DOI:
10.1039/D0RA07865D
(Paper)
RSC Adv., 2020,
10, 42897-42902
Highly effective organic light-emitting diodes containing thermally activated delayed fluorescence emitters with horizontal molecular orientation†
Received
14th September 2020
, Accepted 19th November 2020
First published on 26th November 2020
Abstract
In this study, we report new thermally activated delayed fluorescence (TADF) emitters, AcPYM (10,10′-(pyrimidine-2,5-diylbis(4,1-phenylene))bis(9,9-dimethyl-9,10-dihydroacridine)) and PxPYM (10,10′-(pyrimidine-2,5-diylbis(4,1-phenylene))bis(10H-phenoxazine)), by employing donor units at the 2,5-positions of the pyrimidine acceptor unit. The donor–acceptor–donor (D–A–D) units combined in the linear molecular structure of AcPYM or PxPYM enhanced the horizontally oriented alignment, and the horizontal transition dipole moments were realized by up to 87% in the host matrix. Organic light-emitting diodes (OLEDs) containing AcPYM and PxPYM emitters realized external quantum efficiencies (ηext) of 16.8% for blue and green emissions.
Introduction
After the first research into the organic light-emitting diodes (OLEDs) was reported in 1987,1 significant improvements in OLEDs regarding efficiency, color purity, and lifetime have been achieved, leading to them becoming the next-generation light source for lighting and flexible display applications. Over the past several decades, various studies into organic emitters based on fluorescence and phosphorescence in the visible-light region have been conducted to improve the internal quantum efficiency (ηint).2–7 Even though fluorescence emitters have outstanding reliability and stability, low exciton production efficiency (ηST = 25%) of fluorescence in electrical excitation limits the theoretical ηint to 25%. Phosphorescence emitters can achieve a theoretical ηint of nearly 100% from singlet (S1) and triplet (T1) exciton harvesting through intersystem crossing (ISC) between the S1 and T1 states using heavy transition metals.8–12 However, phosphorescence emitters suffer from a strong exciton annihilation process and a drastic efficiency decrease under the high current density derived from the high density of T1 excitons originating from their long radiative decay time (τ; μs–ms).13
Recently, a thermally activated delayed fluorescence (TADF) emitter that can substitute for fluorescence and phosphorescence emitters in OLEDs due to its effective S1 and T1 excitons harvesting by reverse ISC (RISC) from the T1 to the S1 state has been reported.14–20 The up-conversion through RISC can only arise with a small S1-to-T1 state energy gap (ΔEST < 0.3 eV) in TADF emitters, which is attained by providing less overlapping between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) levels. Furthermore, numerous studies have focused on molecular designs utilizing twisted molecular structures among the donor and acceptor units for reducing ΔEST and achieving efficient intramolecular charge transfer (ICT) based on pure organic derivatives.21,22 Meanwhile, despite the development of various TADF emitters with ηint of nearly 100%, conventional OLEDs have a 20–25% external quantum efficiency (ηext) limitation because of low light out-coupling efficiency (ηout).23–25 External and internal optical modification of OLEDs such as attaching a microlens array, optimizing the layer thickness, and refractive index control of the internal layers have been widely studied to increase ηout.26–38 Another promising approach for enhancing ηout is to introduce horizontally oriented emitters. Previous research by Yokoyama and co-workers demonstrated an increase of ηout in OLEDs by applying horizontally aligned emitters having horizontal transition dipole moment orientation on the substrate.39–41 Furthermore, the orientation degree of the emitters can be controlled by the shape of the molecules: rod- or disk-like structures prefer high degrees of horizontal transition dipole moments for perpendicular light emission.
In this study, we developed two pyrimidine-based TADF emitters: 10,10′-(pyrimidine-2,5-diylbis(4,1-phenylene))bis(9,9-dimethyl-9,10-dihydroacridine) (AcPYM) and 10,10′-(pyrimidine-2,5-diylbis(4,1-phenylene))bis(10H-phenoxazine) (PxPYM) and adopted donor–acceptor–donor (D–A–D) combined linear molecular structures based on a combination of donor units of 9,9-dimethylacridin (Ac) or phenoxazine (Px) and one of the pyrimidine acceptor units. The donor units were introduced at the 2,5-position of the pyrimidine unit to compose a linear D–A–D framework of emitters to induce a horizontal transition dipole moment. These new emitters achieved small ΔEST due to less overlapping between the HOMO and LUMO levels and improved RISC through effective exciton harvesting. Furthermore, a high rate of horizontal molecular orientation of over 80% was observed in angle-dependent photoluminescence (PL) measurements of the host matrix. OLEDs employing horizontally oriented TADF emitters achieved high ηext of 16.8% in blue and green light emissions.
Experimental
Materials
All reagents and solvents were prepared from Alfa Aesar, Sigma-Aldrich, and Tokyo Chemical Industry. Two intermediates, 9,9-dimethyl-10-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)-9,10-dihydroacridine (1) and 10-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)-10H-phenoxazine (2), were synthesized as reported previously.42
Synthesis
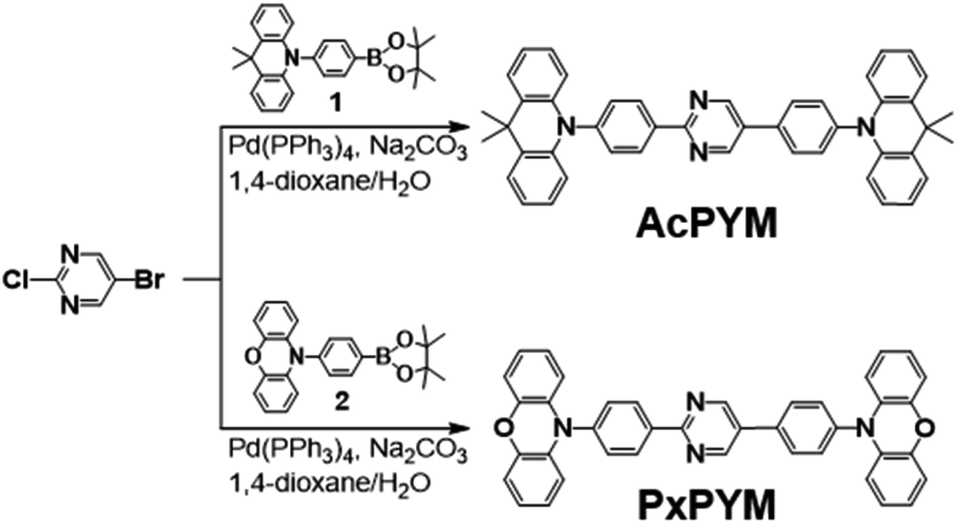
Synthesis of AcPYM. 5-Bromo-2-chloropyrimidine (0.50 g, 2.58 mmol) and 1 (2.34 g, 5.69 mmol) were dissolved in 1,4-dioxane (30 mL) under an N2 atmosphere. Next, sodium carbonate (2.4 M, 15 mL) and tetrakis(triphenylphosphine)-palladium(0) (0.24 g, 0.21 mmol) were added and the mixture refluxed for 12 h. After cooling down to room temperature, the solution was poured into chloroform and distilled water for extraction. The chloroform layer was washed with distilled water several times and dried over magnesium sulfate (MgSO4). The crude product was filtered by using Celite 545 and purified via column chromatography on silica gel (eluent: dichloromethane/hexane, 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v). The product was dried in a vacuum oven to give a white powder (yield = 1.03 g; 62%) and further purified via vacuum train sublimation. 1H NMR (500 MHz, CDCl3): δ 9.20 (s, 2H), 8.79 (d, J = 8.5 Hz, 2H), 7.95 (d, J = 8.5 Hz, 2H), 7.55 (d, J = 8.0 Hz, 4H), 7.49 (t, J = 6.5 Hz, 4H), 7.03–6.93 (m, 8H), 6.38 (d, J = 8.0 Hz, 2H), 6.35 (d, J = 8.0 Hz, 2H). 13C NMR (500 MHz, CDCl3) data are as follows: 155.43, 143.86, 140.73, 132.56, 131.66, 130.82, 130.24, 129.28, 126.44, 125.39, 120.87, 120.73, 36.04, 31.30; Anal. calcd (%) for C46H38N4: C 85.42; H 5.92; N 8.66; found: C 85.32; H 5.92; N 8.68.
:1 v/v). The product was dried in a vacuum oven to give a white powder (yield = 1.03 g; 62%) and further purified via vacuum train sublimation. 1H NMR (500 MHz, CDCl3): δ 9.20 (s, 2H), 8.79 (d, J = 8.5 Hz, 2H), 7.95 (d, J = 8.5 Hz, 2H), 7.55 (d, J = 8.0 Hz, 4H), 7.49 (t, J = 6.5 Hz, 4H), 7.03–6.93 (m, 8H), 6.38 (d, J = 8.0 Hz, 2H), 6.35 (d, J = 8.0 Hz, 2H). 13C NMR (500 MHz, CDCl3) data are as follows: 155.43, 143.86, 140.73, 132.56, 131.66, 130.82, 130.24, 129.28, 126.44, 125.39, 120.87, 120.73, 36.04, 31.30; Anal. calcd (%) for C46H38N4: C 85.42; H 5.92; N 8.66; found: C 85.32; H 5.92; N 8.68.
Synthesis of PxPYM. The synthesis of this compound was similar to AcPYM using 2 (3.07 g; 7.96 mmol). The product was dried in vacuum oven to give a yellow powder (yield = 1.30 g; 60%) and further purified by vacuum train sublimation. 1H NMR (500 MHz, CDCl3): δ 9.14 (s, 2H), 8.74 (d, J = 8 Hz, 2H), 7.90 (d, J = 8.0 Hz, 2H), 7.56 (d, J = 8.5 Hz, 2H), 7.53 (d, J = 8.5 Hz, 2H), 6.74–6.60 (m, 12H), 6.05 (dd, J = 6.5, 1.5 Hz, 2H), 6.02 (dd, J = 6.0, 1.5 Hz, 2H). 13C NMR (500 MHz, CDCl3) data are as follows: 206.91, 155.40, 144.03, 134.10, 132.10, 131.14, 130.97, 129.47, 123.30, 121.69, 121.53, 115.67, 115.56, 113.38, 113.28, 110.73, 110.41, 31.24, 30.92; Anal. calcd (%) for C40H26N4: C 80.79; H 9.42; N 4.41; found: C 80.91; H 9.47; N 4.26.
Results and discussion
Synthesis and thermal properties
AcPYM and PxPYM were synthesized through Suzuki coupling reactions with either Ac or Px, respectively, with the bromine-substituted pyrimidine (Scheme 1).42 The chemical structures of the newly synthesized emitters were analyzed using 1H and 13C NMR spectroscopy, MALDI-TOF mass spectrometry, and elemental analysis. In addition, Differential Scanning Calorimetry (DSC) and thermogravimetric analysis (TGA) was performed to analyze the thermal properties of the emitters (Fig. S4 and S5†); it was observed that the decomposition temperatures (Td) of AcPYM and PxPYM were 435 and 463 °C, respectively.
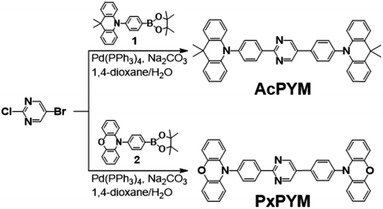 |
| | Scheme 1 Synthesis of AcPYM and PxPYM. | |
Density functional theory (DFT) calculations
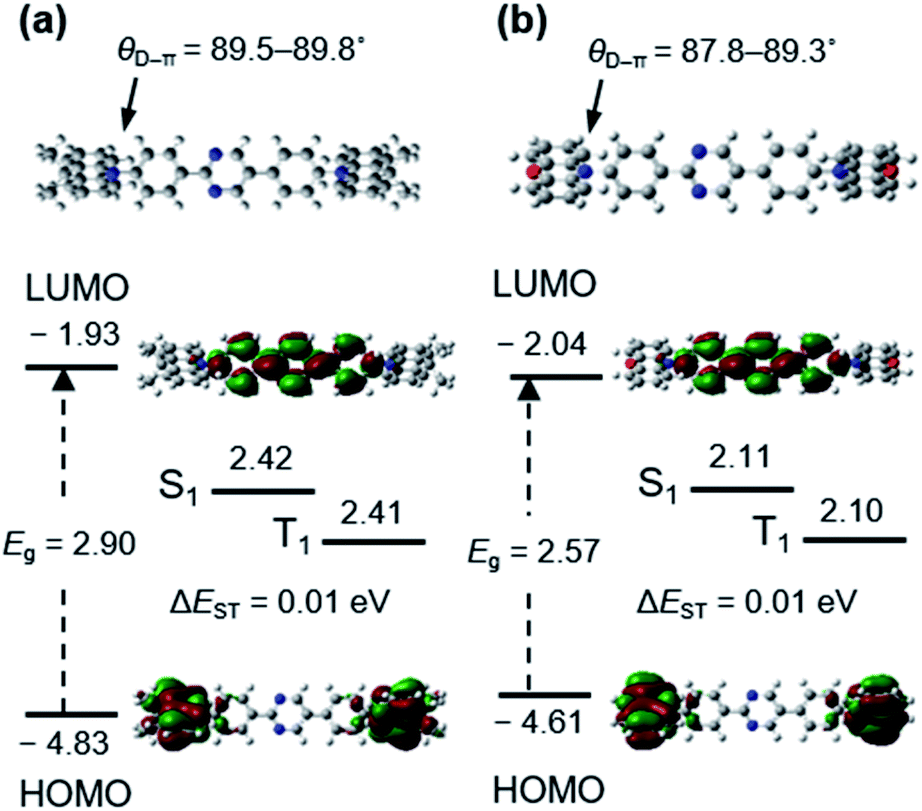
The optimized molecular structures, excited states energy levels, and frontier molecular orbital contributions of AcPYM and PxPYM were calculated via a time-dependent DFT (TD-DFT) at the B3LYP/6-31G(d,p) level (Fig. 1). These emitters have dihedral angles between the phenyl linkers and the Ac or Px units (θD–π) of 89.5–89.8° and 87.8–89.3° for AcPYM and PxPYM, respectively. Moreover, the LUMO and HOMO levels of AcPYM and PxPYM are localized on the pyrimidine acceptor and either the Ac or Px donor units, respectively. This good separation of the frontier molecular orbital distributions of AcPYM and PxPYM originating from the highly sterically hindered structure contributed to the relatively small calculated ΔEST of 0.01 eV.
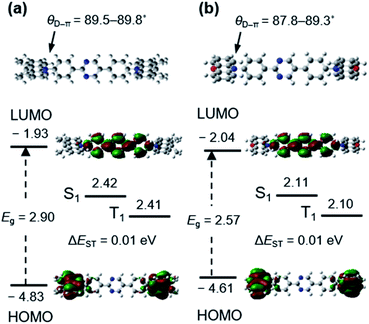 |
| | Fig. 1 The optimized molecular structure, singlet (S1) and triplet (T1) energy levels, and HOMO and LUMO distribution of (a) AcPYM and (b) PxPYM calculated via DFT and TD-DFT at the B3LYP/6-31G(d,p) level. | |
Photophysical properties
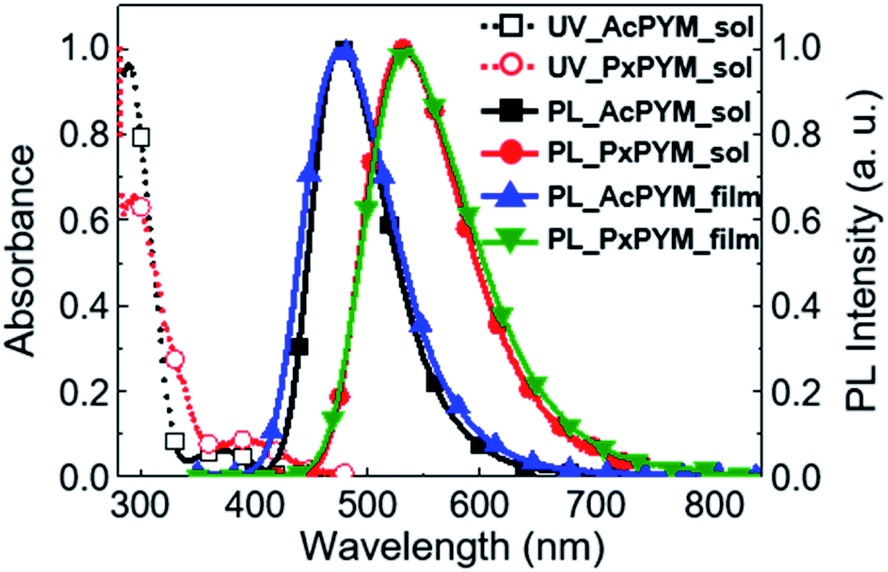
Fig. 2 displays spectra of the ultraviolet-visible (UV-Vis) absorbance and the PL of the emitters in toluene. The broad 375 and 394 nm-centered absorption peaks and the 477 and 532 nm-centered blue and green PL emission peaks (λPL) of AcPYM and PxPYM were observed in the UV-Vis and PL spectra under photoexcitation of oxygen-free toluene, respectively. Furthermore, 6 wt% AcPYM and PxPYM doped in 2,8-bis(diphenyl-phosphoryl)-dibenzo[b,d]thiophene (PPT) host thin films were prepared to confirm the photophysical properties of the emitters in the solid-state. Because of the higher T1 energy level of PPT (ET = 3.1 eV) compare with AcPYM and PxPYM (ET = 2.65 and 2.58 eV, Fig. S7†), the PPT host was chosen to contain the reverse energy transfer between the emitter and the host.17,19,43 As shown in Fig. 2, the 6 wt% AcPYM:PPT-doped film exhibited blue emission with 478 nm λPL, while the 6 wt% PxPYM:PPT-doped film emitted green light at around 537 nm.
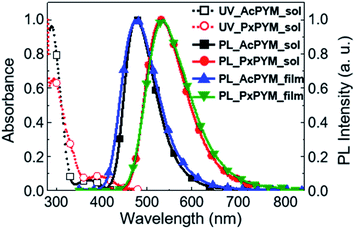 |
| | Fig. 2 UV-Vis absorbance and PL spectra of AcPYM and PxPYM in toluene, and 6 wt% AcPYM and PxPYM:PPT-doped films. | |
The PL quantum yields (PLQY, ΦPL) of the 6 wt% AcPYM- and PxPYM-doped films in PPT hosts were measured using an integrating sphere, with which ΦPL levels of 53.3% and 78.0% were observed. We also examined temperature-dependent transient PL decays to identify the TADF characteristics of the emitters' temperature range from 100 to 300 K using a streak camera (Fig. 3). The delayed component PL intensity increased while the temperature raise from 100 to 300 K, thereby demonstrating that the thermal energy at high temperature accelerated the RISC process.31 Furthermore, 19 and 17 ns of the prompt fluorescence lifetimes (τp) and 486 and 287 μs of the delayed fluorescence lifetimes (τd) were observed in the double-exponential function fitted transient PL decay curves of AcPYM and PxPYM, respectively. ΦPL and the ISC and RISC rate constants are summarized in Table S3.†44
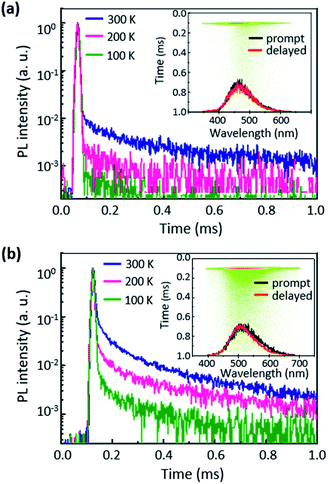 |
| | Fig. 3 Transient decay curves of the (a) 6 wt% AcPYM:PPT- and (b) 6 wt% PxPYM:PPT-doped films (inset: time-resolved PL spectra and streak images). | |
The horizontal alignment of the emitters was confirmed via angle-dependent PL measurements (Fig. 4). To verify the molecular orientation of the AcPYM and PxPYM, the angle dependence of the PL intensity of emitters in the host matrix was estimated to fit the experimental data. Moreover, the horizontal molecular orientation degree was estimated using anisotropy factor Θ calculated from the perpendicularly oriented fraction to the total amount of transition dipole moments:30,39,45
| Θ = [pz]/([px] + [py] + [pz]) |
where [
pz] is vertical, and [
px] and [
py] are horizontal transition dipole moments. A
Θ value of 1/3 means complete isotropy ([
px] = [
py] = [
pz] = 1), and the value of zero means perfectly horizontal ([
px] = [
py] = 1, [
pz] = 0). From the experimental results,
Θ was equal to 0.13 and 0.18 for
AcPYM and
PxPYM:PPT-doped films, respectively, demonstrating that the
AcPYM:PPT-doped film has 87% horizontal transition dipole moments and the
PxPYM:PPT-doped film has 83%. Moreover, the values are higher than those of previously reported emitters having random molecular orientation, indicating that the devices developed using
AcPYM and
PxPYM emitters showed higher
ηext values.
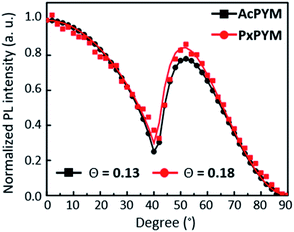 |
| | Fig. 4 Angle-dependent PL spectra of AcPYM and PxPYM (p-polarized light from 15 nm thin film at 480 nm for AcPYM and 525 nm for PxPYM). Θ is an anisotropy factor calculated from the fitted solid lines, while the symbols present the experimental data. | |
Electroluminescence (EL) performance
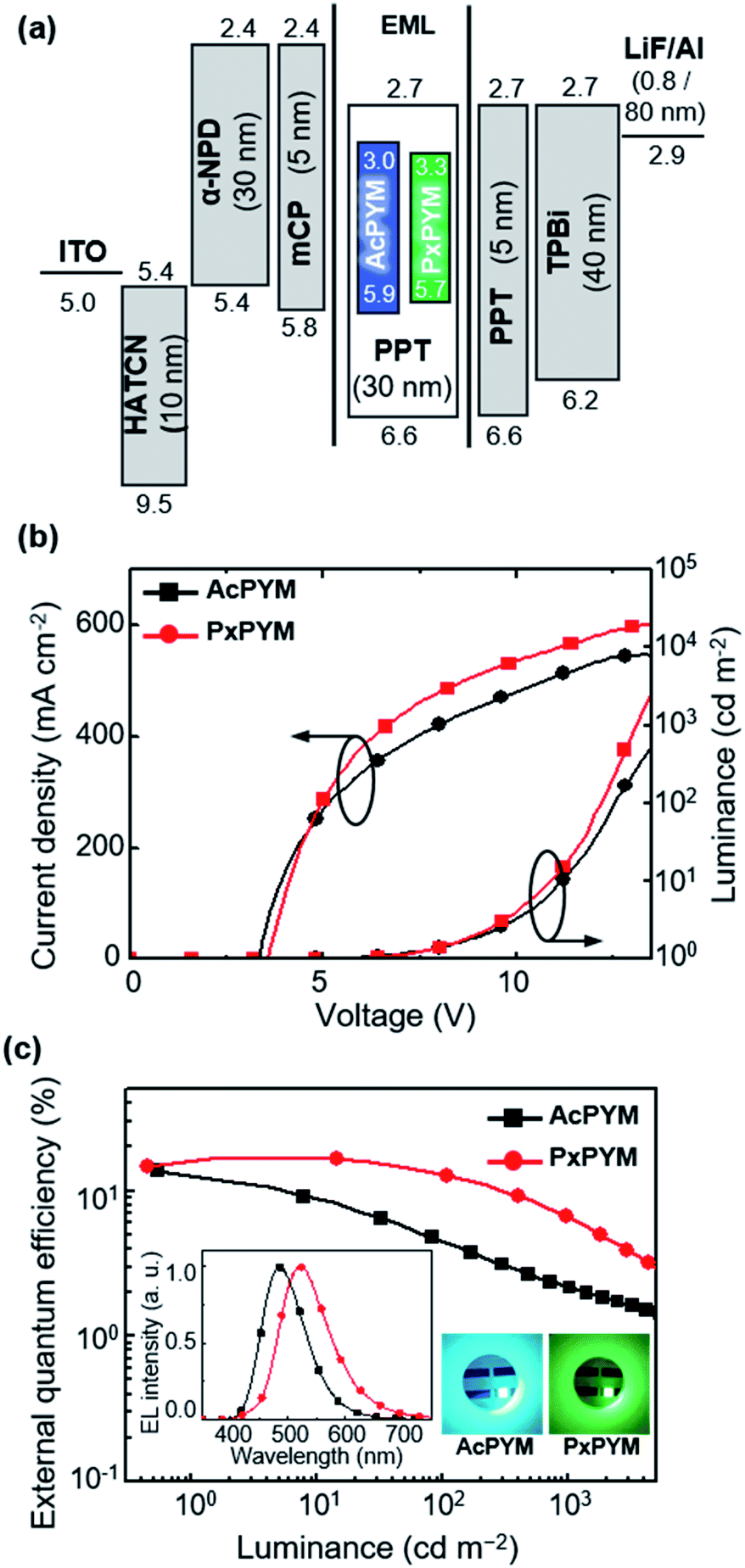
The EL characteristics of TADF-OLEDs using AcPYM and PxPYM emitters in devices of indium tin oxide (ITO)/1,4,5,7,8,11-hexaazatriphenylene-hexacarbonitrile (HATCN, 10 nm)/4,4′-bis[N-(1-naphtyl)-N-phenylamino]-1,1′-biphenyl (α-NPD, 30 nm)/1,3-bis(9-carbazolyl)benzene (mCP, 5 nm)/6 wt% emitter: PPT (30 nm)/PPT (5 nm)/1,3,5-tris(N-phenylbenzimidazol-2-yl)benzene (TPBi, 40 nm)/lithium fluoride (LiF, 0.8 nm)/aluminum (Al, 80 nm) (Fig. 5(a)). To prevent triplet exciton quenching from emitting layer (EML) to hole- and electron-transporting layer in these devices, 5 nm thin layers of mCP and PPT having high T1 energy levels (2.9 and 3.1 eV, respectively) were added as the neighboring layers of the EML.
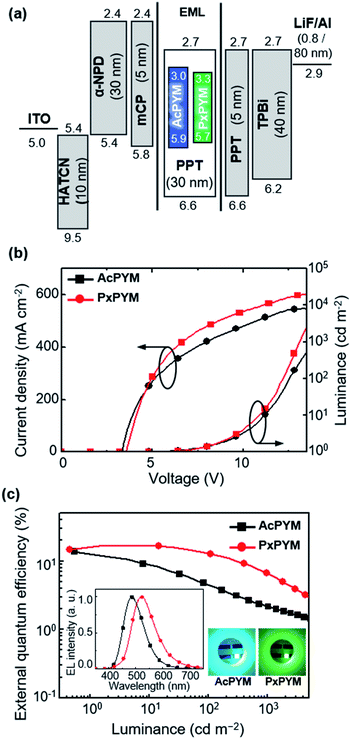 |
| | Fig. 5 (a) Energy level diagrams, (b) current density–voltage–luminance (J–V–L) curves, and (c) external quantum efficiency (ηext) versus luminance plots of the devices employing AcPYM or PxPYM (inset: EL spectra at 10 mA cm−2 and light-emitting images under electrical excitation). | |
The current density–voltage–luminance (J–V–L) properties are displayed in Fig. 5(b). The TADF-OLEDs employing AcPYM and PxPYM as emitters exhibited blue and green EL emission peaks (λEL) at 487 and 524 nm, respectively (Fig. 5(c)). In addition, below a turn-on voltage of 3.6 V for all devices exhibited maximum luminance (Lmax) values of 20370 cd m−2 for AcPYM and 19390 cd m−2 for PxPYM. The AcPYM-based TADF-OLEDs achieved ηext of 11.9%, current efficiency (ηc) of 27.0 cd A−1, and power efficiency (ηp) of 25.0 lm W−1, while the PxPYM-based OLEDs achieved 16.8%, 52.2 cd A−1, and 41.0 lm W−1, respectively (Table 1).
Table 1 EL performances of the devices using AcPYM or PxPYM as the emitter
| Emitter |
λELa (nm) |
Vturn-onb (V) |
Lmaxc (cd m−2) |
ηcd (cd A−1) |
ηpe (lm W−1) |
ηextf (%) |
CIE (x,y)g |
| The peak wavelength of EL emission. Turn-on voltage at 1 cd m−2. maximum luminance. Maximum current efficiency. Maximum power efficiency. External quantum efficiency. Commission Internationale de l'Éclairage (CIE) color coordination. |
| AcPYM |
487 |
3.4 |
20370 |
27.0 |
25.0 |
11.9 (@3.4 V) |
(0.17, 0.32) |
| PxPYM |
524 |
3.6 |
19390 |
52.2 |
41.0 |
16.8 (@4.0 V) |
(0.29, 0.51) |
Conclusions
We designed and synthesized linear D–A–D-structured TADF emitters AcPYM and PxPYM with Ac or Px donor units, respectively, at the 2,5-positions of the pyrimidine acceptor unit. These newly designed TADF emitters realized a small ΔEST, which accelerated the RISC and increased the exciton harvesting efficiency. Moreover, the molecular orientation of the emitters was estimated by angle-dependent PL measurements, indicating that both AcPYM and PxPYM had high rates of horizontal molecular alignment (87% and 83%, respectively). The TADF-OLEDs fabricated using either AcPYM or PxPYM as the emitter exhibited ηext values of 11.9% and 16.8%, respectively, both of which exceed the theoretical ηext evaluated for a random distribution of emitting dipoles. We also believe that our approach clearly presents a move toward horizontally oriented TADF emitters that will inspire the realization of highly effective TADF-OLEDs in the future.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was supported by the Dongguk University Research Fund of 2017 for S. Y. Lee. C. Adachi acknowledges support from the Japan Science and Technology Agency (JST) and ERATO, the Adachi Molecular Exciton Engineering Project, under JST ERATO (grant number: JPMJER1305), Japan, and the International Institute for Carbon Neutral Energy Research (WPI-I2CNER) funded by the Ministry of Education, Culture, Sports, Science & Technology (MEXT).
Notes and references
- C. W. Tang and S. A. VanSlyke, Appl. Phys. Lett., 1987, 51, 913 CrossRef CAS.
- F. B. Dias, K. N. Bourdakos, V. Jankus, K. C. Moss, K. T. Kamatekar, V. Bhalla, J. Santos, M. R. Bryce and A. P. Monkman, Adv. Mater., 2013, 25, 3707 CrossRef CAS.
- G. Schwartz, M. Pfeiffer, S. Reineke, K. Walzer and K. Leo, Adv. Mater., 2007, 19, 3672 CrossRef CAS.
- Q. Wang, J. Ding, D. Ma, Y. Cheng, L. Wang, X. Jing and F. Wang, Adv. Funct. Mater., 2009, 19, 84 CrossRef CAS.
- Y. Sun, N. C. Giebink, H. Kanno, B. Ma, M. E. Thompson and S. R. Forrest, Nature, 2006, 440, 908 CrossRef CAS.
- M. A. Baldo, D. F. O'Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151 CrossRef CAS.
- C. Adachi, M. A. Baldo and S. R. Forrest, J. Appl. Phys., 2000, 87, 8049 CrossRef CAS.
- J. Tagare and S. Vaidyanathan, J. Mater. Chem. C, 2018, 38, 10138 RSC.
- I. S. Park, H. Komiyama and T. Yasuda, Chem. Sci., 2017, 8, 953 RSC.
- H. V. R. Dias, H. V. K. Diyabalanage, M. A. Rawashdeh-Omary, M. A. Franzman and M. A. Omary, J. Am. Chem. Soc., 2003, 125, 12072 CrossRef CAS.
- A. B. Tamayo, B. D. Alleyne, P. I. Djurovich, S. Lamansky, I. Tsyba, N. N. Ho, R. Bau and M. E. Thompson, J. Am. Chem. Soc., 2003, 125, 7377 CrossRef CAS.
- W. Sotoyama, T. Satoh, N. Sawatari and H. Inoue, Appl. Phys. Lett., 2005, 86, 153505 CrossRef.
- S. Reineke, K. Walzer and K. Leo, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 125328 CrossRef.
- S. H. Choi, C. H. Lee, C. Adachi and S. Y. Lee, Dyes Pigm., 2019, 171, 107775 CrossRef CAS.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234 CrossRef CAS.
- Q. Zhang, B. Li, S. Huang, H. Nomura, H. Tanaka and C. Adachi, Nat. Photonics, 2014, 8, 326–332 CrossRef CAS.
- S. Y. Lee, T. Yasuda, H. Nomura and C. Adachi, Appl. Phys. Lett., 2012, 101, 093306 CrossRef.
- C. Adachi, Jpn. J. Appl. Phys., 2014, 53, 060101 CrossRef.
- S. H. Choi, C. H. Lee, C. Adachi and S. Y. Lee, Dyes Pigm., 2020, 172, 107849 CrossRef.
- S. Hirata, Y. Sakai, K. Masui, H. Tanaka, S. Y. Lee, H. Nomura, N. Nakamura, M. Yasumatsu, H. Nakanotani, Q. Zhang, K. Shizu, H. Miyazaki and C. Adachi, Nat. Mater., 2015, 14, 330 CrossRef CAS.
- X. Lv, R. Huang, S. Sun, Q. Zhang, S. Xiang, S. Ye, P. Leng, F.
B. Dias and L. Wang, ACS Appl. Mater. Interfaces, 2019, 11, 10758 CrossRef CAS.
- F. Baraket, B. Pedras, É. Torres, M. J. Brites, M. Dammak and M. N. Berberan-Santos, Dyes Pigm., 2020, 175, 108114 CrossRef CAS.
- G. Méhes, H. Nomura, Q. Zhang, T. Nakagawa and C. Adachi, Angew. Chem., Int. Ed., 2012, 51, 11311 CrossRef.
- S. Möller and S. R. Forrest, J. Appl. Phys., 2002, 91, 3324 CrossRef.
- K. Hong, H. K. Yu, I. Lee, K. Kim, S. Kim and J. L. Lee, Adv. Mater., 2010, 22, 4890 CrossRef CAS.
- S. Reineke, F. Lindner, G. Schwartz, N. Seidler, K. Walzer, B. Lüssem and K. Leo, Nature, 2009, 459, 234 CrossRef CAS.
- Y. Sun and S. R. Forrest, Nat. Photonics, 2008, 2, 483 CrossRef CAS.
- K. B. Choi, S. J. Shin, T. H. Park, H. J. Lee, J. H. Hwang, J. H. Park, B. Y. Hwang, Y. W. Park and B. K. Ju, Org. Electron., 2014, 15, 111 CrossRef CAS.
- M. G. Helander, Z. B. Wang, J. Qiu, M. T. Greiner, D. P. Puzzo, Z. W. Liu and Z. H. Lu, Science, 2011, 332, 944 CrossRef CAS.
- C. Mayr, S. Y. Lee, T. D. Schmidt, T. Yasuda, C. Adachi and W. Brütting, Adv. Funct. Mater., 2014, 24, 5232 CrossRef CAS.
- T. A. Lin, T. Chatterjee, W. L. Tsai, W. K. Lee, M. J. Wu, M. Jiao, K. C. Pan, C. L. Yi, C. L. Chung, K. T. Wong and C. C. Wu, Adv. Mater., 2016, 28, 6976 CrossRef CAS.
- Y. S. Park, S. Lee, K. H. Kim, S. Y. Kim, J. H. Lee and J. J. Kim, Adv. Funct. Mater., 2013, 23, 4914 CrossRef CAS.
- K. H. Kim, E. S. Ahn, J. S. Huh, Y. H. Kim and J. J. Kim, Chem. Mater., 2016, 28, 7505 CrossRef CAS.
- K. H. Kim, J. L. Liao, S. W. Lee, B. Sim, C. K. Moon, G. H. Lee, H. J. Kim, Y. Chi and J. J. Kim, Adv. Mater., 2016, 28, 2526 CrossRef CAS.
- K. H. Kim, J. Y. Baek, C. W. Cheon, C. K. Moon, B. Sim, M. Y. Choi, J. J. Kim and Y. H. Kim, Chem. Commun., 2016, 52, 10956 RSC.
- C. S. Oh, C. K. Moon, J. M. Choi, J. S. Huh, J. J. Kim and J. Y. Lee, Org. Electron., 2017, 42, 337 CrossRef CAS.
- P. Rajamalli, N. Senthikumar, P. Y. Huang, C. C. Ren-Wu, H. W. Lin and C. H. Cheng, J. Am. Chem. Soc., 2017, 139, 10948 CrossRef CAS.
- S. J. Woo, Y. Kim, M. J. Kim, J. Y. Baek, S. K. Kwon, Y. H. Kim and J. J. Kim, Chem. Mater., 2018, 30, 857 CrossRef CAS.
- D. Yokoyama, A. Sakaguchi, M. Suzuki and C. Adachi, Org. Electron., 2009, 10, 127 CrossRef CAS.
- D. Yokoyama, J. Mater. Chem., 2011, 21, 19187 RSC.
- Y. Sakai, M. Shibata and D. Yokoyama, Appl. Phys. Express, 2015, 8, 096601 CrossRef.
- S. Y. Lee, T. Yasuda, I. S. Park and C. Adachi, Dalton Trans., 2015, 44, 8356 RSC.
- Y. S. Tsai, L. A. Hong, F. S. Juang and C. Y. Chen, J. Lumin., 2014, 153, 312 CrossRef CAS.
- K. Goushi, K. Yoshida, K. Sato and C. Adachi, Nat. Photonics, 2012, 6, 253 CrossRef CAS.
- T. D. Schmidt, D. S. Setz, M. Flämmich, J. Frischeisen, D. Michaelis, B. C. Krummacher, N. Danz and W. Brütting, Appl. Phys. Lett., 2011, 99, 163302 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: For synthesis and characterization details of materials. See DOI: 10.1039/d0ra07865d |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
Chihaya Adachi
b,
Chihaya Adachi