
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGreen and facile edge-oxidation of multi-layer graphene by sodium persulfate activated with ferrous ions
Lijing Han†
 a,
Yingxia Zong†b,
Qi Tanga,
Hairui Wanga,
Xiurui Langa,
Lan Caoa and
Chengzhong Zong*a
a,
Yingxia Zong†b,
Qi Tanga,
Hairui Wanga,
Xiurui Langa,
Lan Caoa and
Chengzhong Zong*a
aSchool of Polymer Science and Engineering, Qingdao University of Science and Technology, Qingdao 266042, China. E-mail: polymer@qust.edu.cn
bCollege of Chemical and Molecular Engineering, Qingdao University of Science and Technology, Qingdao 266042, China
First published on 20th August 2020
Abstract
Effective edge oxidation of graphene with high structural integrity is highly desirable yet technically challenging for most practical applications. In this work, we have developed a green and facile strategy to obtain edge-oxidized graphene with good dispersion stability and high electrical conductivity by exploiting high edge reactivity of highly conductive multi-layer graphene and oxidizing radicals (SO4−˙) generated from sodium persulfate (Na2S2O8) with ferrous ion (Fe2+) activation. Owing to high structural integrity of pristine graphene and effective edge oxidation, the obtained edge-oxidized graphene exhibited excellent dispersion stability and satisfactory electrical conductivity (i.e. ≥240 S cm−1). Moreover, the oxidation degree of pristine graphene can be well controlled by adjusting treatment time. The obtained edge-oxidized graphene is expected to find a variety of applications in many fields of anti-static films, energy storage materials, flexible sensors and high-performance nanocomposites.
Introduction
Graphene has attracted considerable attention from both the experimental and theoretical scientific communities in recent years due to its extraordinary properties of high mechanical strength, high thermal and electrical conductivity, and fast electron mobility.1–3 It holds great promise for potential applications in the fields of energy storage systems, nanoelectronics and high-performance composites.4,5 The stable dispersion of graphene in solution makes it possible to prepare macroscopic graphene-based materials, such as films, papers, coatings and functional composites, by using conventional low-cost solution processing techniques, opening up enormous opportunities to use this unique two-dimensional carbon nanostructure for many technological applications.6,7 Compared with graphene organic dispersions, graphene aqueous dispersion has attracted more attentions due to the non-toxic, easy-to-remove and low-cost features of water.8 However, owing to the hydrophobic nature and the strong van der Waals attractions (π–π stacking), the direct dispersion of graphene in water without the assistance of dispersants has generally been considered unattainable.9To fabricate high quality aqueous based graphene, one has to face the dilemma as high quality needs maintaining structural integrity of graphitic structures to the maximum extent while water dispersibility requires introducing lots of chemical groups beneficial for stable dispersion in water onto graphene.10 Therefore, enough modification of graphene without seriously sacrificing structural integrity in order to ensure graphene's good dispersibility in water and other excellent intrinsic properties has been an important topic. Currently, selective edge modification of graphene surface with hydrophilic groups (such as sulfonate groups, carboxyl groups and phenolic hydroxyl groups) has been proved to be a prevailing method to synthesis high quality aqueous based graphene.11–13
Many studies,14–16 mainly from Baek's group, used ball-milling of graphite in the presence of dry ice (solid form of CO2), sulfur trioxide or dry ice/sulfur trioxide mixture to prepare selective edge-modified graphene with different hydrophilic groups. Huang and Wan et al.17 used expanded graphite (EG) as raw materials to prepare carboxyl-functionalized graphene by using ball-milling technique in the presence of oxalic acid. These edge-selectively functionalized graphene with few defects on the graphene basal-plane surfaces exhibited good dispersion stability and high electrical conductivity and held great potential for practical applications in many fields of anti-static films, energy storage materials, flexible sensors and high-performance nanocomposites. Ball-milling is a simple, effective and eco-friendly method to prepare high quality aqueous based graphene. However, this method requires specialized equipment (planetary ball-mill machine), limiting its use. Park and Kim et al.18 developed a new method of edge selective oxidation of graphite by the second oxidation step of the modified Hummers method to avoid oxidation of the basal plane. Subsequently, the edge selectively oxidized graphite was exfoliated into liquid-phase exfoliated edge-selectively oxidized graphene. This method provided an efficient approach to obtain high quality aqueous based graphene, but it depends on the abundant concentrated H2SO4 and KMnO4, which often involves environmental contamination and consuming time during post-treatment process, limiting its industrial commercialization. So far, green and facile preparation of high quality aqueous based graphene with high structural integrity remains a challenging issue.
To the best of our knowledge, no report has ever tried the chemical modification of graphene by oxidizing radicals (SO4−˙) of reasonable oxidability generated from ferrous ion (Fe2+) activated sodium persulfate (Na2S2O8). In this paper, a green and facile edge-oxidation strategy based on sodium persulfate (Na2S2O8) activated with ferrous ion was proposed to enhance hydrophilicity of highly-conductive multi-layer graphene (PG). PG was used as precursor in order to ensure high structural integrity and satisfactory electrical conductivity. After 12–48 h of mixing with Na2S2O8 and FeSO4 solution (continuous addition) at room temperature, the originally hydrophobic PG was afforded with excellent dispersibility and stability in water. By exploiting high edge reactivity of the PG to introduce oxygen-containing groups and acquire good dispersion stability, the sulfate radical based oxidation does not compromise the crystal structure of the PG seriously, which was demonstrated by the structure characterizations and the satisfactory electrical conductivity, providing an evidence for effective edge oxidation. The obtained edge-oxidized graphene is expected to find a variety of applications in many fields of anti-static films, energy storage materials, flexible sensors and high-performance nanocomposites.
Experimental
Raw materials
Sodium persulfate (Na2S2O8) and ferrous sulfate (FeSO4·7H2O) were purchased from Sinopharm Chemical Reagent Co. Ltd., China and used as received without further purification. Highly conductive multi-layer graphene (pristine graphene, PG) was supplied by Qingdao ENE-carbon real new materials Tech. Co. Ltd., China. The PG has graphitic layers less than 10 (about 1–3 nm in thickness), and their average size is about 15 μm, which was measured using a laser particle size analyzer. The water used throughout all experiments was deionized water.Characterization
Zeta potential measurement was performed by dynamic light scattering (DLS; NanoBrook Omni, U.S.) for evaluating surface charge and dispersion stability of the samples in aqueous solution. A Renishaw InVia Reflex Raman system with 512 nm IR-diode laser coupled to an optical microscope was used to record spectra from the samples. X-Ray Photoelectron Spectroscopy (XPS) measurement was carried out using a Thermo ESCALAB 250Xi spectrometer. Fourier transform infrared spectroscopy (FTIR) was recorded by a Bruker Vertex 70 FT-IR spectrometer with the scan range of 400–4000 cm−1 in ATR mode. Thermogravimetric analysis (TGA) was carried out under nitrogen flow with a heating rate of 10 °C min−1 by using an TG209F1 instrument. Electrical conductivity of the samples was measured on a RTS-9 four-point probe resistivity measurement system. Measurements were taken in five different spots of the same sample, and the conductivity was averaged. Samples were prepared by compressing weighted amount of powder in a piston cylinder apparatus at 10 MPa for 5 min.Preparation of edge-oxidized graphene
The edge-oxidation experiments were conducted in a 500 mL round-bottomed flask provided with a constant pressure funnel under magnetic stirring. The stirring speed was fixed at 300 rpm and the temperature of the mixture was kept at 25 ± 2 °C. After 30 g Na2S2O8 was added into a mixture of deionized water (200 mL) and PG (300 mg), the mixed solution was continuously stirred and simultaneously purged with ultrapure nitrogen to remove dissolved oxygen and carbon dioxide. After 15 min, 100 mL of 1 mol L−1 FeSO4 solution was added with continuous diffusion through a constant pressure funnel in 12, 24 and 48 h. Then, the reaction products were washed with deionized water repeatedly to remove the by-products. Finally, graphene aqueous dispersion was obtained by sonicating (ultrasonic cleaner, SB25-12DTD, NingBo Scientz Biotechnology Co., Ltd) at 480 W for 1 h. Graphene powder was obtained by vacuum filtration and completely dried in a 60 °C vacuum oven for one night. As a result, edge-oxidized graphene with various oxidation degrees were obtained and identified as edge-oxidized graphene (EOGr-12, EOGr-24 and EOGr-48) according to the time of adding Fe2+.Results and discussion
Edge oxidation of graphene
Currently, aqueous dispersion of highly conductive multi-layer graphene (PG) is of great importance for practical applications, mainly for those needing high electrical conductivity. To prepare stable and high concentration aqueous dispersion from this intrinsically hydrophobic graphene and simultaneously maintain its high structural integrity, selective edge modification is a better choice considering that the presence of extra dispersants will undermine the properties of graphene. When commercialization, greenness and scalable fabrication are taken into consideration, edge modification approach should exclude involvement of any toxic and hard-to-remove reagent. Bearing these in mind, we propose the ferrous ion activated sodium persulfate based oxidation strategy to introduce oxygen-containing groups onto PG edges and afford it dispersion stability in water. Simultaneously, the high structure integrity and satisfactory electrical conductivity of PG can be well maintained to the maximum extent. This proposal stems mainly from two considerations (1) oxidation is a general way to introduce oxygen functional groups on carbon material;6 (2) sodium persulfate, as an green oxidant, has been drawing an increasing attention, yielding sulfate radicals (SO4−˙) of reasonable oxidability upon ferrous ion activation, which has a long half-life period indicating that it is stable and may be able to disperse in great distance in water.19In our work, the oxidation process, as detailed in the experimental section, starts with the addition of FeSO4 solution into the mixture because at that time, sulfate radicals (SO4−˙), an oxidizing radicals, is generated from sodium persulfate (Na2S2O8) with ferrous ion (Fe2+) activation. Fig. 1a shows the dispersion stability of the pristine and oxidized graphene in aqueous solutions settled after 7 days and 30 days. It can be seen that the EOGr-24 and EOGr-48 aqueous dispersions showed excellent stability without macroscopic aggregations and phase separation after 30 days of storage. However, there was evident change to the suspended state of the EOGr-24 in water and the PG completely precipitated after 7 days of storage. We can conclude that the EOGrs exhibit much better stability than the PG.
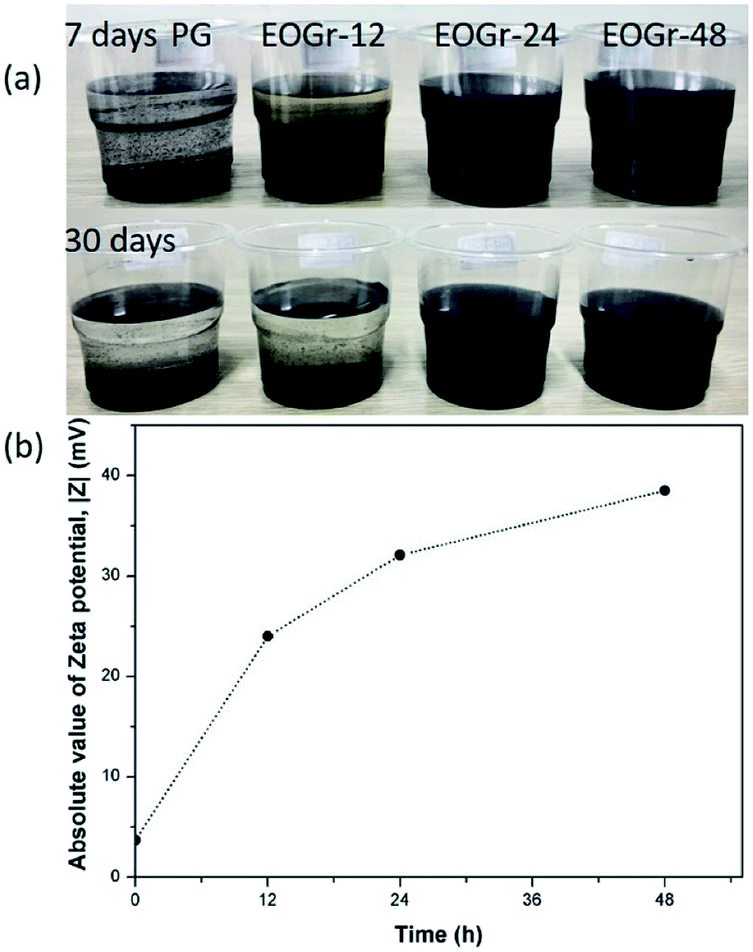 | ||
| Fig. 1 (a) Dispersion stability of the pristine and oxidized graphene (1 mg mL−1) in aqueous solutions settled (top) after 7 days; (bottom) after 30 days. (b) Zeta potential of the pristine and oxidized graphene. | ||
The dispersion stability correlates closely to surface charge of graphene, which can be measured in the absolute value of zeta potential (|Z|).20 In general, a high |Z| value indicates a large amount of surface charges, leading to strong electrostatic repulsion for high dispersion stability. It has been widely accepted that a colloidal system with a |Z| value of over 30 mV generally implies stable dispersing in aqueous solutions, because the electrostatic repulsion between charged nanoparticles is strong enough for maintaining uniform dispersion and high dispersion stability.21
We measured the zeta potential of the pristine and oxidized graphene based on dynamic light scattering in order to find the origin of high dispersion stability of the EOGr and quantitatively assess the effectivity of this method. Fig. 1b shows that the |Z| values obviously increase from 3.7 mV (PG aqueous suspension) to 24.0–38.5 mV (EOGr aqueous dispersions), and the |Z| values of EOGrs gradually increase with increased oxidation time (24.0 mV for EOGr-12, 32.1 mV for EOGr-24, 38.5 mV for EOGr-48). Therefore, the good dispersion stability during 30 days storage can be explained by highly charged surface (high |Z| values) of the dispersed EOGr-24 and EOGr-48, and their long-time stability is predictable. Moreover, the |Z| values are proportional to oxidation time, indicating that the dispersion stability can be controlled by simply adjusting oxidation time.
In conclusion, the selective edge oxidation of the PG in suitable oxidation time can greatly improve its dispersion stability in aqueous solutions. The significant change of surface charge also indicates the successful oxidation of the PG by Na2S2O8 activated with Fe2+.
The sodium persulfate (Na2S2O8) and ferrous sulfate (FeSO4·7H2O) binary-component system is first used for edge-oxidation of pristine graphene to prepare high quality aqueous based graphene by one-step at room temperature. In this system, sulfate radical (SO4−˙), an oxidizing radical of reasonable oxidability, is generated from sodium persulfate (Na2S2O8) with ferrous ion (Fe2+) activation. This strategy does not use strong reductants (such as: N2H4), strong acid (such as: H2SO4 and HNO3) and strong bases (such as: NaOH and KOH) and need extreme rigorous temperature conditions (heat or refrigeration to control the reaction). Our sulfate radical based oxidation strategy is environmentally friendly and is suitable for large scale preparation. From the perspective of greenness, commercialization and scalability, we can clearly identify the advantages of our approach in practical applications.
Structural integrity
In order to verify that sulfate radical selectively oxidize edge of the PG, structural integrity of the oxidized graphene were quantitatively evaluated using Raman spectroscopy. The Raman spectra of graphene before and after treatment are shown in Fig. 2. It can be seen from Fig. 2a that all the samples exhibit two typical characteristic peaks of graphene, i.e., G peak at around 1580 cm−1 and D peak at about 1360 cm−1. It is well known that the G peak related to the in-plane vibration adsorption of sp2 graphitic structure while the D peak is induced by the disorder giving evidences for the presence of either edges or topological defects in the graphene sheet.22 Moreover, the intensity ratio between D peak and G peak (ID/IG) is an important basis to estimate structure integrity of the graphene layer and a higher ID/IG value indicates higher disorder degrees and more defects. In Fig. 2b, the PG shows a low ID/IG value of 0.09, meaning the high structural integrity. When the PG were oxidized by Na2S2O8 activated by Fe2+, the ID/IG values gradually increase from 0.09 to 0.88 with increased treatment time (0.21 for 12 h, 0.28 for 24 h, 0.88 for 48 h), implying high oxidation degrees and significant increase of defect content after oxidation treatment. Notably, in our work, even the ID/IG value as high as 0.88 for the EOGr-48 is still lower than that for GO or reduced GO reported in literature,23 indicating that the EOGr-48 still maintain high structural integrity.10 To provide further evidence for edge selective oxidation, it would be worth considering the D′ defect related band at c. 1620 cm−1 that is typically present on the shoulder of the G band.24 In Fig. 2b, we can see that the ID/ID, ratio of PG does not change obviously in the EOGr samples (1.1 for PG, 1.7 for EOGr-12, 2.2 for EOGr-24, 2.5 for EOGr-48). This implies that the oxidation step has not produced additional basal plane defects but has occurred on the edge of the flakes, that is, EOGrs is edge-selectively oxidized by sulfate radical. In addition, it can be seen clearly from Fig. 2b that the ID/IG values are continuously increased as oxidation time prolonged, implying that the oxidation degree and structural integrity of the edge-oxidized graphene can be controlled by simple adjusting oxidation time.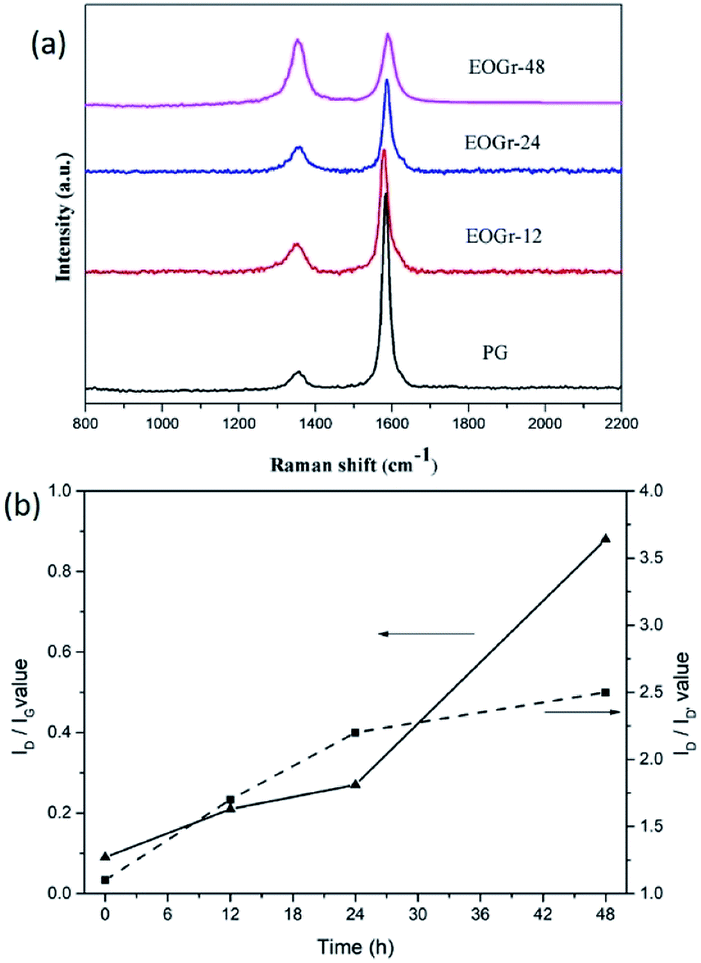 | ||
| Fig. 2 Raman spectra of the pristine and oxidized graphene. | ||
Chemical composition
Chemical composition of graphene before and after treatment was characterized by XPS technique as shown in Fig. 3. We can see from Fig. 3a that the O 1s peaks gradually increase as oxidation time prolonged. We further identified the functional groups by peak-fitting analysis. It can be seen from Fig. 3b, the C 1s spectra of EOGrs can be divided into four peaks around 284.8, 286.0, 287.0 and 289.1 eV, corresponding to the signals of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C/C–C, C–OH/C–O–C, CO and O–CO groups, respectively. This indicates EOGrs have the same type of oxygen-containing groups, which are beneficial to improving their dispersion stability in aqueous solutions, especially carboxyl and hydroxyl groups. In Fig. 3c, the oxygen content increased from 1.5 at% for the PG to 17.5 at% for the EOGr-48, indicating mild degree of oxidation during sulfate radical treatment. Such gradual increment with oxidation time means that the PG has been chemically oxidized by introducing oxygen-containing groups. Nevertheless, even the highest oxygen content for EOGr-48 is still less than that for GO and most of rGO reported in literature,25 revealing that high sp2 graphitic structure for the EOGrs is still well maintained. Furthermore, the FWHM of the 284.8 eV C–C/CC signal is another important basis to estimate the maintenance of a sp2 structure in the oxidized graphene flakes. From the XPS data, we can see that the FWHM of this signal do not increase obviously in the EOGr samples (1.0, 1.0, 1.3 and 1.4 for the PG, EOGr-12, EOGr-24, and EOGr-48), indicating the sp2 structure of the graphene flakes is sustained during oxidation. Thus, the XPS results provided another evidence for the edge-selectively oxidation.
C/C–C, C–OH/C–O–C, CO and O–CO groups, respectively. This indicates EOGrs have the same type of oxygen-containing groups, which are beneficial to improving their dispersion stability in aqueous solutions, especially carboxyl and hydroxyl groups. In Fig. 3c, the oxygen content increased from 1.5 at% for the PG to 17.5 at% for the EOGr-48, indicating mild degree of oxidation during sulfate radical treatment. Such gradual increment with oxidation time means that the PG has been chemically oxidized by introducing oxygen-containing groups. Nevertheless, even the highest oxygen content for EOGr-48 is still less than that for GO and most of rGO reported in literature,25 revealing that high sp2 graphitic structure for the EOGrs is still well maintained. Furthermore, the FWHM of the 284.8 eV C–C/CC signal is another important basis to estimate the maintenance of a sp2 structure in the oxidized graphene flakes. From the XPS data, we can see that the FWHM of this signal do not increase obviously in the EOGr samples (1.0, 1.0, 1.3 and 1.4 for the PG, EOGr-12, EOGr-24, and EOGr-48), indicating the sp2 structure of the graphene flakes is sustained during oxidation. Thus, the XPS results provided another evidence for the edge-selectively oxidation.
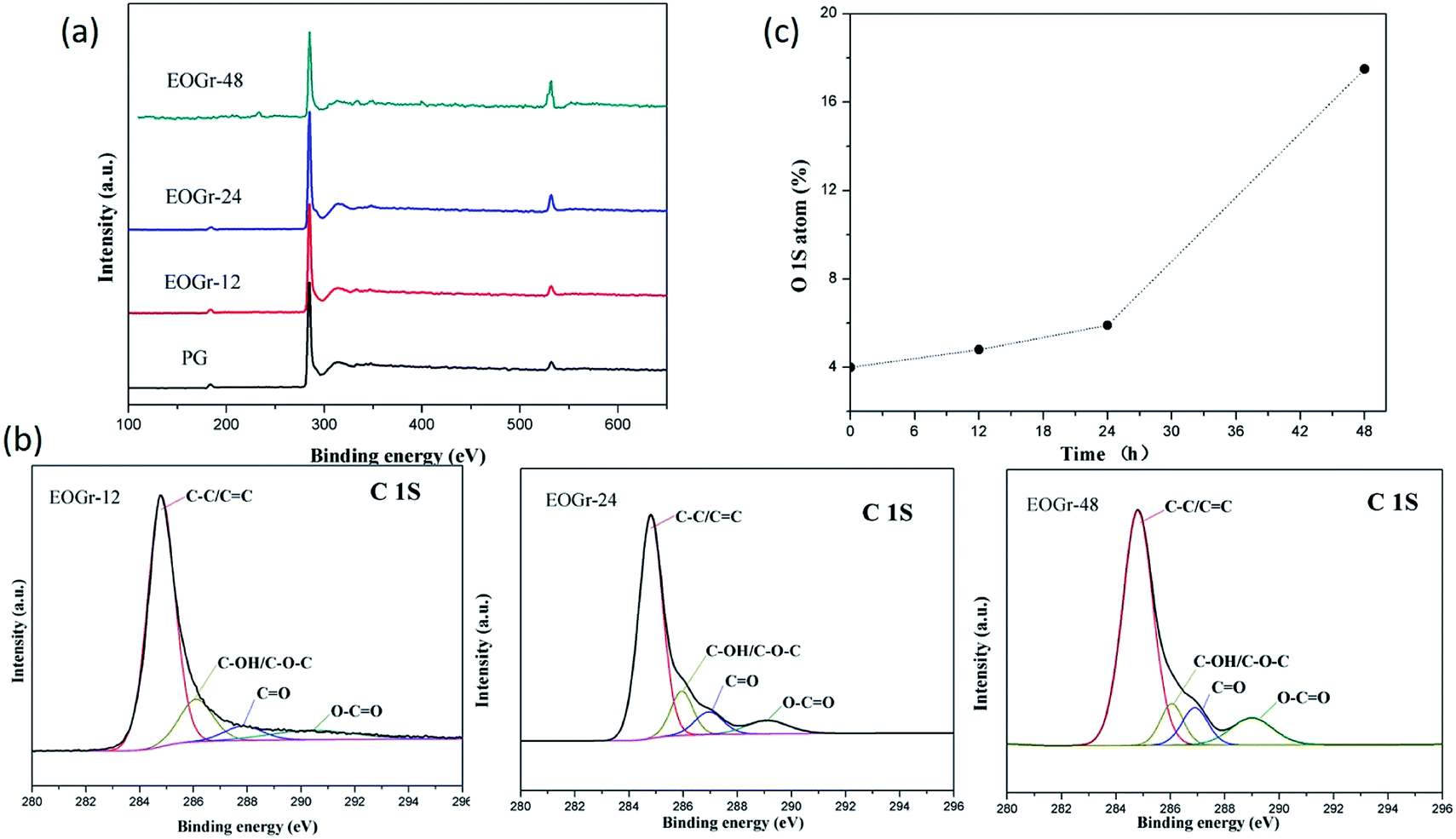 | ||
| Fig. 3 (a) XPS spectra, (b) XPS C 1s peak deconvolution and (c) oxygen atom content and c of the edge-oxidized graphene. | ||
Functional groups
We performed FTIR spectroscopic measurement in ATR absorbance mode to measure functional groups of graphene before and after treatment. It can be clearly seen from Fig. 4 that there are obvious changes at 1730 cm−1, 1537 cm−1 and 1450 cm−1 after sulfate radical treatment, revealing the presence of CO, C–O–H and C–O–C bonds.10 This indicates that the graphene was chemically oxidized, and many oxygen-containing functional groups were introduced, which is consistent with XPS analysis. Therefore, we can conclude that the obviously increased oxygen-containing groups might contribute to the dispersibility of EOGrs in water solutions. In addition, the intensity of adsorption peaks for the oxidized graphene is closely associated with the oxidation time, implying that oxidation degree can be controlled by adjusting oxidation time.
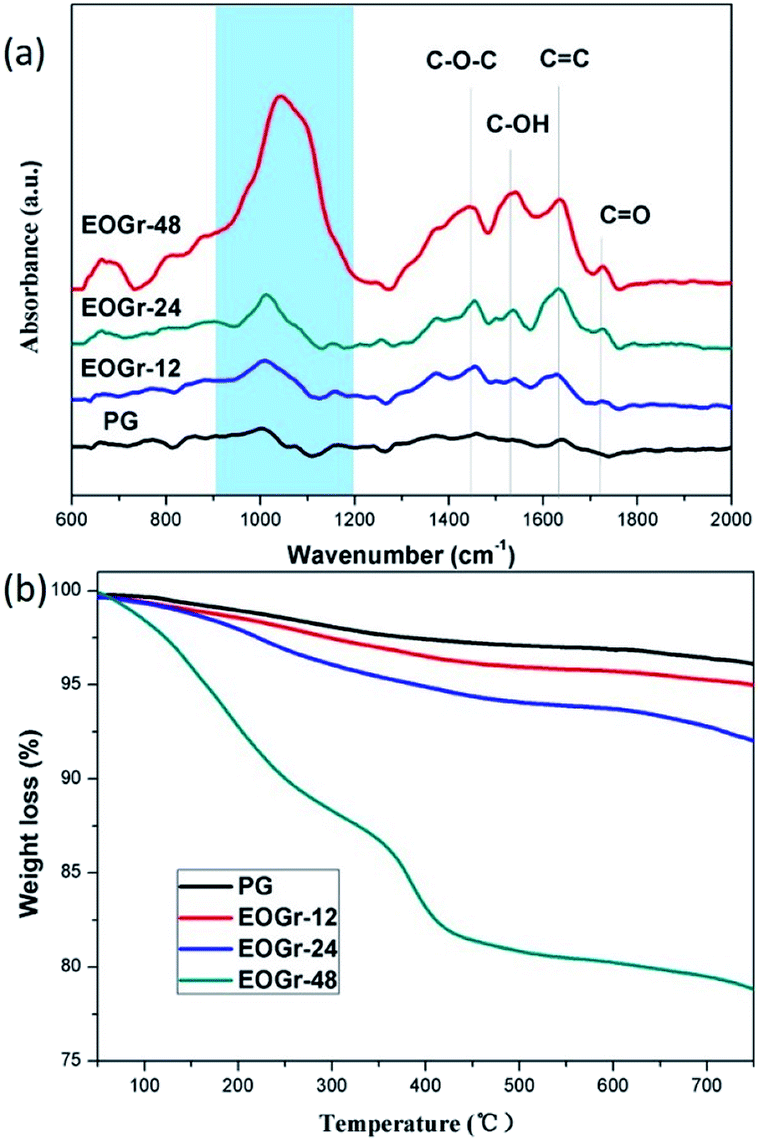 | ||
| Fig. 4 (a) FTIR spectra and (b) TGA of the pristine and oxidized graphene. | ||
The tendency of the degree of oxidation of the EOGrs can also be confirmed by the thermal stability measured by TGA as shown in Fig. 4b. According to previous reports,26 the mass loss of EOGr taking place at the temperature range of 100–300 °C is ascribed to the elimination of hydrophilic but labile oxygenated functional groups, including carboxyl, hydroxyl and epoxy groups, introduced during the sulfate radical treatment. Therefore, the amount of oxygen-containing groups can be analyzed based on the mass loss around 100–300 °C. It can be seen clearly in Fig. 4b that the mass loss increases as oxidation time prolonged, confirming that the content of oxygen-containing groups leading to the improvement of graphene's hydrophilicity increases with the increased oxidation time.
Electrical conductivity
Electrical conductivity of graphene is commonly used to reflect its crystal quality in addition to Raman spectrum. We further investigated electrical property of graphene before and after treatment for establishing a relationship between electrical conductivity, oxidation degree and dispersion stability. Electrical conductivity of the pristine and oxidized graphene is shown in Fig. 5. It can be seen that the PG exhibit the highest electrical conductivity of 450 S cm−1 due to its high structural integrity. For the oxidized graphene, their electrical conductivity gradually decreases with the increased oxidation degrees (405, 390 and 240 S cm−1 for EOGr-12, EOGr-24 and EOGr-48), but the EOGrs maintain the PG's intrinsic conductivity properties to an excellent extent. Such satisfactory electrical conductivity is maily attributed to the high structural integrity of the oxidized graphene, which provides another evidence for effective edge oxidation. We can come to the conclusion that the selective edge oxidation of graphene by Na2S2O8 with Fe2+ activation can effectively introduce oxygen-containing groups onto graphene edges and don't destroy the crystalline structure of graphene heavily, consequently resulting in excellent dispersion stability in aqueous solution and satisfactory electrical conductivity. The EOGr is expected to find a variety of applications in many fields of anti-static films, energy storage materials, flexible sensors and high-performance nanocomposites.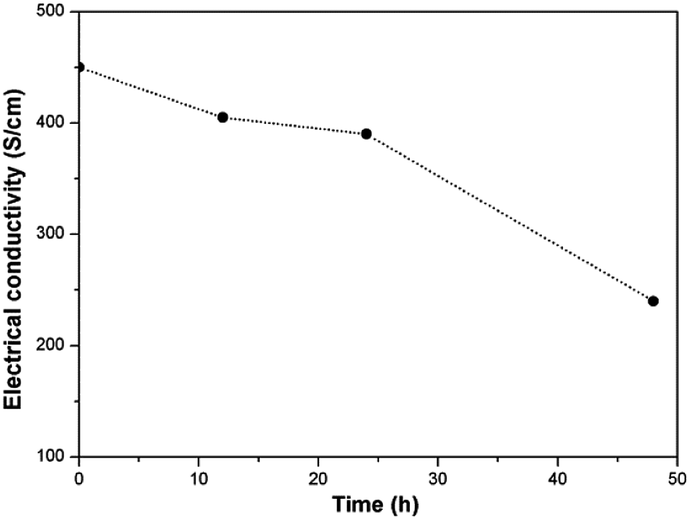 | ||
| Fig. 5 Electrical conductivity of the pristine and oxidized graphene. | ||
Oxidation mechanism
Successful stable dispersing of graphene in water demonstrates that Na2S2O8 activated by Fe2+ is an effective strategy for surface modification. In order to controlling this strategy according to pratical requirements, it's necessary to understand the underlying mechanism. Based on XPS and FTIR results above, Na2S2O8 indeed acts as an oxidant that introduce oxygen-containing groups onto graphene. More importantly, the oxidation is featured by slightly changed structural integrity of initial PG, confirmed by Raman and electrical conductivity results. It is well known that the edges of graphene possess higher reactivity than the graphene lattices, thus they can be easier to be oxidized as active sites in moderate conditions for realizing selective edge modification.27 Therefore, we deduce that the oxidation mainly occurred on the graphene edges.Rational chose of this binary-component system comprised of sodium persulfate and ferrous sulfate is the key point of this strategy. It is well known that Na2S2O8 upon Fe2+ activation can generate highly active radicals (sulfate radical, SO4−˙) at room temperature, as shown in eqn (1).19 The oxidation–reduction potential of sulfate radical (SO4−˙) is 2.6 V exhibiting reasonable oxidability. Furthermore, it has a long half-life period indicating that it is stable and may be able to disperse in great distance in water.
The mechanism of the reaction between PG and SO4−˙ is unclear. We tentatively speculate according to ref. 28 that the whole process (Fig. 6) contains three steps: (1) SO4−˙ is electrophilic, so when PG encountered SO4−˙, it was prone to losing an electron from one of the aromatic rings to be transferred to a radical cation. (2) The cation would react with water molecule and form phenol or vicinal diol structure. (3) The vicinal structure could be further oxidized to produced carboxyl and carbonyl group, or be transferred to ether group by intramolecular dehydration. Therefore, the whole oxidation of the PG can be interpreted as a mild oxidation process that occurs between the edge sp2 carbon and Na2S2O8-derived SO4−˙. This oxidation is featured by increased content of oxygen-containing groups and slightly changed structural integrity of the PG, responsible for the mildly compromised electrical conductivity.
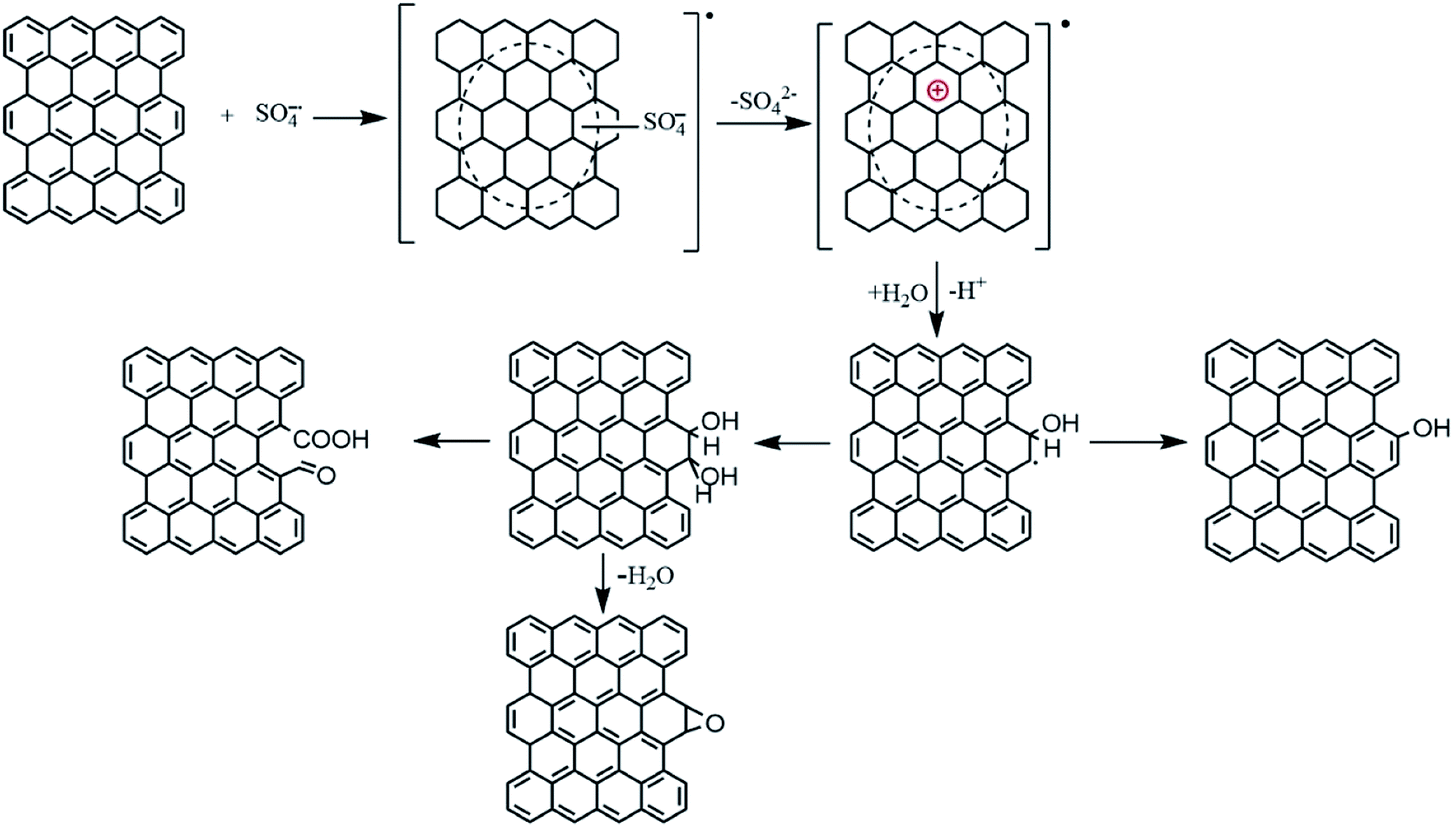 | ||
| Fig. 6 Possible mechanism of edge-selective oxidation of the PG in this study. | ||
It should be noted that according to previous studies,29 Fe2+ could also act as an intrinsic scavenger of SO4−˙, as seen from eqn (2). Therefore, fast addition of Fe2+ might result in consumption of the produced SO4−˙, which react preferentially with Fe2+ ions rather than the PG. Conversely, gradual addition of Fe2+ keeps the in situ concentration of Fe2+ in the mixture much lower, thus more SO4−˙ radicals could react with edge sp2 carbon of graphene instead of Fe2+. Therefore, we can understand easily that the content of oxygen-containing groups leading to the improvement of graphene's hydrophilicity increases with the increased oxidation time.
| Fe2+ + S2O82− → Fe3+ + SO4−˙ + SO42− | (1) |
| Fe2+ + SO4−˙ → Fe3+ + SO42− | (2) |
Conclusions
In this work, we have developed a green and facile edge-oxidation strategy based on a binary-component system composed of sodium persulfate and ferrous sulfate to obtain edge-oxidized graphene with good dispersion stability and high electrical conductivity. By exploiting high edge reactivity of graphene and oxidizing radicals (SO4−˙) generated from sodium persulfate (Na2S2O8) with ferrous ion (Fe2+) activation, the mild oxidation mainly occurs on the graphene edges and does not evidently compromise the crystal structure of graphene. Such selective edge-oxidation and integrated structures are beneficial to obtaining high dispersion stability and electrical conductivity. Moreover, the oxidation degree of graphene can be well controlled by simple adjusting treatment time. The obtained edge-oxidized graphene is expected to find a variety of applications in many fields of anti-static films, energy storage materials, flexible sensors and high-performance nanocomposites.Conflicts of interest
There are no conflicts to declare.Notes and references
- C. Lee, X. Wei, J. W. Kysar and J. Hone, Science, 2008, 321, 385–388 CrossRef CAS PubMed
.
- A. A. Balandin, S. Ghosh, W. Bao, I. Calizo, D. Teweldebrhan, F. Miao and C. N. Lau, Nano Lett., 2008, 8, 902–907 CrossRef CAS PubMed
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, M. I. Katsnelson, I. V. Grigorieva, S. V. Dubonos and A. A. Firsov, Nature, 2005, 438, 197–200 CrossRef CAS PubMed
- H. Wang, C. Wang, E. Matios and W. Li, Nano Lett., 2017, 17, 6808–6815 CrossRef CAS
- H. Wang, G. Xie, M. Fang, Z. Ying, Y. Tong and Y. Zeng, Composites, Part B, 2017, 113, 278–284 CrossRef CAS
- D. Li, M. B. Muller, S. Gilje, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3, 101–105 CrossRef CAS
- S. Park, J. An, R. D. Piner, I. Jung, D. Yang, A. Velamakanni, S. T. Nguyen and R. S. Ruoff, Chem. Mater., 2008, 20, 6592–6594 CrossRef CAS
- F. Withers, H. Yang, L. Britnell, A. P. Rooney, E. Lewis, A. Felten, C. R. Woods, V. Sanchez Romaguera, T. Georgiou, A. Eckmann, Y. J. Kim, S. G. Yeates, S. J. Haigh, A. K. Geim, K. S. Novoselov and C. Casiraghi, Nano Lett., 2014, 14, 3987–3992 CrossRef CAS PubMed
- C. Cheng and D. Li, Adv. Mater., 2013, 25, 13–30 CrossRef CAS
- S. Tian, J. Sun, S. Yang, P. He, G. Wang, Z. Di, G. Ding, X. Xie and M. Jiang, Sci. Rep., 2016, 6, 34127 CrossRef CAS
- G. Zhao, L. Jiang, Y. He, J. Li, H. Dong, X. Wang and W. Hu, Adv. Mater., 2011, 23, 3959–3963 CrossRef CAS
- Y. Si and E. T. Samulski, Nano Lett., 2008, 8, 1679–1682 CrossRef CAS
- J. H. Ding, H. R. Zhao and H. B. Yu, Sci. Rep., 2018, 8, 5567 CrossRef
- I. Y. Jeon, Y. R. Shin, G. J. Sohn, H. J. Choi, S. Y. Bae, J. Mahmood, S. M. Jung, J. M. Seo, M. J. Kim, D. W. Chang, L. Dai and J. B. Baek, Proc. Natl. Acad. Sci. U. S. A, 2012, 109, 5588–5593 CrossRef CAS
- I.-Y. Jeon, H.-J. Choi, S.-M. Jung, J.-M. Seo, M.-J. Kim, L. Dai and J.-B. Baek, J. Am. Chem. Soc., 2012, 135, 1386–1393 CrossRef
- J. Y. Baek, I.-Y. Jeon and J.-B. Baek, J. Mater. Chem. A, 2014, 2, 8690–8695 RSC
- T. Lin, J. Chen, H. Bi, D. Wan, F. Huang, X. Xie and M. Jiang, J. Mater. Chem. A, 2013, 1, 500–504 RSC
- J. Park, Y. S. Kim, J. H. Kang, S. J. Sung, T. Kim and C. R. Park, Nanoscale, 2017, 9, 1699–1708 RSC
- X. Jiang, Y. Wu, P. Wang, H. Li and W. Dong, Environ. Sci. Pollut. Res., 2013, 20, 4947–4953 CrossRef CAS
- D. W. Johnson, B. P. Dobson and K. S. Coleman, Curr. Opin. Colloid Interface Sci., 2015, 20, 367–382 CrossRef CAS
- Z. Sun, V. Nicolosi, D. Rickard, S. D. Bergin, D. Aherne and J. N. Coleman, J. Phys. Chem. C, 2008, 112, 10692–10699 CrossRef CAS
- K. N. Kudin, B. Ozbas, H. C. Schniepp, R. K. Prud'homme, I. A. Aksay and R. Car, Nano Lett., 2008, 8, 36–41 CrossRef CAS
- J. Ma, J. Liu, W. Zhu and W. Qin, Colloids Surf., A, 2018, 538, 79–85 CrossRef CAS
- K. R. Paton, E. Varrla, C. Backes, R. J. Smith, U. Khan, A. O'Neill, C. Boland, M. Lotya, O. M. Istrate, P. King, T. Higgins, S. Barwich, P. May, P. Puczkarski, I. Ahmed, M. Moebius, H. Pettersson, E. Long, J. Coelho, S. E. O'Brien, E. K. McGuire, B. M. Sanchez, G. S. Duesberg, N. McEvoy, T. J. Pennycook, C. Downing, A. Crossley, V. Nicolosi and J. N. Coleman, Nat. Mater., 2014, 13, 624–630 CrossRef CAS
- K. K. H. De Silva, H.-H. Huang and M. Yoshimura, Appl. Surf. Sci., 2018, 447, 338–346 CrossRef CAS
- J. Shen, Y. Hu, M. Shi, X. Lu, C. Qin, C. Li and M. Ye, Chem. Mater., 2009, 21, 3514–3520 CrossRef CAS
- A. Bellunato, H. Arjmandi Tash, Y. Cesa and G. F. Schneider, ChemPhysChem, 2016, 17, 785–801 CrossRef CAS PubMed
- P. Neta, V. Madhavan, H. Zemel and R. W. Fessenden, J. Am. Chem. Soc., 1977, 99, 163–164 CrossRef CAS
- L. W. Matzek and K. E. Carter, Chemosphere, 2016, 151, 178–188 CrossRef CAS PubMed
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |