
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCompartmentalized cross-linked enzymatic nano-aggregates (c-CLEnA) for efficient in-flow biocatalysis†
M. Teresa
De Martino
 a,
Fabio
Tonin
b,
N. Amy
Yewdall
a,
Mona
Abdelghani
a,
David S.
Williams‡
a,
Ulf
Hanefeld
b,
Floris P. J. T.
Rutjes
c,
Loai K. E. A.
Abdelmohsen
*a and
Jan C. M.
van Hest
*a
a,
Fabio
Tonin
b,
N. Amy
Yewdall
a,
Mona
Abdelghani
a,
David S.
Williams‡
a,
Ulf
Hanefeld
b,
Floris P. J. T.
Rutjes
c,
Loai K. E. A.
Abdelmohsen
*a and
Jan C. M.
van Hest
*a
aDepartment of Bio-Organic Chemistry, Institute for Complex Molecular Systems (ICMS) Eindhoven University of Technology, Het Kranenveld 14, 5600 MB Eindhoven, The Netherlands. E-mail: j.c.m.v.hest@tue.nl; l.k.e.a.abdemohsen@tue.nl
bDepartment of Biotechnology, Delft University of Technology, Van der Maasweg 9, 2629 HZ Delft, The Netherlands
cInstitute for Molecules and Materials, Radboud University, Heyendaalseweg 135, 6525 AJ Nijmegen, The Netherlands
First published on 7th February 2020
Abstract
Nano-sized enzyme aggregates, which preserve their catalytic activity are of great interest for flow processes, as these catalytic species show minimal diffusional issues, and are still sizeable enough to be effectively separated from the formed product. The realization of such catalysts is however far from trivial. The stable formation of a micro-to millimeter-sized enzyme aggregate is feasible via the formation of a cross-linked enzyme aggregate (CLEA); however, such a process leads to a rather broad size distribution, which is not always compatible with microflow conditions. Here, we present the design of a compartmentalized templated CLEA (c-CLEnA), inside the nano-cavity of bowl-shaped polymer vesicles, coined stomatocytes. Due to the enzyme preorganization and concentration in the cavity, cross-linking could be performed with substantially lower amount of cross-linking agents, which was highly beneficial for the residual enzyme activity. Our methodology is generally applicable, as demonstrated by using two different cross-linkers (glutaraldehyde and genipin). Moreover, c-CLEnA nanoreactors were designed with Candida antarctica Lipase B (CalB) and Porcine Liver Esterase (PLE), as well as a mixture of glucose oxidase (GOx) and horseradish peroxidase (HRP). Interestingly, when genipin was used as cross-linker, all enzymes preserved their initial activity. Furthermore, as proof of principle, we demonstrated the successful implementation of different c-CLEnAs in a flow reactor in which the c-CLEnA nanoreactors retained their full catalytic function even after ten runs. Such a c-CLEnA nanoreactor represents a significant step forward in the area of in-flow biocatalysis.
Introduction
In recent years microreactor technology for continuous flow catalysis has developed into an attractive alternative for traditional batch processes, because of the higher level of control over mass and heat transfer.1–5 Reactions in-flow are furthermore scalable and offer broader windows of operation and automated optimization protocols.6,7 In case catalytic steps are involved, heterogeneous catalysts are preferred from a processing point of view, as they enable easy separation from the product flow and allow catalyst recycling.8 Catalyst immobilization however can also lead to some disadvantages, such as diffusional limitations and problems associated with catalyst leaching.9,10The current implementation of enzymes in in-flow reactions relies on their immobilization, which mostly involves binding and/or physical adsorption to porous or solid supports (e.g. resin or silica) or encapsulation in an inert carrier, affording a biocatalyst with acceptable performance in-flow.11,12 Indeed, immobilization is advantageous in terms of reusability and stability and is a well-established route for the commercial application of enzyme-based catalysis.10 However, these traditional types of immobilization often affect enzyme performance, provide limited physical protection and leaching remains an important issue which limits the recyclability.13 Leaching can be limited via the formation of cross-linked enzyme aggregates (CLEAs).14 CLEAs provide enhanced stability, with the ability to be recaptured, recycled and reused after filtration or centrifugation processes.12 CLEAs have been studied over the past decade as a green and facile route towards industrial catalysis.10 However, due to their broad distribution of size, such CLEAs are less well-defined regarding enzyme display and are therefore less effectively introduced in microflow systems. In an ideal situation, catalytic systems can operate freely in solution without diffusional barriers, and can still be effectively separated and recovered from the product flow. This can for example be achieved by applying nanoreactors, nanoparticles functionalized with catalytic species that function as homogeneous catalysts in a continuous flow system.15,16 In our group we have much experience with employing copolymeric vesicles or polymersomes, as functional nanoreactors due to their ability to encapsulate enzymes.17–20 Over the past years we have developed two different strategies; we have constructed semi-permeable vesicles in which the enzymes were encapsulated in the lumen,21 and we have created bowl-shaped polymersomes, also known as stomatocytes, in which the enzymes were engulfed in the bowl, or stomach of the particles.22 In both cases the robust polymer membrane provided protection against mechanical forces and enabled physical separation from undesired interactions with other catalytic species. In the case of stomatocytes, diffusional barriers are hardly present, as the stomach is in direct contact with the reaction medium. However, the physical encapsulation of enzymes inside the stomatocytes' cavity still poses a challenge; the encapsulated enzymes are prone to leaching and the nanoreactors are subsequently deactivated upon extended usage.
Herein, we present the formation of nanoreactors that combine the unique structural elements of copolymeric colloidal stomatocytes with the functional elements of CLEAs, displaying the potential of such robust catalytic systems, for the first time, in a flow reactor. Our strategy to achieving this is based on the utility of the stomach of poly(ethylene glycol)-polystyrene (PEG-PS) stomatocytes for in situ formation of compartmentalized cross-linked enzyme nano-aggregates (c-CLEnA) (Fig. 1). Remarkably, the amount of cross-linkers needed was much less compared to the macroscopic CLEA process, which resulted in a much better preservation of catalytic activity. We first demonstrated the feasibility of the c-CLEnA process using the lipase Candida antarctica Lipase B (CalB). Indeed, upon formation of the CalB c-CLEnA nanoreactors the PEG-PS stomatocytes retained their structural integrity and demonstrated enzymatic activity similar to the free enzyme. To demonstrate the generality of this approach, we repeated the process to generate c-CLEnAs with different enzymes, including porcine liver esterase (PLE) and an enzymatic cascade involving glucose oxidase (GOx) and horse radish peroxidase (HRP). Importantly, reactions using the c-CLEnA nanoreactors were successfully implemented in a flow reactor where there was no apparent loss of activity, even after ten cycles.
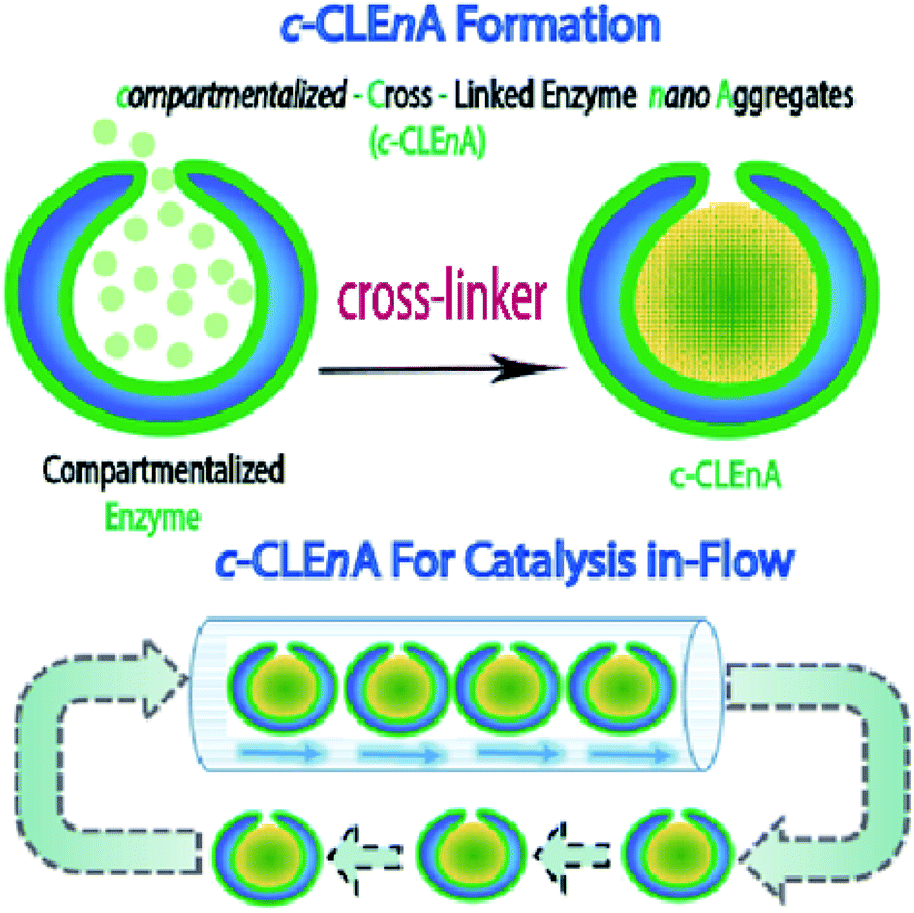 | ||
| Fig. 1 (Top) Formation of compartmentalized-cross-linked enzyme nano-aggregates (c-CLEnA) via cross-linker addition (either glutaraldehyde or genipin). (Bottom) c-CLEnA application and reuse for in-flow catalysis. | ||
Results and discussions
Recently, we presented a robust platform for the compartmentalization of enzymes in bowl-shaped stomatocyte nanoparticles.18,22 Such stomatocytes were formed through osmotic-induced shape transformation of spherical polymer vesicles (polymersomes) that were comprised of poly(ethylene glycol)-block-poly(styrene) (PEG-PS) copolymers. Stomatocytes are able to encapsulate one, or more enzymatic species within their inner compartment, with relatively high encapsulation efficiency ranging from 8 to 35%, dependent on the initial feed and the enzyme properties. The high local concentration of catalyst and the easy accessibility of the substrate toward the cavity, makes stomatocytes an interesting tool in catalytic aqueous processes. In our present strategy, we utilized PEG-PS stomatocytes to entrap proteins, which were subsequently turned into cross-linked enzyme nano-aggregates (CLEnA) under the action of glutaraldehyde (Fig. 2A) or genipin. | ||
| Fig. 2 Transmission electron microscopy (TEM) images of (A and B) CalB loaded stomatocytes with narrow neck. (C and D) Stomatocytes compartmentalizing cross-linked CalB; CalB c-CLEnA. Scale bars correspond to 200 nm. | ||
In the first instance, glutaraldehyde was used to validate our ability to cross-link enzymes insides the cavity of the stomatocytes. To this end, formation of c-CLEnA was initiated by encapsulation of the 34 kDa lipase CalB within PEG-PS stomatocytes under relatively mild conditions, whereby CalB (12 mg mL−1) was added to open neck stomatocytes (0.5 mL, 10 mg mL−1), and the enzyme was encapsulated in the stomatocytes' cavity by narrowing the neck via a shape change process induced by a small aliquot of organic solvent and subsequent washing with salt solution (Fig. S1 and S2†).18,22 Afterwards, stomatocytes were dispersed in phosphate-buffered saline (PBS), and glutaraldehyde (0.1 mL, 150 mM) was slowly added. Employing this CLEA-based enzyme cross-linking strategy, in which lysine residues are chemically bonded, templated formation of CLEnA was performed in the stomach of the PEG-PS stomatocytes. The resulting c-CLEnAs maintained their unique morphological characteristics (Fig. 2B) with increasing internal density (due to CLEnA formation). This was also confirmed via analysis of the shape factor, ρ (the ratio between radius of gyration, Rg, and hydrodynamic radius, Rh), where the ρ value of stomatocytes compartmentalizing CalB CLEnA was ca. 0.73 (Fig. S3†), indicative of the formation of a solid sphere.22,23 In order to characterize the nature of the CLEnA formed during this process, stomatocytes were unloaded via adding an excess of organic solvent, which dissolved the polymer shell so that the templated CalB particles could be characterized. Dynamic Light Scattering (Fig. S4†) analysis showed formation of CLEnA particles of ca. 45 nm. This size was larger than the neck size of the PEG-PS stomatocytes (ca. 5 nm, Fig. 2A), ensuring better entrapment than the free CalB enzymes. Furthermore, from SDS-PAGE gel analysis it was evident that no monomeric enzyme existed (Fig. S5†), as only a high molecular weight species was observed, which showed that the cross-linking process proceeded with high efficiency. When CalB was free in solution, similar conditions of cross-linking led to formation of CLEAs, however, with less control over the size of the structures and a broad size distribution ranging from 10 to 100 μm, as confirmed by scanning electron microscopy (Fig. S6†).
To demonstrate the ability of the formed c-CLEnA to function as catalytic system we measured the activity of the CalB-mediated hydrolysis of p-nitrophenyl acetate (pNPA). The activity of c-CLEnA was superior to that of the free enzyme or the encapsulated CalB into stomatocytes prior to cross-linking (Fig. S7†). Although this seems remarkable, as activity is normally diminished due to cross-linking with glutaraldehyde, CalB is a known exception to this rule.24,25 In order to confirm the generality of our methodology, we set out to cross-link a different enzyme, PLE, employing the same conditions as for CalB (0.1 mL of 150 mM of glutaraldehyde, ESI† 2.3 and 2.4). However, these initial conditions had a strong negative impact on PLE activity (Fig. S8A†). Through careful tuning of the amount of glutaraldehyde employed, we were able to induce the formation of PLE c-CLEnAs under milder conditions by using 50 mM of glutaraldehyde instead of 150 mM. These milder conditions of cross-linking led to a higher VMAX (Fig. 3A and S8B†). It is worth to mention that 50 mM glutaraldehyde was not enough to cross-link PLE present in a free enzyme solution when the same ratio enzyme/glutaraldehyde was used and even the addition of 75 mM and 100 mM of glutaraldehyde led only to partial cross-linking of the free enzyme (Fig. S9†). This highlights the advantageous effect of enzyme compartmentalization as it increases the enzyme local concentration and facilitates the cross-linking process. Since genipin is known to be a milder cross-linker and is widely used in applications in medicine and food technology,26–28 we set out to cross-link PLE using genipin instead of glutaraldehyde – aiming at fully preserving its activity. Gratifyingly, when genipin was used (30–50 mM), the activity of PLE c-CLEnA was maintained and no decrease of enzyme activity was observed (Fig. S8C and D†). Notably, the addition of 30 mM of genipin has a positive impact on the activity (Fig. 3C and S8D†). Besides, the aspect of the PLE nano-aggregates and the shape of the stomatocyte nanoreactor was not affected by genipin (Fig. S10 and S18†). Furthermore, to demonstrate that this method is not restricted to single enzyme systems we generated a dual enzyme c-CLEnA where glucose oxidase (GOx) and horseradish peroxidase (HRP) were co-encapsulated in stomatocytes prior to cross-linking. The presence of both enzymes within the stomatocyte was confirmed by SDS-PAGE (Fig. S11†). Again, cross-linking inside the stomatocytes proved to be a more efficient process than cross-linking of a free enzyme solution at same concentrations. The activity of the dual-functional c-CLEnA was confirmed with the peroxidation of Amplex Red, and compared with the activity of stomatocytes loaded with uncross-linked GOx/HRP and with the free enzymes (Fig. S12†). Similarly to the case of PLE, the activity was dependent on the concentration of cross-linker, and when using 30 mM of glutaraldehyde the native activity was preserved (Fig. 3B and S12A†). Again, when genipin was used as cross-linker the native activity of GOx/HRP was not affected (Fig. S12B†).
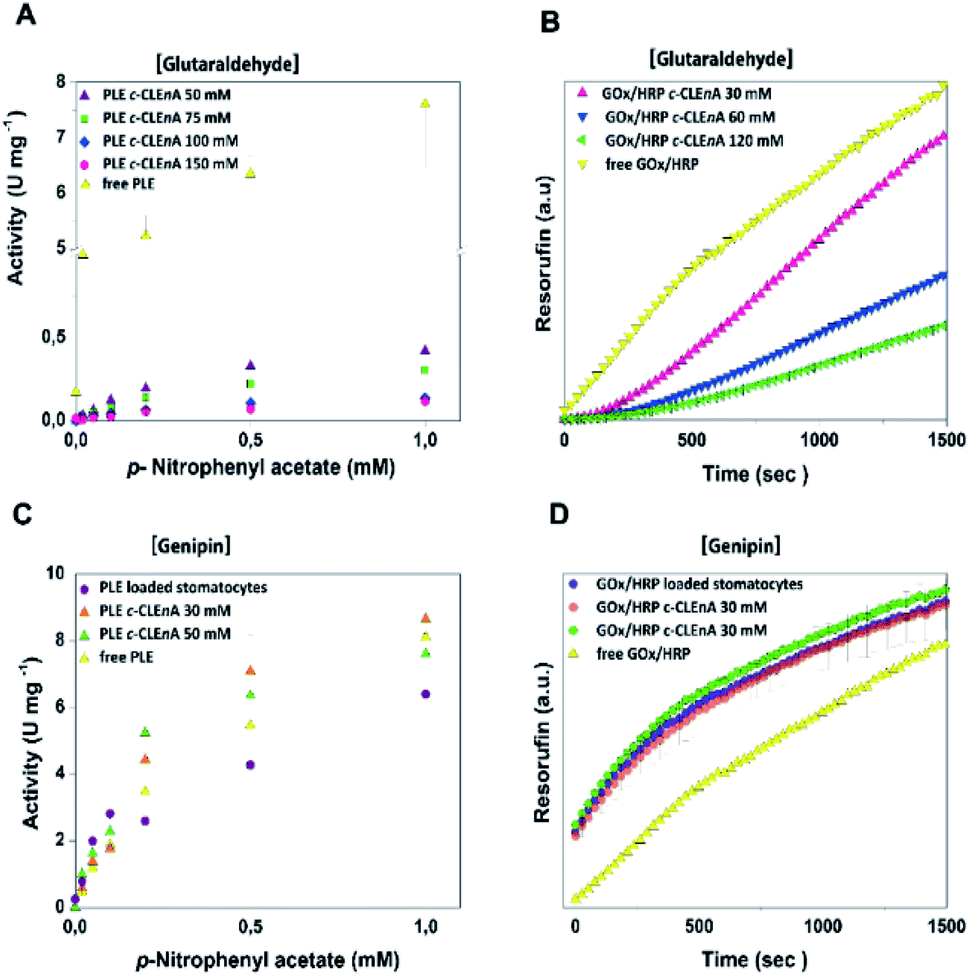 | ||
| Fig. 3 (A) Activity of PLE c-CLEnA formed at different concentrations of glutaraldehyde. An increase in activity is observed when glutaraldehyde concentrations are decreased. The absorbance of the product is measured at 405 nm at different concentration of p-NPA. (B) Activity of GOx/HRP c-CLEnA formed at different concentrations of glutaraldehyde. Resorufin formation is measured at a (D)-glucose concentration of 20 mM. Please note the difference in enzymatic activity of both PLE and GOx/HRP when glutaraldehyde (A and B) and genipin (C and D) were used for c-CLEnA formation. | ||
Finally, the performance of CalB c-CLEnA was evaluated in a flow setup. We first set out to optimize the enzyme loading for maximum efficiency in-flow. Three different enzyme feeds were added to different stomatocyte batches prior to the encapsulation process. When 12 mg mL−1 of CalB was initially added, an encapsulation efficiency of 14% of the initial feed was reached, whilst 33% and 8% were achieved when 16 mg mL−1 and 6 mg mL−1 were added, proving that a good control over the loading efficiency could be achieved by simply varying the initial enzyme concentration. We next set out to test the capacity of stomatocytes in-flow. CalB loaded stomatocytes and CalB c-CLEnA (with 33%, 14% and 8% encapsulation efficiency) were immobilized into a flow reactor (Fig. S13†), equipped with an internal membrane to retain the polymeric vesicles. Catalytic tests were performed after which the stomatocytes or the c-CLEnA were recollected from the membrane outlet, and washed by spin filtration before the next catalytic cycle (ESI 2.9†). When the different non-cross-linked CalB loaded stomatocytes were fed in the flow reactor, after the first catalytic run (∼13 min, 0.3 mL min−1), a clear loss in activity was observed (Fig. S14†). This loss in activity was most severe with the 8% loaded stomatocytes, which only retained approximately 60% of the initial catalytic efficiency, after the second run. A clear loss in activity was observed in all non-crosslinked stomatocyte samples over the course of 5 runs of the flow experiments (Fig. 4A and S14†). It was evident that the loss of activity was related to significant leaching of the enzyme during the reaction. Due to the small size of the protein, the entrapped enzyme was partially flushed away from the stomatocytes' cavity due to the washing and reloading process (ESI 2.9†). In contrast, when in the same process c-CLEnA was employed, no loss in activity, over the course of five runs, was observed (Fig. 4A). Even when CalB c-CLEnA were reused in-flow for ten reaction runs in flow, still no leaching or loss in activity was detected (Fig. 4B), confirming their potential for continuous flow applications. Moreover, after ten runs in flow (flow rate 0.6 mL min−1) there was no apparent change in particle morphology, with fully intact stomatocytes being observed by scanning electron microscopy (SEM) (Fig. S15†). Due to the strong negative effect of glutaraldehyde on PLE enzyme, PLE c-CLEnA was formed using genipin for all studies in-flow (Fig. 4C and D). Similar to CalB c-CLEnA, PLE c-CLEnA was recycled for ten runs in flow, however, ca. 20% decrease in activity was observed. In comparison to PLE loaded stomatocytes, the decrease in activity of PLE c-CLEnA is not remarkable. Such loss of activity can be attributed to the highly sensitive nature of PLE. Another plausible reason is due to the mild cross-linking effect of genipin, which might lead to the presence of small fraction of non-cross-linked PLE. After several cycles, such non cross-linked PLE would be able to escape the stomatocyte, leading to this decrease in activity. That notwithstanding, this experiment is a direct proof of the applicability of our approach toward more sensitive enzymes – a promising platform for enzymatic catalysis in-flow.
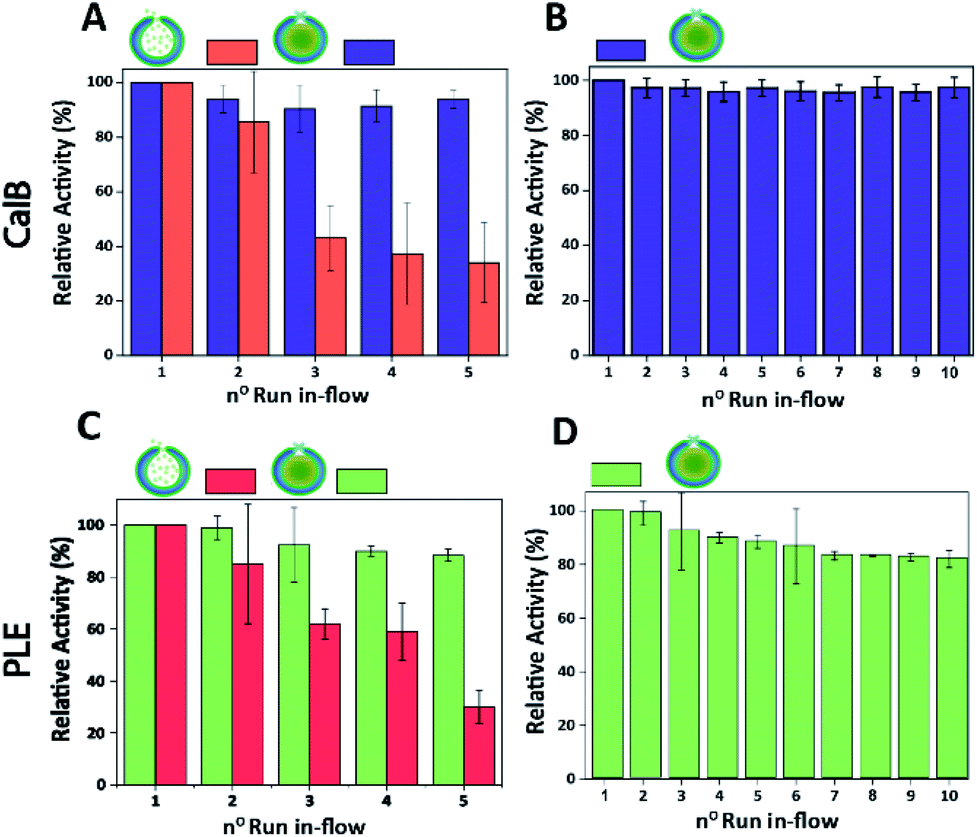 | ||
| Fig. 4 (A) Comparison between the relative activity of CalB loaded stomatocytes (with non-cross-linked CalB) and CalB c-CLEnA (both with 33% encapsulation efficiency) during five runs in-flow. (B) Relative activity of CalB c-CLEnA (with 33% encapsulation efficiency) during ten runs in-flow. (C) Comparison between the relative activity of PLE loaded stomatocytes (with non-cross-linked PLE) and PLE c-CLEnA (both with 21% encapsulation efficiency) during five runs in-flow. (D) Relative activity of PLE c-CLEnA (with 21% encapsulation efficiency) during ten runs in-flow. | ||
Conclusions
In summary, we have shown a novel and robust methodology to construct nano-sized cross-linked enzyme aggregates (c-CLEnA) via their templated formation in the cavity of bowl-shaped polymeric compartments, or stomatocytes. The preorganization of enzymes in the stomatocyte cavity resulted in a high local concentration, which therefore required a substantially smaller amount of cross-linking agent to obtain stable cross-linked systems. In comparison to previously reported CLEA approaches, our c-CLEnA combines the beneficial aspects of a cross-linked enzyme system with the well-defined nanometer sized dimensions of the nanoreactors. The milder cross-linking conditions enabled a better preservation of enzyme activity, proving the general advantages of our method for applications in biocatalysis. Our cross-linking methodology worked equally well for single enzymes and enzyme mixtures. The robustness of the c-CLEnA systems was tested in a micro-reactor flow set-up, in which the c-CLEnA could be reused for ten times without losing any activity, in clear contrast to the controls. This conceptually new approach allows the mild cross-linking of enzymes into well-defined nano-sized particles, which can find their application in flow chemistry and other processes in which catalytic nano-aggregates are beneficial.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge support from Horizon 2020 FET-Open program 737266 – ONE-FLOW. The Dutch Ministry of Education, Culture and Science (Gravitation program 024.001.035), the ERC Advanced Grant (Artisym 694120) and The European Union's Horizon 2020 research and Innovation programme Marie Skłodowska-Curie Innovative Training Networks (ITN) Nanomed, under Grant No 676137. A. J. Lopes is acknowledged for assistance with the preliminary experiments.References
- A. Adamo, R. L. Beingessner, M. Behnam, J. Chen, T. F. Jamison, K. F. Jensen, J. C. M. Monbaliu, A. S. Myerson, E. M. Revalor, D. R. Snead, T. Stelzer, N. Weeranoppanant, S. Y. Wong and P. Zhang, Science, 2016, 352, 61–67 CrossRef CAS PubMed.
- M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796–11893 CrossRef CAS PubMed.
- S. V. Ley, Chem. Rec., 2012, 12, 378–390 CrossRef CAS PubMed.
- D. T. McQuade and P. H. Seeberger, J. Org. Chem., 2013, 78, 6384–6389 CrossRef CAS PubMed.
- B. Gutmann, D. Cantillo and C. O. Kappe, Angew. Chem., Int. Ed., 2015, 54, 6688–6728 CrossRef CAS PubMed.
- M. U. Kopp, A. J. De Mello and A. Manz, Science, 1998, 280, 1046–1048 CrossRef CAS PubMed.
- J. C. Pastre, D. L. Browne and S. V. Ley, Chem. Soc. Rev., 2013, 42, 8849–8869 RSC.
- J. Britton, S. Majumdar and G. A. Weiss, Chem. Soc. Rev., 2018, 47, 5891–5918 RSC.
- S.-H. Jun, J. Lee, B. C. Kim, J. E. Lee, J. Joo, H. Park, J. H. Lee, S.-M. Lee, D. Lee, S. Kim, Y.-M. Koo, C. H. Shin, S. W. Kim, T. Hyeon and J. Kim, Chem. Mater., 2012, 24, 924–929 CrossRef CAS.
- R. A. Sheldon and S. van Pelt, Chem. Soc. Rev., 2013, 42, 6223–6235 RSC.
- I. Denčić, S. De Vaan, T. Noël, J. Meuldijk, M. De Croon and V. Hessel, Ind. Eng. Chem. Res., 2013, 52, 10951–10960 CrossRef.
- R. A. Sheldon, Appl. Microbiol. Biotechnol., 2011, 92, 467–477 CrossRef CAS PubMed.
- K. Ariga, Q. Ji, T. Mori, M. Naito, Y. Yamauchi, H. Abe and J. P. Hill, Chem. Soc. Rev., 2013, 42, 6322 RSC.
- I. Matijošyte, I. W. C. E. Arends, S. de Vries and R. A. Sheldon, J. Mol. Catal. B: Enzym., 2010, 62, 142–148 CrossRef.
- S. H. Petrosko, R. Johnson, H. White and C. A. Mirkin, J. Am. Chem. Soc., 2016, 138, 7443–7445 CrossRef CAS PubMed.
- H.-P. M. De Hoog, I. W. C. E. Arends, A. E. Rowan, J. J. L. M. Cornelissen and R. J. M. Nolte, Nanoscale, 2010, 2, 709 RSC.
- R. J. R. W. Peters, M. Marguet, S. Marais, M. W. Fraaije, J. C. M. van Hest and S. Lecommandoux, Angew. Chem., Int. Ed., 2014, 53, 146–150 CrossRef CAS PubMed.
- M. Nijemeisland, L. K. E. A. Abdelmohsen, W. T. S. Huck, D. A. Wilson and J. C. M. Van Hest, ACS Cent. Sci., 2016, 2, 843–849 CrossRef CAS PubMed.
- M. T. De Martino, L. K. E. A. Abdelmohsen, F. P. J. T. Rutjes and J. C. M. van Hest, Beilstein J. Org. Chem., 2018, 14, 716–733 CrossRef CAS PubMed.
- S. F. M. Van Dongen, M. Nallani, J. J. L. M. Cornelissen, R. J. M. Nolte and J. C. M. Van Hest, Chem.–Eur. J., 2009, 15, 1107–1114 CrossRef CAS PubMed.
- L. M. P. E. Van Oppen, L. K. E. A. Abdelmohsen, S. E. Van Emst-De Vries, P. L. W. Welzen, D. A. Wilson, J. A. M. Smeitink, W. J. H. Koopman, R. Brock, P. H. G. M. Willems, D. S. Williams and J. C. M. Van Hest, ACS Cent. Sci., 2018, 4, 917–928 CrossRef CAS.
- L. K. E. A. Abdelmohsen, M. Nijemeisland, G. M. Pawar, G.-J. A. Janssen, R. J. M. Nolte, J. C. M. van Hest and D. A. Wilson, ACS Nano, 2016, 10, 2652–2660 CrossRef CAS PubMed.
- O. Stauch, R. Schubert, G. Savin and W. Burchard, Biomacromolecules, 2002, 3, 565–578 CrossRef CAS PubMed.
- T. Zisis, P. L. Freddolino, P. Turunen, M. C. F. van Teeseling, A. E. Rowan and K. G. Blank, Biochemistry, 2015, 54, 5969–5979 CrossRef CAS PubMed.
- B. Stauch, S. J. Fisher and M. Cianci, J. Lipid Res., 2015, 56, 2348–2358 CrossRef CAS PubMed.
- Y. Beldengrün, J. Aragon, S. F. Prazeres, G. Montalvo, J. Miras and J. Esquena, Langmuir, 2018, 34, 9731–9743 CrossRef PubMed.
- S. H. Chiou, T. C. Hung, R. Giridhar and W. T. Wu, Prep. Biochem. Biotechnol., 2007, 37, 265–275 CrossRef CAS PubMed.
- B. Hu, L. Zhang, R. Liang, F. Chen, L. He and X. Zeng, J. Agric. Food Chem., 2015, 63, 2033–2040 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc05420k |
| ‡ Present address: Department of Chemistry, College of Science, Swansea University, Singleton campus, Swansea SA2 8PP, United Kingdom. |
| This journal is © The Royal Society of Chemistry 2020 |