | Calkene | 144.1 | 99.9 | 111.1 | 75.0 | 72.5 | 72.4 |
| 76.7 | 66.4 | 75.2 | 68.4 | 68.8 | |||
| CNMe2 | 49.5 | 53.0 | 52.5 | 53.9 | 45.9 | 55.4 | |
| 49.3 | 52.8 | 53.8 | 55.2 | ||||
| δ 31P | 56.5 | 57.3 | 58.1 | 58.8 | 63.2 | 65.3 | |

Single crystals of complexes 3, 5, 6 and 7 suitable for X-ray diffraction analysis were obtained.11 Overall, the four complexes adopt similar structures in the solid state (Fig. 2): the SbF6− counter anion is ion separated and the P^N ligand chelates gold. In all complexes, the gold centre is in trigonal planar environment and the alkene sits in the coordination plane of gold despite the ensuing steric shielding.12 This arrangement ensures optimal overlap between the HOMO of the [(P^N)Au+] fragment (in-plane d-type orbital) and the π*(alkene) orbital. The presence of substantial Au → alkene back-donation is also apparent from the elongation of the CC bond upon coordination to gold (by ∼0.06 Å compared to the free alkene). For comparison with related gold(I) alkene complexes, see Table S4.†11 In all cases, the alkene is η2-coordinated to gold, and complexes 3, 6 and 7 present quasi-symmetric structures with almost identical Au–C bond lengths (Table 2). As expected from the electron-rich and polarized character of DHP, complex 5 adopts a slipped coordination mode (Au–C 2.193(3) and 2.276(3) Å), but the alkene remains η2-coordinated. This situation contrasts with the highly dissymmetric structures (terminal σ-coordination rather than side-on π-coordination) generally observed in the few previously reported gold(I) complexes featuring electron-rich alkenes (Fig. S28†).7 Within the series of π-complexes 3, 5, 6 and 7, little changes are observed in the Au–P bond length [min.–max. values = 2.3032(2) and 2.3393(3) Å], whereas the Au–N bond length varies from 2.234(6) Å in complex 7 with the most electron-poor alkene NPM, to 2.505(2) Å in complex 5 presenting the most electron-rich alkene DHP. This variation attests the adaptative behavior of the P^N ligand.13 The N → Au interaction strengthens/weakens to accommodate the electronic demand at gold depending on the alkene.
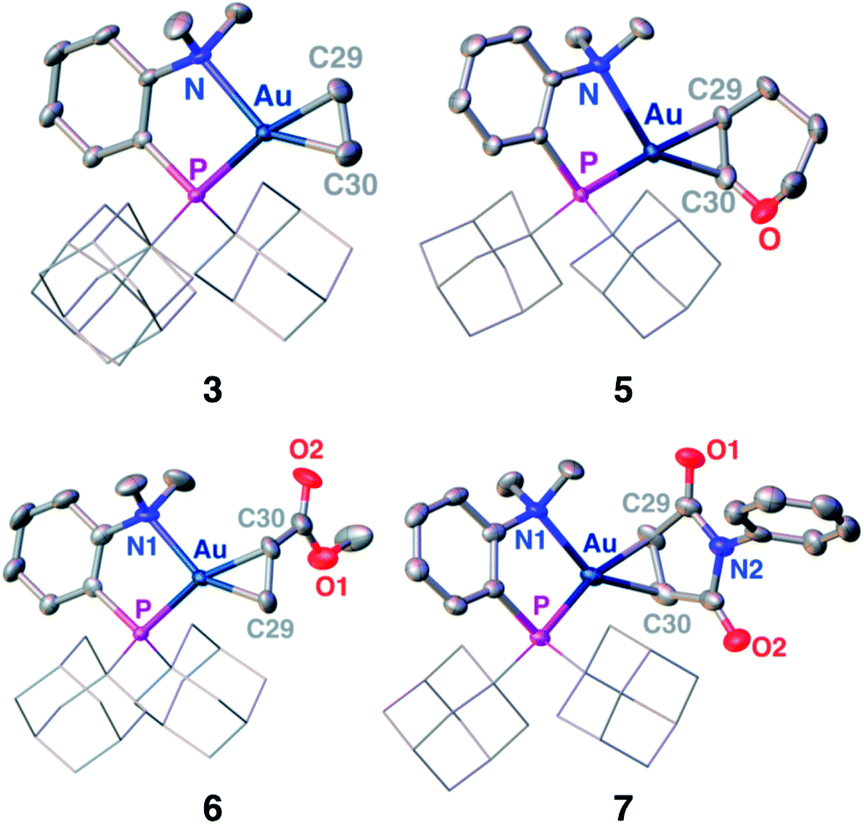 | ||
| Fig. 2 Molecular structures of complexes 3 (ethylene), 5 (DHP), 6 (MA) and 7 (NPM). Hydrogen atoms, counter anions and solvate molecules are omitted; the adamantyl groups are simplified for clarity. | ||


Synthesis and characterization of (P^P)Au(I) π-complexes
To assess the role of the ancillary ligand on the ability of gold(I) to form stable π-complexes with a variety of alkenes, we then turned to the P^P chelating o-carboranyl diphosphine ligand for which a stable styrene complex (8) analogous to the P^N complex 1 was previously prepared.10 The (P^P)AuCl precursor was reacted with AgSbF6 in the presence of an excess of ethylene (9), DHP (10), MA (11) and NPM (12) in dichloromethane (Scheme 2).11 The coordination of ethylene to the [(P^P)Au+] fragment was readily established thanks to 1H NMR spectroscopy: a new singlet appeared at 4.47 ppm, close to that found for the corresponding P^N ethylene complex 3 (4.10 ppm). Complex 9 was isolated as a white solid in good yield (84%). As for the P^P styrene complex, no sign of ethylene dissociation was observed either in solution or under vacuum. Complex 9 was characterized by X-ray diffraction, showing the side-on in plane coordination of ethylene. In contrast with that observed with the MeDalphos ligand, electron-rich and electron-poor alkenes did not form stable complexes with the o-carboranyl diphosphine. With DHP, complex 10 was detected in small amounts by 1H NMR spectroscopy at the beginning of the reaction (Fig. S25†), but it rapidly degrades with oligomerization of DHP. Better results were obtained with the electron-poor alkenes MA and NPM. Complexes 11 and 12 formed quantitatively and could be unambiguously characterized by 1H and 31P NMR in the presence of an excess of alkene (Fig. S26 and S27†), but attempts at isolation proved unsuccessful due to decomposition. These results highlight the faculty of the heteroleptic P^N ligand to accommodate all types of alkenes thanks to its adaptative behavior. The o-carboranyl diphosphine ligand is apparently less flexible electronically and forms isolable gold(I) π-complexes only with unbiased alkenes.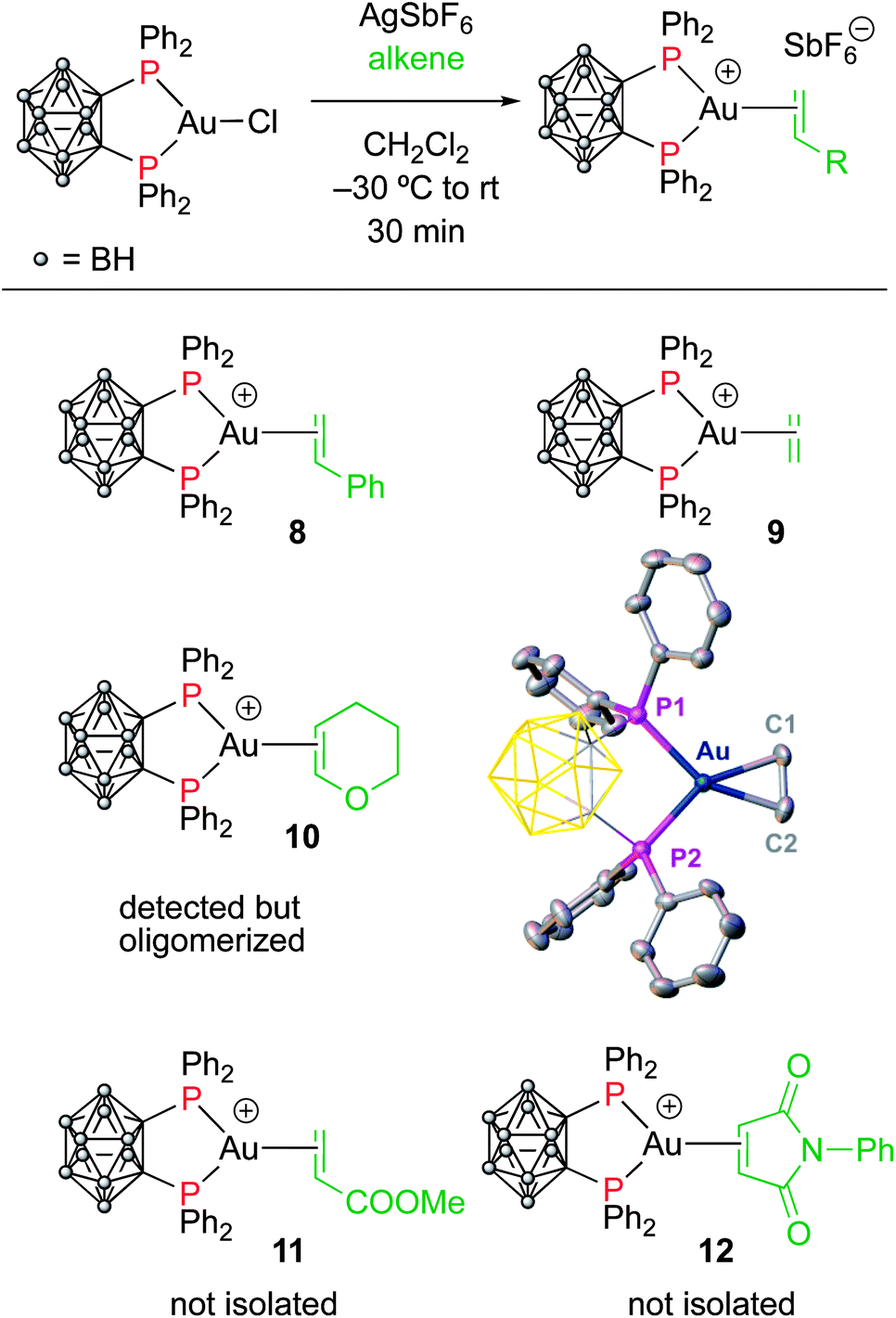 | ||
| Scheme 2 Synthesis of the (P^P) gold(I) complexes 8–12. Molecular structure of complex 9. Hydrogen atoms, counter anions and solvate molecules are omitted; the carboranyl group is simplified for clarity. Selected bond lengths (Å) and angles (°): Au–P1 2.371(2), Au–P2 2.368(2), Au–C1 2.176(9), Au–C2 2.175(10), C1–C2 1.365(15), P1–Au–P2 91.32(7). | ||
Theoretical investigation of (P^N)Au(I) π-complexes
With the aim to better understand the ability of the P^N ligand to accommodate the π-coordination of a wide range of alkenes at gold, a detailed computational study was conducted. To the best of our knowledge, very few computational studies have been performed on tridentate gold(I) alkene complexes. The bonding situation within [(2,2′-bipy)Au(ethylene)+]8b was investigated in 2006 and very recently, calculations were carried out on [(phenanthroline)Au(norbornene)+] complexes.14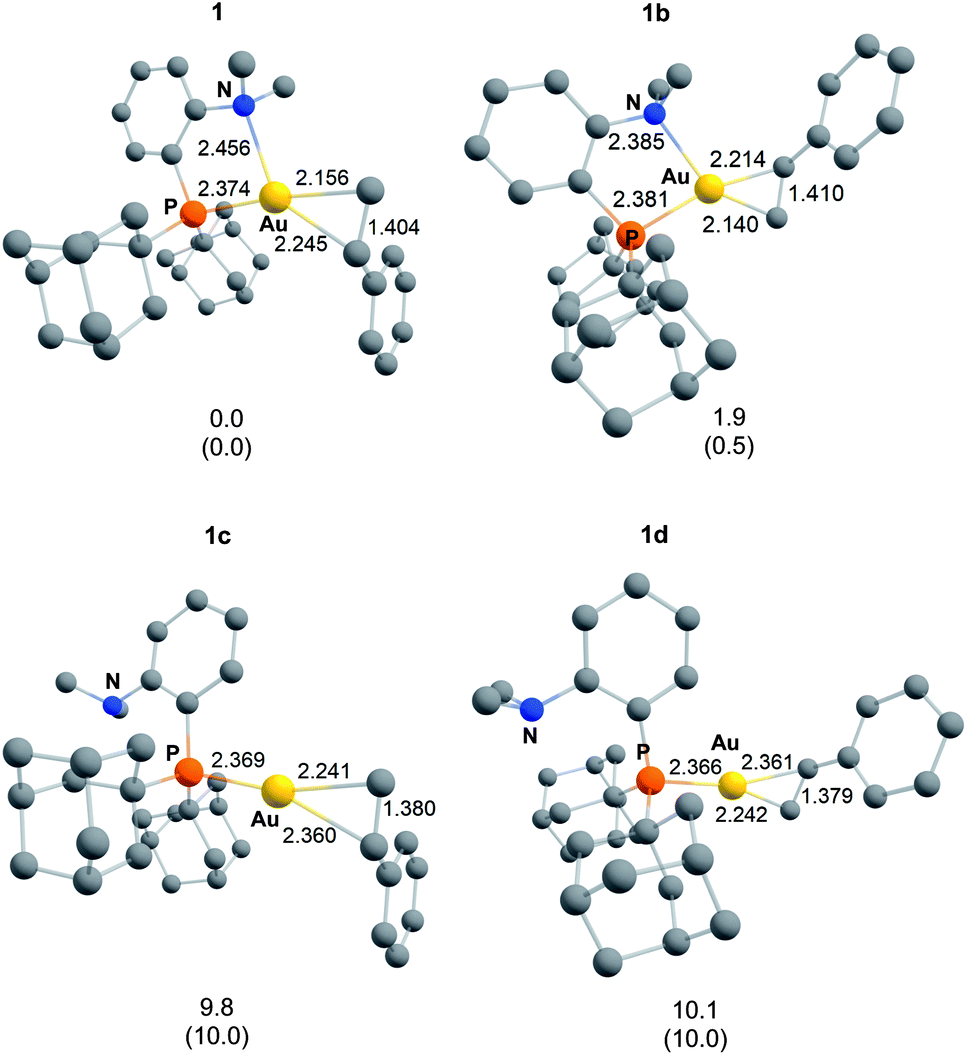 | ||
| Fig. 3 Energy minima located on the potential energy surface of the [(P^N)Au(styrene)+] complex 1. Bond distances in Å, relative energies from the ground-state structure 1 in kcal mol−1 (ΔG values, with ΔE values in brackets). Calculations performed at the SMD-(dichloromethane)-B3PW91/SDD+f(Au),6-31G** (other atoms) level of theory. | ||
|
|
|
|
|
|
|
|
|---|---|---|---|---|---|---|
|
a Difference between the two Au–C bond lengths.
b Difference between CC bond length in the gold complex and in the free alkene.
c Bond indexes [WBI: Wiberg and δ: Bader].
d Stabilizing energy ΔE(2) at 2nd order perturbation theory in kcal mol−1.
e Donation term (d) and back-donation term (b).
f Density in e bohr−3.
g Charge transfer (CT) from Hirshfeld charges.
|
||||||
| Geometrical parameters | ||||||
| Au–C | 2.219 | 2.156 | 2.159 | 2.158 | 2.171 | 2.157 |
| 2.371 | 2.245 | 2.215 | 2.151 | 2.122 | 2.128 | |
| Δ(Au–C)a | 0.152 | 0.089 | 0.056 | 0.007 | 0.049 | 0.029 |
| CC |
1.394 | 1.404 | 1.402 | 1.406 | 1.417 | 1.421 |
| (CC olefin) |
(1.339) | (1.339) | (1.334) | (1.331) | (1.335) | (1.334) |
| Δ(CC)b |
0.055 | 0.065 | 0.068 | 0.075 | 0.082 | 0.087 |
| Au–N | 2.688 | 2.456 | 2.457 | 2.360 | 2.333 | 2.269 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||||
| NBO analysis | ||||||
| WBI(CC)c |
1.477 | 1.475 | 1.501 | 1.489 | 1.437 | 1.341 |
| πCC → LP*(Au)d | 119.8 | 143.7 | 158.2 | 175.0 | 168.1 | 178.3 |
|
23.0 | 36.7 | 38.9 | 45.1 | 45.1 | 48.9 |
| WBI(Au–N)c | 0.048 | 0.085 | 0.093 | 0.125 | 0.138 | 0.167 |
| LP(N) → Aud | 10.8 | 20.0 | 22.0 | 32.4 | 34.0 | 44.0 |
|
||||||
| CDA analysis | ||||||
| d(CC → Au)e | 0.411 | 0.416 | 0.529 | 0.477 | 0.485 | 0.399 |
| b(Au → CC)e | 0.146 | 0.211 | 0.230 | 0.262 | 0.274 | 0.323 |
| d/b | 2.82 | 1.97 | 2.30 | 1.82 | 1.77 | 1.24 |
|
||||||
| AIM analysis | ||||||
|
ρ(CC)f |
0.313 | 0.305 | 0.307 | 0.303 | 0.297 | 0.297 |
|
δ(CC)c |
1.29 | 1.31 | 1.32 | 1.32 | 1.26 | 1.22 |
| ρ(Au–N)f | 0.035 | 0.054 | 0.053 | 0.064 | 0.068 | 0.078 |
| δ(Au–N)c | 0.269 | 0.383 | 0.385 | 0.448 | 0.474 | 0.516 |
|
||||||
| Hirshfeld charge | ||||||
| CT(CC → Au)g | 0.16 | 0.06 | 0.05 | −0.003 | −0.06 | −0.12 |


NBO (Natural Bond Orbital), CDA (Charge Decomposition Analysis) and AIM (Atoms-In-Molecules) approaches were used to thoroughly analyse the bonding situation in complex 1 (Table 3), referring to the dicoordinate forms and free alkene when appropriate.11 Special attention was given to the Au/alkene interaction and N → Au coordination. CDA indicates significant contribution of back-donation in 1 (d/b = 1.97), much more than in the dicoordinate forms (d/b = 3.78). This is in line with the in-plane arrangement of the alkene [the bent P–Au–N fragment formed by chelation features a high-energy occupied d(Au) orbital of suitable geometry for back-donation to the 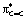 (alkene) orbital].13,15 The
(alkene) orbital].13,15 The 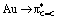 back-donation is also apparent in the NBO analysis as a donor–acceptor interaction with noticeable stabilizing energy (36.7 kcal mol−1). Consistently, the CC double bond is substantially weakened upon coordination to gold, as apparent from the Wiberg bond index (WBI) at 1.475 in 1 (versus 1.900 in free styrene) and from the electron density at the bond-critical point (BCP) ρ(CC) = 0.305 au in 1 (versus 0.341 in free styrene). Back-donation roughly compensates donation so that overall, the alkene to gold charge transfer is small (0.06e as deduced from Hirshfeld charges). NBO and AIM also provide evidence for significant N → Au interaction in 1: a donor–acceptor interaction between N and Au is found at the second order perturbation level in NBO, with a delocalization energy of 20 kcal mol−1, and a BCP with non-negligible electron density [ρ(N–Au) = 0.05 au] is found in AIM.
back-donation is also apparent in the NBO analysis as a donor–acceptor interaction with noticeable stabilizing energy (36.7 kcal mol−1). Consistently, the CC double bond is substantially weakened upon coordination to gold, as apparent from the Wiberg bond index (WBI) at 1.475 in 1 (versus 1.900 in free styrene) and from the electron density at the bond-critical point (BCP) ρ(CC) = 0.305 au in 1 (versus 0.341 in free styrene). Back-donation roughly compensates donation so that overall, the alkene to gold charge transfer is small (0.06e as deduced from Hirshfeld charges). NBO and AIM also provide evidence for significant N → Au interaction in 1: a donor–acceptor interaction between N and Au is found at the second order perturbation level in NBO, with a delocalization energy of 20 kcal mol−1, and a BCP with non-negligible electron density [ρ(N–Au) = 0.05 au] is found in AIM.
As for the styrene complex 1, the alkene is in-plane side-on coordinated to gold and the P^N ligand is chelating. The ground-state structures correspond to those observed experimentally, and their optimized geometries reproduce well those determined cristallographically. All alkenes are coordinated to gold in a quasi-symmetric manner [Δ(Au–C) ≤ 0.056 Å], the largest deviations between the two Au–C bond lengths being observed for styrene (0.089 Å) and for the electron-rich DHP (0.152 Å). Coordination of the alkene to gold induces noticeable elongation of the CC double bond, which increases from 0.055 Å for DHP, to 0.087 Å for NPM. The N–Au distance also varies significantly in the series (by about 20%, from 2.269 Å for NPM, to 2.688 Å or DHP), in line with that observed cristallographically.
CDA indicates marked differences in the Au/alkene interaction as well, with the d/b ratio varying from 2.8 for DHP to 1.2 for NPM. This evolution is mainly driven by the 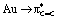 back-donation term (πCC → Au donation varies less), in line with the energy of the frontier molecular orbitals of the alkenes [the π(CC) orbital varies less in energy than the π*(CC), Fig. S32†]. The variation of the d/b ratio is accompanied by an inversion of the alkene → Au charge transfer, from 0.16e for DHP to −0.12e for NPM. According to NBO and AIM, the weakening of the CC double bond upon coordination to gold increases from electron-rich to electron-poor alkenes, consistent with the strengthening of
back-donation term (πCC → Au donation varies less), in line with the energy of the frontier molecular orbitals of the alkenes [the π(CC) orbital varies less in energy than the π*(CC), Fig. S32†]. The variation of the d/b ratio is accompanied by an inversion of the alkene → Au charge transfer, from 0.16e for DHP to −0.12e for NPM. According to NBO and AIM, the weakening of the CC double bond upon coordination to gold increases from electron-rich to electron-poor alkenes, consistent with the strengthening of 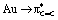 back-donation (and decrease of the d/b ratio): the Wiberg bond index decreases from 1.477 for DHP, to 1.341 for NPM, while the electron density at the bond-critical point ρ(CC) decreases from 0.313 (DHP) to 0.297 (NPM).
back-donation (and decrease of the d/b ratio): the Wiberg bond index decreases from 1.477 for DHP, to 1.341 for NPM, while the electron density at the bond-critical point ρ(CC) decreases from 0.313 (DHP) to 0.297 (NPM).
It is also noteworthy that the P^N ligand strongly responds to the electronic requirements at gold by strengthening/weakening the N → Au coordination. The delocalization energy associated with the donor–acceptor interaction (as found in the NBO analysis) increases from 10.8 kcal mol−1 for DHP, to 44.0 kcal mol−1 for NPM. In parallel, AIM calculations show an increase of the electron density at the bond critical point ρ(N–Au) from 0.269 for DHP, to 0.516 for NPM.
Parametrization of the P^N ligand response as a function of the alkene coordination to gold
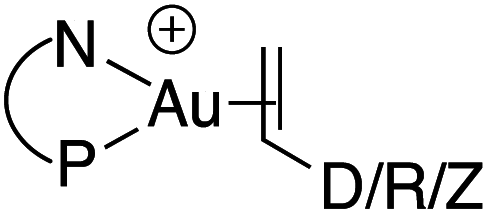
Having in hand a range of experimental and theoretical data on a series of π-alkene complexes gave us the possibility to analyse in detail how the P^N ligand responds to the electronics of the alkene coordinated to gold. We looked in particular for correlations between the P^N ligand behaviour and the Au/alkene interaction.16 Based on the previous detailed analyses, four descriptors were chosen to account for the P^N ligand coordination: the Au–N distance,17 the 1H NMR chemical shift of the NMe2 group, the electron density at the N–Au bond critical point ρ(N–Au) (AIM) and the 31P NMR chemical shift. For the Au/alkene interaction, three parameters were used: the d/b ratio (CDA), the difference in the electron density at the CC bond critical point between the coordinated and free alkene Δρ(CC) (AIM) and the charge transfer from the alkene to the [(P^N)Au+] fragment (Hirshfeld charges).
A very similar and general trend is observed for all the examined sets of descriptors (Fig. 4): linear relationships are found between the P^N ligand behaviour and the alkene coordination to gold. Regression factors are generally above 0.92 and all are above 0.84. Most noteworthy is the intimate correlation between the strength of N → Au coordination and the Au/alkene interaction. These linear correlations highlight the progressive response of the P^N ligand to the alkene electronics. Thanks to its hemilability, the P^N ligand displays non-innocent and adaptative character which is key to accommodate the π-coordination of a wide range of alkenes at gold, from electron-rich to electron-poor ones.
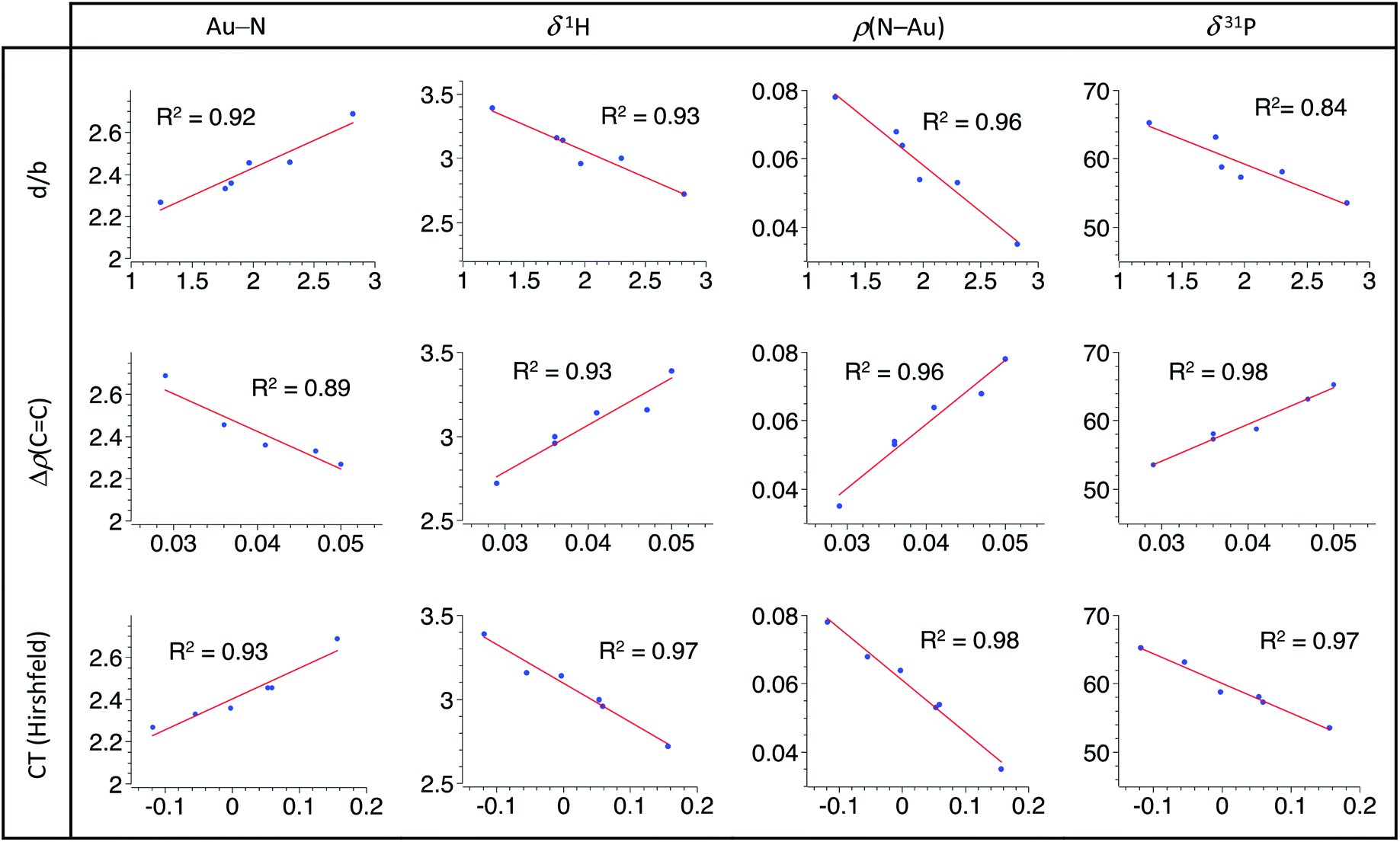 | ||
| Fig. 4 Correlation graphs accounting for the response of the P^N ligand (as monitored by the N⋯Au bond length in Å, the 1H NMR chemical shift of the NMe2 group in ppm, the electron density at the N–Au bond critical point in au, the 31P NMR chemical shift in ppm) to the alkene coordination to gold (as characterized by the d/b ratio, the difference in the electron density at the CC bond critical point between the coordinated and free alkene in au, the charge transfer from the alkene to the [(P^N)Au+] fragment in e), including linear regressions and associated R-factors. | ||
Catalytic relevance of the (P^N)Au(I) π-complexes
Finally, we sought to show that the gold(I) π-complexes isolated thanks to the P^N ligand exhibit reactivity typical of transient π-alkene gold(I) complexes and are catalytically relevant. To this end, the intermolecular hydroarylation of alkenes with indoles, as pioneered by M.-K. Wong and C.-M. Che,18 was chosen.19 In our previous study, MeDalphos gold(I) chloride was shown to catalyze the hydroarylation of styrene to give indoles 13a,b and the role of the gold(I) π-complex as key intermediate was supported.10 Here, π-systems with distinct electronic properties, MA and NPM as electron-poor alkenes and DHP as electron-rich alkene, were tested (Scheme 3).11 In all cases, the hydroarylation reaction proceeded and complete selectivity for C3-alkylation of the indole was observed. The obtained compounds 13 result from nucleophilic attack of the indole to the most electrophilic position of the π-coordinated alkene, resulting in Markovnikov or anti-Markovnikov products depending on the substrate.20 In the case of DHP, the double addition products 13e,f were quantitatively obtained. They result from facile ring-opening of the first-formed hydroarylation product upon addition of a second indole molecule.21 The fate of the hydroarylation reaction of DHP was unambiguously established by using 6-nitroindole, for which the resulting product 13g could be characterized by X-ray diffraction.11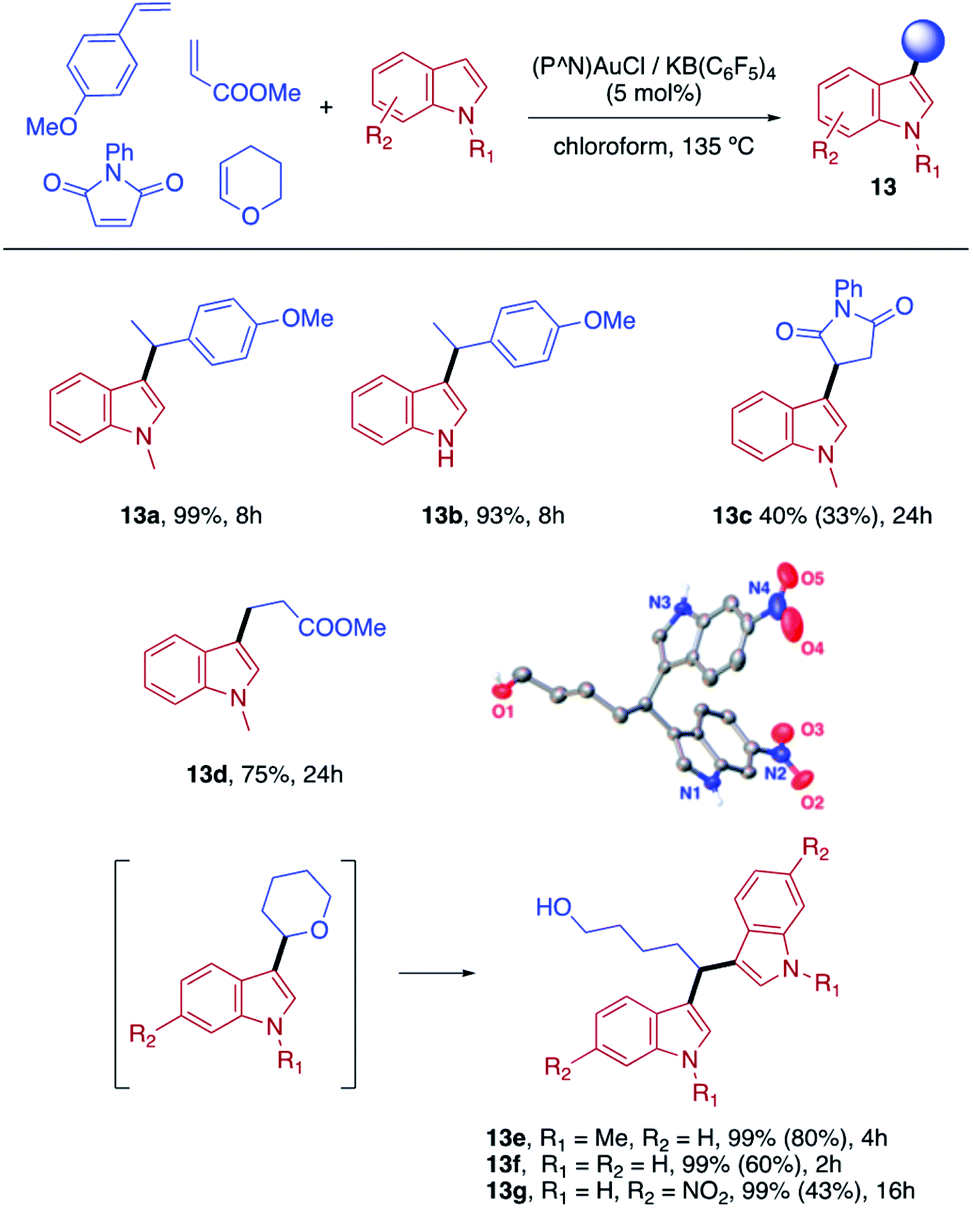 | ||
| Scheme 3 Hydroarylation of alkenes with indoles catalyzed by the (P^N) gold(I) complex. Conversions determined by 1H NMR spectroscopy; isolated yields after column chromatography in brackets. Crystallographic structure of compound 13g as an insert. | ||
Conclusions
In summary, the P^N ligand MeDalphos was found to enable access to stable gold(I) π-complexes with a wide range of alkenes, from unbiased to electron-rich as well as electron-poor alkenes. Precedents of gold complexes deriving from electron-rich alkenes are rare,7 and in the examples reported so far, the CC bond tends to coordinate in a terminal or highly dissymmetric fashion. Here, dihydropyrane (DHP) was found to coordinate to the [(P^N)Au+] fragment more unsymmetrically than the other alkenes. Yet it remains side-on coordinated, with weakened but not negligible Au → alkene back-donation. To the best of our knowledge, the methylacrylate (MA) and N-phenylmaleimide (NPM) adducts are the first examples of gold π-complexes with electron-poor alkenes.22
The P^N ligand displays adaptative character and accommodates the electron demand at gold as a function of the coordinated alkene by weakening/strengthening the N → Au coordination. This “chameleon” behaviour enables to access stable π-complexes with various degrees of alkene → Au donation/Au → alkene back-donation, CC bond weakening and alkene to gold charge transfer. Detailed examination of the structural, electronic and spectroscopic data collected experimentally and computationally for the series of π-complexes reveal direct and linear response of the P^N ligand to the Au/alkene coordination.
These results highlight a new feature of the MeDalphos ligand framework in gold chemistry, besides its ability to trigger Au(I)/Au(III) two-electron redox transformation13,23 and to support hydrogen bonding to gold(I).24 It is also noteworthy that the isolated P^N gold(I) π-complexes are reactive and amenable to catalysis, as substantiated by the chemo and regio-selective alkylation of indoles. The use of bidentate ligands in π-activation of alkenes at gold may be very interesting to circumvent the inherent difficulty associated with monodentate ligands and two-coordinate linear complexes when dealing with asymmetric catalysis using chiral ligands.25
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Centre National de la Recherche Scientifique and the Université de Toulouse is gratefully acknowledged. M. N. thanks the Fonds National Suisse de la Recherche Scientifique for an Early Postdoc Mobility fellowship. A. T. thanks Spanish MINECO for a mobility fellowship (BES-2014-067770 and EEBB-I-18-12874). S. Massou and P. Lavedan (ICT) are is acknowledged for his assistance with NMR experiments. The “Direction du Numérique” of the Université de Pau et des Pays de l'Adour, MCIA (Mésocentre de Calcul Intensif Aquitain) and CINES under allocation A005080045 made by Grand Equipement National de Calcul Intensif (GENCI) are acknowledged for computational facilities.Notes and references
- B. Hammer and J. K. Norskov, Nature, 1995, 376, 238–240 CrossRef CAS.
- (a) G. Dyker, Angew. Chem., Int. Ed., 2000, 39, 4237–4239 CrossRef CAS; (b) A. S. K. Hashmi, Gold Bull., 2004, 37, 51–65 CrossRef CAS; (c) A. Hoffman-Röder and N. Krause, Org. Biomol. Chem., 2005, 3, 387–391 RSC; (d) A. S. K. Hashmi and F. D. Toste, Modern Gold Catalyzed Synthesis, Wiley-VCH, 2012 CrossRef; (e) D. Toste and V. Michelet, Gold Catalysis: An Homogeneous Approach, Imperial College Press, 2014 Search PubMed; (f) L. M. Slaughter, Homogeneous Gold Catalysis, Springer, 2015 CrossRef.
- (a) C. Winter and N. Krause, Chem. Rev., 2011, 111, 1994–2009 CrossRef PubMed; (b) M. Chiarucci and M. Bandini, Beilstein J. Org. Chem., 2013, 9, 2586–2614 CrossRef PubMed; (c) C. Obrados and A. M. Echavarren, Chem. Commun., 2014, 50, 16–28 RSC; (d) R. Doral and A. M. Echavarren, Chem. Rev., 2015, 115, 9028–9072 CrossRef PubMed; (e) C. J. V. Halliday and M. Lynam, Dalton Trans., 2016, 45, 12611–12626 RSC; (f) J. L. Mascareñas, I. Varela and F. López, Acc. Chem. Res., 2019, 52, 465–479 CrossRef PubMed.
- (a) H. Schmidbaur and A. Schier, Organometallics, 2010, 29, 2–23 CrossRef CAS; (b) M. A. Cinellu in Modern Gold Catalyzed Synthesis, Wiley-VCH, Weinheim, 2012, pp. 175–199 Search PubMed; (c) R. E. M. Brooner and R. A. Widenhoefer, Angew. Chem., Int. Ed., 2013, 52, 11714–11724 CrossRef CAS PubMed; (d) A. C. Jones, Top. Curr. Chem., 2015, 357, 133–166 CrossRef CAS PubMed.
- (a) D. B. Dell'Amico, F. Calderazzo, R. Dantona, J. Strähle and H. Weiss, Organometallics, 1987, 6, 1207–1210 CrossRef; (b) R. M. Davila, R. J. Staples and J. P. Fackler Jr, Organometallics, 1994, 13, 418–420 CrossRef CAS; (c) T. J. Brown, M. G. Dickens and R. A. Widenhoefer, J. Am. Chem. Soc., 2009, 131, 6350–6351 CrossRef CAS PubMed; (d) T. N. Hooper, M. Green, J. E. McGrady, J. R. Patel and C. A. Russell, Chem. Commun., 2009, 3877–3879 RSC.
- Gold(III) π complexes are also known, but rare, see: (a) N. Salvjani, D. Rosca, M. Schormann and M. Bochmann, Angew. Chem., Int. Ed., 2013, 52, 874–877 CrossRef PubMed; (b) E. Langseth, M. L. Scheuermann, D. Balcells, W. Kaminsky, K. I. Goldberg, O. Eisenstein, R. H. Heyn and M. Tilset, Angew. Chem., Int. Ed., 2013, 52, 1660–1663 CrossRef CAS PubMed; (c) C. Blons, A. Amgoune and D. Bourissou, Dalton Trans., 2018, 47, 10388–10393 RSC.
- (a) A. Fürstner, M. Alcarazo, R. Goddard and C. W. Lehmann, Angew. Chem., Int. Ed., 2008, 47, 3210–3214 CrossRef PubMed; (b) R. A. Sanguramath, T. N. Hooper, C. P. Butts, M. Green, J. E. McGrady and C. A. Russell, Angew. Chem., Int. Ed., 2011, 50, 7592–7595 CrossRef CAS PubMed; (c) Y. Zhu, C. S. Day and A. C. Jones, Organometallics, 2012, 31, 7332–7335 CrossRef CAS; (d) M. Sriram, Y. Zhu, A. M. Camp, C. S. Day and A. C. Jones, Organometallics, 2014, 33, 4157–4164 CrossRef CAS; (e) A. Zhdanko and M. E. Maier, Angew. Chem., Int. Ed., 2014, 53, 7760–7764 CrossRef CAS PubMed.
- (a) M. A. Cinellu, G. Minghetti, S. Stoccoro, A. Zucca and M. Manassero, Chem. Commun., 2004, 1618–1619 RSC; (b) M. A. Cinellu, G. Minghetti, F. Cocco, S. Stoccoro, A. Zucca, M. Manassero and M. Arca, Dalton Trans., 2006, 5703–5716 RSC; (c) J. Wu and H. V. R. Dias, Angew. Chem., Int. Ed., 2004, 46, 7814–7816 Search PubMed; (d) J. A. Flores and H. V. R. Dias, Inorg. Chem., 2008, 47, 4448–4450 CrossRef CAS PubMed; (e) H. V. R. Dias and J. Wu, Organometallics, 2012, 31, 1511–1517 CrossRef CAS; (f) S. G. Ridlen, J. Wu, N. V. Kulkarni and H. V. R. Dias, Eur. J. Inorg. Chem., 2016, 2573–2580 CrossRef CAS; (g) K. Klimovica, K. Kirschbaum and O. Daugulis, Organometallics, 2016, 35, 2938–2943 CrossRef CAS PubMed; (h) M. J. Harper, C. J. Arthur, J. Crosby, E. J. Emmet, R. L. Falconer, A. J. Fensham-Smith, P. J. Gates, T. Leman, J. E. McGrady, J. F. Bower and C. A. Russell, J. Am. Chem. Soc., 2018, 140, 4440–4445 CrossRef CAS PubMed; (i) Y. Yang, P. Antoni, M. Zimmer, K. Sekine, F. F. Mulks, L. Hu, L. Zhang, M. Rudolph, F. Rominger and A. S. K Hashmi, Angew. Chem., Int. Ed., 2019, 58, 5129–5133 CrossRef CAS PubMed.
- M. A. Gimeno and A. Laguna, Chem. Rev., 1997, 97, 511–522 CrossRef CAS PubMed.
- M. Navarro, A. Toledo, M. Joost, A. Amgoune, S. Mallet-Ladeira and D. Bourissou, Chem. Commun., 2019, 55, 7974–7977 RSC.
- See the ESI for details.†.
- Only one form is found for the non-symmetric complexes 1, 5 and 6. The Ph groups of styrene and the O atom of DHP sit in the cis position to the PAd2 group in 1 and 5, while the CO2Me group of MA sits in the cis position to the NMe2 group in 6. These are also the only forms present in solution for complexes 1 and 6, as supported by 1H–15N HSQC NMR experiments.11.
- We have previously shown that the MeDalphos ligand enables oxidative addition to gold and promote Au(I)/Au(III) cycling thanks to its hemilabile character: (a) A. Zeineddine, L. Éstevez, S. Mallet-Ladeira, K. Miqueu, A. Amgoune and D. Bourissou, Nat. Commun., 2017, 8, 565–572 CrossRef PubMed; (b) J. Rodríguez, A. Zeineddine, E. D. Sosa-Carrizo, K. Miqueu, N. Saffon-Merceron, A. Amgoune and D. Bourissou, Chem. Sci., 2019, 10, 7183–7192 RSC.
- M. A. Cinellu, M. Arca, F. Ortu, S. Stoccoro, A. Zucca, A. Pintus and L. Maiore, Eur. J. Inorg. Chem., 2019, 4784–4795 CrossRef CAS.
- (a) M. Joost, A. Zeineddine, L. Estévez, S. Mallet-Ladeira, K. Miqueu, A. Amgoune and D. Bourissou, J. Am. Chem. Soc., 2014, 136, 14654–14657 CrossRef CAS PubMed; (b) M. Joost, L. Estévez, S. Mallet-Ladeira, K. Miqueu, A. Amgoune and D. Bourissou, Angew. Chem., Int. Ed., 2014, 53, 14512–14516 CrossRef CAS PubMed; (c) M. Joost, L. Estévez, K. Miqueu, A. Amgoune and D. Bourissou, Angew. Chem., Int. Ed., 2015, 54, 5236–5240 CrossRef CAS PubMed.
- For recent reviews on parametrizing ligand properties and interrogation of structure/function relationship in transition metal chemistry, see: (a) M. S. Sigman, K. C. Harper, E. N. Bess and A. Milo, Acc. Chem. Res., 2016, 49, 1292–1301 CrossRef CAS PubMed; (b) D. J. Durand and N. Fey, Chem. Rev., 2019, 119, 6561–6594 CrossRef CAS PubMed.
- The value for the DFT-optimized structure was used for all complexes so that the correlation covers all alkenes, including hex-1-ene..
- M.-Z. Wang, M.-K. Wong and C.-M. Che, Chem.–Eur. J., 2008, 14, 8353–8364 CrossRef CAS PubMed.
- For a review on gold-catalyzed functionalization of indoles, see: V. Pirovano, Eur. J. Org. Chem., 2018, 1925–1945 CrossRef CAS.
- For a recent theoretical study of the regioselectivity of nucleophilic addition to dicoordinate π-alkene gold(I) complexes, see: A. Couce-Rios, A. Lledós, I. Fernández and G. Ujaque, ACS Catal., 2019, 9, 848–858 CrossRef CAS.
- J. S. Yadav, B. V. S. Reddy, G. Satheesh, A. Prabhakar and A. C. Kunwar, Tetrahedron Lett., 2003, 44, 2221–2224 CrossRef CAS.
- For gold-catalyzed transformations involving electron-poor alkenes as substrates, see: (a) F. Medina, C. Michon and F. Agbossou-Niedercorn, Eur. J. Org. Chem., 2012, 6218–6227 CrossRef CAS; (b) V. Pirovano, D. Facoetti, M. Dell'Acqua, E. Della Fontana, G. Abbiati and E. Rossi, Org. Lett., 2013, 15, 3812–3815 CrossRef CAS PubMed; (c) X. Hu, D. Martin, M. Melaimi and G. Bertrand, J. Am. Chem. Soc., 2014, 136, 13594–13597 CrossRef CAS PubMed.
- (a) M. O. Akram, A. Das, I. Chakrabarty and N. T. Patil, Org. Lett., 2019, 21, 8101–8105 CrossRef CAS PubMed; (b) J. Rodríguez, N. Adet, N. Saffon-Merceron and D. Bourissou, Chem. Commun., 2020, 56, 94–97 RSC.
- (a) M. Rigoulet, S. Massou, E. D. Sosa Carrizo, S. Mallet-Ladeira, A. Amgoune, K. Miqueu and D. Bourissou, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 46–51 CrossRef CAS PubMed; (b) H. Schmidbaur, Angew. Chem., Int. Ed., 2019, 58, 5806–5809 CrossRef CAS PubMed.
- (a) F. López and J. L. Mascareñas, Beilstein J. Org. Chem., 2013, 9, 2250–2264 CrossRef PubMed; (b) Y.-M. Wang, A. D. Lackner and F. D. Toste, Acc. Chem. Res., 2014, 47, 889–901 CrossRef CAS PubMed; (c) W. Zi and F. D. Toste, Chem. Soc. Rev., 2016, 45, 4567–4589 RSC; (d) Y. Li, W. Li and J. Zhang, Chem.–Eur. J., 2017, 23, 467–512 CrossRef CAS PubMed; (e) J. Rodríguez and D. Bourissou, Angew. Chem., Int. Ed., 2018, 57, 386–388 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and computational details, analytical data and NMR spectra. CCDC 1967901 (3), 1967904 (5), 1967902 (6), 1967903 (7), 1967905 (9) and 1967906 (13g). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc06398f |
| This journal is © The Royal Society of Chemistry 2020 |