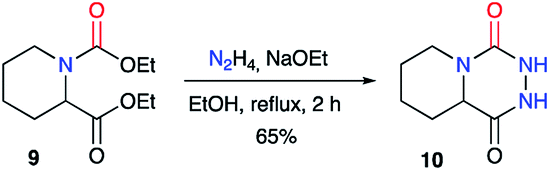DOI:
10.1039/D0RA09457A
(Review Article)
RSC Adv., 2020,
10, 43358-43370
Three cheers for nitrogen: aza-DKPs, the aza analogues of 2,5-diketopiperazines
Received
6th November 2020
, Accepted 10th November 2020
First published on 7th December 2020
Abstract
Nitrogen-containing heterocycles represent a major source of pharmacological probes and drug candidates. To extend their molecular diversity and their potential biological activities, it is of importance to design and synthesize new N-heterocyclic scaffolds. Therefore, aza-diketopiperazines (aza-DKPs), the aza analogues of well-known 2,5-diketopiperazines (DKPs), emerged as a promising new scaffold. Although the first synthesis of an aza-DKP dates from 1951, significant developments have been made during the last decade. This feature article summarizes the different synthetic strategies to access and functionalise aza-DKPs. Their biological properties and potential applications in medicinal chemistry and drug discovery are discussed as well.
1. Introduction
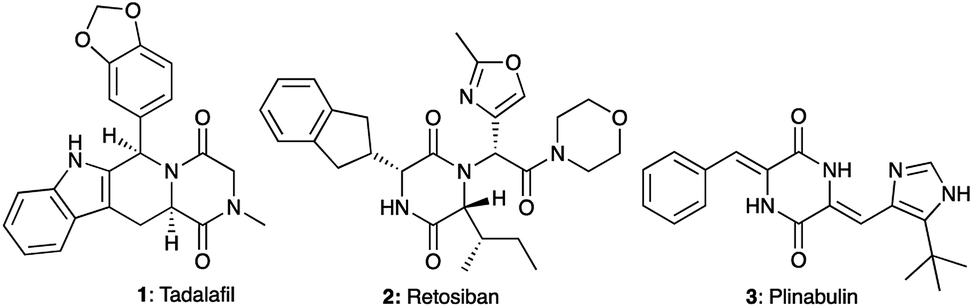
A large majority of FDA-approved drugs contain a heterocycle in their structure.1,2 Amongst these heterocycles, 2,5-diketopiperazines (DKPs) represent a privileged scaffold in medicinal chemistry3–7 as well as in organic synthesis.5–8 Obtained from the double condensation of two amino acids, DKPs are the smallest cyclic peptides with nonetheless different properties from their linear counterparts.9 At the same time, DKPs contain a six membered ring like many drug molecules, unlike the macrocycles that arise from the cyclization of larger peptides. Due to the ubiquitous availability of amino acids, DKPs are highly represented among natural bioactive molecules extracted from plants, fungi, bacteria and mammals as well as synthetically prepared in the laboratory.10,11 The privileged nature of the DKP scaffold can be seen from the biological activities associated with DKPs against different targets including GPCRs such as the GABAergic,12 serotoninergic 5HT1A,13 and oxytocin receptors and enzymes such as phosphodiesterase 5 (PDE5). Several DKP compounds (Fig. 1) have advanced to clinical development, among which the PDE5 inhibitor tadalafil (Cialis™) 1 (ref. 14) received FDA approval for the treatment of erectile dysfunction. Meanwhile, retosiban 2 (ref. 15–17) and plinabulin 3 (ref. 18) reached Phase III clinical trials for preterm labour and cancer respectively.
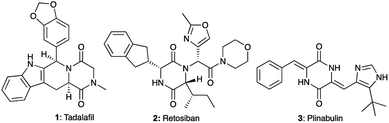 |
| | Fig. 1 Examples of bioactive compounds with a DKP skeleton: tadalafil (PDE5 inhibitor), retosiban (oxytocin receptor antagonist) and plinabulin (tubulin polymerisation inhibitor). | |
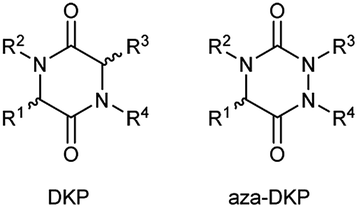
The DKP scaffold can be conveniently diversified at four different positions, with substituents oriented along different directions in space, by using a variety of natural and unnatural amino acid building blocks. Furthermore, the availability of chiral amino acids (often as both enantiomers) enables the chirality at the two stereocenters to be fixed. However, the concept of privileged scaffolds19,20 shows limits intrinsically linked with the use of over-represented structures, thus reducing molecular diversity and patentability. Synthetic chemists then reoriented towards rarer and unprivileged scaffolds with physicochemical properties that are compatible with the discovery of new drug candidates and overcome the disadvantages mentioned above. Indeed, as demonstrated by Pitt's group at Celltech,21 many heterocycles are yet to be synthesised: amongst the 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 heterocyclic structures theoretically generated as potentially of interest for medicinal chemists, only 1700 had been synthesised with around 5 to 10 new scaffolds being published each year. Based on these considerations, our laboratory in collaboration with A. Ganesan focused on 1,2,4-triazine-3,6-diones or aza-diketopiperazines (aza-DKPs) as underprivileged analogues of DKPs with potentially similar or better properties. These compounds can be envisioned as arising from DKPs through scaffold hopping with the replacement of a sp3 carbon atom by a sp2/sp3 nitrogen atom (Fig. 2). Compared to the DKP, there are now three nitrogen atoms in the ring system (hence the title of our review) with the opportunity to functionalise these positions through the choice of starting materials or late stage reactions after formation of the heterocycle.
000 heterocyclic structures theoretically generated as potentially of interest for medicinal chemists, only 1700 had been synthesised with around 5 to 10 new scaffolds being published each year. Based on these considerations, our laboratory in collaboration with A. Ganesan focused on 1,2,4-triazine-3,6-diones or aza-diketopiperazines (aza-DKPs) as underprivileged analogues of DKPs with potentially similar or better properties. These compounds can be envisioned as arising from DKPs through scaffold hopping with the replacement of a sp3 carbon atom by a sp2/sp3 nitrogen atom (Fig. 2). Compared to the DKP, there are now three nitrogen atoms in the ring system (hence the title of our review) with the opportunity to functionalise these positions through the choice of starting materials or late stage reactions after formation of the heterocycle.
 |
| | Fig. 2 Comparison between DKPs and aza-DKPs. | |
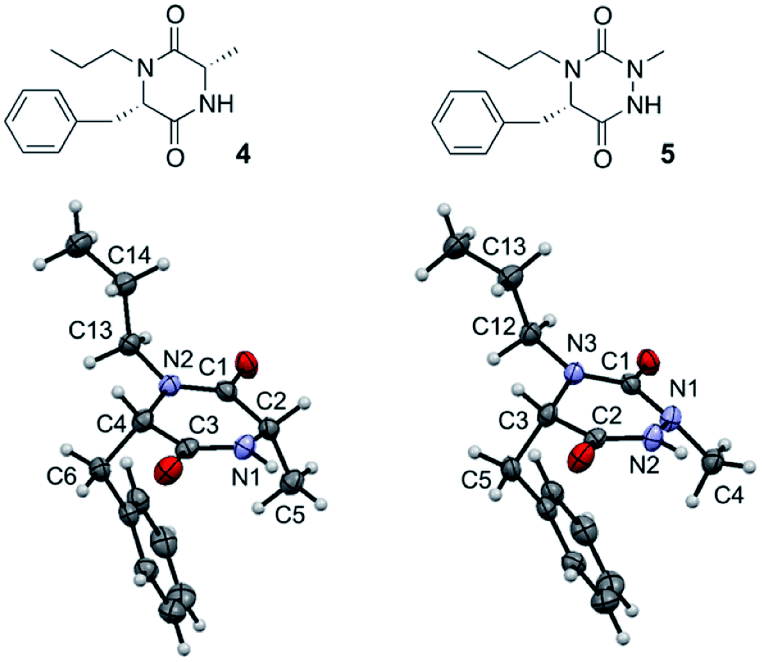
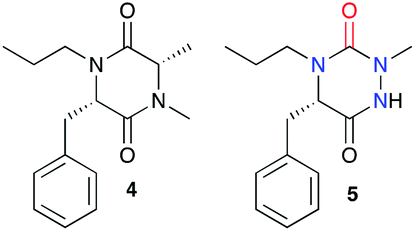
Such a modification involving the replacement of a peptide α-carbon by an extra nitrogen has already been studied in the backbone of linear polypeptides to give so called ‘aza-peptides’. These modified peptides showed a flattening of the spatial arrangement of the substituents linked to the new nitrogen atom while having the potential to exhibit adaptive chirality.22,23 This structural difference was also observed by us in the comparison between a DKP and its aza-DKP analogue by X-ray crystallography (Fig. 3).24 The DKP 4 exhibits a general planar conformation of the ring, in this case with dihedral angles between C1–C2 and N1–C3 or C1–N2 and C4–C3 being close to 20°. On the other hand, the aza-DKP 5, with the introduction of the third sp2/sp3 nitrogen atom, displays a similar behaviour between C1–N1 and N2–C2 with an angle of 19°. However, the dihedral angle between C2–C3 and N3–C1 is here importantly deviated towards a less planar arrangement with a dihedral angle of 37°. Furthermore, NMR experiments performed in CDCl3 and DMSO-d6 and at several temperatures showed a relatively small variation of the different chemical shifts confirming the rigidity of this new scaffold in solution. Thus, this new heterocycle exhibits similar drug-like properties to a DKP (low molecular weight, 4 diversity points, in agreement with Lipinski's rules25) while orienting slightly differently its substituents in space offering different possible interactions with biological targets.
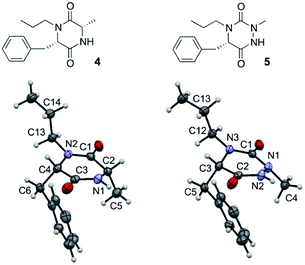 |
| | Fig. 3 X-ray crystallographic structure of DKP 4 (CCDC 1520134) and its aza-DKP analogue 5 (CCDC 1520133). | |
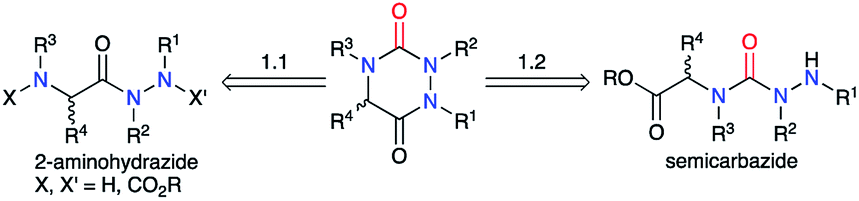
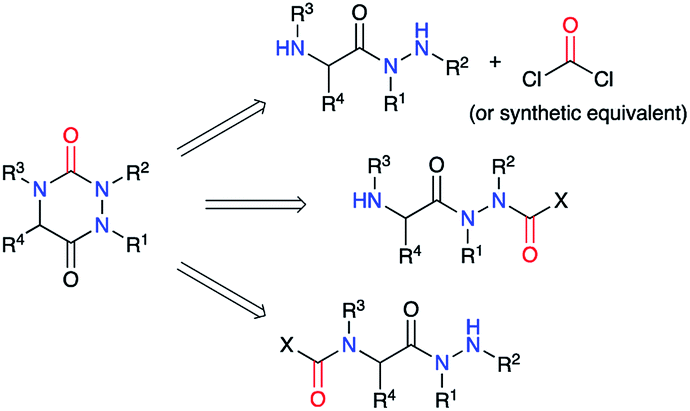
A review of the literature showed that in 2020, only about 300 molecules exhibiting an aza-DKP skeleton were found in the Scifinder® database whereas around 40000 DKPs were described, making aza-DKPs an underprivileged scaffold by comparison. To date, no reviews concerning this family of compounds have been published. Thus, we gathered all the publications dealing with the syntheses and properties of aza-DKPs. We first classified the different synthetic strategies according to the precursor involved in the cyclisation: the semicarbazide or the 2-aminohydrazide (Scheme 1). We then listed the different methods for the introduction of the different substituents at the four available ring positions either during the synthesis or via post-functionalisation. The final part of our review focuses on the physicochemical properties and biological activities of aza-DKPs.
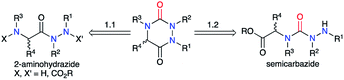 |
| | Scheme 1 Retrosynthesis of an aza-DKP. | |
2. Synthetic strategies
2.1. Via a cyclisation of a 2-aminohydrazide
This strategy was based on the use of a 2-aminohydrazide derivative and the introduction or the formation of a suitable functional group (carbamate, carbazate or isocyanate) leading, through a cyclisation, to the urea moiety present in an aza-DKP (Scheme 2).
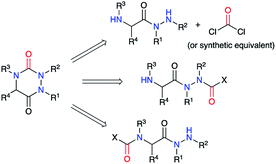 |
| | Scheme 2 Aza-DKP retrosynthesis via the cyclisation of a 2-aminoacetohydrazine. | |
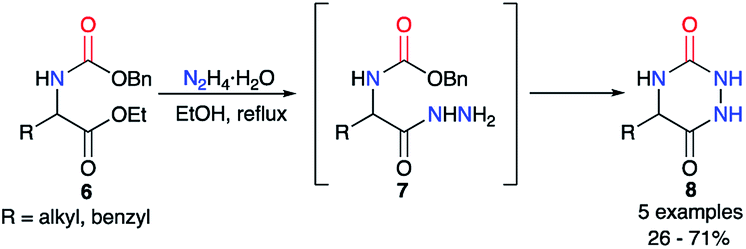
The first synthesis to obtain aza-diketopiperazines was first published by Schlögl and Korger in 1951,26 and was performed in two steps from an amino acid precursor in racemic or enantiopure series. The carbamate derivatives of the amino acid 6 was treated with hydrazine monohydrate to form in situ the 2-aminoacetohydrazide derivatives 7, which then cyclised onto the benzyl carbamate through an addition/elimination process to give the desired triazines 8 in yields ranging from 26 to 71% (Scheme 3). Nonetheless, the authors noted a total loss of optical activity in the final aza-DKP when enantiopure amino acids (L-leucine and L-tyrosine) were used. This racemisation could be explained by the use of the hydrazine solution at high temperatures for several hours.
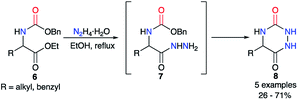 |
| | Scheme 3 First aza-DKP synthesis. | |
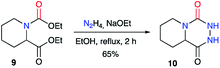
A similar approach was used by Winterfield and Nair on the racemic 1,2-dicarboxylated piperidine 9, which was treated with anhydrous hydrazine and sodium ethoxide to obtain the first example of a bicyclic aza-DKP 10 in 65% yield (Scheme 4).27
 |
| | Scheme 4 Bicyclic aza-DKP first example. | |
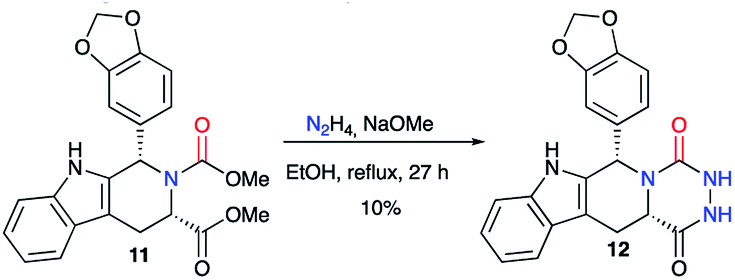
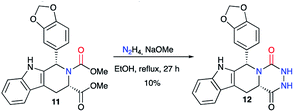
This method was also employed in a patent published in 2002 regarding the synthesis of tadalafil analogues as PDE5 inhibitors.28 Thus, from the enantiopure dicarboxylated precursor 11 and hydrazine, aza-DKP 12 was synthesised as a tadalafil analogue with a 10% yield (Scheme 5).
 |
| | Scheme 5 Synthesis of an aza-DKP analogue of tadalafil. | |
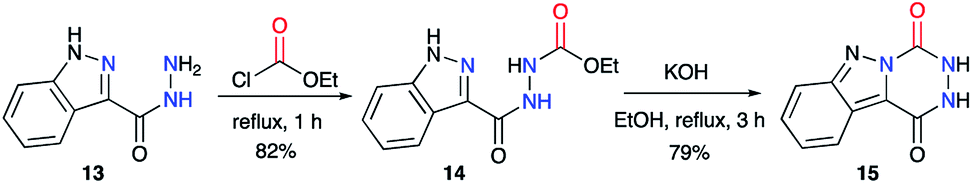
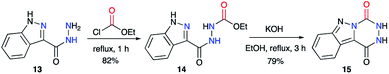
This strategy was also described by the team of Robba,29–31 to obtain aza-DKPs fused to diverse N-heterocycles. Starting from carboxy-3-indazole hydrazide 13 and ethyl chloroformate, the acyclic precursor 14 was obtained (Scheme 6).29 Then, upon treatment with KOH, 14 cyclised to the corresponding triazino-1,2,4-[4,5-b]indazole 15 with a yield of 79%. It can be noted here that in the final product, the 1-H-indazole heterocyclic moiety is replaced by a 2-H-indazole moiety, usually less stable by a few kcal mol−1.32
 |
| | Scheme 6 Triazino-1,2,4-[4,5-b]indazole synthesis from a 2-aminoacetohydrazine. | |
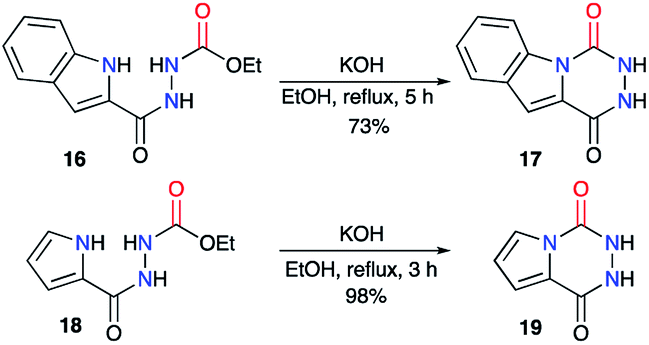
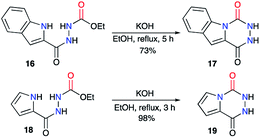
The same authors then extended their strategy to the synthesis of other fused aza-DKPs 17 and 19 involving heterocycles such as indoles30 or pyrroles31 (Scheme 7).
 |
| | Scheme 7 Syntheses of aza-DKPs fused with N-heterocycles. | |
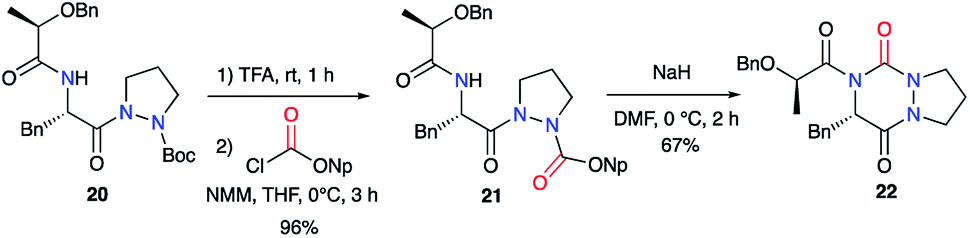
Pinnen et al. used the monocyclic precursor 20 and first switched the Boc-protecting group with a p-nitrophenoxycarbonyl by treating 20 sequentially with TFA and the corresponding aryl chloroformate. The amide function of 21 was then deprotonated by NaH leading after cyclisation on the activated carbazate to the lactic acid-derived bicyclic aza-DKP 22 (Scheme 8).33
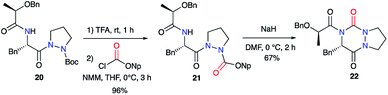 |
| | Scheme 8 Acylated aza-DKP synthesis via an amine cyclisation on an activated carbazate. | |
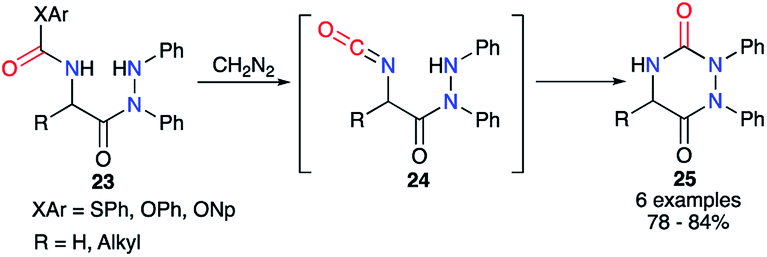
From the 2-aminoacetohydrazide carbamate derivative 23, Baudet et al. generated in situ upon treatment with diazomethane the isocyanate of an activated carbamate derived from phenol, 4-nitrophenol or thiophenol (Scheme 9).34 After formation of the isocyanate intermediates 24, the cyclisation occurred through the attack of the hydrazine on the carbonyl leading to the aza-DKPs 25 with yields between 78 and 84%.
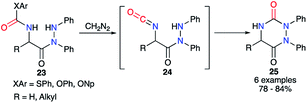 |
| | Scheme 9 Cyclisation via a masked isocyanate for the synthesis of aza-DKPs. | |
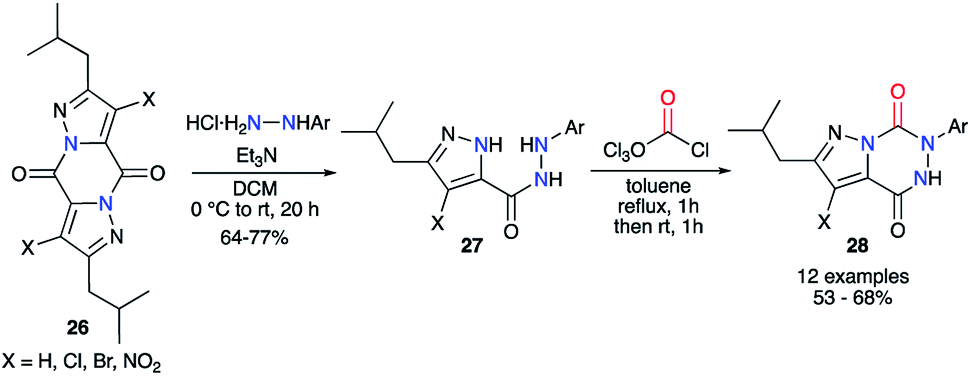
This strategy was also performed in one single step from a 2-aminoacetohydrazide using phosgene or one of its synthetic analogues. Baraldi and co-workers have been the first to use diphosgene for the synthesis of aza-DKPs. It allowed a quick cyclisation from precursors 27 via the double attack of both nucleophilic nitrogen atoms to obtain a series of pyrazolo[1,5-d]-1,2,4-triazines 28 with good yields between 53 and 68% (Scheme 10).35
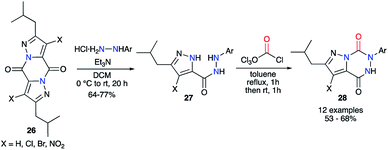 |
| | Scheme 10 Aza-DKP synthesis using diphosgene. | |
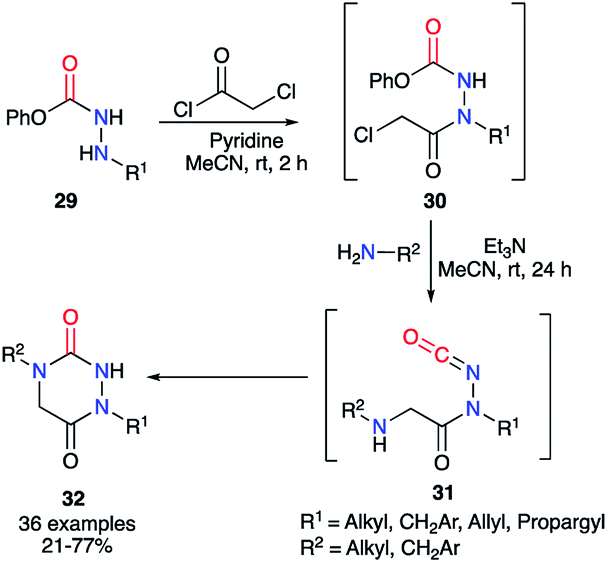
Finally, the method described by Beauchemin et al. in 2015 showed the possibility to synthesise a whole series of disubstituted aza-DKPs 32 through a one-pot sequence in which 30 are not isolated (Scheme 11).36 The phenol-derived carbazates 29 were first acylated by chloroacetyl chloride catalysed by pyridine, which cannot trigger the formation of the isocyanate. Then, the corresponding primary amine and triethylamine were added to the reaction mixture, the latter being basic enough to trigger the isocyanates 31 formation and the cyclisation, leading to the aza-DKPs 32 with global yields of 21 to 77%. It should be noted that this method was also performed on substrates with nucleophilic functions present on the hydrazine or the amine (alcohol, phenol, aniline, pyridine, thiophene, indole…) with yields from 33 to 73%.
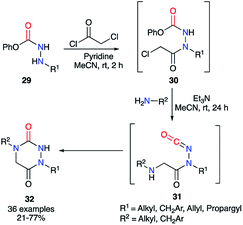 |
| | Scheme 11 Aza-DKP synthesis with one formal step by Beauchemin et al. | |
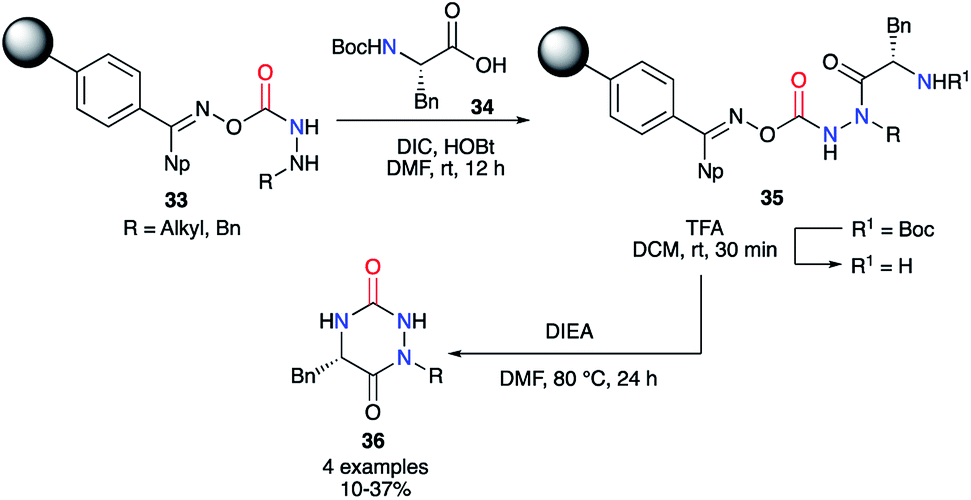
This strategy was also used on solid phase by the team of Scialdone in 1999.37 After fixing the desired hydrazine on a Phoxime® resin, the peptide coupling was performed with DIC and HOBt to add phenylalanine and to obtain the 2-aminoacetohydrazide precursors 35. After N-Boc deprotection with TFA, treatment with Hünig's base allowed isolation of the desired aza-DKPs 36 with low yields between 10 and 37% (Scheme 12).
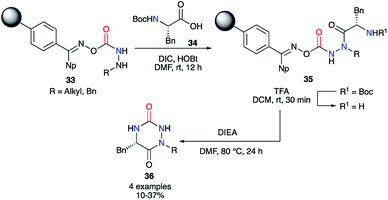 |
| | Scheme 12 First example of an aza-DKP synthesis on a Phoxime® resin. | |
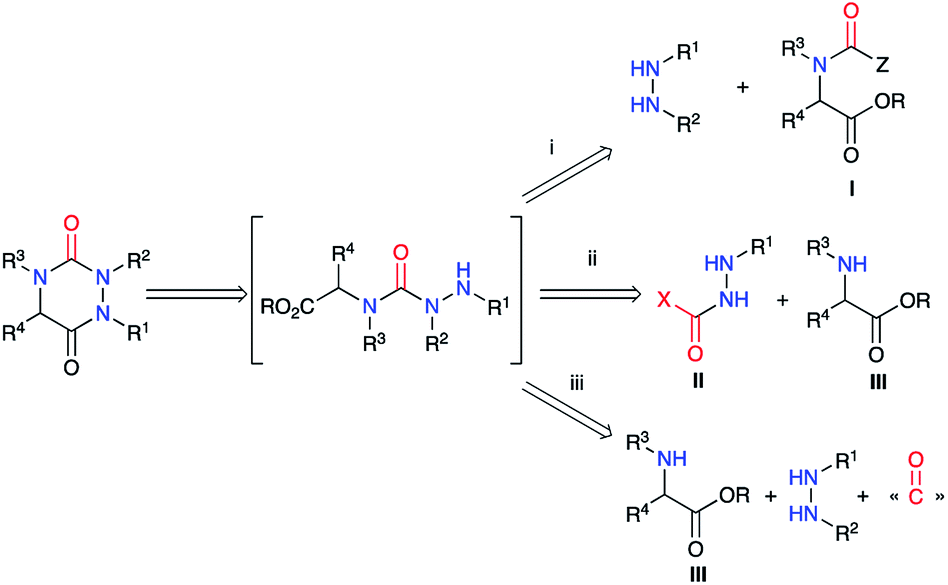
2.2. Via cyclisation of a semicarbazide
The second strategy is based on a semicarbazide (isolated or not) that can be obtained by one of these three following routes (Scheme 13): (i) reaction of a hydrazine with an activated N-carboxylated amino acid derivative I (as an isocyanate or α-lactam); (ii) reaction of an activated carbazate II with an amino acid derivative III; (iii) reaction of an amino acid derivative III with a hydrazine and a carbonyl source. The cyclisation of the semicarbazide through an amide bond formation affords the corresponding aza-DKPs.
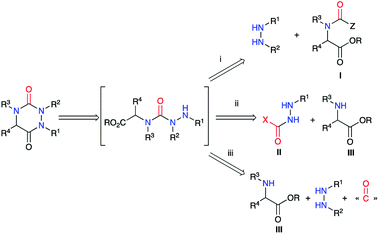 |
| | Scheme 13 Aza-DKP retrosynthesis via a cyclisation of a semicarbazide. | |
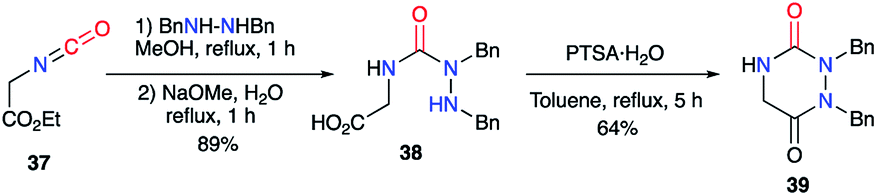
2.2.1. From a hydrazine and a dicarbonylated substrate. Ethyl isocyanoacetate 37 was used as a nitrogen-containing and dicarbonylated derivative in a synthesis described by Schwan in 1983.38 By reacting this isocyanate with a symmetrically disubstituted hydrazine, the author obtained the semicarbazide 38, which then cyclised under acidic catalysis, leading to the desired aza-DKP 39 (Scheme 14).
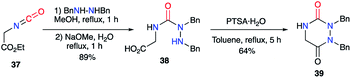 |
| | Scheme 14 Aza-DKP synthesis from an isocyanate. | |
Furthermore, Hoffman et al. used α-lactams as partners in the reaction with hydrazines. The α-lactam is obtained in situ from a N-(mesyloxy)malonamide by treatment with Hünig's base. Then, this intermediate may easily react by ring-opening with different amines, even poorly nucleophilic ones such as dicyclohexylamine, or hydrazines, leading to ureas or semicarbazides respectively.39 Thus, different N-(mesyloxy)malonamides 40 and diversely substituted hydrazines were used to obtain aza-DKPs 43 with yields between 52 and 92% (Scheme 15).40
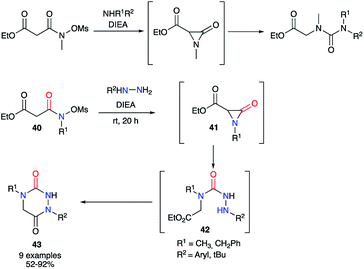 |
| | Scheme 15 Amines reaction with an α-lactam; application to the synthesis of aza-DKPs. | |
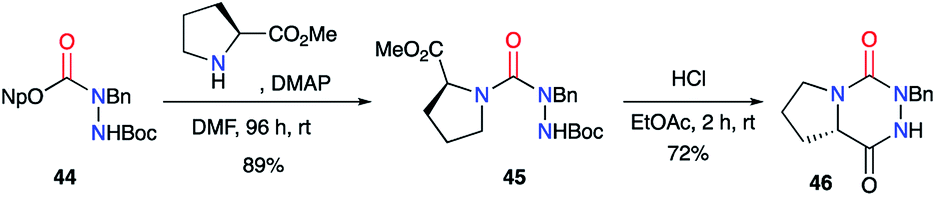
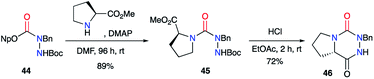
2.2.2. From activated carbazates and amino acids. An alternative method to the previous one involves activated carbazates as masked isocyanates for the formation of semicarbazides. Pinnen et al.41 used DMAP as a nucleophilic catalyst to activate the 4-nitrophenyl-carbazate derivative 44 and thus generate the isocyanate at room temperature. The latter, in the presence of proline methyl ester, led to the desired semicarbazide 45 in 89% yield after 96 hours (Scheme 16). The cyclisation was performed under HCl catalysis and gave the final triazine 46 with a yield of 72%.
 |
| | Scheme 16 Aza-DKP synthesis from an activated carbazate. | |
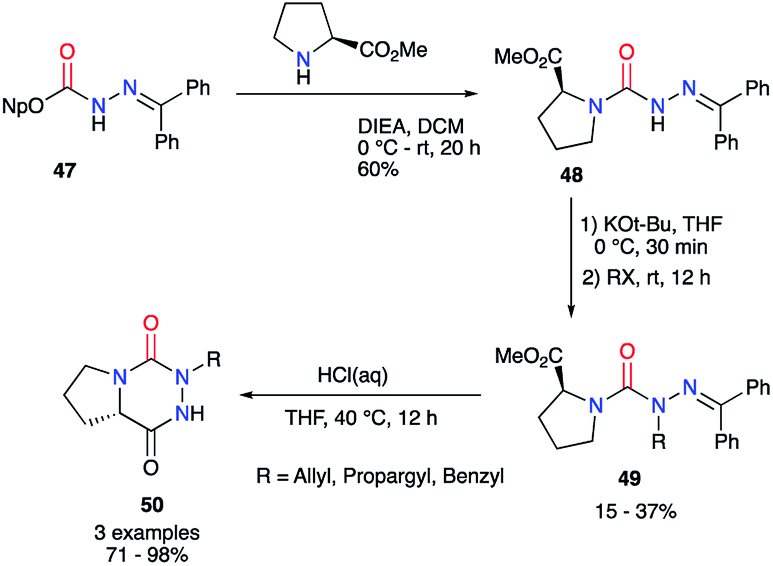
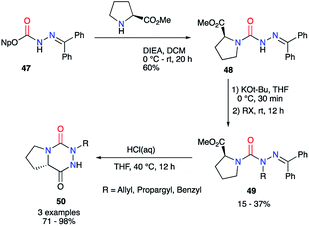
From the activated imino carbazate 47 and using Hünig's base, Lubell et al. obtained the corresponding semicarbazide 48 with a 60% yield while lowering reaction time to 20 hours (Scheme 17).42 This intermediate was then N-alkylated in classical conditions with moderate yields to give the desired aza-DKPs 50 after the deprotection of the hydrazone and HCl-catalysed cyclisation.
 |
| | Scheme 17 Aza-DKP synthesis using an activated carbazate. | |
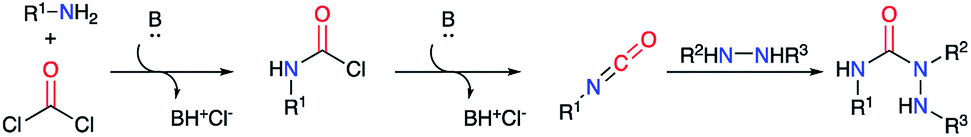
2.2.3. From hydrazines, amino acids and a phosgene equivalent. The reaction between phosgene (COCl2) and a primary amine represents a highly efficient method to form an isocyanate in situ by action of a base. This method enables the preparation of a large diversity of isocyanates for a straightforward access to compounds of interest such as semicarbazides (Scheme 18).
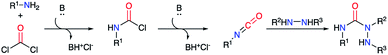 |
| | Scheme 18 Semicarbazide synthesis using phosgene. | |
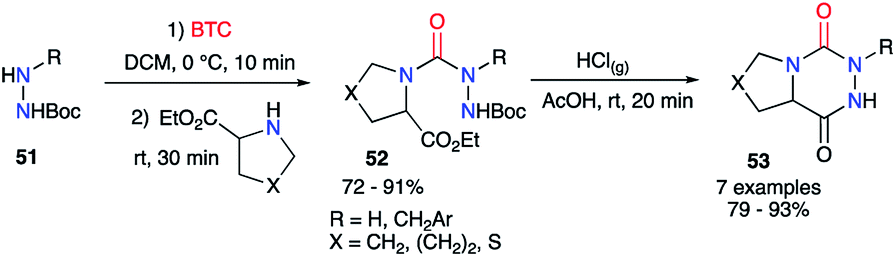
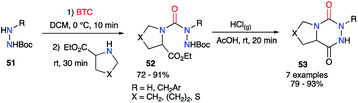
However, as phosgene is a highly reactive and toxic gas, less hazardous synthetic analogues were developed to replace it, such as liquid diphosgene (TCF, trichloromethyl chloroformate) or solid triphosgene (BTC, bis(trichloromethyl) carbonate). During the synthesis of aza-DKPs from hydrazines and cyclic amino acids, Obreza and Urleb used this kind of species to introduce the carbonyl group (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) between two nitrogen atoms of the two partners (Scheme 19).43 Triphosgene was used and the corresponding semicarbazides 52 were isolated in excellent yields. After acid-catalysed Boc-deprotection, the desired bicyclic aza-DKPs 53 were obtained by cyclisation through an amidation process.
O) between two nitrogen atoms of the two partners (Scheme 19).43 Triphosgene was used and the corresponding semicarbazides 52 were isolated in excellent yields. After acid-catalysed Boc-deprotection, the desired bicyclic aza-DKPs 53 were obtained by cyclisation through an amidation process.
 |
| | Scheme 19 Synthesis of bicyclic aza-DKPs using triphosgene. | |
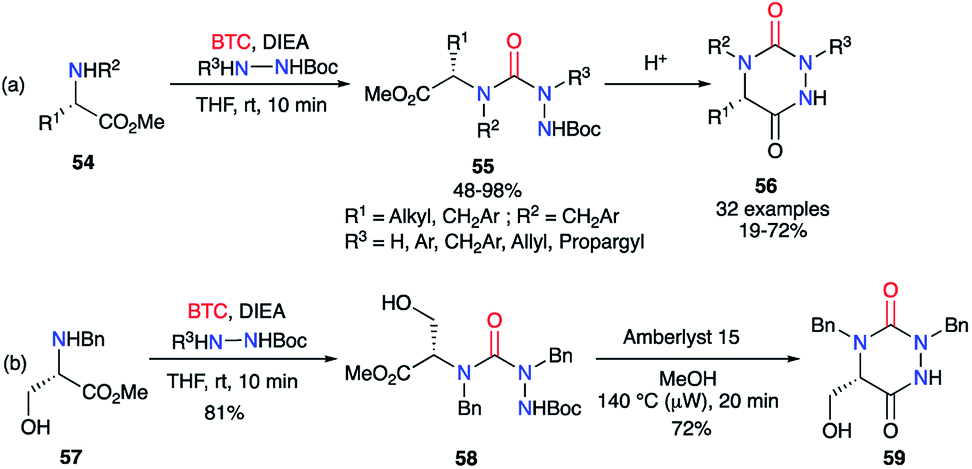
Based on the same strategy, several aza-DKPs 56 were synthesised from enantiopure amino acids by Ganesan's group in collaboration with our laboratory.44,45 First, the synthesis was performed in two steps by isolating the intermediary semicarbazide 55 in high yields (Scheme 20). This intermediate was then cyclised with PPTS or the ion exchange resin Amberlyst 15, depending on the amino acid employed. These conditions allowed the L-serine methyl ester 57 to be used as a precursor, without protecting the hydroxyl function, and the corresponding aza-DKP 59 was obtained in excellent yields (81 and 72%) without any undesired side-products.
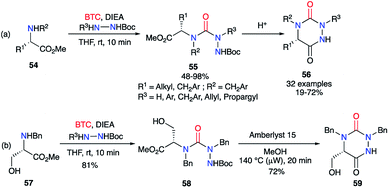 |
| | Scheme 20 (a) Aza-DKPs synthesis from enantiopure amino acids; (b) aza-DKP synthesis from the unprotected L-serine. | |
Subsequently, a one-pot version of this strategy was developed (without isolating the semicarbazide intermediates 61) using various amino acids (Scheme 21). The crude mixture obtained after the first step was directly treated with TFA in water to give after cyclisation the desired aza-DKPs 62 with yields between 27 and 70%.
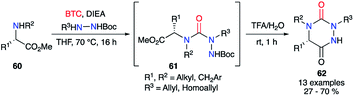 |
| | Scheme 21 One-pot synthesis of aza-DKPs. | |
Compared to the original racemizing reaction conditions described by Schlögl,26 the milder method enabled access to chiral semicarbazides and their corresponding triazines. Although, no evidence regarding the absence of a partial racemisation has been described, this strategy still represents a significant step towards the synthesis of chiral aza-DKPs.
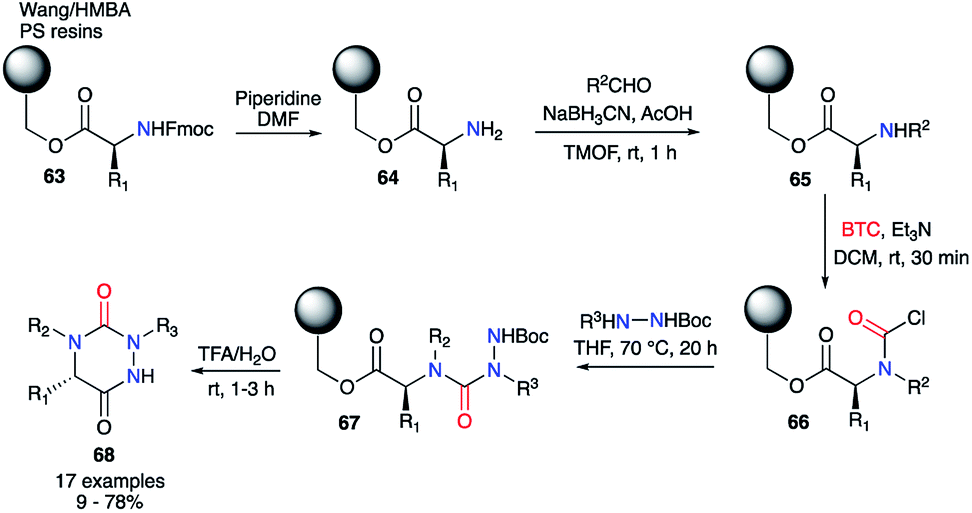
Furthermore, the same authors transferred this strategy towards solid phase synthesis (Scheme 22).44 This method allowed a rapid synthesis of diverse derivatives without intermediate purification. Wang or HMBA resins were selected in order to start the synthesis with the desired amino acid directly attached to the solid support. After Fmoc-deprotection and reductive amination, the amino acids 65 was acylated with triphosgene leading to the corresponding carbamoyl chlorides 66. This intermediate was then reacted with different tert-butyl carbazate derivatives to obtain the desired semicarbazides 67. Upon treatment with TFA in water, the semicarbazides led to the corresponding aza-DKPs 68 in solution with yields between 9 and 78% for Wang resin and 53 to 93% for HMBA resin.
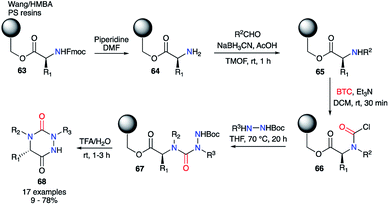 |
| | Scheme 22 Aza-DKP synthesis on Wang or HMBA resin (PS = polystyrene). | |
3. Aza-DKP structural variation
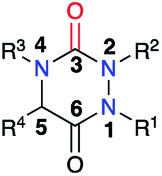
The variation of the substituents at the four positions of an aza-DKP structure (Fig. 4) may be performed either during their synthesis through the different strategies presented previously or by post-functionalisation.
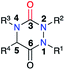 |
| | Fig. 4 Numbering of a tetrasubstituted aza-DKP. | |
3.1. Variation during the synthesis
This strategy is based on the introduction of the desired substituent during the aza-DKP synthesis, by selecting the suitable reactive partners, according to the strategy employed (cf. § 2).
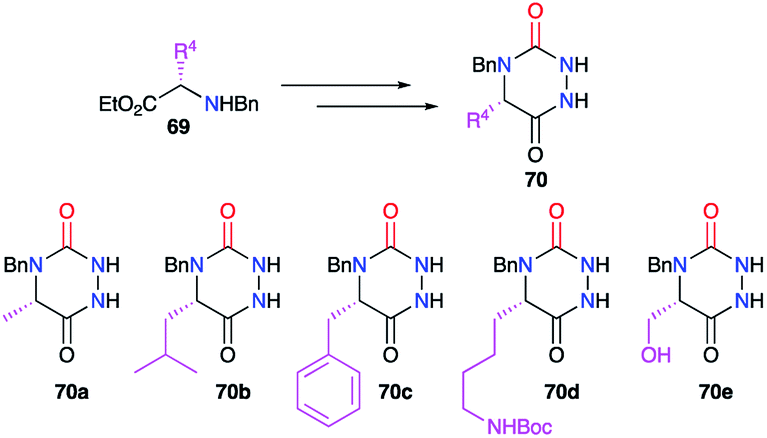
3.1.1. Variation at positions 4 and 5. The first category of variations can be performed at positions 4 and 5 of the heterocycle. The position 5 represents a key element for the structural diversification of aza-DKPs. The substituent placed at this position can originate from natural enantiopure amino acids used as precursors in several strategies (cf. § 2.2.3., Schemes 20 and 21).44,45 Thus, different groups (alkyl, benzyl, or functionalised chains) have been varied at position 5 of an aza-DKP to obtain 70a–e with the conservation of the amino acid enantiopurity (Scheme 23).
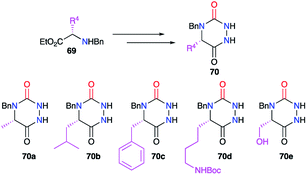 |
| | Scheme 23 Variation of the substituent at position 5 with amino acids. | |
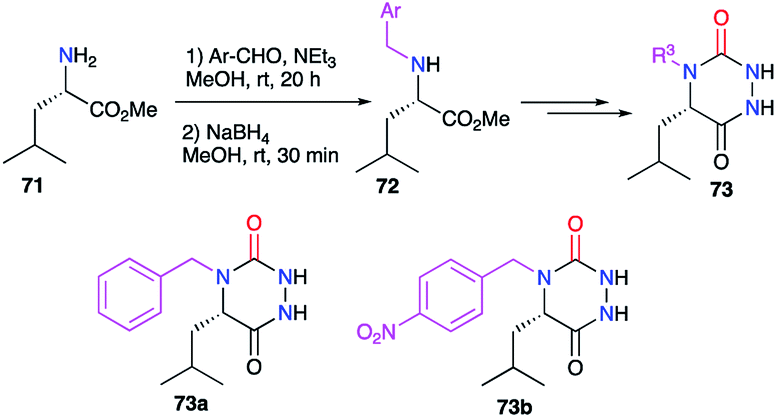
The substituent at position 4, linked to the nitrogen, was also modified during the syntheses of aza-DKPs. For instance, benzyl groups have been introduced at this position (cf. § 2.2.3., Scheme 19), from N-benzylated amino acids previously obtained by a reductive amination on 71 (Scheme 24).44
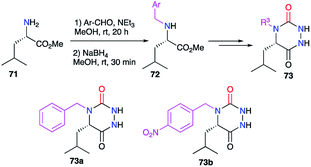 |
| | Scheme 24 Variation of the substituent at position 4 with a reductive amination. | |
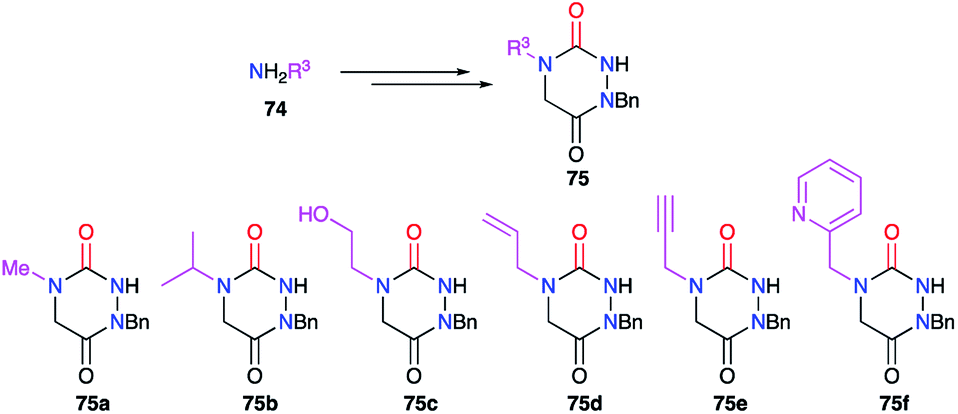
This substituent was also diversified thanks to another strategy introducing a primary amine bearing this substituent before cyclisation (cf. § 2.1., Scheme 11).36 Indeed, a large number of aza-DKPs (75) have been synthesised from commercially available primary amines (Scheme 25).
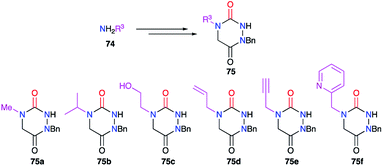 |
| | Scheme 25 Variation of the substituent at position 4 with primary amines. | |
In the bicyclic aza-DKP series, positions 4 and 5 can also be linked together. Structures such as 77 can be obtained from suitable cyclic amino acid precursors 76 such as L-proline, pipecolinic acid or thiaproline (Scheme 26). The different methods used to synthesise such bicyclic systems were previously detailed (cf. § 2.1., Scheme 4; § 2.2.2., Schemes 16 & 17; § 2.2.3., Schemes 19–22).27,41–45
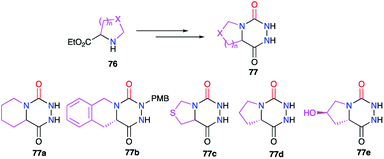 |
| | Scheme 26 Bicyclic aza-DKPs: Variation of substituents at positions 4 and 5. | |
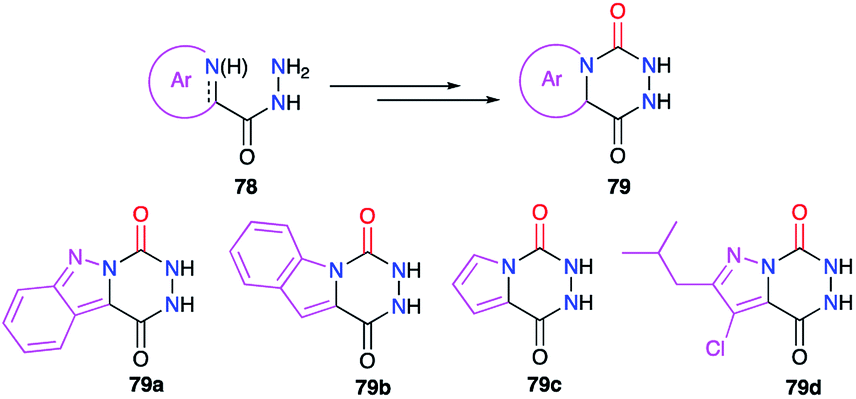
Finally, bicyclic aza-DKPs 79 fused to N-heterocycles have also been obtained (Scheme 27). The precursors used in these cases included the corresponding heterocycle (indazole, pyrrole or pyrazole) in their structure (cf. § 2.1., Schemes 6, 7 and 10).29–31,35
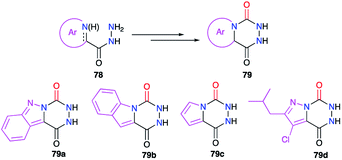 |
| | Scheme 27 Aza-DKPs fused with N-heterocycles. | |
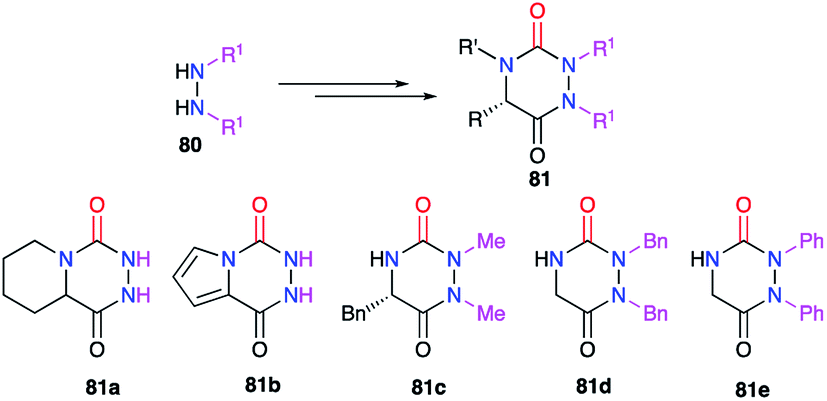
3.1.2. Variation at positions 1 and 2. Positions 1 and 2 relate to the substituents linked to the nitrogen atoms of the hydrazine moiety. Amongst the synthetic methods previously described, several included a reaction between hydrazine and a carboxylated derivative. To avoid any regioselectivity issues, symmetric hydrazines 80 were first used (cf. 2.1., Schemes 3–7 & 9; cf. § 2.2.1, Scheme 14).26–31,34,38 Thus several aza-DKPs 81 having identical substituents on nitrogen atoms N1 and N2 were obtained (Scheme 28).
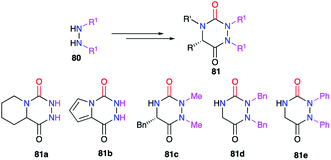 |
| | Scheme 28 Identical substituents at positions 1 and 2 from symmetrical disubstituted hydrazines. | |
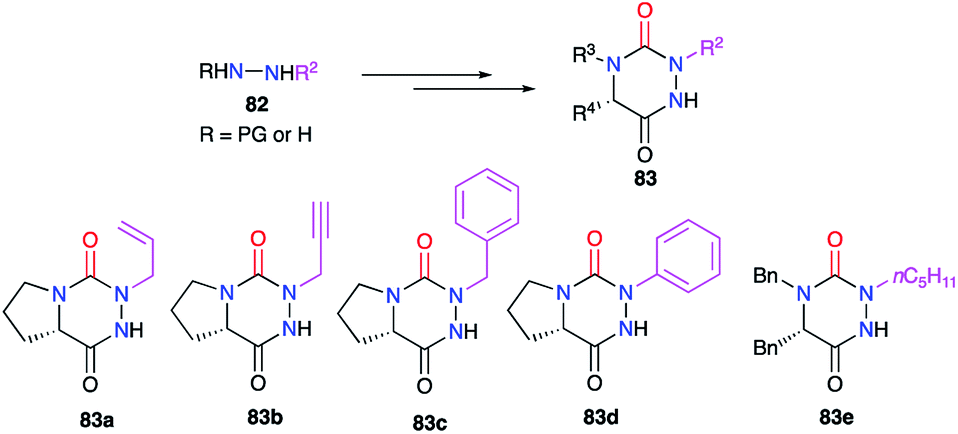
The modification of the substituent at position 2 was also performed through different strategies, either by using monosubstituted hydrazines or 1,2-disubstituted hydrazines with one nitrogen atom being deactivated by a protecting group. For instance, the phenyl group was introduced by phenylhydrazine, the regioselectivity being controlled by deactivation of the N-phenyl nitrogen through mesomeric effect (cf. § 2.1., Scheme 10).35 Using a particular hydrazine bearing on one side the desired substituent while the other is deactivated by a Boc protecting group, the formation of the semicarbazide precursor of the desired triazine was regioselective (cf. § 2.2.3., Schemes 20 and 21).44,45 Another method used a hydrazone-protection for one of the nitrogen atoms while the other was deprotonated in order to introduce the desired substituent by N-alkylation (cf. § 2.2.2., Scheme 17).42 This way alkyl, allyl, propargyl, benzyl, or phenyl groups have been introduced at position 2 during the synthesis of the corresponding aza-DKPs 83 (Scheme 29).
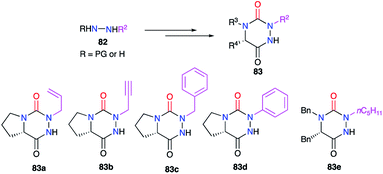 |
| | Scheme 29 Substituents variation at position 2. | |
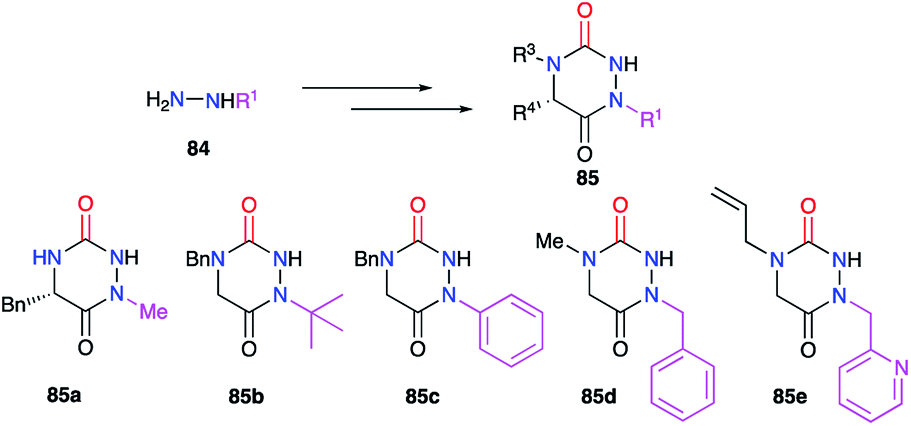
Finally, the substituent at position 1 can also be varied using monosubstituted hydrazines 84. In a similar way, the regioselectivity of the reaction involving hydrazine can be controlled by decreasing the nucleophilicity of the nitrogen bearing the desired substituent by steric or electronic effects (cf. § 2.1., Schemes 11 and 12; cf. § 2.2.1., Scheme 15).36,37,40 Thereby, several groups (alkyl, benzyl, aryl) have been introduced during the synthesis of the corresponding aza-DKPs 85 on nitrogen N1 (Scheme 30).
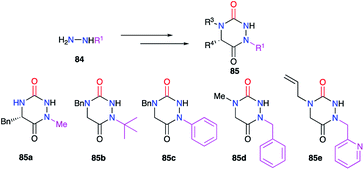 |
| | Scheme 30 Substituents variation at position 1. | |
Table 1 below summarizes the variations at the four positions of an aza-DKP, according to the precursor involved and the strategy employed, with the corresponding references from the literature.
Table 1 Synthetic strategies for the variation of the substituents at the four positions of an aza-DKP
3.2. Variation by post-functionalization
This strategy is based on the introduction or the modification of the substituents after the cyclisation step and has been developed by our team for the generation of aza-DKPs that cannot be easily obtained otherwise.
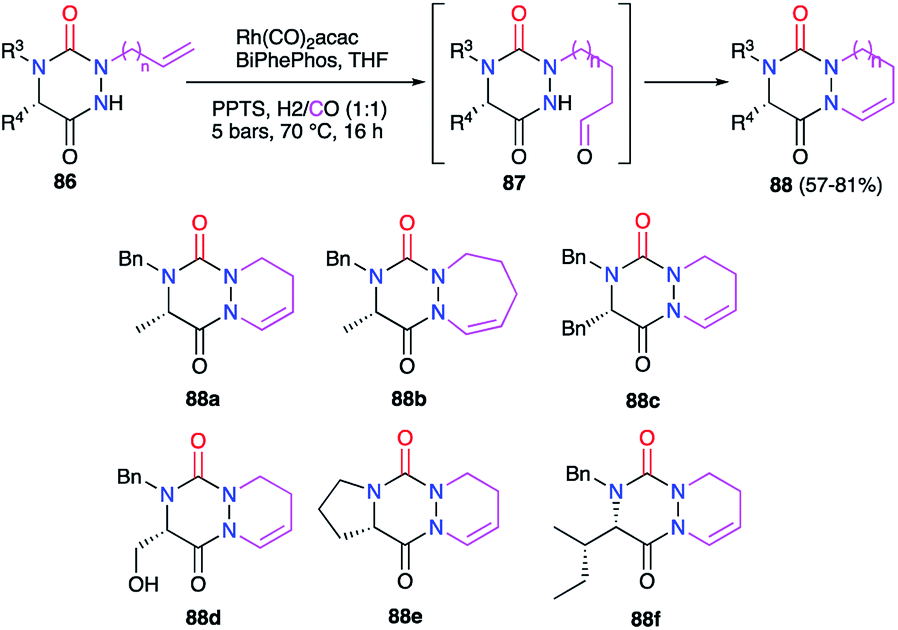
3.2.1. Modification of the substituent in 1- and/or 2-positions. Aza-DKPs 86 bearing either an allyl or a homoallyl group at position 2 were prepared in our laboratory (cf. § 2.2.3., Scheme 21). The double bond present in these structures could then be modified by a rhodium-catalysed hydroformylation, introducing regioselectively a formyl group at the terminal position. The aldehyde intermediate obtained was then engaged in a cyclisation process through an intramolecular condensation reaction on the nitrogen atom N1 leading to the corresponding bicyclic aza-DKPs 88 with yields between 57 and 81% (Scheme 31).45 This cyclohydrocarbonylation strategy allowed the synthesis of a new series of bicyclic aza-DKPs fused with 6- or 7-membered rings on the hydrazine moiety. Furthermore, this process resulted in the formation of a cyclic enamide, with the double bond offering new opportunities for post-synthetic transformations.
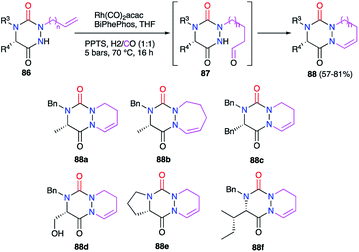 |
| | Scheme 31 Bicyclic aza-DKP formation via cyclohydroformylation. | |
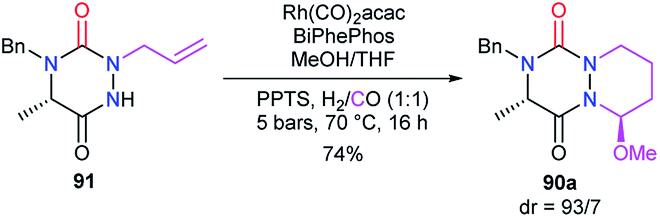
The reactivity of the enamide was at first exploited to introduce functions at the α-position to the nitrogen. For instance, when reacted with a nucleophile such as methanol in acidic medium (CSA), aza-DKPs 89 led to the corresponding chiral hemiaminal ethers 90 with yields between 61 and 86%. Diastereomeric ratios were higher than 93/7 for 90a–d and about 37/63 for aza-DKP 90e fused with a 7-membered ring (Scheme 32).45 Aza-DKP 90a was also obtained through a one-pot process, from the N-allyl aza-DKP 91, in cyclohydrocarbonylation conditions with methanol as a co-solvent. The yield as well as the diastereomeric ratio obtained in this case (74%, 93/7) were similar to the one obtained in the two-step process (Scheme 33).45
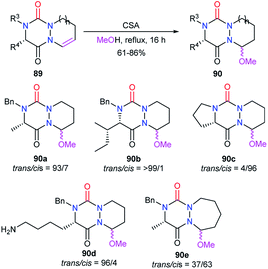 |
| | Scheme 32 Introduction of a methoxy group on a bicyclic aza-DKP. | |
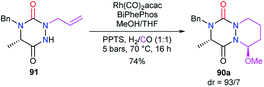 |
| | Scheme 33 Domino cyclohydrocarbonylation/MeOH addition for the synthesis of bicyclic aza-DKP 90a. | |
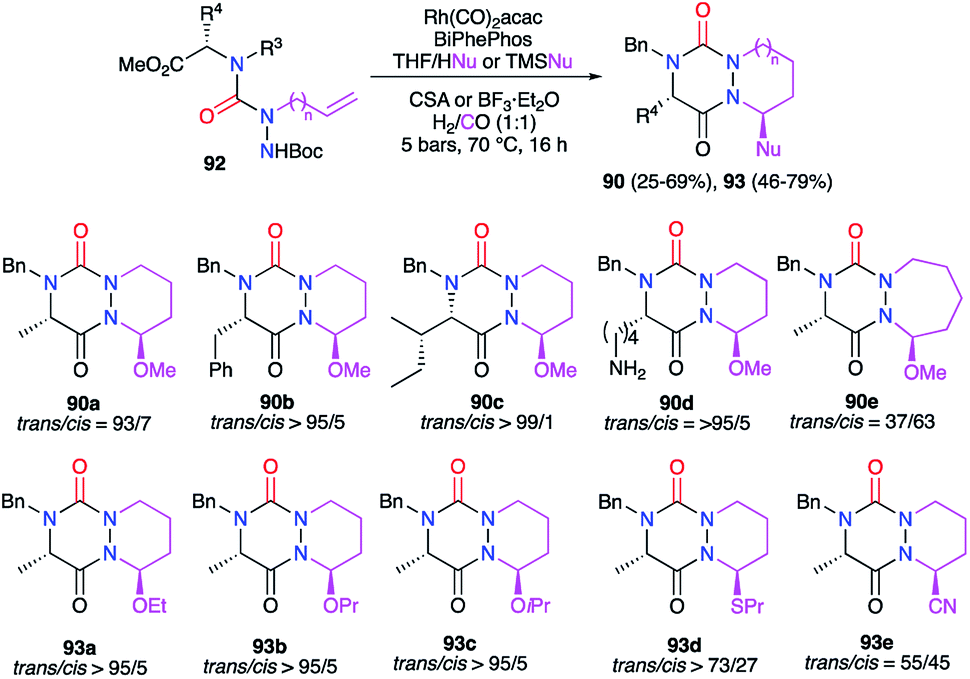
A multicomponent process including the formation of the triazine ring was also developed to obtain aza-DKPs. The linear precursor placed in cyclohydrocarbonylation conditions in the presence of a nucleophile (ROH, TMSCN, nPrSH) and a Brønsted (CSA) or Lewis acid (BF3), semicarbazides 92 led directly to the corresponding aza-DKPs 90 and 93 (Scheme 34).46
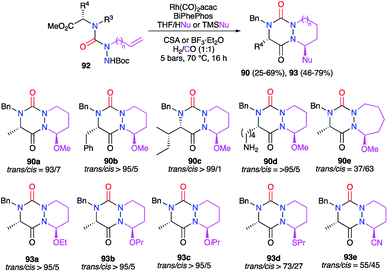 |
| | Scheme 34 One-pot synthesis of bicyclic functionalised aza-DKPs. | |
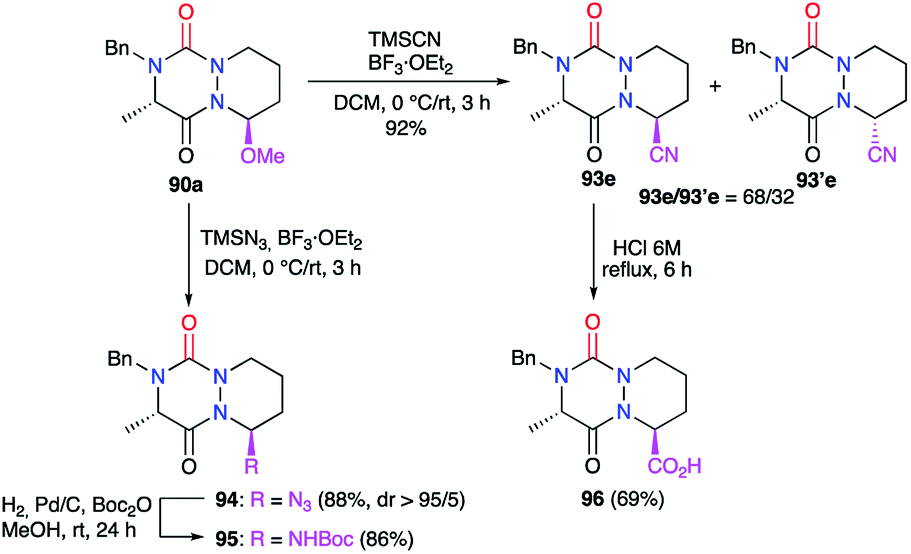
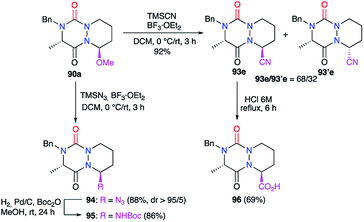
Treatment of the hemiaminal ether 90a with other nucleophiles under BF3·Et2O catalysis allowed the introduction of several functions at the α-position to the nitrogen (Scheme 35).45 The use of trimethylsilyl cyanide (TMSCN) for instance allowed the introduction of a nitrile group at this position with a yield of 92% and a modest diastereomeric ratio of 68/32. The acidic hydrolysis of the major diastereomer gave the corresponding carboxylic acid 96 in 69% yield. In similar conditions, trimethylsilyl azide (TMSN3) allowed the addition of an azide function at this position in 88% yield and an excellent diastereomeric ratio (>95/5). The catalytic hydrogenation of 94 in the presence of di-tert-butyl dicarbonate (Boc2O) led to the corresponding carbamate 95 with a yield of 86%.
 |
| | Scheme 35 Functionalisation of a bicyclic aza-DKP with other nucleophiles. | |
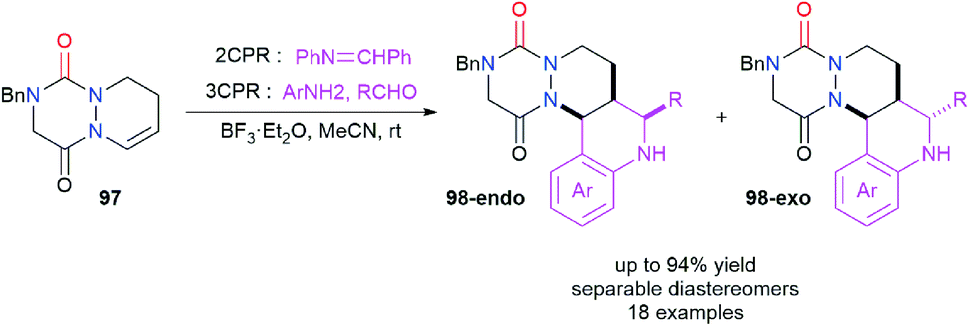
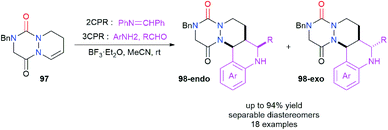
Aza-DKP 97 was also studied as an endocyclic enamide partner in the Povarov reaction in order to perform a formal difunctionalisation of the double bond (Scheme 36).47 This reaction was catalysed by BF3·Et2O and was performed either with the iminodiene partner (2CPR) or directly with an aniline derivative and an aldehyde via a three component Povarov reaction (3CPR). Aliphatic and electron-deficient aromatic aldehydes were rather successfully used while electron poor or rich anilines could also be employed for this reaction. The Povarov adducts 98-endo and 98-exo were obtained in yields between 18 and 94% and diastereomeric ratios between 50/50 and >99/1, the diastereomers being separable by reversed-phase chromatography. The mechanistic aspects of this reaction were studied computationally.
 |
| | Scheme 36 Quinoline formation via a Povarov reaction on a endocyclic enamide. | |
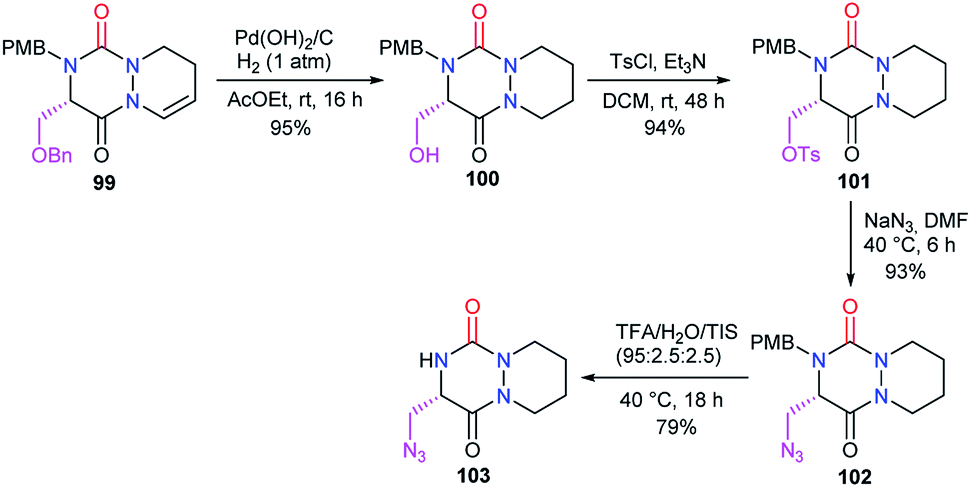
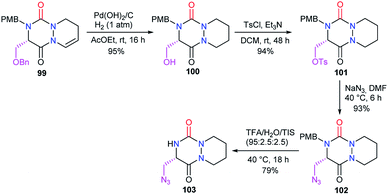
3.2.2. Modification of the substituent in 5-position. The structural modification of the substituent in the 5-position of the aza-DKP has so far only been studied by our laboratory. From the serine-derived aza-DKP 99, palladium-catalysed hydrogenation and hydrogenolysis allowed us to obtain directly aza-DKP 100. The hydroxyl was substituted by an azide via a tosylate, leading after acid-promoted deprotection to the aza-DKP 103 with a good yield (Scheme 37).48
 |
| | Scheme 37 Hydroxyl substitution with an azide at position 5 of an aza-DKP. | |
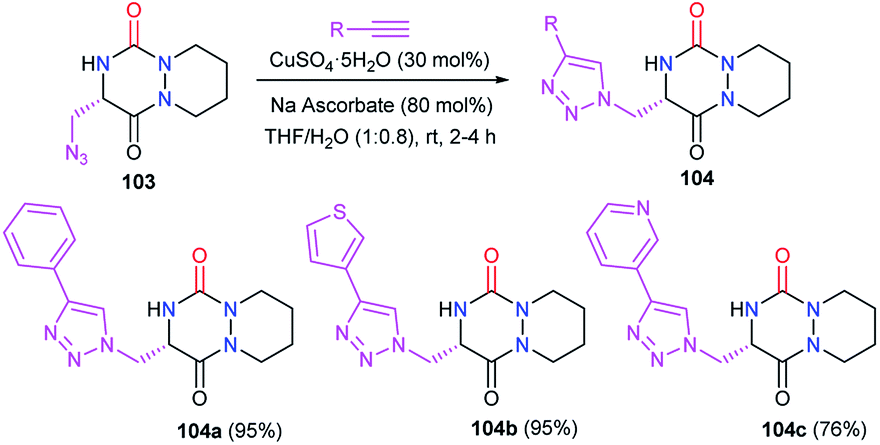
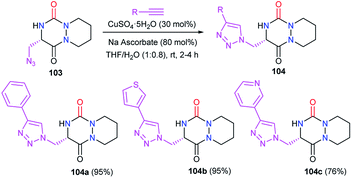
Afterwards, the azide moiety on the aza-DKP 103 was transformed through copper(I)-catalysed cycloaddition with several terminal alkynes, into the corresponding triazoles 104 with high yields (Scheme 38).
 |
| | Scheme 38 Triazole formation at position 5 via Cu-catalysed [3 + 2] cycloaddition. | |
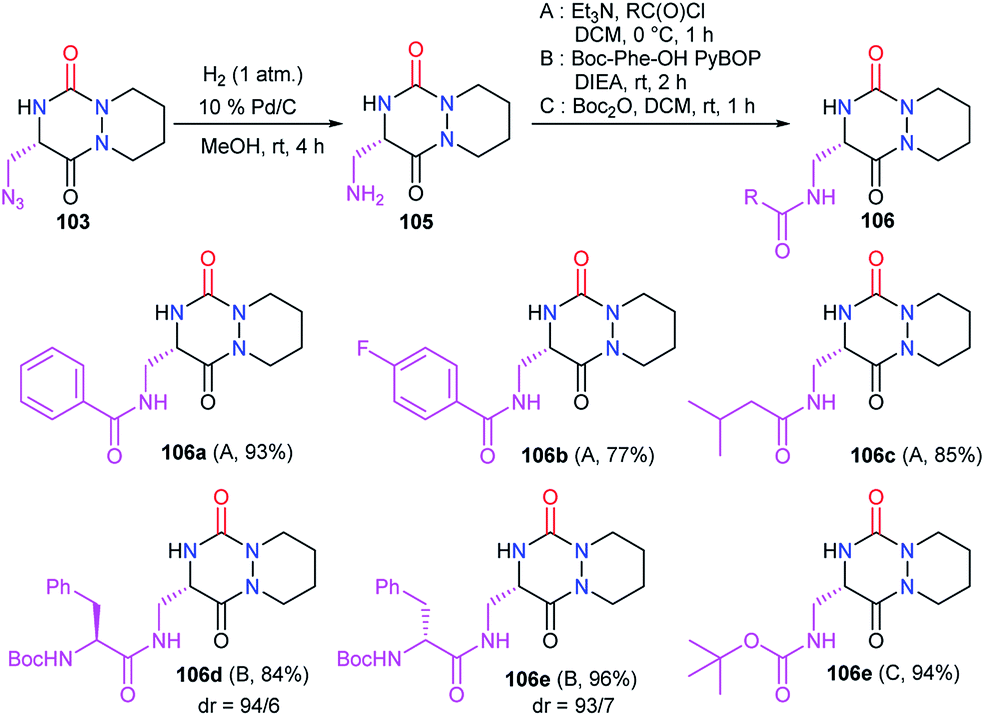
Starting from the same precursor 103, the azide function was also reduced to the amine via palladium-catalysed hydrogenation. The resulting aza-DKP 105 was then N-acylated using different methods (Scheme 39). Using acyl chlorides led to the corresponding amides 106a–c with yields between 77 and 93% (Scheme 39). Peptide coupling conditions allowed access to aza-DKPs 106d–e coupled with L- and D-phenylalanine with respective yields and diastereomeric ratio of 84% (dr = 94/6) and 96% (dr = 93/7). A slight partial racemisation had occurred during the synthesis of the azide 103, as the second diastereomer was observed. Moreover, the treatment of amine 105 with Boc2O led to the corresponding carbamate 106f in 94% yield.48
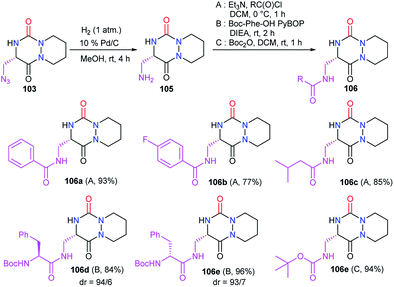 |
| | Scheme 39 Azide reduction and amine functionalisation. | |
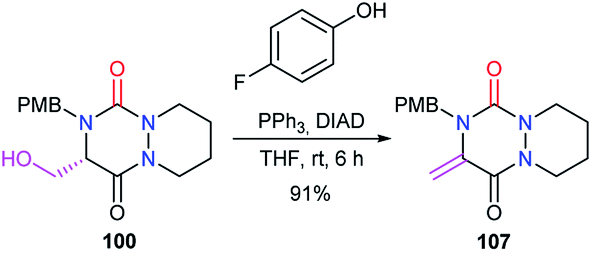
Finally, under Mitsunobu conditions, aza-DKP 100 previously obtained was dehydrated to give the corresponding product 107 with an exocyclic double bond, in 91% yield (Scheme 40).48
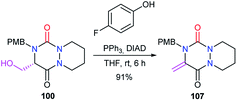 |
| | Scheme 40 Aza-DKP dehydration at position 5. | |
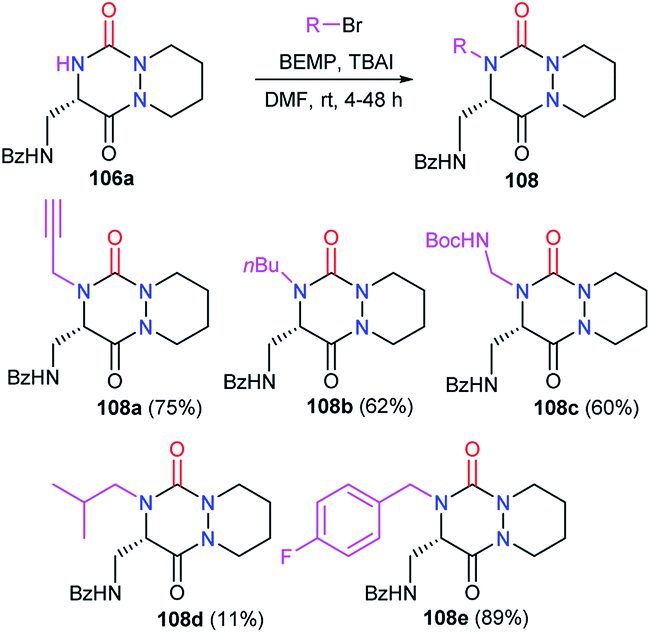
3.2.3. Introduction of a substituent in 4-position. Studies from our laboratory also focused on the post-functionalisation of the position 4 of the heterocycle. The alkylation of the aza-DKP 106a with bromide derivatives was performed in the presence of a phosphazene-type, non-nucleophilic, strong base (BEMP) and allowed us to introduce several alkyl, propargyl and benzylic substituents on the position 4 of the enantiopure aza-DKPs 108 with yields between 11 and 89% (Scheme 41).48
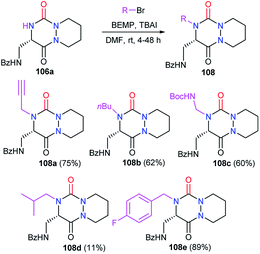 |
| | Scheme 41 N-Alkylation at position 4. | |
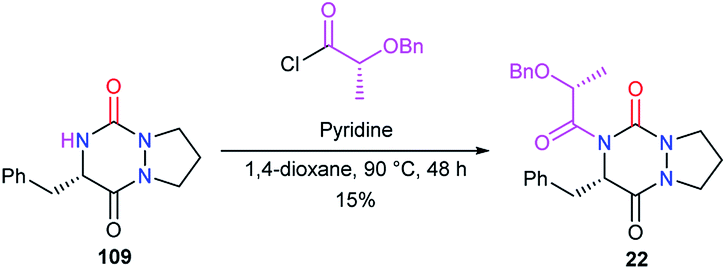
To perform the N-acylation in 4-position of the bicyclic aza-DKP 109, Pinnen et al. used the acyl chloride derived from O-benzyl lactic acid, in the presence of pyridine (Scheme 42).33 This method led to aza-DKP 22 in only 15% yield, which was lower than that obtained by the same authors when the acyl group was introduced before the cyclisation step (cf. Scheme 8).
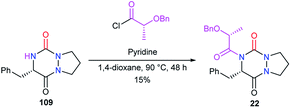 |
| | Scheme 42 N-Acylation at position 4 with an acyl chloride. | |
4. Aza-DKP physicochemical properties and biological activities
Although studies focused on aza-DKPs’ physicochemical and biological properties are less extensive compared to their more common DKP counterparts, nevertheless they have shown promising results.
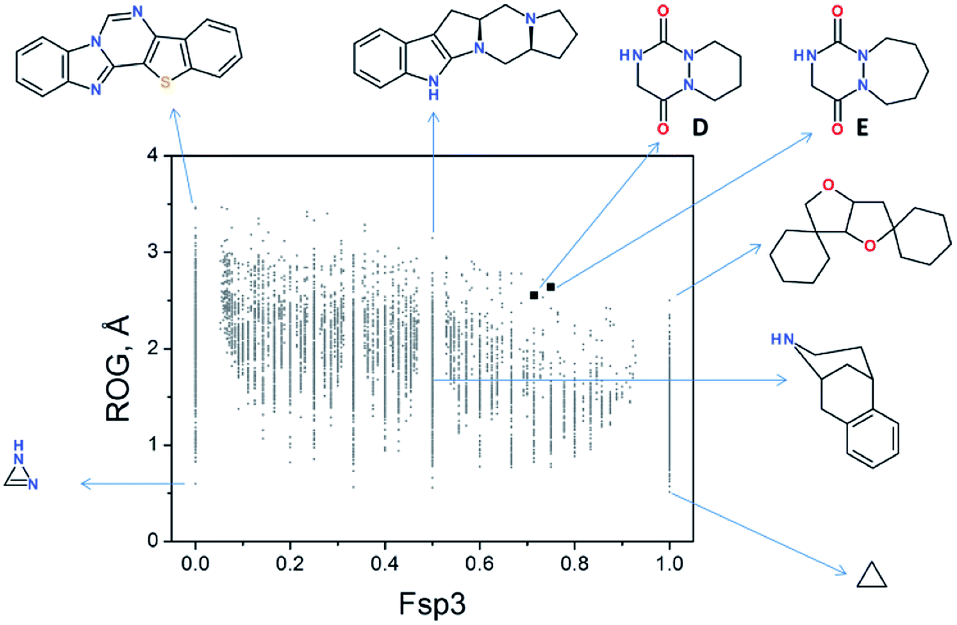
Recently, we evaluated the chemical space occupied by aza-DKPs and their “drug-likeness” properties by determining the quantitative estimate of the drug-likeness (QED).46 Thus, the radius of gyration (ROG) was plotted as a function of the fraction of sp3-hybridised carbons (Fsp3) of aza-DKP skeletons D and E and compared with more than 9000 unique structures described in the literature (Fig. 5).46 This analysis showed that both structures D and E were found in an under-populated area of the graph, demonstrating that a new chemical space can be explored with these new structures. Furthermore, both D and E exhibited high Fsp3 values (∼0.75) which allows a larger panel of decorating substituents around the scaffold while keeping this value high enough to improve their probability to successfully go through the different stages of clinical trials.49
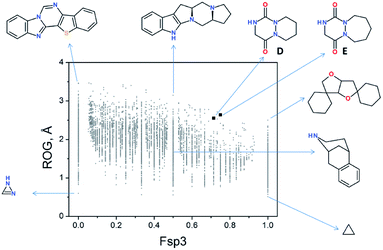 |
| | Fig. 5 2D plot of current drug-like scaffold space (reproduced from Org. Biomol. Chem., 2016, 14, 8859). | |
Our group also compared the physicochemical properties of aza-DKP 5 and its DKP equivalent 4. The aza-DKP studied exhibits a 16-fold higher aqueous solubility in PBS than the corresponding DKP (Table 2).46 Compound 5 also shows a lower logD 7.4 than 4 enabling the introduction of apolar substituents around the core during the drug discovery process. Finally, although both structures exhibited similar stability in PBS or mouse plasma, a higher metabolic stability was observed for aza-DKP 5 in mouse liver microsomes (83% vs. 51% of remaining molecules after 30 minutes in the presence of microsomes). These observations, if extrapolated, show better drug-like properties for these heterocycles compared with DKPs, a family of molecules already well known in medicinal chemistry.5
Table 2 Physicochemical properties comparison of an aza-DKP and its DKP equivalent
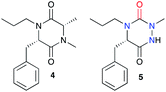 |
| Compound |
Aza-DKP 5 |
DKP 4 |
| Solubility (mM) |
16.5 ± 0.6 |
1.0 ± 0.1 |
| LogD |
1.1 ± 0.1 |
1.4 ± 0.1 |
| Stability (%) |
PBS (25 °C, 24 h) |
>98 |
>98 |
| Plasma (37 °C, 6 h) |
>98 |
>98 |
| Microsomes (37 °C, 30 min) |
83 ± 3 |
51 ± 1 |
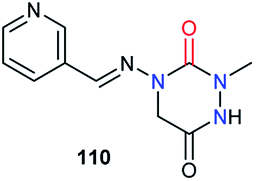
Regarding biological activities, only three studies including aza-DKPs have been reported to date. A team at Ciba–Geigy first described an aza-DKP 110, in a patent about pyridine derivatives as pesticides, without specifying its activity (Fig. 6).50
 |
| | Fig. 6 Aza-DKP structure with potential crop protection activity. | |
Another patent published by a team at Lilly in 2002, focusing on tadalafil analogues, also synthesised aza-DKP 112 (Fig. 7).28 This molecule exhibited an inhibitory activity against PDE5, lower than that of tadalafil 1 (113 nM vs. 5 nM).51 Nevertheless, this example shows that the DKP to aza-DKP switch did not lead to a dramatic reduction of the activity profile, and may be compensated by pharmacokinetic gains such as aqueous solubility or stability.
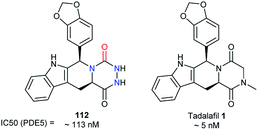 |
| | Fig. 7 Structures and activities of tadalafil and an aza-DKP analogue. | |
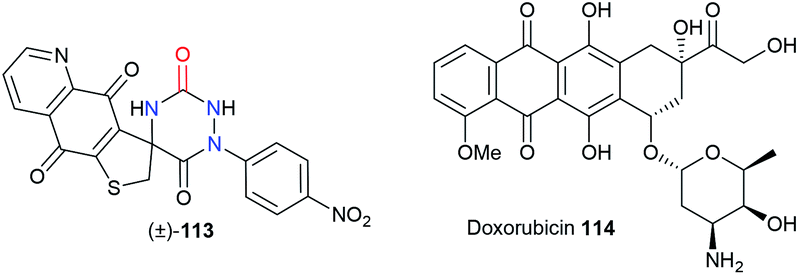
Finally, both the groups of Bolognese and Lavecchia focused on an aza-DKP series as potential anti-cancer drugs (Table 3).52 The spiro-fused aza-DKP (±)-113 emerged from this series as the most promising example with its high activity against several cancer lines (CCRF-CEM, CCRF-SB, MT-4) especially when compared with doxorubicin 114.
Table 3 Structures and cytotoxicity of an aza-DKP candidate 113 compared to doxorubicin 114
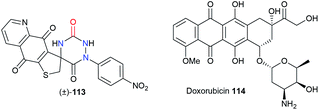
|
| Compound |
IC50 (μM) |
| CCRF-CEM |
CCRF_SB |
MT-4 |
HT-29 |
| rac-113 |
0.002 |
0.002 |
0.005 |
1.01 |
| 114 |
0.02 |
0.03 |
0.01 |
0.01 |
| Compound |
IC50 (μM) |
| MCF-7 |
HeLa |
ACHN |
SF-268 |
XF-498 |
| rac-113 |
0.55 |
0.61 |
0.65 |
3.55 |
4.80 |
| 114 |
0.05 |
0.07 |
0.04 |
0.2 |
0.16 |
5. Conclusions
In this review we highlight the interest of aza-DKPs as potential alternative to the well-known DKPs. Several different synthetic strategies have been developed since the first example published by Schlögl and Korger in 1951. Many variations around the aza-DKP core have also been made during their synthesis or by post-functionalisation to reach the corresponding mono-, di-, tri- or tetra-substituted aza-DKPs. Therefore a wide variety of these structures is currently available occupying a new drug-like scaffold space. These 3D-shaped heterocycles with one or more sp3-hybridized carbons including stereocenters display promising physicochemical and biological properties. Thus, the replacement of one carbon-stereogenic center of a DKP by a nitrogen atom results in a significant increase of their water solubility and microsomal stability. The different accesses to chiral aza-DKPs starting from amino acid precursors combined with the range of substitution patterns available around the ring, should fuel further interest in the synthesis of these triazines and the investigation of their potential in chemical biology and drug discovery.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the LabEx MEDALIS (ANR-10-LABX-0034), the Centre National de la Recherche Scientifique (CNRS), the University of Strasbourg. T. M. was supported by a fellowship from the Ecole Normale Supérieure Paris-Saclay.
Notes and references
- P. Das, M. D. Delost, M. H. Qureshi, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2019, 62, 4265 CrossRef CAS.
- E. Vitaku, D. T Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257 CrossRef CAS.
- C. Cornacchia, I. Cacciatore, L. Baldassarre, A. Mollica, F. Feliciani and F. Pinnen, Mini-Rev. Med. Chem., 2012, 12, 2 CrossRef CAS.
- Y. Wang, P. Wang, H. Ma and W. Zhu, Expert Opin. Ther. Pat., 2013, 23, 1 CrossRef.
- A. D. Borthwick, Chem. Rev., 2012, 112, 3641 CrossRef CAS.
- M. Jida, D. Tourwé and S. Ballet, RSC Adv., 2014, 4, 38159 RSC.
- R. Colombo, M. Mingozzi, L. Belvisi, D. Arosio, U. Piarulli, N. Carenini, P. Perego, N. Zaffaroni, M. De Cesare, V. Castiglioni, E. Scanziani and C. Gennari, J. Med. Chem., 2012, 55, 10460 CrossRef CAS.
- J. F. González, I. Ortín, E. de la Cuesta and J. C. Menéndez, Chem. Soc. Rev., 2012, 41, 6902 RSC.
- P. G. Sammes, Naturally occurring 2,5-dioxopiperazines and related compounds, in Fortschritte der Chemie Organischer Naturstoffe, ed. W. Herz, H. Grisebach and G. W. Kirby, Springer-Verlag, Vienna, 1975, pp. 51–118 Search PubMed.
- K. McCleland, P. J. Milne, F. R. Lucieto, C. Frost, S. C. Brauns, M. Van De Venter, J. Du Plessis and K. Dyason, J. Pharm. Pharmacol., 2004, 56, 1143 CrossRef CAS.
- M. B. Martins and I. Carvalho, Tetrahedron, 2007, 63, 9923 CrossRef CAS.
- M. Imamura and C. Prasad, Peptides, 2003, 24, 445 CrossRef CAS.
- M. L. Lopez-Rodriguez, J. Morcillo, M. E. Fernandez, E. Porras, L. Orensanz, M. E. Beneytez, J. Manzanares and J. A. Fuentes, J. Med. Chem., 2001, 44, 186 CrossRef CAS.
- A. Daugan, P. Grondin, C. Ruault, A.-C. Le Monnier de Gouville, H. Coste, J. M. Linget, J. Kirilovksy, F. Hyafil and R. Labaudinière, J. Med. Chem., 2003, 46, 4533 CrossRef CAS; U. Gresser and C. H. Gleiter, Eur. J. Med. Res., 2002, 7, 435 Search PubMed; K. Hatzimouratidis, Ther. Adv. Urol., 2004, 6, 135 CrossRef; N. S. Ahmed, Drug Dev. Res., 2008, 1 Search PubMed.
- P. G. Wyatt, M. J. Allen, A. D. Borthwick, D. E. Davies, A. M. Exall, R. J. D. Hatley, W. R. Irving, D. G. Livermore, N. D. Miller, F. Nerozzi, S. L. Sollis and A. K. Szardenings, Bioorg. Med. Chem. Lett., 2005, 15, 2579 CrossRef CAS.
- J. Liddle, M. J. Allen, A. D. Borthwick, D. P. Brooks, D. E. Davis, R. M. Edwards, A. M. Exall, C. Hamlett, W. R. Irving, A. M. Mason, G. P. McCafferty, F. Nerozzi, S. Peace, J. Philp, D. Pollard, M. A. Pullen, S. S. Shabbir, S. L. Sollis, T. D. Westfall, P. M. Woolard, C. Wu and D. M. B. Hickey, Bioorg. Med. Chem. Lett., 2008, 18, 90 CrossRef CAS.
- A. D. Borthwick and J. Liddle, Med. Res. Rev., 2011, 31, 576 CrossRef CAS.
- R. S. Heist, O. R. Aren, A. C. Mita, J. Polikoff, L. Bazhenova, G. K. Lloyd, W. Mikrut, W. Reich, M. A. Spear and L. Huang, J. Clin. Oncol., 2014, 15, 8054 Search PubMed.
- A. A. Patchett and R. P. Nargund, Annu. Rep. Med. Chem., 2000, 35, 289 CAS.
- M. E. Welsch, S. A. Snyder and B. R. Stockwell, Curr. Opin. Chem. Biol., 2010, 14, 347 CrossRef CAS.
- W. R. Pitt, D. M. Parry, B. G. Perry and C. R. Groom, J. Med. Chem., 2009, 52, 2952 CrossRef CAS.
- C. Didierjean, V. Del Duca, E. Benedettin, A. Aubry, M. Zouikri, M. Marraud and G. Boussard, J. Pept. Res., 1997, 50, 451 CrossRef CAS.
- S. Bouayad-Gervais and W. D. Lubell, Molecules, 2013, 18, 14739 CrossRef CAS.
- P. Regenass, D. Bosc, S. Riché, P. Gizzi, M. Hibert, L. Karmazin, A. Ganesan and D. Bonnet, J. Org. Chem., 2017, 6, 3239 CrossRef.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 1997, 23, 3 CrossRef CAS.
- K. Schlögl and G. Korger, Monatsh. Chem., 1951, 82, 799 CrossRef.
- K. Winterfield and G. Nair, Arch. Pharm., 1971, 71, 216 CrossRef.
- M. Orme, J. S. Sawyer and L. M. Schultze, Fused heterocyclic derivatives as phosphodiesterase inhibitors, WO200210166A1, 2002.
- M. Robba, J.-C. Lancelot, D. Maume and A. Rabaron, J. Heterocycl. Chem., 1978, 7, 1159 CrossRef.
- D. Maume, J.-C. Lancelot and M. Robba, J. Heterocycl. Chem., 1979, 6, 1217 CrossRef.
- J.-C. Lancelot, D. Maume and M. Robba, J. Heterocycl. Chem., 1981, 18, 743 CrossRef CAS.
- J. Catalán, J. L. G. de Paz and J. Elguero, J. Chem. Soc., Perkin Trans. 2, 1996, 1, 57 RSC.
- F. Pinnen, G. Luisi, A. Calcagni, G. Lucente, E. Gavuzza and S. Cerrini, J. Chem. Soc., Perkin Trans. 1, 1994, 12, 1611 RSC.
- P. Baudet and M. Calin, Helv. Chim. Acta, 1969, 1, 282 CrossRef.
- P. G. Baraldi, B. Cacciari, R. Romagnoli and G. Spalluto, Synthesis, 1999, 1999, 453 CrossRef; P. G. Baraldi, B. Cacciari, R. Romagnoli and G. Spalluto, Arzneim.-Forsch./Drug Res., 1999, 12, 997 Search PubMed.
- R. A. Ivanovitch, J.-F. Vincent-Rocan, E. B. Elkaeed and A. M. Beauchemin, Org. Lett., 2015, 17, 4898 CrossRef.
- Y. Hamuro, W. J. Marshall and M. A. Scialdone, J. Comb. Chem., 1999, 1, 163 CrossRef CAS.
- T. Schwan, J. Heterocycl. Chem., 1983, 20, 547 CrossRef CAS.
- R. V. Hoffman, N. K. Nayyar and W. Chen, J. Org. Chem., 1995, 13, 4121 CrossRef.
- R. V. Hoffman, N. K. Nayyar and W. Chen, J. Org. Chem., 1995, 60, 5992 CrossRef CAS; R. V. Hoffman, M. M. Reddy, C. M. Klumas and F. Cervantes-Lee, J. Org. Chem., 1998, 63, 9128 CrossRef.
- F. Pinnen, G. Luisi, G. Lucente, E. Gavuzza and S. Cerrini, J. Chem. Soc., Perkin Trans. 1, 1993, 7, 819 RSC.
- C. B. Bourguet, C. Proulx, S. Klocek, D. Sabatino and W. D. Lubell, J. Pept. Sci., 2010, 16, 284 CrossRef CAS.
- A. Obreza and U. Urleb, Synth. Commun., 2003, 33, 1011 CrossRef CAS.
- D. Bonnet, J.-F. Margathe, S. Radford, E. Pfimlin, S. Riché, P. Doman, M. Hibert and A. Ganesan, ACS Comb. Sci., 2012, 14, 323 CrossRef CAS.
- P. Regenass, J.-F. Margathe, A. Mann, J. Suffert, M. Hibert, N. Girard and D. Bonnet, Chem. Commun., 2014, 50, 9657 RSC; P. Regenass, Doctoral thesis, Laboratoire d'Innovation Thérapeutique, Faculté de Pharmacie, 2015.
- P. Regenass, S. Riché, F. Péron, D. Rognan, M. Hibert, N. Girard and D. Bonnet, Org. Biomol. Chem., 2016, 14, 8859 RSC.
- T. Maujean, N. Girard, A. Ganessan, M. Gulea and D. Bonnet, Eur. J. Org. Chem., 2020 DOI:10.1002/ejoc.202001339.
- F. Péron, S. Riché, B. Lesur, M. Hibert, P. Breton, J.-M. Fourquez, N. Girard and D. Bonnet, ACS Omega, 2018, 3, 15182 CrossRef.
- F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752 CrossRef CAS.
- H. Szczepanski, H. Kristinsson, P. Maienfisch and J. Ehrenfreund, Pyridine derivatives as pesticides, WO95/18123, 1995.
- Y.-Y. Huang, Z. Li, Y.-H. Cai, L.-J. Feng, Y. Wu, X. Li and H.-B. Luo, J. Chem. Inf. Model., 2013, 11, 3044 CrossRef.
- A. Bolognese, G. Correale, M. Manfra, A. Esposito, E. Novellino and A. Lavecchia, J. Med. Chem., 2008, 51, 8148 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Nicolas Girard
a,
Nicolas Girard