
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe effect of saccharides on equilibrium swelling of thermo-responsive gels†
A. D. Drozdov * and
J. deClaville Christiansen
* and
J. deClaville Christiansen
Department of Materials and Production, Aalborg University, Fibigerstraede 16, Aalborg 9220, Denmark. E-mail: aleksey@m-tech.aau.dk
First published on 20th August 2020
Abstract
Mechanical and optical properties of thermo-responsive (TR) gels change drastically at their volume phase transition temperature. As the critical temperature is strongly affected by the presence of small amounts of additives in aqueous solutions, TR gels can be employed as sensors for detection and recognition of multiple analytes (from specific ions to hazardous biochemicals to pathogenic proteins) and actuators for biomedical applications. A simplified mean-field model is developed for equilibrium swelling of TR gels in aqueous solutions of additives. Its advantage is that the model involves a relatively small (compared with the conventional approaches) number of material constants and accounts for changes in the thermo-mechanical response at transition from the swollen to collapsed state. The ability of the model to describe experimental swelling diagrams and to predict the influence of additives on the equilibrium degree of swelling and the volume phase transition temperature of TR gels is confirmed by comparison of observations on poly(N-isopropylacrylamide) gel in aqueous solutions of saccharides (glucose, sucrose and galactose) with results of numerical analysis.
1 Introduction
Hydrogels are three-dimensional networks of polymer chains bridged by covalent and/or physical junctions. Thermo-responsive (TR) gels form a special group of hydrogels whose equilibrium degree of swelling, as well as mechanical and optical properties are strongly affected by temperature.1 Equilibrium and transient swelling of TR gels in various solvents and their mixtures has recently attracted substantial attention due to a wide area of potential applications of these materials in technology2 and medicine.3Two types of TR hydrogels are conventionally distinguished. Gels of the LCST (low critical solution temperature)-type swell noticeably in water at temperatures T below their volume phase transition temperature Tc and shrink (collapse) at temperatures above Tc.4 Gels of the UCST (upper critical solution temperature)-type immersed into water shrink at temperatures below Tc and swell at T > Tc.5 Pronounced changes in degree of swelling Q at the critical temperature Tc are accompanied by morphological transformations of TR gels (a homogeneous micro-structure of a swollen gel becomes strongly inhomogeneous in the collapsed state due to phase separation into polymer-poor and polymer-rich domains).6
Temperature is not the only trigger causing abrupt changes in degree of swelling of TR hydrogels. Similar changes are observed when relatively small amounts of additives (starch,7 saccharides,8 phenols,9 inorganic salts,10 surfactants,11 oligomers12) are dissolved in water. The ability of TR gels to recognize host molecules make them suitable for applications as chemical and biochemical sensors.13 Development of hydrogel-based sensors for analytical, chemical and biomedical applications has recently become a focus of attention.14–17 Design of high-speed sensors with robust responsiveness to additives and optimization of their properties require adequate models for the thermo-mechanical behavior of TR gels in aqueous solutions.
Conventional models for swelling of TR gels in water are grounded on the Flory–Rehner concept18 that presumes the specific Helmholtz free energy of a gel Ψ to consist of three parts: (i) the specific free energy of water not interacting with the polymer network Ψ1, (ii) the specific strain energy of the network not interacting with water Ψ2, and (iii) the specific energy of interaction between water molecules and segments of chains Ψint. In the Flory–Rehner theory, the function Ψint is adopted in the form
Ψint = kBT0(Cf![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) lnϕf + χCfϕn), lnϕf + χCfϕn),
| (1) |
When the FH parameter χ is treated as a function of temperature T only, this approach results in rather poor agreement with experimental swelling diagrams.19 To improve the quality of fitting, this coefficient is traditionally replaced with an effective FH parameter χeff, which is presumed to depend on two arguments, T and ϕn. Adopting an analytical expression for χeff (as a polynomial20 or rational21 function of ϕn with temperature-dependent coefficients), an acceptable agreement can be reached between experimental data and results of simulation.
Two shortcomings of this approach are to be mentioned: (i) observations22,23 show that the volume phase transition temperature Tc is a characteristic parameter for a TR gel (this temperature is practically unaffected by preparation conditions and volume fractions of monomers and cross-linker in a pre-gel solution). However, the model (1) does not involve Tc as a parameter and does not permit it to be determined in simulation. Another approach to the description of swelling of TR gels was suggested in ref. 24 and 25. It is based on the Landau theory of phase transition and allow Tc to be found by fitting equilibrium swelling diagrams. (ii) Experimental data26,27 demonstrate that the elastic moduli of TR gels increase strongly (by an order of magnitude) at temperatures above Tc. This growth (driven by aggregation of hydrophobic segments into clusters that serve as physical junctions between chains in the network23) is disregarded in the models based on eqn (1). An explicit account for the evolution of shear moduli of TR gels with temperature was performed in ref. 28 and 29.
To study equilibrium swelling of a TR gel in a mixture of two solvents (solvent-1 and solvent-2), the Flory–Rehner concept is generalized by including all terms describing interactions between segments of chains and solvent-1 and solvent-2 molecules into eqn (1). The following expression is adopted for the specific free energy Ψint:30,31
|
Ψint = kBT0[(C1lnϕ1 + C2lnϕ2) + (χ13C1ϕn + χ23C2ϕn + χ12C1ϕ2)],
| (2) |
Observations in equilibrium swelling tests are reproduced poorly when the coefficients χij are treated as functions of temperature T only.32 To ameliorate the accuracy of fitting, three approaches were suggested: (i) introduction of prescribed dependencies of the coefficients χij on volume fractions of constituents,33 (ii) extension of eqn (2) to account for size and shape of solvent molecules and segments of chains, as well as for attractive and repulsive interactions between them,34 and (iii) inclusion of higher-order terms with respect to ϕ1, ϕ2, ϕn into eqn (2).35 Although these refinements improve the quality of matching experimental data, they lead to a strong increase in the number of adjustable parameters, which makes questionable the ability of the models to predict the thermo-mechanical behavior of TR gels in mixtures of solvents.
The objective of this study is threefold: (i) to develop a model with a reasonably small number of material constants for equilibrium swelling of TR gels in aqueous solutions of additives that accounts for changes in the micro-structure of the polymer network and its elastic moduli at transition from the swollen to collapsed state and allows the critical temperature Tc to be determined explicitly, (ii) to find adjustable parameters in the governing equations by matching equilibrium swelling diagrams on covalently cross-linked poly(N-isopropylacrylamide) (PNIPA) gel in water and aqueous solutions of glucose, galactose and sucrose at various temperatures and molar fractions of saccharides, (iii) to verify the ability of the model to predict observations by comparing results of simulation with experimental data in independent tests.
We focus on equilibrium swelling of LCST-type covalently cross-linked thermo-responsive gels in aqueous solutions with relatively low volume fractions of additives (below the concentration at which reentrant swelling starts36). Analysis of the physical mechanisms for the co-solvency and co-nonsolvency phenomena in TR gels in mixed solvents37 is beyond the scope of this work. Unlike previous studies, we do not confine ourselves to the response of TR gels at a fixed temperature. Temperature T and volume fraction of additive in the bath ϕ2bath are treated as independent parameters. This allows the critical temperature Tc(ϕ2bath) and the critical concentration of additive ϕ2cbath(T) to be predicted at which the volume phase transition occurs.
A strong decay in the equilibrium degree of swelling at the critical temperature Tc for a TR gel in pure water is conventionally explained by pronounced changes in the effective hydrophilicity of chains involving hydrophilic and hydrophobic segments.38,39 At temperatures below Tc, each hydrophobic segment is surrounded by a cage-like structure formed by water molecules bridged by hydrogen bonds.40 When temperature grows, clustered water molecules are destabilized by thermal fluctuations. Breakage of cages formed by water molecules induces agglomeration of hydrophobic segments and formation of hydrophobic aggregates from which water molecules are expelled. At temperatures above Tc, most cages are broken, and the structure of a TR gel becomes inhomogeneous: it consists of a number of deswollen hydrophobic aggregates bridged by hydrophilic segments and separated by nano-channels in which water molecules are located.41
The similarity between the equilibrium swelling diagrams on TR gels in pure water, Q(T), and in aqueous solution of additives at a fixed temperature, Q(ϕbath), see Fig. 1 below, can be explained by the same mechanism of destabilization of cage-like structures surrounding hydrophobic segments. In swelling tests with increasing temperature, breakage of cages is induced by thermal fluctuations, whereas in experiments with growing concentration of additives, it is caused by complexation of additives with water molecules (formation of molecular complexes linked by hydrogen bonds).42 When water molecules leave the cage-like structures around hydrophobic segments to form complexes with molecules of additives, these structures are destroyed. As a result, the “naked” segments aggregate into hydrophobic clusters, the polymer chains dehydrate, and the network collapses.43–45
 | ||
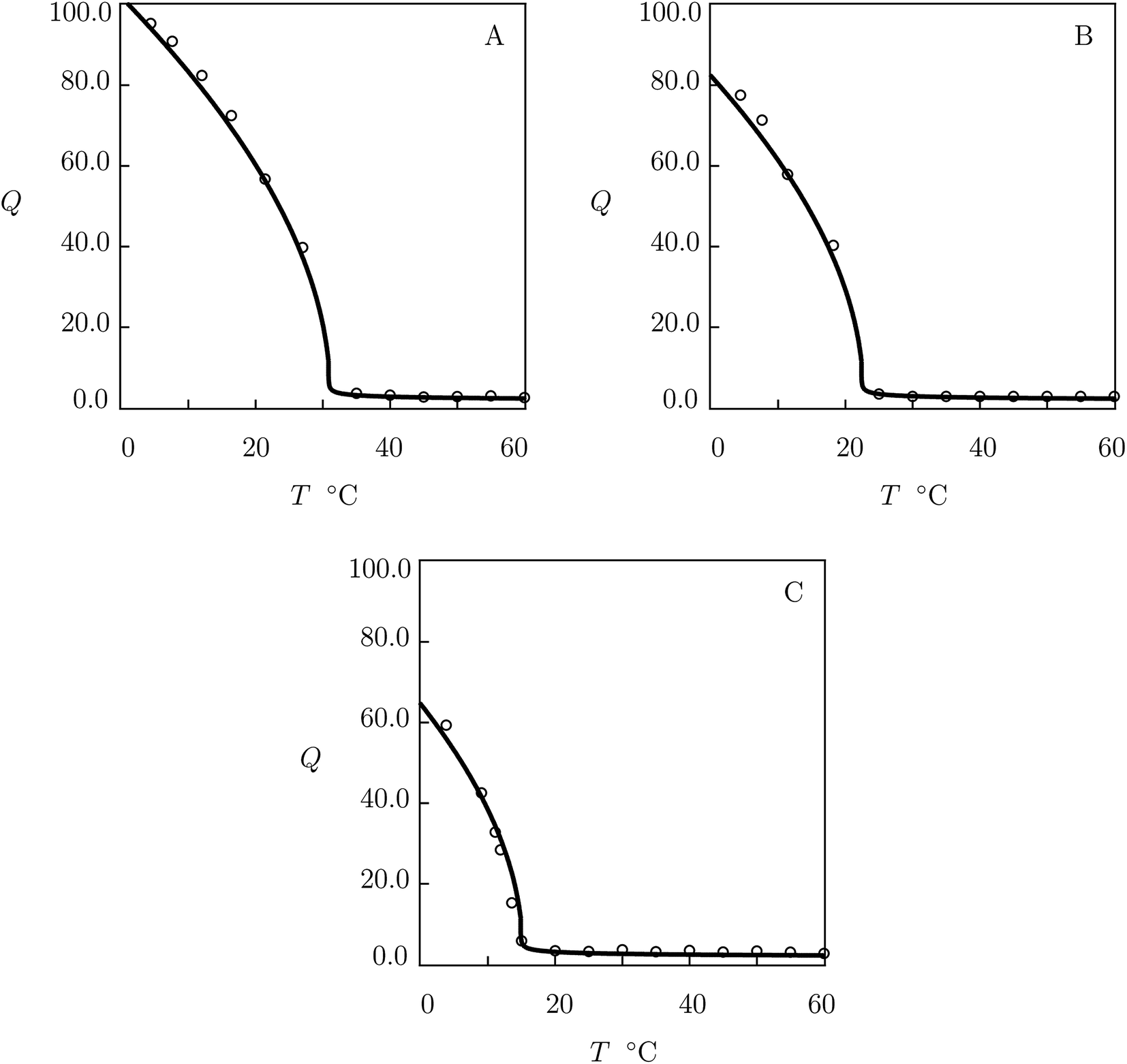
| Fig. 1 (A) Degree of swelling Q versus temperature T. (B–D) Degree of swelling Q versus volume fraction of saccharides in the bath ϕ2bath. Circles: experimental data8 on PNIPA gel in pure water (A) and in aqueous solutions of glucose (B), galactose (C) and sucrose (D) at temperature T = 20 °C. Solid lines: results of simulation. | ||
 | ||
| Fig. 2 Degree of swelling Q versus temperature T. Circles: experimental data8 on PNIPA gel in aqueous solutions of glucose with volume fractions ϕ2bath = 0.031 (A), 0.122 (B) and 0.188 (C). Solid lines: predictions of the model. | ||
The novelty of our study consists in the following: (i) a model is developed for the mechanical response and equilibrium swelling of TR gels in aqueous solutions of additives. Unlike the conventional approach, we do not introduce phenomenological dependencies of the FH parameters χij on volume fractions of polymer chains or cosolvent molecules. A pronounced decay in degree of swelling when temperatures and/or concentration of additives exceed their critical values is described by taking into account an increase in the elastic modulus of a gel caused by aggregation of hydrophobic segments. (ii) An advantage of the model is that it involves a small number of material constants with transparent physical meaning. In particular, to describe equilibrium swelling of a TR gel in an aqueous solution of an additive, only one extra coefficient is introduced in the governing equations. This allows the model to be applied not only for the description of experimental data, but also for prediction of equilibrium swelling diagrams on TR gels (see Fig. 3 and 4 below). (iii) Adjustable parameters are determined by fitting observations on PNIPA gel is aqueous solutions of saccharides. Application of TR gels with boronic acid derivatives for continuous glucose monitoring has recently attracted noticeable attention.46,47 The proposed approach can be helpful for the development of insulin-regulatory systems as interactions between glucose molecules and segments of PNIPA chains are conventionally disregarded in their analysis.
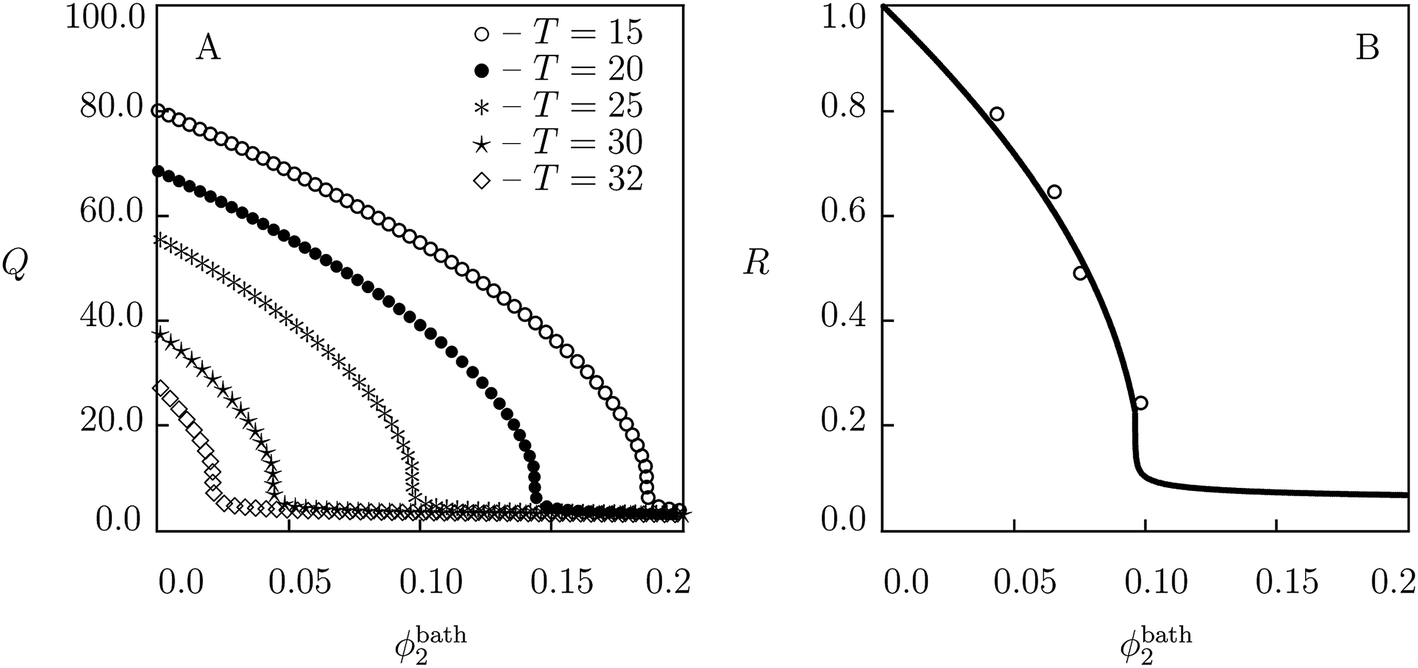 | ||
| Fig. 3 (A) Degree of swelling Q versus volume fraction of glucose in the bath ϕ2bath. Symbols: predictions of the model for PNIPA gel in aqueous solution of glucose at various temperatures T °C. (B) The ratio of masses R = M(ϕ2bath)/M(0) of a gel sample versus volume fraction of glucose in the bath ϕ2bath. Solid line: predictions of the model for PNIPA gel in aqueous solution of glucose at temperature T = 25 °C. Circles: experimental data.64 | ||
 | ||
| Fig. 4 (A) Degree of swelling Q versus temperature T. Symbols: predictions of the model for PNIPA gel in aqueous solutions of glucose with various volume fractions ϕ2bath. (B) Volume phase transition temperature Tc versus volume fraction of glucose in the bath ϕ2bath. Solid line: predictions of the model. Symbols: experimental data (○ – ref. 65, ● – ref. 66). | ||
The exposition is organized as follows. Equilibrium swelling of TR gels in pure water is discussed in Section 2. The model is extended in Section 3 for the analysis of swelling in solutions of additives. The governing equations are verified in Section 4, where results of simulation are compared with experimental data. Concluding remarks are formulated in Section 5. A detailed derivation of the governing equations is provided in ESI.†
2 Swelling of TR gels in pure water
A TR gel is modeled as a two-phase medium composed of solid (an equivalent polymer network) and fluid (water) constituents. The solid and fluid phases are treated as immiscible (mass exchange between the phases is disregarded) interpenetrating (any elementary volume contains both phases) continua.The initial configuration of a gel coincides with that of an undeformed dry specimen at some temperature T0 < Tc. Adopting the affinity hypothesis, we suppose that macro-deformation of a gel coincides with deformation of its polymer network. Transformation of the initial configuration into the actual configuration at an arbitrary temperature T is described by the deformation gradient F. Disregarding thermally induced volume expansion, we write the molecular incompressibility condition in the form
|
detF = 1 + Cfv,
| (3) |
The polymer network involves two components. The first network with covalent bonds is build under cross-linking polymerization of a pre-gel solution. The other network with physical junctions is formed at temperatures T > Tc due to aggregation of hydrophobic segments belonging to different chains. Disregarding the viscoelastic phenomena, we treat both networks as permanent.
For the covalently cross-linked network, transformation of the initial state into the reference (stress-free) state is described by the deformation gradient f1. Presuming the network to be isotropic, we set
 | (4) |
Keeping in mind that all water molecules are expelled from hydrophobic aggregates, we suppose that the reference state of the network with physical bonds coincides with the initial (dry) state of the gel.
Applying the multiplicative decomposition formula, we find the deformation gradients for elastic deformation of the networks

 | (5) |
The Helmholtz free energy (per unit volume in the initial state) Ψ equals the sum of the specific energies of fluid and solid constituents not interacting with each other and the energy of their interaction,
| Ψ = Ψ1 + Ψ2 + Ψint, | (6) |
The specific energy of water reads
| Ψ1 = μ0Cf, | (7) |
The specific energy of the polymer network (consisting of two parts with chemical and physical bonds) is given by
 | (8) |
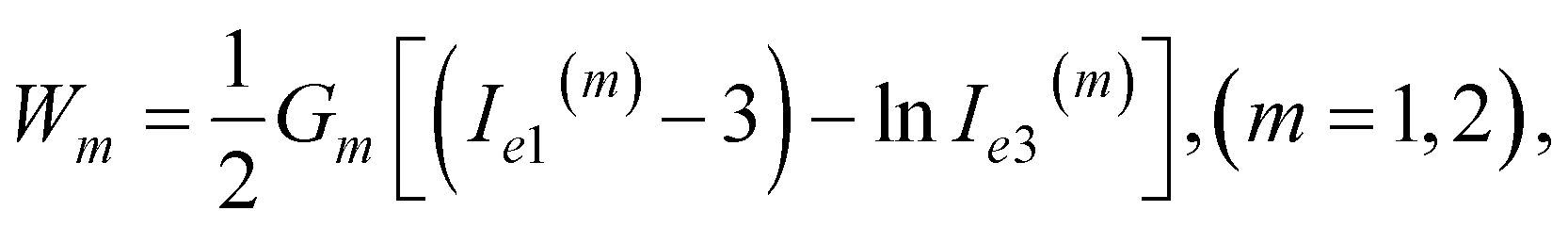 | (9) |
Confining ourselves to the analysis of swelling in a rather narrow interval of temperatures near the volume phase transition temperature Tc, we treat the modulus G1 of the covalently cross-linked network as a constant. The modulus G2 of the network with physical bonds vanishes below Tc and grows with the difference T − Tc above the volume phase transition temperature. The following relation is adopted for the function G2(T):
 | (10) |
 of PNIPA gel in water and NaCl solution together with their fit by eqn (10) with the material parameters listed in Table S-1.†
of PNIPA gel in water and NaCl solution together with their fit by eqn (10) with the material parameters listed in Table S-1.†
Introducing an order parameter
 | (11) |
 | (12) |
Bearing in mind that G2 is proportional to concentration of physical bonds (aggregates from which molecules of water are expelled) that bridge polymer chains in the network above Tc, eqn (12) can be treated as a simple kinetic relation for aggregation of hydrophobic segments. More sophisticated relations for the kinetics of aggregation were discussed in ref. 51 and 52.
The specific energy of mixing of water molecules with segments of chains is adopted in the Flory–Huggins form (1), where ϕf and ϕn are given by
 | (13) |
The following expression is accepted for the FH parameter χ:
|
χ = χ0 + χ1T(T < Tc), χ = χmax(T > Tc),
| (14) |
| χmax = χ0 + χ1Tc. | (15) |
Eqn (14) means that breakage of cage-like structures formed by water molecules surrounding hydrophobic segments leads to an increase in the effective hydrophobicity of chains (characterized by the parameter χ) at temperatures T below Tc. When T exceeds the volume phase transition temperature Tc, further growth of χ is prohibited by formation of aggregates of hydrophobic segments.
Applying the free energy imbalance inequality, we find that under equilibrium swelling of a TR gel, its degree of swelling Q = Cfv obeys the nonlinear equation
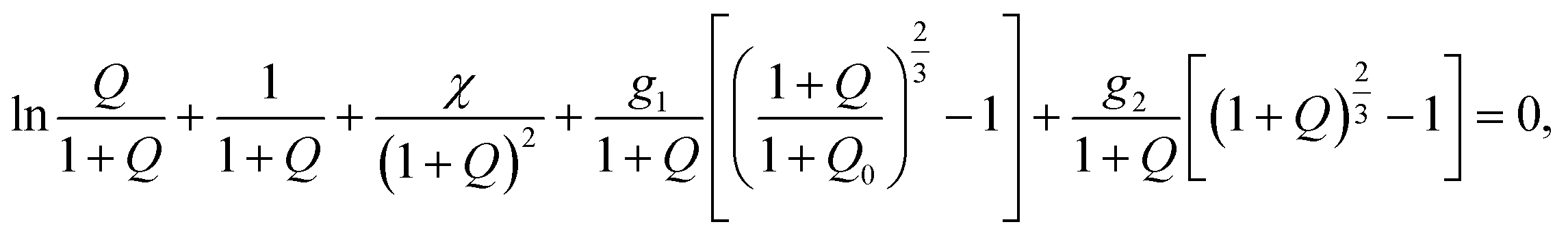 | (16) |
stand for the dimensionless shear moduli. Derivation of eqn (16) is given in Appendix A (ESI).†

Eqn (16) together with eqn (14) for the Flory–Huggins parameter χ and eqn (10) for the shear modulus G2 provides governing relations for the equilibrium degree of swelling Q at an arbitrary temperature T. These equations involve seven adjustable parameters: (i) g1 stands for the dimensionless shear modulus of the polymer network with covalent bonds, (ii) Q0 denotes degree of swelling of a gel in its stress-free state, (iii) χ0, χ1 characterize temperature-induced changes in the FH parameter χ below the critical temperature Tc, (iv) χmax is the FH parameter above the volume phase transition temperature, and (v) Ḡ2, β2 describe the kinetics of aggregation of hydrophobic segments of chains above the critical temperature. Given material constants, the critical temperature Tc is found from eqn (15).
To confirm the ability of the model to describe equilibrium swelling diagrams, we fit experimental data53 on PNIPA gel prepared by irradiation cross-linking of an aqueous solution of NIPA chains with the irradiation dose Γ = 20 kGy. Observations in equilibrium water uptake tests are depicted in Fig. S-2A.† The data are matched by means of a two-step procedure. At the first step, observations below Tc (where g2 = 0) are fitted. We set Q0 = 12.81 (which means that the reference state coincides with the as-prepared state54), determine χ from eqn (16), and approximate the experimental dependence χ(T) by means of eqn (14), where χ0 and χ1 are calculated by the least-squares technique. The dimensionless elastic modulus g1 is found from the condition of the best-fit of the dependence χ(T). The coefficient χmax is determined from eqn (15). At the other step, the data above Tc are matched by means of two parameters, β2 and ḡ2, with ḡ2 = Ḡ2v/(kBT0).
Fig. S-2A† demonstrates good agreement between the observations and results of simulation with the material constants listed in Table S-2.† Fig. S-2B† shows that eqn (14) describes correctly an increase in χ with temperature below the critical temperature Tc. An advantage of the governing eqn (10), (14) and (16) compared with previous models for swelling of TR gels (see, for example,55,56 where similar observations are approximated) is that it correctly describes equilibrium swelling diagrams by means of a rather limited set of adjustable parameters.
3 Swelling of TR gels in solutions of additives
Our aim now is to describe how degree of swelling of TR gels is affected by the presence of additives in aqueous solutions.Experimental data show that volume phase transition in a TR gel immersed in water occurs at a critical temperature Tc which is practically independent of preparation conditions and concentrations of monomers and cross-linkers.22 This observation serves as a basis for treatment of Tc as a parameter characterizing the transition point, see eqn (12), and the function (9) (proportional to the difference T − Tc) as the order parameter η for volume phase transition.
Under swelling of a TR gel in a mixture of water with another solvent, the temperature Tc is strongly affected by the chemistry and concentration of the cosolvent. As Tc cannot be used as a criterion for the volume phase transition, three questions arise: (i) which parameter can replace Tc as a measure for coil-to-globule transition in polymer chains, (ii) how to express the order parameter η (that describes the kinetics of aggregation of hydrophobic segments) in terms of this quantity, and (iii) how to define χ in eqn (16) in order to account for the influence of cosolvents or additives. The main requirement for appropriate answers to these questions is that they should result in eqn (10), (14) and (16) when volume fraction of additives in the bath ϕ2bath vanishes.
We introduce the coefficient χmax as a criterion for the volume phase transition in TR gels in binary mixtures. In other words, we suppose that some “equivalent” FH parameter χeq exists (treated as a function of temperature, volume fraction of additive in the gel, etc.) such that
| χeq < χmax |
| χeq ≥ χmax |
It follows from eqn (14) that χeq = χ0 + χ1T for a TR gel in pure water. This equality together with eqn (15) implies that when a TR gel collapses in pure water,

Combining this equality with eqn (11), we introduce the order parameter η by the formula
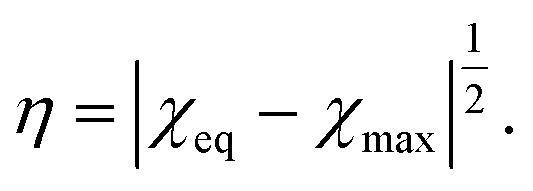 | (17) |
According to eqn (12), the shear modulus G2 of the polymer network with physical bonds obeys the differential equation
 | (18) |
 . The solution of eqn (18) reads
. The solution of eqn (18) reads
 | (19) |
To derive an expression for χeq of a TR gel in an aqueous solution, we transform eqn (2) for the specific energy of interaction between segments of chains and molecules of solvent-1 (water) and solvent-2 (additives). Bearing in mind that
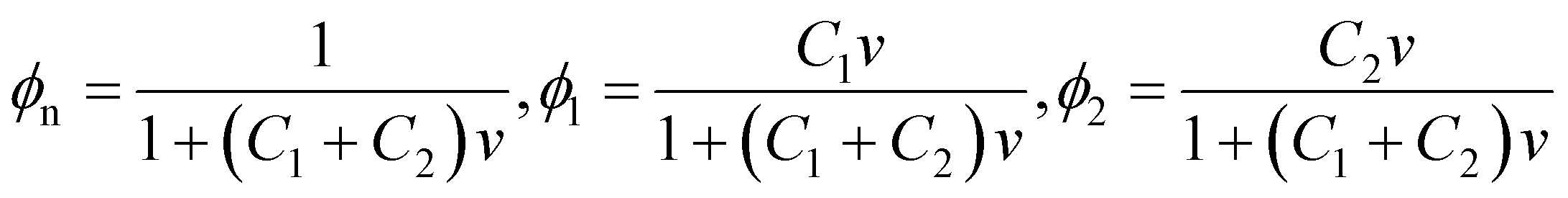 | (20) |
 | (21) |
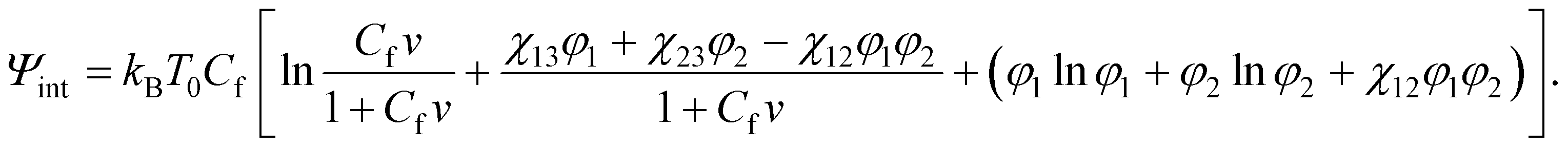 | (22) |
Measurements of concentration of additives in PNIPA gels immersed in aqueous solutions of saccharides7,8 reveal that the partitioning coefficient
 | (23) |
 | (24) |
 | (25) |
| χ* = χ13ϕ1bath + χ23ϕ2bath − χ12ϕ1bathϕ2bath. | (26) |
When a TR gel is in the swollen state, and the specific energy of interaction between segments of chains and solvent-1 and solvent-2 molecules is given by eqn (25), its equilibrium degree of swelling Q obeys the equation
 | (27) |
Comparison of eqn (16) and (27) implies that under condition (24), the equilibrium degree of swelling Q of a TR gel in an aqueous solution of additives obeys eqn (16), where χ is given by an extension of eqn (14),
| χ = χeq(χeq < χmax), χ = χmax(χeq > χmax), | (28) |
It follows from eqn (26) that
| χeq = χ13(1 − ϕ2bath) + χ23ϕ2bath − χ12ϕ2bath(1 − ϕ2bath). |
At small volume fractions of additives in an aqueous solution, ϕ2bath ≪ 1, this relation is simplified
| χeq = χ13(1 − ϕ2bath) + (χ23 − χ12)ϕ2bath. | (29) |
The coefficient χ13 in eqn (29) describes interactions between water molecules and segments of chains. This quantity coincides with the FH parameter χ in eqn (1), and its dependence on temperature T is given by eqn (14).
The coefficient χ12 accounts for interactions between water molecules and molecules of an additive. Although this parameter depends slightly on temperature,59 we disregard this dependence, as an appropriate activation energy is lower (by at least an order of magnitude) than the enthalpy characterizing the effect of T on χ13 (see Fig. S-3,† where experimental data60 on aqueous solutions of glucose and sucrose are presented).
Observations on equilibrium swelling of PNIPA gels in water/ethanol and water/methanol mixtures20,61,62 show that at high concentrations of cosolvents, Q becomes independent of temperature, which implies that χ23 is temperature-independent for these additives. This conclusion is explained in ref. 63 by the fact that intensity of thermal fluctuations in the interval of temperatures under investigation (between 0 °C and Tc) is insufficient to activate oscillations of heavy alcohol molecules (their molecular mass exceeds that of water by several times) and to induce breakage of cage-like structures formed by these molecules around hydrophobic segments of chains. The same argument can be applied to the saccharides under consideration as their molecular mass exceeds mass of water molecules by an order of magnitude.
Based on this reasoning, we disregard the effect of temperature on χ12 and χ23, and find from eqn (14) and (28) that
| χeq = (χ0 + χ1T)(1 − ϕ2bath) + Kϕ2bath, | (30) |
| K = χ23 − χ12. | (31) |
It follows from eqn (30) that K is the only extra constant required to describe equilibrium swelling of a TR gel in an aqueous solution of additives. The other material constants are determined by fitting observations in water uptake tests at various temperatures.
Combination of eqn (28) and (30) results in the formula for the volume phase transition temperature Tc of a TR gel in an aqueous solution
 | (32) |
The governing eqn (16), (19), (28), (30) together with eqn (32) for the critical temperature Tc express the unknown parameters Q and Tc in terms of the volume fraction of additives in the bath ϕ2bath. These relations are based on the assumption (24) that volume fractions of additives in the bath and in the fluid phase of the gel coincide. Eqn (24) is confirmed by observations7,8 on PNIPA gels in aqueous solutions of saccharides with relatively low concentrations of additives at which the gels remain in the swollen state. Our aim now is to prove that condition (24) is fulfilled, and the partition coefficient P is close to unity (independently of the type of additive) when degree of swelling Q is sufficiently large.
For this purpose, we study equilibrium swelling of a TR gel with the energy of interaction between segments of chains and molecules of solvent-1 and solvent-2 described by eqn (2). A detailed derivation of the governing equations for this gel is provided in Appendix C (ESI).† Under the condition that the volume fraction of additives in the bath ϕ2bath is small compared with unity, our analysis implies that the partition coefficient P reads
 | (33) |
4 Fitting of observations
To demonstrate the ability of the model to describe equilibrium swelling diagrams on TR gels in aqueous solutions of saccharides and to predict changes in degree of swelling Q and critical temperature Tc induced by an increase in volume fraction of additives in the bath, we study observations in swelling tests on PNIPA gel in solutions of monosaccharides (D-glucose and D-galactose) and disaccharide (sucrose).The gel is prepared by free radical cross-linking polymerization (24 h at 5 °C) of an aqueous solution of N-isopropylacrylamide (NIPA) monomers (0.7 M) by using N,N′-methylenebis(acrylamide) (BIS, 7.0 mM) as a cross-linker, ammonium persulfate (APS) as an initiator, and N′,N′,N′,N′-tetramethylethylenediamine (TEMED) as an accelerator.8
We begin with fitting experimental data in equilibrium swelling tests on PNIPA gel in pure water (Fig. 1A). First, observations at temperatures T < Tc are approximated by using the universal coefficients χ0, χ1 reported in Table S-2.† Parameters Q0 and g1 are found from the best-fit condition for the experimental dependence χ(T) below Tc. The coefficient χmax is determined from measurements of Tc. The quantities β2 and ḡ2 are found by matching the experimental data above Tc. Adjustable parameters in the governing equations are determined by the method of nonlinear regression from the condition of minimum for the function
| ∑(Qexp − Qsim)2, |
We proceed with fitting equilibrium swelling diagrams in aqueous solutions of glucose, galactose and sucrose at T = 20 °C (Fig. 1B, C and D). Each set of data is approximated separately by means of the only parameter K. Given a volume phase transition temperature Tc for PNIPA gel in an aqueous solution of additive with volume fraction ϕ2bath, this coefficient is calculated from eqn (32). The best-fit values of K are collected in Table S-4.†
To examine the accuracy of predictions of swelling diagrams, we present experimental data in equilibrium swelling tests on PNIPA gel in solutions with three molar fractions of additives (0.3, 1.2 and 2.0 mol L−1) together with results of simulation with the material parameters reported in Table S-3 and S-4.† Observations and results of numerical analysis for swelling tests on PNIPA gel in solutions of glucose are depicted in Fig. 2. Similar results for solutions of galactose and sucrose are reported in Fig. S-4 and S-5.† These figures confirm the ability of the model to predict experimental data.
To evaluate the effect of temperature T on equilibrium swelling curves, simulation is conducted of the governing equations for swelling of PNIPA gel in solutions of glucose with various volume fractions ϕ2bath at temperatures T = 15, 20, 25, 30 and 32 °C. Results of numerical analysis are reported in Fig. 3A. This figure shows that the growth of temperature induces a pronounced shift of the volume phase transition point to smaller volume fractions of the additive. To confirm this conclusion, results of the numerical analysis at temperature T = 25 °C are re-plotted in the dimensionless form in Fig. 3B together with experimental data64 on PNIPA gel with the same preparation conditions and a similar composition (the only difference is in the molar fraction of cross-linker: 18.4 mM (ref. 64) compared with 7 mM (ref. 8)).
To examine the influence of volume fraction of additives ϕ2bath on swelling diagrams, simulation of the governing equations is performed for PNIPA gel immersed in solutions of glucose, galactose and sucrose with ϕ2bath ranging from 0 to 0.3. Results of numerical analysis are reported in Fig. 4A (glucose), Fig. S-6A† (galactose) and Fig. S-7A† (sucrose), where equilibrium degree of swelling Q is plotted versus temperature T. These figures demonstrate that the presence of saccharides in aqueous solutions leads to a noticeable shift of the critical temperature Tc to smaller values. The decay in the volume phase transition temperature Tc with ϕ2bath is illustrated in Fig. 4B (glucose), Fig. S-6B† (galactose) and Fig. S-7B† (sucrose), where predictions of eqn (32) are compared with experimental data.65–67 Good agreement is demonstrated between predictions of the model and experimental data for the critical temperature of PNIPA gel in solutions of glucose and galactose. Slight deviations (less than 2 °C, that is the width of the volume phase transition interval for PNIPA gel) between the data and results of simulation in Fig. S-7C† may be explained by the non-standard technique used in ref. 67 for measurements of Tc.
The coefficient K in Table S-4† can be connected with hydration number n of additives in aqueous solutions. As the latter parameter is model-dependent (its value for saccharides is strongly affected by the method used to find n experimentally), two sets of data are reported in Fig. S-8A and S-8B.† In Fig. S-8A,† K is plotted versus n determined from measurements of density and sound velocity of aqueous solutions (parameter n is taken from ref. 68 for glucose and galactose and from ref. 69 for sucrose). In Fig. S-8B,† the same K is depicted versus n found by means of terahertz reflection spectroscopy.70 In both cases, linear dependencies are revealed between the K values calculated by matching swelling diagrams on PNIPA gel in aqueous solutions of saccharides and their hydration numbers n. To demonstrate that these dependencies have some physical meaning, we calculate K for mannose from the graph in Fig. S-8A,† and apply it to predict (by means of eqn (32)) the effect of volume fraction of mannose ϕ2bath on the critical temperature Tc of PNIPA gel in mannose solution. Results of simulation are presented in Fig. S-8C† together with experimental data taken from ref. 65. An acceptable agreement is shown between the observations and predictions of the model.
Keeping in mind that the hydration number characterizes interaction between molecules of water and molecules of an additive, the proportionality between the coefficient K and hydration number n means that the parameter χ12 in eqn (31) (that accounts for this interaction in the Flory–Rehner model) plays the key role in the volume phase transition of TR gels. This conclusion is in accord with the concept of complexation of cosolvent molecules,43–45 according to which hydration of polymer chains requires formation of cage-like structures of water molecules surrounding hydrophobic segments. When water molecules leave these structures to form complexes with molecules of an additive, the chains dehydrate and collapse.
5 Concluding remarks
A mean-field model is developed for equilibrium swelling of thermo-responsive gels in aqueous solutions of additives. Three issues are worth to be mentioning that distinguish our model from the conventional approach to swelling of TR gels in binary mixtures: (i) an increase in the shear modulus of a gel in the collapsed state (driven by aggregation of hydrophobic segments into clusters from which solvent molecules are expelled) is taken into account, see eqn (10) and (19), (ii) the governing equations allow the volume phase transition temperature Tc to be determined explicitly as a function of volume fraction of additive, see eqn (32), and (iii) to reduce the number of adjustable parameters, the entire set of FH coefficients χij is not employed in the analysis (although connection (31) is established between these quantities and coefficient K in the model).The model is grounded on the following assumptions: (i) hydrophobicity of polymer chains in a network is characterized by the only scalar coefficient χeq. In the swollen state of a gel, evolution of this quantity with temperature and volume fraction of additive is governed by semi-phenomenological eqn (30). Volume phase transition in a TR gel occurs when the equivalent FH parameter χeq reaches its critical value χmax. (ii) In the collapsed state, the FH parameter adopts its ultimate value χmax, and the kinetics of association of hydrophobic segments into clusters is governed by the order parameter (17). (iii) In the simplified model, volume fraction of additives in the fluid phase of a gel is presumed to coincide with that in the bath, see eqn (24). This hypothesis is confirmed by experimental data, and its accuracy is verified analytically, see eqn (33).
The model for the thermo-mechanical response and equilibrium swelling of a TR gel in a solution of additives is derived by means of the free energy imbalance inequality (Fig. S-8†). The governing eqn (16), (19), (28) and (30) involve eight material constants with transparent physical meaning: (i) g1 stands for the dimensionless shear modulus of the polymer network with covalent bonds, (ii) Q0 denotes degree of swelling of a gel in the stress-free state, (iii) χ0, χ1 characterize the effect of temperature on the equivalent FH parameter χeq, (iv) K accounts for evolution of χeq with volume fraction of additives, (v) χmax is the ultimate value of the equivalent FH parameter characterizing volume phase transition in the gel, and (vi) ḡ2 and β describe the kinetics of aggregation of hydrophobic segments of chains into clusters in the collapsed state. The critical temperature Tc is found from eqn (32).
The coefficients χ0, χ1 and χmax are treated as “universal,” which means that their values are determined by chemical structure of monomers only. Parameters Q0, ḡ2 and β are considered as “semi-universal”: these quantities are affected by the type and concentration of cross-linker, but are independent of temperature and volume fraction of additives. The coefficient K is the only parameter that accounts for the chemistry of additives.
The model is applied to the analysis of equilibrium swelling of PNIPA gel in aqueous solutions on saccharides at various temperatures T. Fig. 1 demonstrates good agreement between the experimental swelling diagrams and results of simulation. The ability of the model to predict the effect of temperature on the equilibrium degree of swelling of PNIPA gel in solutions of glucose, galactose and sucrose with various concentrations is confirmed by Fig. 2, 3, S-4 and S-5.† Its ability to predict changes in the volume phase transition temperature Tc with volume fraction of saccharides in the bath is illustrated in Fig. 4, S-6–S-8.†
Conflict of interest
There are no conflicts of interest to declare.Acknowledgements
Financial support by Innovationsfonden (Innovation Fund Denmark, project 9091-00010B) is gratefully acknowledged.References
- A. Gandhi, A. Paul, S. O. Sen and K. K. Sen, Asian J. Pharm. Sci., 2015, 10, 99–107 CrossRef.
- M. A. Cohen-Stuart, W. T. Huck, J. Genzer, M. Muller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef CAS PubMed.
- P. Zarrintaj, M. Jouyandeh, M. R. Ganjali, B. S. Hadavand, M. Mozafari, S. S. Sheiko, M. Vatankhah-Varnoosfaderani, T. J. Gutierrez and M. R. Saeb, Eur. Polym. J., 2019, 117, 402–423 CrossRef CAS.
- Y. Hirokawa and T. Tanaka, J. Chem. Phys., 1984, 81, 6379–6380 CrossRef.
- K. K. Bansal, P. K. Upadhyay, G. K. Saraogi, A. Rosling and J. M. Rosenholm, eXPRESS Polym. Lett., 2019, 13, 974–992 CrossRef CAS.
- M. Shibayama, Polym. J., 2011, 43, 18–34 CrossRef CAS.
- T. Ishidao, I.-S. Song, N. Ohtani, K. Sato, Y. Iwai and Y. Arai, Fluid Phase Equilib., 1997, 136, 163–171 CrossRef CAS.
- H. Kawasaki, S. Sasaki, H. Maeda, S. Mihara, M. Tokita and T. Komai, J. Phys. Chem., 1996, 100, 16282–16284 CrossRef CAS.
- S. Koga, S. Sasaki and H. Maeda, J. Phys. Chem. B, 2001, 105, 4105–4110 CrossRef CAS.
- M. Annaka, K. Motokawa, S. Sasaki, T. Nakahira, H. Kawasaki, H. Maeda, Y. Amo and Y. Tominaga, J. Chem. Phys., 2000, 113, 5980–5985 CrossRef CAS.
- E. Kokufuta, S. Nakaizumi, S. Ito and T. Tanaka, Macromolecules, 1995, 28, 1704–1708 CrossRef CAS.
- T. Ishidao, M. Akagi, H. Sugimoto, Y. Iwai and Y. Arai, Macromolecules, 1993, 26, 7361–7362 CrossRef CAS.
- E. Kokufuta and T. Tanaka, Macromolecules, 1991, 24, 1605–1607 CrossRef CAS.
- H. R. Culver, J. R. Clegg and N. A. Peppas, Acc. Chem. Res., 2017, 50, 170–178 CrossRef CAS PubMed.
- I. Y. Jung, J. S. Kim, B. R. Choi, K. Lee and H. Lee, Adv. Healthcare Mater., 2017, 6, 1601475 CrossRef PubMed.
- L. Hu, Q. Zhang, X. Li and M. J. Serpe, Mater. Horiz., 2019, 6, 1774–1793 RSC.
- J. R. Clegg and N. A. Peppas, Soft Matter, 2020, 16, 856–869 RSC.
- P. J. Flory and J. Rehner, J. Chem. Phys., 1943, 11, 521–526 CrossRef CAS.
- N. R. Richbourg and N. A. Peppas, Prog. Polym. Sci., 2020, 105, 101243 CrossRef CAS.
- S. Hirotsu, J. Chem. Phys., 1988, 88, 427–431 CrossRef CAS.
- Y. C. Bae, J. J. Shim, D. S. Soane and J. M. Prausnitz, J. Appl. Polym. Sci., 1993, 47, 1193–1206 CrossRef CAS.
- M. Shibayama and T. Tanaka, Adv. Polym. Sci., 1993, 109, 1–62 CrossRef CAS.
- H. Kojima, Polym. J., 2018, 50, 411–418 CrossRef CAS.
- A. D. Drozdov, Eur. Phys. J. E, 2014, 37, 93 CrossRef CAS PubMed.
- A. D. Drozdov, Acta Mech., 2015, 226, 1283–1303 CrossRef.
- L. Hanykova, J. Spevacek, M. Radecki, A. Zhigunov, J. Stastna, H. Valentova and Z. Sedlakova, Colloid Polym. Sci., 2015, 293, 709–720 CrossRef CAS.
- M. Lehmann, P. Krause, V. Miruchna and R. von Klitzing, Colloid Polym. Sci., 2019, 297, 633–640 CrossRef CAS.
- A. D. Drozdov and J. deClaville Christiansen, Polymer, 2017, 132, 164–173 CrossRef CAS.
- A. D. Drozdov, Int. J. Eng. Sci., 2018, 128, 79–100 CrossRef CAS.
- A. R. Shultz and P. J. Flory, J. Polym. Sci., 1955, 15, 231–242 CrossRef CAS.
- O. Okeowo and J. R. Dorgan, Macromolecules, 2006, 39, 8193–8202 CrossRef CAS.
- R. Xiao, J. Qian and S. Qu, Int. J. Appl. Mech. Eng., 2019, 11, 1950050 CrossRef.
- R. Koningsveld and L. A. Kleintjens, Macromolecules, 1971, 4, 637–641 CrossRef CAS.
- D. S. Abrams and J. M. Prausnitz, AlChE J., 1975, 21, 116–128 CrossRef CAS.
- Q. Xin, C. Peng, H. Liu and Y. Hu, Ind. Eng. Chem. Res., 2008, 47, 9678–9686 CrossRef CAS.
- S. Katayama, Y. Hirokawa and T. Tanaka, Macromolecules, 1984, 17, 2641–2643 CrossRef CAS.
- D. Mukherji, C. M. Marques and K. Kremer, J. Phys.: Condens. Matter, 2018, 30, 024002 CrossRef PubMed.
- V. Aseyev, H. Tenhu and F. M. Winnik, Adv. Polym. Sci., 2011, 242, 29–89 CrossRef CAS.
- A. Halperin, M. Kroger and F. M. Winnik, Angew. Chem., Int. Ed., 2015, 54, 15342–15367 CrossRef CAS PubMed.
- Y. Ono and T. Shikata, J. Am. Chem. Soc., 2006, 128, 10030–10031 CrossRef CAS PubMed.
- D. Kurzbach, M. J. N. Junk and D. Hinderberger, Macromol. Rapid Commun., 2013, 34, 119–134 CrossRef CAS PubMed.
- L. Comez, M. Paolantoni, P. Sassi, S. Corezzi, A. Morresi and D. Fioretto, Soft Matter, 2016, 12, 5501–5514 RSC.
- G. Zhang and C. Wu, J. Am. Chem. Soc., 2001, 123, 1376–1380 CrossRef CAS.
- F. Tanaka, T. Koga and F. M. Winnik, Phys. Rev. Lett., 2008, 101, 028302 CrossRef PubMed.
- T. Zuo, C. Ma, G. Jiao, Z. Han, S. Xiao, H. Liang, L. Hong, D. Bowron, A. Soper, C. C. Han and H. Cheng, Macromolecules, 2019, 52, 457–464 CrossRef CAS.
- W. L. A. Brooks and B. S. Sumerlin, Chem. Rev., 2016, 116, 1375–1397 CrossRef CAS PubMed.
- B. J. van Enter and E. von Hauff, Chem. Commun., 2018, 54, 5032–5045 RSC.
- A. D. Drozdov and J. de. C. Christiansen, Int. J. Solids Struct., 2013, 50, 3570–3585 CrossRef CAS.
- A. D. Drozdov and J. deClaville Christiansen, Acta Mech., 2018, 229, 5067–5092 CrossRef.
- A. Ikehata and H. Ushiki, Polymer, 2002, 43, 2089–2094 CrossRef CAS.
- A. M. Morris, M. A. Watzky and R. G. Finke, Biochim. Biophys. Acta, 2009, 1794, 375–397 CrossRef CAS PubMed.
- A. S. Buchelnikov, V. P. Evstigneev and M. P. Evstigneev, Phys. Chem. Chem. Phys., 2019, 21, 7717–7731 RSC.
- T. Norisuye, Y. Kida, N. Masui, Q. Tran-Cong-Miyata, Y. Maekawa, M. Yoshida and M. Shibayama, Macromolecules, 2003, 36, 6202–6212 CrossRef CAS.
- M. Shibayama, Y. Shirotani, H. Hirose and S. Nomura, Macromolecules, 1997, 30, 7307–7312 CrossRef CAS.
- K. Poschlad and S. Enders, J. Chem. Thermodyn., 2011, 43, 262–269 CrossRef CAS.
- H. E. Yang and Y. C. Bae, J. Polym. Sci., Part B: Polym. Phys., 2017, 55, 455–463 CrossRef CAS.
- W. H. Stockmayer, L. D. Moore, M. Fixman and B. N. Epstein, J. Polym. Sci., 1955, 16, 517–530 CrossRef CAS.
- Z. S. Petrovic, W. J. MacKnight, R. Koningsveld and K. Dusek, Macromolecules, 1987, 20, 1088–1096 CrossRef CAS.
- R. G. M. van der Sman, Food Funct., 2017, 8, 360–371 RSC.
- P. K. Banipal, N. Aggarwal and T. S. Banipal, J. Chem. Eng. Data, 2016, 61, 1992–2001 CrossRef CAS.
- F. M. Winnik, H. Ringsdorf and J. Venzmer, Macromolecules, 1990, 23, 2415–2416 CrossRef CAS.
- S. Hirotsu, T. Okajima and T. Yamamoto, Macromolecules, 1995, 28, 775–777 CrossRef CAS.
- F. Rodriguez-Ropero, T. Hajari and N. F. A. van der Vegt, Mechanism of polymer collapse in miscible good solvents, J. Phys. Chem. B, 2015, 119, 15780–15788 CrossRef CAS PubMed.
- N. Manukovsky, A. Shpigelman, R. Edelman and Y. D. Livney, J. Polym. Sci., Part B: Polym. Phys., 2011, 49, 523–530 CrossRef CAS.
- A. Shpigelman, I. Portnaya, O. Ramon and Y. D. Livney, J. Polym. Sci., Part B: Polym. Phys., 2008, 46, 2307–2318 CrossRef CAS.
- A. Shpigelman, Y. Paz, O. Ramon and Y. D. Livney, Colloid Polym. Sci., 2011, 289, 281–290 CrossRef CAS.
- P. Narang, S. B. Vepuri, P. Venkatesu and M. E. Soliman, J. Colloid Interface Sci., 2017, 504, 417–428 CrossRef CAS PubMed.
- R. Edelman, I. Kusner, R. Kisiliak, S. Srebnik and Y. D. Livney, Food Hydrocolloids, 2015, 48, 27–37 CrossRef CAS.
- M. Starzak, S. D. Peacock and M. Mathlouthi, Crit. Rev. Food Sci. Nutr., 2000, 40, 327–367 CrossRef CAS PubMed.
- K. Shiraga, Y. Ogawa, N. Kondo, A. Irisawa and M. Imamura, Food Chem., 2013, 140, 315–320 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra05845a |
| This journal is © The Royal Society of Chemistry 2020 |