
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical investigation of adsorption of graphene oxide at an interface between two immiscible electrolyte solutions†
Haiyan Qiua,
Tao Jianga,
Xiaoyuan Wanga,
Lin Zhua,
Qingwei Wanga,
Yun Zhao a,
Jianjian Geb and
Yong Chen*a
a,
Jianjian Geb and
Yong Chen*a
aSchool of Chemical and Environmental Engineering, Shanghai Institute of Technology, Shanghai 201418, China. E-mail: yongchen@sit.edu.cn
bSchool of Science, Shanghai Institute of Technology, Shanghai 201418, China
First published on 8th July 2020
Abstract
Graphene oxide (GO) has been recognized as an amphiphilic molecule or a soft colloidal particle with the ability to adsorb and assemble at the liquid/liquid (L/L) interface. However, most extant works concerning the adsorption behaviors of GO at the L/L interface have been limited to the non-polarized L/L interface. Here, we studied what would happen if GO nanosheets met with a polarizable L/L interface, namely an interface between two immiscible electrolyte solutions (ITIES). On one hand, the adsorption behavior of GO nanosheets at the L/L interface was electrochemically investigated firstly by using cyclic voltammetry (CV) and alternating current voltammetry (ACV). On the other hand, the influence of the adsorbed GO layers at the L/L interface on the ion transfer reactions was studied by employing ion-transfer voltammetry of TEA+ and ClO4− selected as probe ions. Capacitance measurements show that the interfacial capacitance increases greatly in the presence of GO nanosheets inside the aqueous phase, which can be attributed to the increases of interfacial corrugation and charge density induced by the parallel adsorption and assembly of GO at the L/L interface. In addition, it is found that the application of an interfacial potential difference by external polarization can promote the adsorption of GO at the L/L interface. Moreover, the ion-transfer voltammetric results further demonstrate that the GO layers formed at the interface can suppress the ion transfer reactions due to interfacial blocking and charge screening, as well as the hindrance effect induced by the GO layers. All the results with insights into the interfacial behavior of GO under polarization with an external electric field enable understanding the adsorption behavior of GO at the L/L interface more comprehensively.
1. Introduction
As a kind of fascinating derivative of graphene, graphene oxide (GO) has attracted much attention because of its wide potential applications in the fields of electrochemistry, energy, biomedicine etc.1–8 Due to its distinctive chemical structure composed of hydrophilic edges and a more hydrophobic basal plane, GO can be viewed as an amphiphilic molecule or a soft colloidal particle acting as a molecular or colloidal surfactant with the ability to adsorb and assemble at various interfaces.9–24 Since Huang et al. have systematically reported the interfacial adsorption of GO at various interfaces including liquid/liquid interfaces,9 the adsorption of GO at liquid/liquid (abbreviated herein as L/L) interfaces, or so-called oil/water interfaces, has received a great deal of interest over the last decade9–24 because L/L interfaces have been proven to be a defect free support or platform for the study of adsorption and assembly of various nanomaterials including two-dimensional (2D) nanosheets.9–25 In the case of GO, it can self-assemble generate paper-like films13–15 and Pickering emulsions9,16–24 by spontaneous or induced adsorption and enrichment at the L/L interface.In general, it has been found that the adsorption behavior of GO at L/L interface is closely related to its amphilicity, which can be greatly affected by the conditions of aqueous phase.14–17,24 For instance, the degree of ionization of the edge –COOH group of GO can be tuned by pH because the GO nanosheets with abundant –COOH groups on their edges can make GO nanosheets more hydrophilic.17 However, most extant works concerning the adsorption behavior of GO at the L/L interface have been limited to the non-polarized L/L interface.9–24 Although all previous reports on the adsorption of GO at the non-polarized L/L interface have illuminated the key role of amphilicity of GO for its adsorption at the interface,9–17,24 there has been rare report on the adsorption behavior of GO at the polarized L/L interfaces. In view of the chargeability and the charge screening of GO,9,14–17,24 it is urgent to further reveal its interfacial adsorption behavior at the polarized L/L interfaces under the external electric field.
When appropriate electrolytes dissolved in both contacting phases of water and oil, a special class of polarizable L/L interface can be formed, namely the interface formed between two immiscible electrolyte solutions (ITIES).26–42 Recently, increasing interests have arisen on the question what would happen if nanomaterials meet with the polarizable L/L interface under the external electric field.27–41 Several groups have demonstrated that such a polarizable L/L interface can act as a highly reproducible and defect-free scaffold and allow the application of external electric field to drive the adsorption and self-assembly of various nanomaterials to the interface including 0D nanoparticles, such as Au,27–29 SiO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 30 and TiO231 nanoparticles, as well as 1D carbon nanotube,32,33 Mo2C nanowire,34 2D graphene,35,36 WS2 and MoS2,37 etc. Those studies have demonstrated that electrochemical methods conducted at the polarizable L/L interface can not only in situ monitor the adsorption and self-assembly processes of nanomaterials, such as cyclic voltammetry (CV) and alternating current voltammetry (ACV),28,30 but also investigate the influence of adsorbed nanomaterials on the electron transfer (ET) and ion transfer (IT) behaviors,30,32 as well as the catalytic reactions occurring at the L/L interface.29,33–37 In spite of the abundant literatures on the adsorption of GO nanosheets at L/L interface studied by employing various methods including the in situ measurement of interface tension etc.,9–24 the electrochemical investigation on the adsorption behavior of GO at the polarizable L/L interface under the external electric field has not been reported until now.
30 and TiO231 nanoparticles, as well as 1D carbon nanotube,32,33 Mo2C nanowire,34 2D graphene,35,36 WS2 and MoS2,37 etc. Those studies have demonstrated that electrochemical methods conducted at the polarizable L/L interface can not only in situ monitor the adsorption and self-assembly processes of nanomaterials, such as cyclic voltammetry (CV) and alternating current voltammetry (ACV),28,30 but also investigate the influence of adsorbed nanomaterials on the electron transfer (ET) and ion transfer (IT) behaviors,30,32 as well as the catalytic reactions occurring at the L/L interface.29,33–37 In spite of the abundant literatures on the adsorption of GO nanosheets at L/L interface studied by employing various methods including the in situ measurement of interface tension etc.,9–24 the electrochemical investigation on the adsorption behavior of GO at the polarizable L/L interface under the external electric field has not been reported until now.
Herein, we investigated the interfacial adsorption behavior of GO nanosheets at the polarizable L/L interface by using electrochemical methods including CV and ACV for the first time. A polarizable L/L interface widely studied in the field of L/L interface electrochemistry,28–32,38–41 namely water/1,2-dichloroethane (W/DCE) interface, was used in this work. In addition, the influence of the adsorbed GO layer on the ion-transfer behaviors was also preliminarily studied by employing ion-transfer voltammetry of TEA+ and ClO4− selected as probe ions at the W/DCE interface on the basis of our previous work on the ion transfer reactions occurring at the membrane-modified L/L interface.38–41 The main object of current work is to develop more in situ method to investigate the interfacial adsorption behavior of GO and to reveal the adsorption mechanism of GO at the polarizable L/L interface under the external electric field, as well as its impact on the ion transfer reactions at the GO-modified W/DCE interface as shown as Scheme 1. Considering that GO is viewed as a promising drug nanocarrier for the drug delivery system (DDS) in biomedicine3–8 and the L/L interface can act as a simple model to mimic semi-biomembrane,26,38–41 this work should help to understand more comprehensively not only the adsorption behavior of GO at the L/L interface, but also the ion transfer reactions occurring at the GO-based nano–bio interface when GO is applied as drug nanocarrier in the biomedicine.
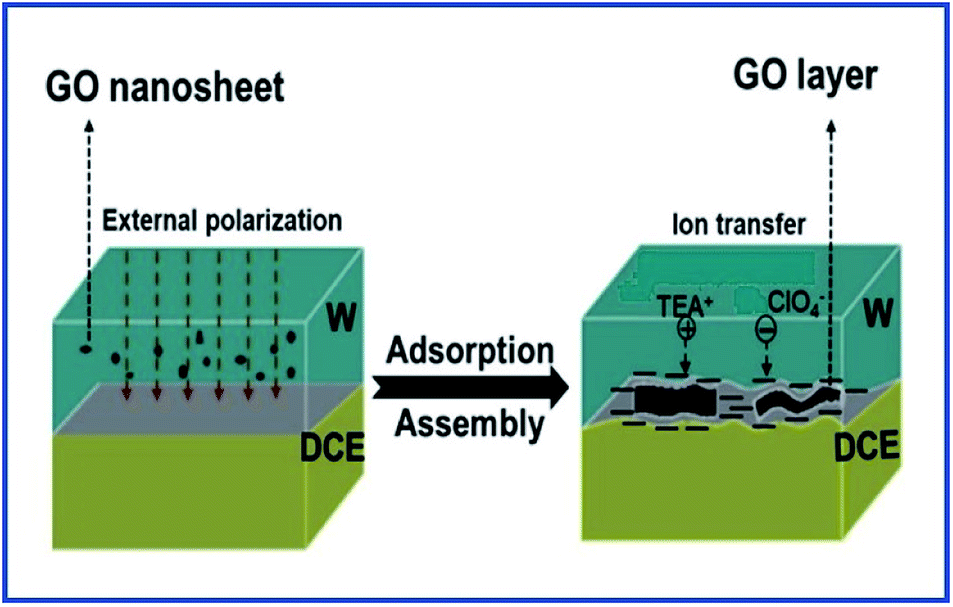 | ||
| Scheme 1 Schematic of the adsorption and assembly of GO at the W/DCE interface under external polarization and the ion transfer across the GO-layers-modified W/DCE interface. | ||
2. Experimental
2.1 Chemicals & materials
Graphene oxide was obtained from Nanjing XFNANO materials Tech Co., Ltd in a concentrated aqueous solution (2 g L−1) with an average size of 50 to 200 nm of a single layer of GO nanosheets, which was characterized by AFM and shown as Fig. S1.† GO dispersion solution used in the experiments was prepared by dilution using distilled water or pH buffer solution and then ultrasonic shaking for 15 minutes. GO membrane was also obtained from Nanjing XFNANO materials Tech Co., Ltd with a thickness of ∼20 μm. The chemicals listed were all used as received. Bis(triphenylphosphoranylidene)ammonium chloride (BTPPACl) and sodium tetraphenylboron (NaTPB) were obtained from Sigma-Aldrich; 1,2-dichloroethane (DCE), lithium chloride, tetraethylammonium chloride (TEACl), sodium perchlorate monohydrate, sodium phosphate dibasic dodecahydrate from Sinopharm Chemical Reagent Co; sodium dihydrogenorthophosphate, acetic acid, sodium acetate, hydrochloric acid and lithium hydroxide from Adamas. The aqueous supporting electrolyte solutions with different pH values and fixed ionic strength was prepared on the basis of previous reports.42,43 An aqueous buffer solution with an ionic strength of 0.1 M (adjusted with LiCl) with different pH values was used as follows: 0.1 or 0.01 M HCl for a chosen pH of 1.0 or 2.0, 20 mM acetate buffer for a chosen pH between 3.0 and 6.0, 20 mM phosphate buffer for pH of 7.4 and 1.0 × 10−5 or 1.0 × 10−4 M LiOH for a pH of 9.0 or 10.0. The ionic strengths of the different buffer solutions used in this work are almost around 0.10 except the aqueous solution with pH of 1.0 (ionic strength ∼ 0.20). In addition, the cations of supporting electrolyte and buffer solution are used as one valence cations including H+, Li+ and Na+ in order to avoid the effect of cation valence on the stability of the GO dispersion. An organic supporting electrolyte, BTPPATPB, was prepared using a previously reported method.412.2 Experimental methods
The L/L interface electrochemical cell was prepared by adding an equal volume of GO dispersion solution on top of the organic electrolyte solution (DCE). The electrochemical experiments were carried out in a custom-made electrochemical cell with a geometric cross-section of 2.27 cm2 including two sets of Ag/AgCl and Pt wires as reference and counter electrodes for both of aqueous and organic phases. The composition of different electrochemical cells is outlined as follows, where the X are 0 or 0.1 in cells 1 and 4, as well as 0–0.1 in cell 2, respectively (Scheme 2).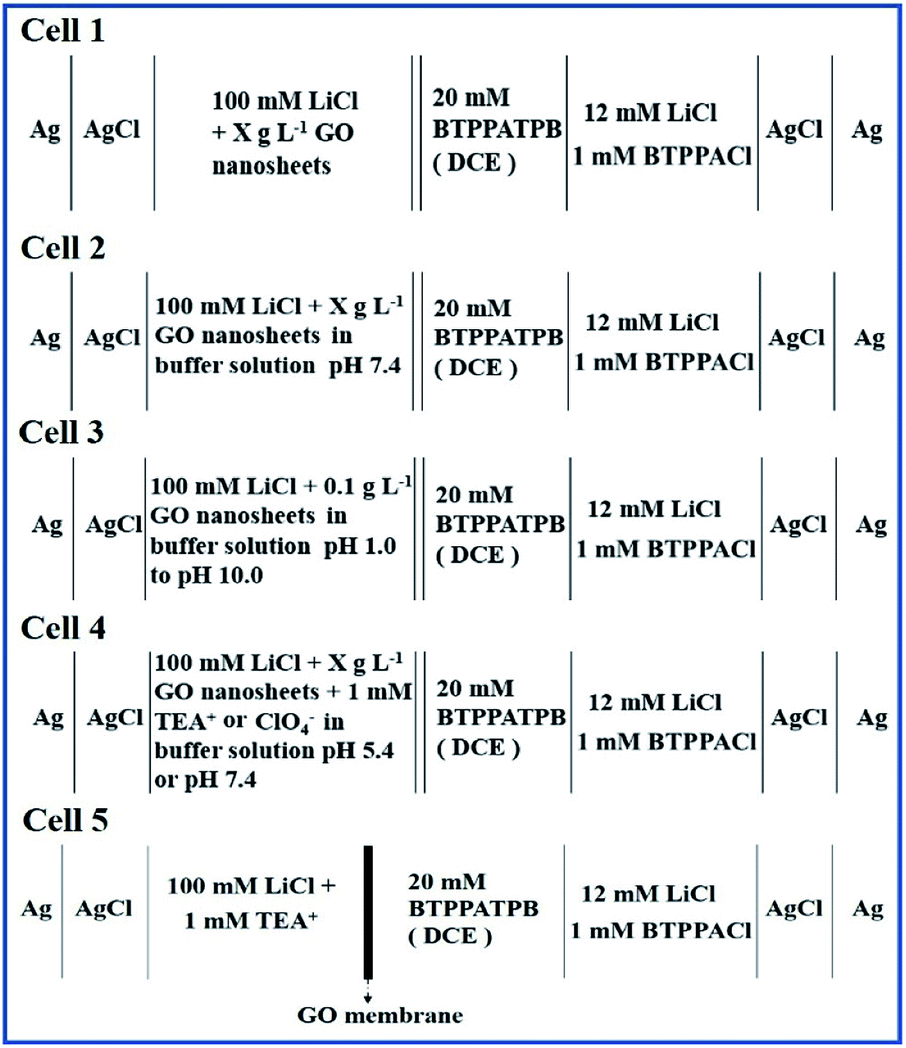 | ||
| Scheme 2 The electrochemical cells and their compositions used in the studies on the adsorption of GO (cells 1–3), as well as the ion transfer reactions occurring at the GO-layers-modified W/DCE interface (cell 4) and the GO-membrane-modified W/DCE interface (cell 5). | ||
CV and ACV were performed on a CHI 660D electrochemical workstation (CHI, USA) by using all above cells in a CHI202 shielded box (CHI, USA) at room temperatures (25 ± 2 °C). The influence of the presence or absence of GO nanosheets in the aqueous phase on the ion transfer reactions was studied by CV. The monitoring of interfacial capacitance was employed by ACV on the basis of the methods reported previously28,30 and the parameters for the ACV experiments were as follows: modulation time: 0.33 s, interval time: 0.7 s, frequency: 6 Hz, step potential: 5 mV, amplitude: 5 mV. The GO membrane-modified W/DCE interface was also polarized using four-electrode potentiostat (CHI660D) with homemade four-electrode electrochemical cell (cell 5), which was constructed by using the similar method as our previous reports.38–41 Optical images of GO interfacial layers were recorded with a transparent reflection metallographic microscope (VM4000M/dyj-950) connected to an industrial digital camera (UCMOS10M-U2). SEM images were obtained by using a FEI Quanta200 FEG operated under high-vacuum state with an accelerating voltage of 15 keV. XPS was performed by using an ESCALAB 250Xi X-ray photoelectron spectrometer (Termo Fisher Scientific). AFM was conducted by Shimadzu SPM-9700 Atomic Force Microscope. The zeta potentials of the GO dispersions at different pH values were measured using a Malvern Zetasizer Nano ZS at 25 °C.
3. Results and discussion
The interfacial adsorption of GO at the W/DCE interface under still-standing without an external electric field was firstly characterized by using optical microscopy when both of aqueous phase (W) and organic phase (DCE) contain electrolytes, namely LiCl and BTPPATPB. As shown as (Fig. 1a and b), it is obvious that the adsorption and assembly phenomenon of GO can occur at the W/DCE interface (Fig. 1a), and some stacked GO layers can be observed at the W/DCE interface after 48 hours (Fig. 1b). However, some suspended GO solids were found at the inner surface of glass bottle and a little GO layers with smaller size could be observed at the W/DCE interface when only aqueous phase contains electrolyte LiCl (Fig. S2a and b†). If only organic phase contains electrolyte BTPPATPB, a few stacked GO layers could be observed at the W/DCE interface and most of GO nanosheets float in the aqueous phase after 48 hours (Fig. S2c and d†), indicating that the adsorption of GO indeed occurs at the W/DCE interface. In addition, it is worth to be noted that the DCE phase is slightly brown when it contains electrolyte BTPPATPB as shown as Fig. 1a and S2c,† which is not obvious in Fig. S2b† when the DCE phase contains no electrolyte BTPPATPB.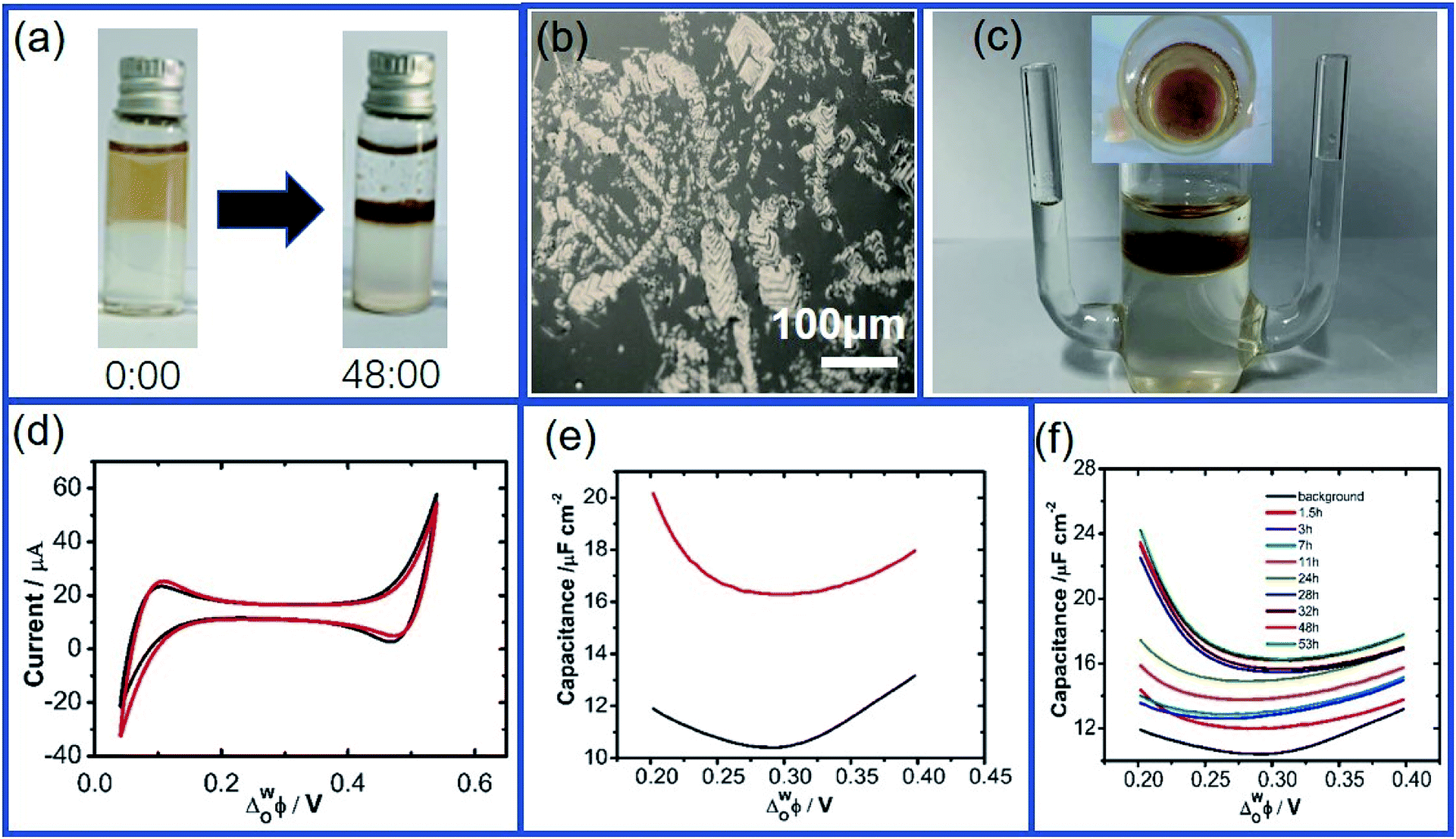 | ||
| Fig. 1 The side-viewed photograph (a) and the top-viewed optical microscopy (b) for the adsorption of GO at the W/DCE interface without an external electric field; (c) the side-viewed and the top-viewed (the inset) photographs of electrochemical cell for the GO-layers-modified W/DCE interface; (d) cyclic voltammograms at a scan rate of 5 mV s−1 obtained with (red line) or without (black line) GO in the aqueous phase; (e) the potential dependence of the interfacial capacitance for the blank cell (black line) and the presence of GO (0.1 g L−1) (red line) at 48 h; (f) the changes of ACV with time. | ||
All above results of still-standing experiments implies that GO could transfer across the W/DCE interface and the electrolytes in both of phases, LiCl and BTPPATPB, could affect the adsorption behavior of GO at the W/DCE interface. So far, only the impact of aqueous salts on the adsorption of GO at the L/L interface has been investigated.16,18 As demonstrated by Gao et al.,16 the salt in water can influence the state of the GO dispersion because both the carboxylic acid and hydroxyl groups in GO are candidates for charge screening when the salt is added. The adsorption phenomena as observed above indicate that the electrolyte BTPPATPB in organic phase can also affect the adsorption behavior of GO at the L/L interface, which may be attributed to the possible π–π interaction between the residual conjugate domains in GO and the aromatic ions of organic electrolyte, BTPPA+ and TPB−. On the basis of previous report,32 XPS spectra of pure GO and the GO layers obtained from the W/DCE interface of Fig. 1a were characterized and shown as Fig. S3a–d.† In addition, the element contents of pure GO and as-obtained GO layers are listed in Table S1.† According to Fig. S3a–d and Table S1,† it can be found that the XPS spectrum of pure GO only presents both signals of C and O elements and their contents are 70.3% (C 1s) and 29.7% (O 1s). As a comparison, the XPS spectrum of GO layers extracted from the W/DCE interface presents more signals of elements, including C, O, B, N and P. The contents of those elements are 69.42% (C 1s), 26.08% (O 1s), 1.74% (B 1s), 1.76% (N 1s) and 1.00% (P 2p). Moreover, the B/N ratio is evaluated about 1:1, which is consistent with the stoichiometric composition of BTPPATPB, indicating that aromatic cation and anion in DCE phase, namely BTPPA+ and TPB−, can indeed adsorb on the GO layers. However, the P/N ratio is lower than the stoichiometric composition of BTPPA+, which could be due to the possible overlap of the peaks of B and P, resulting in that it is difficult to accurately subtract the contribution of the P signal as reported previously.32 Based on all above XPS results and analyses, it can be inferred that the charge screening property of GO should be indeed available for the aromatic ions in organic phase, namely BTPPA+ and TPB−, due to the π–π stacking interaction between organic electrolyte and GO as shown as Fig. S3e,† which is similar to the previous report on the interaction between aromatic molecules and GO16,24 or organic electrolyte and SWCNTs.32
As for the adsorption of GO at the W/DCE interface under the external electric field, Fig. 1c and d respectively show a typical four-electrode electrochemical cell used in this work and the cyclic voltammograms (CVs) obtained by using cell 1. As shown as Fig. 1c, it was observed that the DCE phase was also slightly brown after CV although most of GO nanosheets assembled and remained on the aqueous side of the interface, which further indicates that it is possible for the transfer of GO across the W/DCE interface. Since L/L interface can be considered as a simple model for mimicking semi-biomembrane, it has been demonstrated from the studies on the transfer of GO at bio-nano interface that GO nanosheets can transfer into the cell membranes.44 Fig. 1d and the changes of CVs with time (Fig. S4†) shows that the presence of GO nanosheets has little effect on the CVs without additional faradaic current resulting from the stacked GO layers formed at the interface, suggesting that no ion-transfer happened within the potential window (PW), which is limited by the ion-transfer of background electrolyte ions on the positive and negative ends of PW, respectively. Since ACV is more sensitive to interfacial phenomena than CV,28,30 the interface adsorption with GO was further characterized by the capacitance measurement using ACV. Fig. 1e showed the ACV curves for the potential dependence of the interfacial capacitance (Cinter) recorded within the potential region where no ion-transfer was observed in the CVs of Fig. 1d. The significant increase of capacitance can be seen in Fig. 1e within the whole potential range of 0.2–0.4 V and in the Fig. 1f corresponding to the changes of ACV curves with time. It is found from Fig. 1f that the minimum capacitance values (Cmin) of those ACV curves increased with time from 10.40 μF cm−2 (background) to 16.28 μF cm−2 (48 h) and almost kept constant after 48 h, indicating that the adsorption and assembly of GO nanosheets at W/DCE interface could reach stable around 48 h. In addition, the potential for the minimum capacitance (ECmin) corresponding to the potential of zero charge (PZC) shifted from 0.294 V (background) to 0.302 V (48 h). The increase of Cinter and the positive shift of ECmin as observed above almost agree with the previous reports on the adsorption of negatively-charged nanoparticles at the polarizable L/L interface,28,30 indicating that the adsorption of GO indeed occurs at the W/DCE interface.
Fig. 2a and b further show the CV and ACV curves obtained at the W/DCE interface when the aqueous phase contains GO with different concentrations (0–0.1 g L−1) under pH ∼ 7.4 (cell 2). As shown as Fig. 2a, the increase of GO concentration in the aqueous phase has also little effect on the CVs and the increases in the GO concentration only resulted in very little increase in the capacitive current without additional faradaic current, while the significant increase of Cmin with the GO concentration can be seen in the ACV curves (Fig. 2b), which increased from 11.97 (0 g L−1) to 26.43 μF cm−2 (0.1 g L−1). In addition, the values of ECmin also shifted to higher potential values with the increase of GO concentration from 0.294 V (0 g L−1) to 0.306 V (0.1 g L−1). The increase of Cinter and the positive shift of ECmin with the increase of GO concentration observed above also agree with those previous electrochemical studies on the interfacial adsorption behavior of nanoparticles upon polarization at the W/DCE interface.28,30 For example, Girault et al.28 and Herzog et al.30 have respectively reported that the adsorption of negatively-charged Au and SiO2 nanoparticles at the W/DCE interface can lead to an increase of Cinter and the positive shift of ECmin, which can be ascribed to the increases of the charge density and the interfacial corrugation induced by the adsorption of negatively-charged nanoparticles at the interface as compared with the Gouy–Chapman capacitance for a flat interface.45
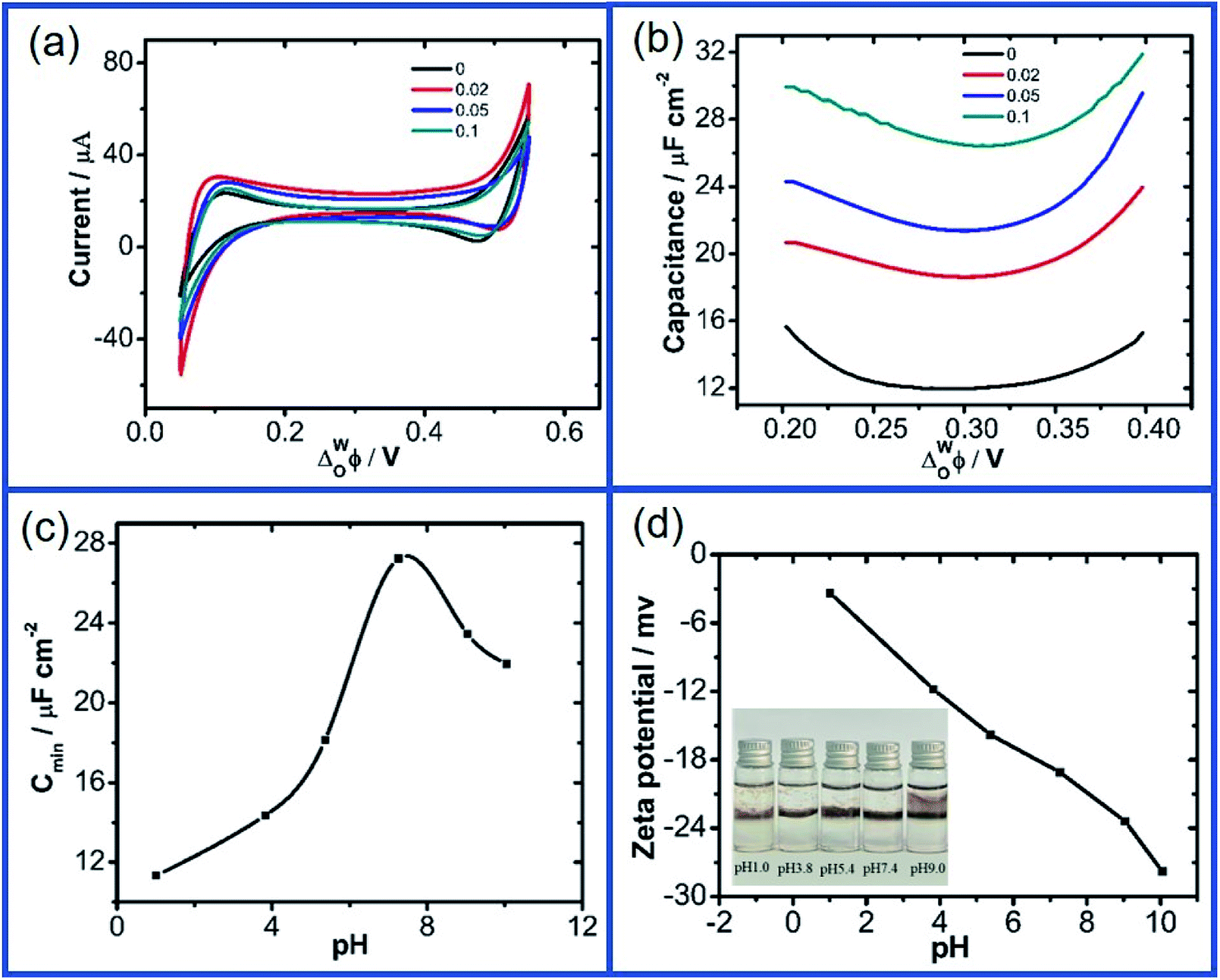 | ||
| Fig. 2 (a) Cyclic voltammograms at a 5 mV s−1 scan rate and (b) potential dependence of the interfacial capacitance obtained under different GO concentrations of 0, 0.02 g L−1, 0.05 g L−1 and 0.1 g L−1 (cell 2); (c) the changes of minimum capacitance (Cmin) and (d) zeta potentials of GO of 0.1 g L−1 dispersed in water under different pHs (1.0, 3.8, 5.4, 7.4, 9.0, 10.0) (cell 3). The inset in (d) is the photograph for the adsorption of GO at the W/DCE interface under different pH conditions. | ||
As introduced before, the adsorption of GO at the non-polarized L/L interface are closely related to its amphilicity, which can be tuned by the pH.9,16–18 Fig. 2c shows the dependence between Cmin and the pH of aqueous phase measured by using cell 3. It is found that Cmin increased with pH from 11.33 (pH ∼ 1.0) to 27.24 μF cm−2 (pH ∼ 7.4), but became smaller after pH is further increased into 10.0, which could be attributed to the pH-relative amphilicity of GO9,16–18 resulting in the changes of the charge property and the adsorption behavior of GO with pH. As shown as Fig. 2d, the zeta potentials of GO aqueous phase increase with pH from −3.37 mV at pH ∼ 1.0 to −27.8 mV at pH ∼ 10.0.
Generally, as for the nanomaterial-modified L/L interface, the increase of its capacitance can result from the higher counter ion surface charge density imposed by the charge density at the surface of charged nanomaterials, and the increase of the interfacial corrugation induced by the modification of nanomaterials.27–31 Therefore, the change of interface capacitance of GO-modified L/L interface should mainly depend on the adsorption process and charge status of GO at the L/L interface. According to the dependence between the zeta potential of GO aqueous solution and pH (Fig. 2d), GO nanosheets are negatively charged within the pH range of 1.0–10.0. Thus, the adsorption of negatively charged GO nanosheets at the W/DCE interface can result in the increase of the excess charge in the electric double layer, which is similar to the increase of the capacitance observed at the capacitance measurements for the adsorption of negatively charged nanoparticles at the L/L interface. Meanwhile, the increase of capacitance of L/L interface can result from the stronger interface corrugation induced by the adsorption of nanoparticles at the L/L interface, because the increase of corrugation can lead to an increase in capacitance with comparison to the Gouy–Chapman capacitance for a flat interface and the charging the interface gives rise to larger “dynamic corrugation” that immediately leads to increased values of capacitance.28,30
As for the positive shift of ECmin as observed in this work, it should be closely related to the polarization-induced assembly of the negatively nanomaterials at the L/L interface boundary as reported previously.27–31 It has been demonstrated that anion or negatively charged nanoparticles can adsorb when the inner potential of the aqueous phase is biased negatively with respect to the organic phase, which can induce the accumulation of anions or negatively-charged nanoparticles in the diffuse layer of the aqueous side of the interface and the increase of interface capacitance as discussed above.27,31 If the Galvani potential difference is polarized to more positive potential, which can lead to the desorption of GO and the corresponding decrease of interface capacitance. In other words, the minimum of capacitance depends on the adsorption capacity of GO nanosheets at the interface, which leads to the shift of the minimum of capacitance to more positive potentials with increasing GO concentrations.
Interestingly, Cmin increased with pH from 11.33 (pH ∼ 1.0) to 27.24 μF cm−2 (pH ∼ 7.4), but became smaller after pH is further increased into 10.0, which should be closely related to the GO chemistry in aqueous solution on the basis of a relatively unconventional structure of GO, that is dynamic structural model (DSM) developed by Dimiev et al.46,47 To our best knowledge, two conventional structural models of GO have been proposed until now. One is the Lerf–Klinowski (LK) model,48 namely GO consists of two different randomly distributed domains, namely the areas of pure graphene with sp2-hybridized carbon atoms and the areas of oxidized and thus sp3-hybridized carbon atoms. The oxidized GO domains contain epoxy and hydroxyl functional groups, while carboxyl and hydroxyl groups terminate the flake edges. The other is the Szabo–Dekany (SD) model,49 which represents GO as a periodic ribbon-like structure of aromatic and nonaromatic domains, as well as suggests that ketones and quinones are formed where C–C bonds have been cleaved. Until now, most of the recently published GO-related studies employ exclusively the LK model to interpret experimental data, but it is difficult to understand the high acidity of GO aqueous solutions on the basis of the LK models.46 As demonstrated by Dimiev et al.,46,47 the DSM considers GO as a system, constantly changing its chemical structure due to the interaction of GO with water, which can result in C–C bond cleavage, formation of vinylogous carboxylic acids, and the generation of protons. In addition, an electrical double layer formed at the GO interface in aqueous solutions plays an important role in the observed GO chemistry, which is also related to the salt in water.
In the case of the capacitance of GO-modified W/DCE interface (abbreviated as CGO-W/DCE), more comprehensive analyses of all above phenomena of the change of CGO-W/DCE should be done on the basis of DSM. For example, three key points derived from the DSM and chemistry of GO should be considered. One is that dynamic structure means GO can react with water, resulting in the cleavage of its C–C bond and the constant change of its structure, which can be intensified by the addition of strong bases. The second is that deoxygenation of GO can occur in alkaline conditions, in other words, GO reduction can occur in alkaline conditions because OH− can play the role to facilitate the ionization of existing acidic groups and nucleophilic attack at active sites on GO. The third is the negative electrical charge built up on a GO platform can not only be affected by pH but also by the salt in water. According to the DSM of GO and those key points as mentioned above, it can be inferred that the CGO-W/DCE should be closely related with the constant change of chemical structure of GO due to its interaction with water. If the aqueous phase only contains electrolyte LiCl and GO nanosheets, the CGO-W/DCE changes with time and remains constant after around 48 hours, which should be closely to not only the interfacial adsorption process of GO but also the constant interaction of GO with water, resulting in the dynamic structure of W/DCE interface, including the constant increase of interface charge and corrugation induced by the negatively-charged products of GO before both processes of adsorption and reaction reach their equilibriums.
Additionally, on the basis of the DSM of GO and the pH-relative adsorption and assembly of GO, the stronger increase of Cmin observed within in the pH region of 3.8–7.4 should be mainly due to that the reaction extent of GO with water increases with pH and the excess Na+ from buffer solution, because Na+ in aqueous phase can play as a feedstock for building an electrical double layer, which can facilitate higher negative charge on GO flakes and accelerate the reaction between GO and water.46,47 All above additional reactions can result in the constant increase of interface charge although the images of the optical microscopy and the SEM (Fig. 3) show that the GO layers formed at pH ∼ 7.4 are looser and smaller than those obtained under pH ∼ 5.4. In spite of the modest increase in negative charge when pH is increased from 5.4 to 7.4, it is worth to noticed that more winkles can be observed at the surface of GO layers obtained under pH = 7.4 than that obtained under pH = 5.4, which is expected to result in much larger interfacial corrugation induced by the modification of GO. When pH is increased from 7.4 to 10.0, OH− in aqueous phase can facilitate the ionization of existing acidic groups and nucleophilic attack at active sites on GO,46 which can affect the adsorption and assembly of GO at the W/DCE interface. Indeed, Fig. 2d shows that higher pH results in larger zeta potentials of GO aqueous phase. Meanwhile, the photograph for the adsorption of GO at the W/DCE interface under different pH conditions (the inset of Fig. 2d) clearly demonstrate that higher pH (9.0) is unfavorable for the adsorption of GO nanosheets at the W/DCE interface. Therefore, it can be inferred that the changes of adsorption behavior and the charge status of GO with pH at the L/L interface could be main reason that the values of Cmin decreased with the increase of pH after pH is larger than 7.4. However, some questions about the GO chemistry and structure in water have still been unsolved so far, such as numerous possible scenarios for the reactions involving in water because of the complexity of the GO structure,46,47 as well as the unclear mechanism and rate of those reactions, which reflects that the GO-modified L/L interface is much more complex than those L/L interfaces modified by 0D nanoparticles.
 | ||
| Fig. 3 The optical microscopy (a and d) and SEM images (b, c and e, f) of the GO layers formed at the W/DCE interface under pH of 5.4 (a–c) and 7.4 (d–f). | ||
Noticeable, it is found that the increase magnitudes of capacitance induced by the adsorption of GO nanosheets as observed in Fig. 1e and 2b are higher than those obtained by using Au and SiO2 nanoparticles,28,30 which implies that the interfacial adsorption behavior of GO nanosheets could be different from those nanoparticles at the polarizable L/L interface because the adsorption behaviors of nanomaterials at the L/L interface are also closely related to their dimension.11,12 It is well-known that 0D rigid and hydrophilic Au and TiO2 nanoparticles entrap at the L/L interface with contact angle close to 90°.12,28,30 Whereas, as a kind of 2D soft and amphiphilic nanomaterial, GO nanosheets turn to adsorb with an orientation parallel to the interface.9–24 Considering the distinct properties of amphiphilicity, chargeability, high specific surface and charge-screening of GO from those 0D nanoparticles,1–3,9–12 it is possible that the parallel adsorption of negatively-charged soft 2D GO nanosheets at L/L interface could result in higher charge density and stronger dynamic interface-corrugation as shown as Scheme 1, which leads to the greater increase of interfacial capacitance than that induced by nanoparticles.28,30 On one hand, the charge screening property of GO is available for both of electrolytes in two phase as discussed above, which could lead to higher charge density of GO-layers-modified L/L interface than of nanoparticle-modified L/L interface. On the other hand, as shown as Fig. 3, the optical microscopy images (Fig. 3a and d) and the SEM images (Fig. 3b, c and e, f) clearly show that the GO nanosheets indeed adsorb and assembly in parallel at the W/DCE interface to form some overpacked layers with different size and some wrinkles are visible on those layers, which is similar to the phenomena observed previously at the non-polarized L/L interface.14,16 In addition, the DSM and chemistry of GO as discussed above should be another reason for the greater increase of interfacial capacitance than that induced by those nanoparticles in addition to the distinct properties of amphiphilicity, chargeability, high specific surface and charge-screening of GO from nanoparticles, because those 0D nanoparticles studied at the L/L interface by the measurement of interfacial capacitance almost can not react with water as GO.27–31
Furthermore, previous work on kinetic analysis of the adsorption and assembly processes of GO nanosheets at the non-polarized L/L interface has demonstrated that GO transfer in aqueous phase is slow.13,14,18 In order to improve the interfacial adsorption of GO, the solvent-assisted13 or surfactant-induced18 methods have been explored to enhance the adsorption and self-assembly of GO at the non-polarized L/L interface. In view of the polarizability of W/DCE interface and the negatively-charged property of GO, it is necessary to further explore the possibility of electrochemical-assisted adsorption of GO at the W/DCE interface. Fig. 4a and b respectively show the dependence between minimum capacitance (Cmin) and time without the external pre-polarization and with the external pre-polarization at 0.2 V where no ion transfer reaction takes place. It was found that the minimum capacitance values increased with time from 12.05 to 16.28 μF cm−2 without the external pre-polarization and from 14.07 to 16.22 μF cm−2 with the external pre-polarization. Although the final interfacial capacitance values obtained with or without pre-polarization are almost equal, the stabilization time of GO adsorption and assembly at W/DCE interface is dramatically decreased from 48 h (without pre-polarization) to 24 h (with pre-polarization). The decrease of stabilization time of GO adsorption as observed in Fig. 4b indicates that the application of an interfacial potential difference induced by the external polarization of W/DCE interface can promote the adsorption and assembly of GO at the interface, which should be ascribed to the improvement of diffusion rate of negatively charged GO in aqueous phase under external electric field since the diffusion of GO to the interface is usually the rate-determining step.14 Thus, the electrochemistry-assisted method developed herein can play similar role as those solvent-assisted or surfactant-induced methods to enhance the diffusion and adsorption of GO to the L/L interface.
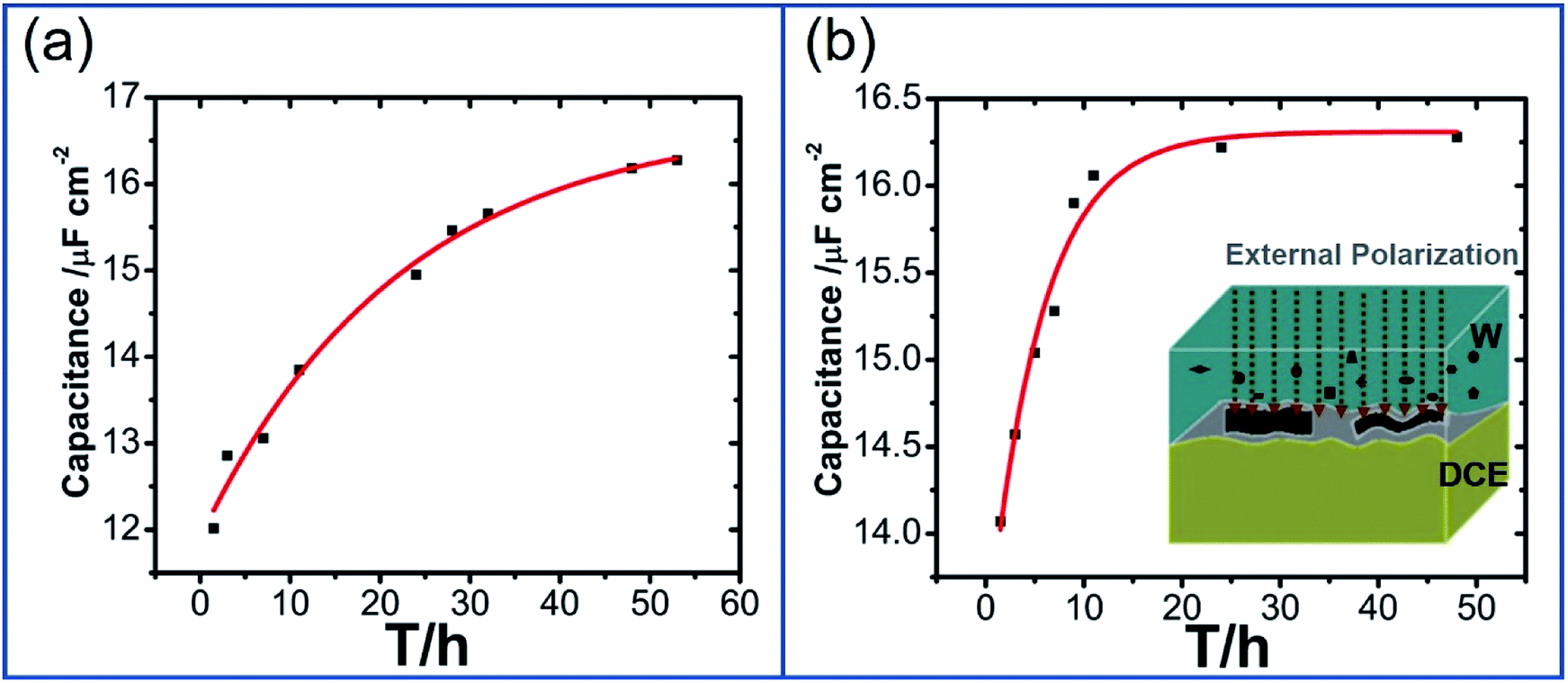 | ||
| Fig. 4 The dependence between minimum capacitance (Cmin) and time (a) without the external pre-polarization and (b) with the external pre-polarization at 0.2 V for 24 hours; the insets in (b) are the scheme for the adsorption of GO at the W/DCE interface with pre-polarization. | ||
Recent reports have demonstrated that the presence of nanomaterials can affect the ion transfer behaviors at the L/L interface.30,32 For example, single-wall carbon nanotubes adsorbed at the W/DCE interface can make the ion-transfer more difficult.32 Considering the different adsorption and assembly behaviors of GO under different pH, the influence of the adsorbed GO layer on ion transfer across the W/DCE interface was preliminarily investigated by employing the voltammetric transfer of TEA+ and ClO4− under pH ∼ 5.4 and ∼7.4 (cell 4), which are respectively close to the representative pH conditions of cancer and normal cells.3–8 Fig. 5 shows the CVs for the ion transfer of TEA+ (Fig. 5a) and ClO4− (Fig. 5b) performed in the absence of GO and then repeated in the presence of GO under different pHs. It can be seen that the responses of both ions were similar to those obtained in the absence of the interfacial GO layers, but there was only a small increase in peak separation and a slight reduction in the peak current magnitudes, especially under pH ∼ 5.4, indicating that the GO layers formed at the interface can suppress the ion transfer of both of ions, which is also related to the aqueous pH value as illustrated as Fig. 5c and d due to the pH-relative adsorption of GO and the different morphology of GO layers obtained under pH ∼ 5.4 and ∼7.4 as discussed above. According to the linear relationships between the peak currents for the ion transfer of TEA+ and ClO4− from W to DCE and the square root of scan rate (Fig. S5†), as well as the Randles–Ševčik equation,38–41 the diffusion coefficients (Dw) of each ion in the aqueous phase under pH ∼ 5.4 and ∼7.4 at the GO-layers-modified W/DCE interface were calculated and listed in Table S2.† It is found that the 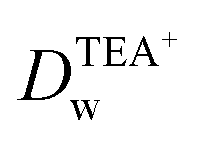 and
and 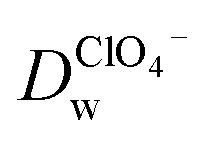 evaluated under pH ∼ 5.4 are smaller than those values obtained under pH ∼ 7.4. As demonstrated in our previous studies on the ion transfer reactions occurring at the L/L interface modified by porous membranes,38–41 the diffusion coefficients of ions can be affected by the presence of membranes, which is attributed to the hindrance effect of nanochannels on the transport of molecules or ions. Additionally,
evaluated under pH ∼ 5.4 are smaller than those values obtained under pH ∼ 7.4. As demonstrated in our previous studies on the ion transfer reactions occurring at the L/L interface modified by porous membranes,38–41 the diffusion coefficients of ions can be affected by the presence of membranes, which is attributed to the hindrance effect of nanochannels on the transport of molecules or ions. Additionally, 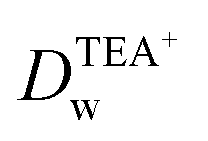 and is higher than that
and is higher than that 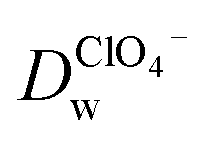 and the peak current corresponding to the ion transfer of TEA+ from aqueous phase to organic phase is also larger than the value obtained from the ion transfer of ClO4−. Higher diffusion coefficient and larger peak current of cation with comparison to the values of anion demonstrated that the negatively charged GO layers adsorbed at the W/DCE interface presents charge-selectivity. In other words, it could be easier for cations than anions to transfer across the W/DCE interface modified by the negatively-charged GO layers.
and the peak current corresponding to the ion transfer of TEA+ from aqueous phase to organic phase is also larger than the value obtained from the ion transfer of ClO4−. Higher diffusion coefficient and larger peak current of cation with comparison to the values of anion demonstrated that the negatively charged GO layers adsorbed at the W/DCE interface presents charge-selectivity. In other words, it could be easier for cations than anions to transfer across the W/DCE interface modified by the negatively-charged GO layers.
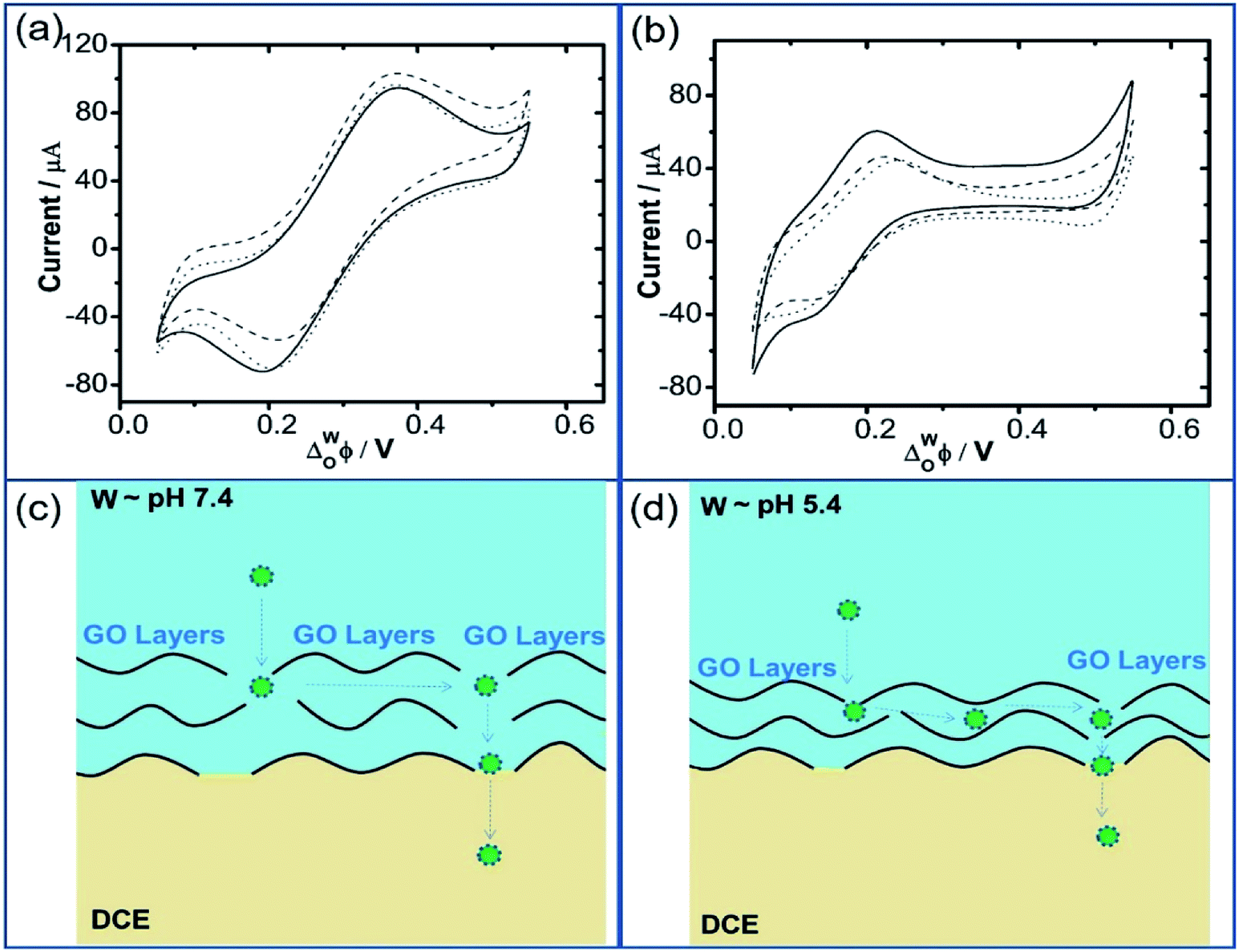 | ||
| Fig. 5 Cyclic voltammograms for the transfer of 1 mM of (a) TEA+ and (b) ClO4− in the absence (solid line) and in the presence of GO (0.1 g L−1) at pH ∼ 5.4 (dashed line) and ∼7.4 (dotted line); schemes for the ion transfer across the GO-layers-modified W/DCE interface under (c) pH ∼ 5.4 and (d) pH ∼ 7.4 by using cell 4. | ||
In order to further clarify the impact of GO on the ion transfer reactions at the L/L interface, the ion transfer of TEA+ occurring at the GO-membrane-modified W/DCE interface is further investigated. As shown as Fig. 6, the potential window (PW) obtained at the GO-membrane-modified W/DCE interface (0.0–0.65 V) became a little more broader than that obtained at the GO-layer-modified W/DCE interface (0.05–0.55 V), which indicates that the ion transfer of background electrolyte ions on the positive and negative ends of PW become more difficult. The possible reason for the broader PW obtained at the GO-membrane-modified W/DCE interface should be ascribed to that the commercial GO membrane is more compact than the GO layers obtained herein, which could result in stronger hindrance effect of GO membrane on ion transfer reactions occurring at the L/L interface as reported in the previous studies on the ion transfer across the membrane-modified W/DCE interfaces.38–41 In addition, the IP of TEA+ obtained at the GO-membrane-modified W/DCE (12.69 ± 0.02 μA) is much smaller than the values obtained above at the GO-layers-modified W/DCE interface (Table S2†). Moreover, the 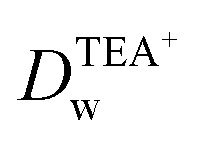 is evaluated about (7.83 ± 0.03) × 10−6 cm2 s−1, which is also smaller than those values obtained at the GO-layers-modified W/DCE interface (Table S2†) and the value reported previously,38 indicating that the commercial GO membrane with more compact structure can results in stronger hindrance effect on the ion transfer reactions than that observed at the GO-layers-modified W/DCE interface. Based on all above ion-transfer voltammetric results, it can be demonstrate that the interfacial adsorption and assembly of GO can indeed make ion transfer reactions more difficult at the W/DCE interface, which should be due to the interfacial blocking and the charge screening, as well as the hindrance effect induced by the GO layers at the W/DCE interface.
is evaluated about (7.83 ± 0.03) × 10−6 cm2 s−1, which is also smaller than those values obtained at the GO-layers-modified W/DCE interface (Table S2†) and the value reported previously,38 indicating that the commercial GO membrane with more compact structure can results in stronger hindrance effect on the ion transfer reactions than that observed at the GO-layers-modified W/DCE interface. Based on all above ion-transfer voltammetric results, it can be demonstrate that the interfacial adsorption and assembly of GO can indeed make ion transfer reactions more difficult at the W/DCE interface, which should be due to the interfacial blocking and the charge screening, as well as the hindrance effect induced by the GO layers at the W/DCE interface.
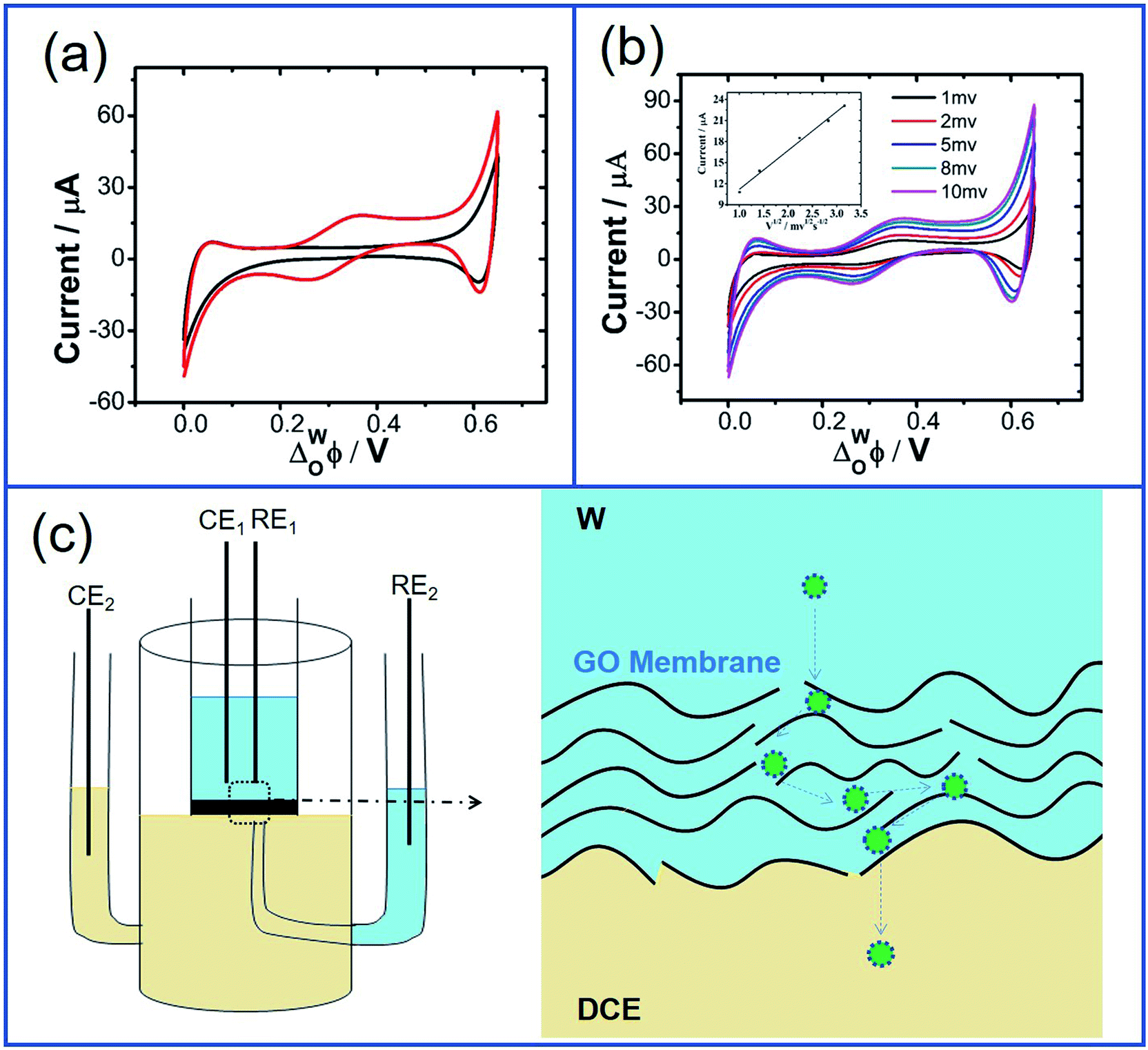 | ||
| Fig. 6 (a) Cyclic voltammograms for the background (black line) and the ion transfer of 1 mM TEA+ (red line) across the GO-membrane-modified W/DCE interface at the scan rate of 5 mV s−1 and (b) different scan rates (1, 2, 5, 8, 10 mV s−1) and the inset in (b) is the corresponding linear relationship between the peak current for ion transfer of TEA+ from W to DCE and the square roots of the different scan rates; (c) is the scheme for the ion transfer across the GO-membrane-modified W/DCE interface in a four-electrode electrochemical cell. | ||
This work with insights into the adsorption of GO at a polarizable L/L interface can not only provide a novel method to study the GO adsorption at the L/L interface, but also help to understand the adsorption behavior of GO at the L/L interface more comprehensively, especially the adsorption mechanism of GO at the L/L interface under external electric field and its impacts on the ion transfer reactions occurring at the L/L interface, which is significant for the further studies on the GO-based nano–bio interface when GO is applied as drug nanocarrier in the biomedicine in view of that GO can be applied as a promising drug nanocarrier for the drug delivery system in biomedicine3–8 and the L/L interface can act as a simple model to mimic semi-biomembrane.38–41 For example, the pH-relative adsorption behavior of GO at the L/L interface implies that the adsorption behaviors of GO-based drug nanocarriers at the membrane surface of normal cells could be different from that of cancer cells because the pH environment of tumor tissues (pH ∼ 4.0–5.5) are drastically different from those of normal tissues (pH ∼ 7.0).3–8 In addition, the pH- and charge-relative ion transfer behaviors observed at the GO-modified L/L interface implies that the ionizable drug transfer of GO-based drug delivery system across the nano–bio interface might be affected by the pH of cell environment and the valence state of GO. In order to understand the desorption and transport GO at the nano–bio interface more comprehensively, the GO-based drug delivery system should be further investigated at the polarizable L/L interface in the future.
4. Conclusions
In the present study, we demonstrated the use of CV and ACV to monitor the interfacial adsorption and assembly of GO at W/DCE interface. It was found that the interfacial capacitance value greatly increased upon addition of GO nanosheets to the aqueous phase. This behavior can be ascribed to the increases of interfacial charge density and corrugation induced by the interfacial adsorption and assembly of GO. In addition, it is found that the adsorption behavior of GO at W/DCE interface is closely related to the aqueous pH value. Moreover, ion-transfer voltammetric results further demonstrate that the GO layer formed at the interface can suppress the transfer of both of model ions, TEA+ and ClO4−, which could be ascribed to the interfacial blocking and the charge screening, as well as the hindrance effect induced by the GO layers at the W/DCE interface. This work helps to further understand not only the adsorption behavior of GO at the L/L interface, but also the GO-based nano–bio interface when GO is applied as drug nanocarrier in the biomedicine more comprehensively. In view of the special DSM and chemistry of GO in water, GO-modified L/L interface seems to be more complex dynamic interface than those L/L interfaces modified by nanoparticles, which should be further investigated more in detail by using versatile polarizable L/L systems.Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
This work was supported by the National Science Foundation of China (Grant No. 21605103 and 21005049) and the Natural Science Foundation of Shanghai, China (No. 14ZR1440900), China.References
- D. Chen, H. Feng and J. Li, Chem. Rev., 2012, 112, 6027–6053 CrossRef CAS PubMed.
- U. R. Farooqui, A. L. Ahmad and N. A. Hamid, Renewable Sustainable Energy Rev., 2018, 82, 714–733 CrossRef CAS.
- M. Li, Z. Luo and Y. Zhao, Sci. China: Chem., 2018, 61, 1214–1226 CrossRef CAS.
- S. Bullo, K. Buskaran, R. Baby, D. Dorniani, S. Fakurazi and M. Z. Hussein, Pharm. Res., 2019, 36, 91 CrossRef PubMed.
- H. Tiwaria, N. Karkia, M. Palb, S. Basakc, R. K. Vermad, R. Bale, N. D. Kandpalf, G. Bishta and N. G. Sahooa, Colloids Surf., B, 2019, 178, 452–459 CrossRef PubMed.
- H. Zhang, Y. Li, Z. Pan, Y. Chen, Z. Fan, H. Tian, S. Zhou, Y. Zhang, J. Shang, B. Jiang, F. Wang, F. Luo and Z. Hou, Mol. Pharmaceutics, 2019, 16, 1982–1998 CrossRef CAS PubMed.
- N. Ma, A. Song, Z. Li and Y. Luan, ACS Biomater. Sci. Eng., 2019, 5, 1384–1391 CrossRef CAS.
- T. T. Phama, H. T. Nguyena, C. D. Phunga, S. Pathaka, S. Regmia, D. H. Haa, J. O. Kima, C. S. Yonga, S. K. Kimb, J. E. Choic, S. Yookd, J. B. Parke and J. H. Jeonga, J. Ind. Eng. Chem., 2019, 76, 310–317 CrossRef.
- J. Kim, L. J. Cote, F. Kim, W. Yuan, K. R. Shull and J. Huang, J. Am. Chem. Soc., 2010, 132, 8180–8186 CrossRef CAS PubMed.
- J. Shao, W. Lv and Q. Yang, Adv. Mater., 2014, 26, 5586–5612 CrossRef CAS PubMed.
- L. Hu, M. Chen, X. Fang and L. Wu, Chem. Soc. Rev., 2012, 41, 1350–1362 RSC.
- S. G. Booth and R. A. W. Dryfe, J. Phys. Chem. C, 2015, 119, 23295–23309 CrossRef CAS.
- F. Chen, S. Liu, J. Shen, L. Wei, A. Liu, M. B. Chan-Park and Y. Chen, Langmuir, 2011, 27, 9174–9181 CrossRef CAS PubMed.
- Z. Sun, T. Feng and T. P. Russell, Langmuir, 2013, 29, 13407–13413 CrossRef CAS PubMed.
- D. Chen, Z. Sun, T. P. Russell and L. Jin, Langmuir, 2017, 33, 8961–8969 CrossRef CAS PubMed.
- Y. He, F. Wu, X. Sun, R. Li, Y. Guo, C. Li, L. Zhang, F. Xing, W. Wang and J. Gao, ACS Appl. Mater. Interfaces, 2013, 5, 4843–4855 CrossRef CAS PubMed.
- P. Sharma, D. J. Borah and M. R. Das, ChemPhysChem, 2014, 15, 4019–4025 CrossRef CAS PubMed.
- T. M. McCoy, S. A. Holt, A. M. Rozario, T. D. M. Bell and R. F. Tabor, Adv. Mater. Interfaces, 2017, 4, 170083 Search PubMed.
- C. F. d. Matos, S. Holakoeid, N. D. Yamamotoa, M. L. M. Roccod, A. J. G. Zarbinc and L. S. Romana, Org. Electron., 2019, 75, 105440 CrossRef.
- Q. Luo, Y. Wang, Z. Chen, P. Wei, E. Yoo and E. Pentzer, ACS Appl. Mater. Interfaces, 2019, 11, 9612–9620 CrossRef CAS PubMed.
- T. Lan, H. Zeng and T. Tang, J. Phys. Chem. C, 2019, 123, 22989–22999 CrossRef CAS.
- M. M. Gudarzi and F. Sharif, Soft Mater., 2011, 7, 3432–3440 RSC.
- H. Jahandideh, P. Ganjeh-Anzabi, S. L. Bryant and M. Trifkovic, Langmuir, 2018, 34, 12870–12881 CrossRef CAS PubMed.
- H. Yang, J. Li and X. Zeng, ACS Appl. Nano Mater., 2018, 1, 2763–2773 CrossRef CAS.
- S. Shi, B. Qian, X. Wu, H. Sun, H. Wang, H. Zhang, Z. Yu and T. P. Russell, Angew. Chem., Int. Ed., 2019, 58, 18171–18176 CrossRef CAS PubMed.
- S. Liu, Q. Li and Y. Shao, Chem. Soc. Rev., 2011, 40, 2236–2253 RSC.
- B. Su, J. P. Abid, D. J. Fermín, H. H. Girault, H. Hoffmannova, P. Krtil and Z. Samec, J. Am. Chem. Soc., 2004, 126, 915–919 CrossRef CAS PubMed.
- N. Younan, M. Hojeij, L. Ribeaucourt and H. H. Girault, Electrochem. Commun., 2010, 12, 912–915 CrossRef CAS.
- E. Smirnov, P. Peljo, M. D. Scanlon and H. H. Girault, Electrochim. Acta, 2016, 197, 362–373 CrossRef CAS.
- M. C. Collins, M. Hebrant and G. Herzog, Electrochim. Acta, 2018, 282, 155–162 CrossRef CAS.
- H. Jensen, D. J. Fermín, J. E. Moser and H. H. Girault, J. Phys. Chem. B, 2002, 106, 10908–10914 CrossRef CAS.
- A. K. Rabiu, P. S. Toth, A. N. J. Rodgers and R. A. W. Dryfe, ChemistryOpen, 2017, 6, 57–63 CrossRef CAS PubMed.
- P. S. Toth, A. N. J. Rodgers, A. K. Rabiu and R. A. W. Dryfe, Electrochem. Commun., 2015, 50, 6–10 CrossRef CAS.
- X. Bian, M. D. Scanlon, S. Wang, L. Liao, Y. Tang, B. Liu and H. H. Girault, Chem. Sci., 2013, 4, 3432 RSC.
- A. N. J. Rodgers and R. A. W. Dryfe, ChemElectroChem, 2016, 3, 472–479 CrossRef CAS.
- P. S. Toth, Q. M. Ramasse, M. Velický and R. A. W. Dryfe, Chem. Sci., 2015, 6, 1316–1323 RSC.
- W. Hirunpinyopas, A. N. J. Rodgers, S. D. Worrall, M. A. Bissett and R. A. W. Dryfe, ChemNanoMat, 2017, 3, 428–435 CrossRef CAS.
- Y. Chen, S. Bian, K. Gao, Y. Cao, H. Wu, C. Liu, X. Jiang and X. Sun, J. Membr. Sci., 2014, 457, 9–18 CrossRef CAS.
- X. Jiang, K. Gao, D. Hu, H. Wang, S. Bian and Y. Chen, Analyst, 2015, 140, 2823–2833 RSC.
- K. Gao, X. Jiang, D. Hu, S. Bian, M. Wang and Y. Chen, Chin. Chem. Lett., 2015, 26, 285–288 CrossRef CAS.
- D. Hu, H. Wang, K. Gao, X. Jiang, M. Wang, Y. Long and Y. Chen, RSC Adv., 2014, 4, 57035–57040 RSC.
- S. Jeshycka, E. M. Kim and H. J. Lee, Electrochim. Acta, 2018, 282, 964–972 CrossRef CAS.
- S. Jeshycka, H. Y. Han and H. J. Lee, Electrochim. Acta, 2017, 245, 211–218 CrossRef CAS.
- P. Chen, H. Yue, X. Zhai, Z. Huang, G. Ma, W. Wei and L. Yan, Sci. Adv., 2019, 5, eaaw3192 CrossRef PubMed.
- L. I. Daikhin and M. Urbakh, J. Electroanal. Chem., 2003, 560, 59–67 CrossRef CAS.
- A. M. Dimiev, L. B. Alemany and J. M. Tour, ACS Nano, 2013, 7, 576–588 CrossRef CAS PubMed.
- A. M. Dimiev and S. Eigler, Graphene Oxide, John Wiley & Sons, American, 2016 Search PubMed.
- A. Lerf, H. He, M. Forster and J. Klinowski, J. Phys. Chem., 1998, 102, 4477–4482 CrossRef CAS.
- T. Szabo, Ot. Berkesi, P. Forgo, K. Josepovits, Y. Sanakis, D. Petridis and I. Dekany, Chem. Mater., 2006, 18, 2740–2749 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra02560g |
| This journal is © The Royal Society of Chemistry 2020 |