
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDependence on co-adsorbed water in the reforming reaction of ethanol on a Rh(111) surface†
Yu-Yao Hsiaa,
Po-Cheng Chienb,
Lu-Hsin Leea,
Yu-Ling Laic,
Li-Chung Yuc,
Yao-Jane Hsu c,
Jeng-Han Wang*b and
Meng-Fan Luo*a
c,
Jeng-Han Wang*b and
Meng-Fan Luo*a
aDepartment of Physics, National Central University, No. 300 Jhongda Road, Jhongli District, Taoyuan 32054, Taiwan. E-mail: mfl28@phy.ncu.edu.tw
bDepartment of Chemistry, National Taiwan Normal University, No. 88, Sec. 4, Ting-Zhou Road, Taipei, Taiwan. E-mail: jenghan@ntnu.edu.tw
cNational Synchrotron Radiation Research Center, 101 Hsin-Ann Road, Hsinchu Science Park, Hsinchu 30076, Taiwan
First published on 7th May 2020
Abstract
We have studied the reforming reaction of ethanol co-adsorbed with atomic oxygen (O*, * denotes adspecies) and deuterated water (D2O*) on a Rh(111) surface, with varied surface probe techniques under UHV conditions and with density-functional-theory calculations. Adsorbed ethanol molecules were found to penetrate readily through pre-adsorbed water, even up to eight overlayers, to react at the Rh surface; they decomposed at a probability promoted by the water overlayers. The production probabilities of H2, CO, CH2CH2 and CH4 continued to increase with co-adsorbed D2O*, up to two D2O overlayers, despite separate increasing rates; above two D2O overlayers, those of H2, CO and CH2CH2 were approximately saturated while that of CH4 decreased. The increased (or saturated) production probabilities are rationalized with an increased (saturated) concentration of surface hydroxyl (OD*, formed by O* abstracting D from D2O*), whose intermolecular hydrogen bonding with adsorbed ethanol facilitates proton transfer from ethanol to OD* and thus enhances the reaction probability. The decreasing behavior of CH4 could also involve the competition for H* with the formation of H2 and HDO.
1. Introduction
As an efficient approach to produce hydrogen for use in fuel-cells, the reforming reaction of ethanol has drawn considerable attention.1 Ethanol has advantages of low toxicity, high availability, high hydrogen density and ease of handling and storage; it can be readily extracted from fermentation of biomass like sugarcane and corn.2–4 Among various reforming reactions, oxidative steam reforming of ethanol (OSR, C2H5OH + (3 − x)H2O + xO2 → (6 − 2x)H2 + 2CO2) is promising, because its hydrogen yield and exothermicity can be balanced by controlling molar ratios of reagents (ethanol, steam and oxygen).4,5 The mechanism of OSR of ethanol has thus been widely investigated. Earlier mechanistic studies find that the reaction is initiated with scission of O–H bond of adsorbed ethanol, forming surface ethoxy (CH3CH2O*, * denoting adspecies);6–9 either C–Hα or C–Hβ bond is sequentially cleaved, producing surface acetaldehyde (CH3CHO*) and oxametallacycle (CH2CH2O*), respectively. The surface acetaldehyde ultimately leads to the production of CH3CHO, CH4, CH3COOH, CO and CO2, while the surface oxametallacycle to CH2CH2 and CO.6,7,10The reagent oxygen (molecular) is dissociated into atomic oxygen (O*) on catalyst surfaces; the O* promotes the decomposition probability of ethanol and could also alter the reaction path toward acetaldehyde, as indicated on Rh(100) and Rh(111) surfaces.11–13 This alteration highly promotes the production of H2, along with side products CO, CH4 and H2O. With increased oxygen content, the reaction path shifts further to acetate (CH3COO*) intermediates; the production of H2 is suppressed but that of CO2 is highly promoted.13 The reagent water (steam) in OSR is typically regarded as another supplier of reagent oxygen or an assistance to the side process—water–gas-shift reaction—of the reforming reaction. Preceding studies on a Rh(111) surface showed comparable effects of hydroxyl (OH*, from dissociated H2O*) and O*; the OH* further enhanced the reaction probability of ethanol on the Rh surfaces pre-covered with O* but affected little the reaction path.13 Nevertheless, how this effect evolves with the quantities of adsorbed water is not clarified. This issue becomes critical as the advantages of OSR depend largely on the molar ratios of its reagents. The present study aims to remedy this lack of knowledge and to shed light on detailed mechanisms.
We have studied the reactions of ethanol co-adsorbed with O* and deuterated water (D2O*) on a Rh(111) single crystal under ultrahigh vacuum (UHV) conditions. The Rh(111) substrate, as a model system, was chosen because Rh-based catalysts become the most promising catalyst in the reforming reaction6,14–19 and (111) facets typically make up a great fraction of the surface of the Rh catalysts.20–23 Temperature-programmed desorption (TPD) and synchrotron-based photoelectron spectroscopy (PES) were applied to probe the catalyzed reactions, and density-functional-theory (DFT) modelling to illuminate the picture how ethanol interacts with co-adsorbed water. The results show that the reactions of ethanol adsorbed on the Rh surface pre-covered with O* and molecular water proceeded despite adsorbed water increased up to eight overlayers. The reactions persisted as the pre-adsorbed water did not obstruct completely the adsorption of ethanol; besides, the adsorbed ethanol diffused, through exchanging positions with the pre-adsorbed water, toward the Rh surface to react. Furthermore, the decomposition probability was evidently enhanced. The production probabilities of all species, including H2, CO, CH2CH2 and CH4, were increased with co-adsorbed water, up to two water overlayers; above two water overlayers, those of H2, CO and CH2CH2 exhibited a trend of saturation while that of CH4 decreased. The behavior is strongly correlated with the concentration of surface OD*. We discussed in detail the mechanisms with our DFT simulations.
2. Methods
2.1 Experimental section
Our experiments were conducted in UHV chambers at a base pressure 4 × 10−10 torr. The Rh(111) single crystal, polished to a roughness <10 nm and an orientation accuracy <0.1°, was purchased from MaTeck GmbH. Before each experiment, alternative cycles of sputtering and subsequent annealing (900 K) were conducted to clean the crystal surface. We confirmed the cleanliness of the crystal surface with surface probe techniques such as low-energy electron diffraction and Auger electron spectroscopy. The crystal was then quenched to desired temperatures for adsorption: molecular oxygen (O2) at 300 K, deuterated water (D2O) and ethanol at 120 K. The adsorption was performed with a doser pointing toward the crystal, at a background pressure 5 × 10−8 to 5 × 10−9 torr. Adsorbed O2 on Rh(111) at 300 K was dissociated into atomic oxygen (O*). The deuterated water (purchased from Merck, 99.8%) was further purified by several freeze–pump–thaw cycles before the adsorption experiments. Their exposures were reported in Langmuir units (1.0 L = 10−6 torr s). We collected TPD spectra with a quadruple mass spectrometer (Hiden) to monitor various masses and by ramping the sample at a rate of 3 K s−1; we shielded and placed the spectrometer near the crystal surface (about 2 mm). The PES experiments were conducted at the BL09A2 beamline (U5 spectroscopy) at National Synchrotron Radiation Research Center in Taiwan.24 The photon beam had a fixed energy 600 eV and was incident normal to the surface; emitted photoelectrons were detected at an angle 58° off from the surface normal. The energy resolution attained 0.1 eV. All PES spectra shown in the current work were normalized to their photon flux. The binding energy (BE) indicated in the spectra is referred to the bulk Rh 3d5/2 at 307.1 eV.2.2 Computational section
Our computations were performed with Vienna Ab initio Simulation Package (VASP),25–27 a DFT-based computational package with a 3D periodic boundary condition. The computational level was at GGA-PAW, the generalized gradient approximation28 with Perdew–Wang 1991 formulation29 utilized for the exchange-correlation function. The valence electrons were treated by plane waves with a maximal kinetic energy (cutoff energy) of 600 eV; the core electrons were treated by the cost-effective pseudopotentials implemented in VASP, the projector-augmented wave method (PAW). The integration in the Brillouin-Zone (BZ) was sampled by the Monkhorst–Pack scheme30 with the k-point at 0.05 × 2 (1/Å) interval in the reciprocal space. For the structural optimizations and energetic calculations of stable adsorptions, we applied quasi-Newton method with an energetic convergence of 1 × 10−4 eV and a gradient convergence of 1 × 10−2 eV Å−1; those of transition states were utilized by Nudged Elastic Band method31 at the same convergence criterions. The chosen convergence condition has been widely applied in previous studies;21,32,33 a convergence test, with a more strict convergence condition (1 × 10−6 eV and 1 × 10−3 eV Å−1), had also been performed to justify the present calculations.34 The vibrational analysis, with the finite displacement approach at the Γ point,35,36 was utilized to confirm the optimized local minimums (without imaginary frequency) and apply zero-point energy (ZPE) corrections on the DFT computed energies.The Rh(111) surface was constructed with a Rh slab consisting of five layers of 4 × 4 surface units and equivalent five-layer distant vacuum space to avoid artificial interaction between separate Rh slabs; the bottom two Rh layers were fixed at the computed lattice constants to represent the semi-infinite bulk crystal beneath the surface and the top three layers were free to relax. The adspecies, such as water, ethanol and their fragments, were then placed on the Rh surface for optimization of their adsorption structures and related energies.
3. Results and discussion
3.1 TPD and PES experiments
The reactions of ethanol were monitored primarily with TPD. We compared the TPD spectra from ethanol on Rh(111) pre-covered with O* at 0.08 ML and water at varied coverages to investigate quantitatively the effect of water on the reactions. Adsorbed water molecules alter the OSR reaction of ethanol because they are dissociated into OH*. The dissociation on Rh(111) is largely assisted by pre-adsorbed O*.37 Our previous work showed, in line with other studies,37 that water adsorbed on Rh(111) pre-adsorbed with 0.08 monolayer (ML) O* (denoted as Rh(111)O*(0.08 ML)) yielded a maximal production of OH*,13 so we examined the present effect on Rh(111)O*(0.08 ML). We used deuterated water (D2O), instead of typical water (H2O), for our TPD measurements. These isotopic variants behavior similarly, since their adsorption energies, activation energies for dissociation and their interaction with ethanol are determined by their electronic structures, rather than their isotopic properties. Adsorbed D2O, unlike H2O, contributed no TPD signals of H2 and H2O, two major products from decomposed ethanol on Rh(111)O*(0.08 ML), but gave clear, separate D2 (or DH) and D2O (or DHO) signals. The use of D2O avoids mixing signals from different processes and thus permits ready identification of the role of water in the ethanol reaction.We noted in the series of TPD experiments that adsorbed ethanol penetrated readily through pre-adsorbed water overlayers to react at the Rh(111)O*(0.08 ML) surface. Fig. 1a shows the D2O (m/z = 20 u) TPD spectra from Rh(111)O*(0.08 ML) exposed to D2O of varied amounts (denoted as Rh(111)D2O*/O*(0.08 ML)). 0.3 L D2O adsorbed on Rh(111)O*(0.08 ML) at 120 K gave a single desorption feature around 195 K (the bottom in Fig. 1a), assigned to desorbing sub-monolayer D2O from the surface. The desorption temperature of the sub-monolayer or monolayer D2O on Rh(111)O*(0.08 ML) is higher than that on Rh(111) (about 170 K, Fig. S1†), because of the formation of a hydrogen-bonded network of D2O* and OD*37 and also the interaction of D2O* with O*. The desorbing D2O came from two channels: D2O* in the D2O*–OD* hydrogen bonded network and that from disproportionation of OD* (2OD* → D2O* + O*).37 As the exposure of D2O increased, the monolayer feature was enhanced; above 1.0 L, the monolayer feature remained similar whereas an additional feature grew about 150 K (top and second in Fig. 1a), which is assigned to the desorption of multilayer D2O. As the integrated intensity of the D2O desorption feature increased almost linearly with the exposure and as the desorption feature of 1.0 L D2O corresponds about to a full monolayer D2O, the sticking coefficient of D2O onto the sample at 120 K is nearly 1; 1.0 L D2O yielded about a single water overlayer on either Rh(111)O*(0.08 ML) or Rh(111)D2O*/O*(0.08 ML). The D2O TPD spectra altered significantly when ethanol was adsorbed atop Rh(111)D2O*/O*(0.08 ML). For Rh(111)D2O*(0.3 L)/O*(0.08 ML) and Rh(111)D2O*(0.5 L)/O*(0.08 ML) exposed to ethanol (the third and bottom in Fig. 1b), the monolayer feature of D2O, about 195 K, attenuated, in comparison to those without ethanol (the third and bottom in Fig. 1a), while the multilayer one, about 150 K, emerged. At higher D2O coverages (the first and second in Fig. 1b), the multilayer feature became obviously enhanced whereas the monolayer one remained smaller than its counterparts without ethanol (the first and second in Fig. 1a). Nevertheless, the integrated intensities of the D2O lines with and without co-adsorbed ethanol were similar, as plotted in Fig. 1c. The comparison implies that the diminished monolayer D2O was compensated by the increased multilayer D2O – a fraction of the first overlayer D2O on Rh(111) migrated to the multilayer region and desorbed. The migration was induced because the adsorbed ethanol diffused toward the Rh surface and exchanged position with the underneath D2O. The involvement of D2O in the ethanol reaction is reflected on systematically increased DHO desorption signals, which result from surface OD* (from D2O* + O* → 2OD*, discussed below) abstracting H from ethanol and desorbing as DHO. On such ethanol on Rh(111)D2O*/O*(0.08 ML), O* were entirely consumed and no trace of it was observed with increased temperature,13 contrasting ethanol and D2O separately adsorbed on Rh(111)O*(0.08 ML).13,37 Details are explained in ESI (Fig. S2†) and DFT calculations below.
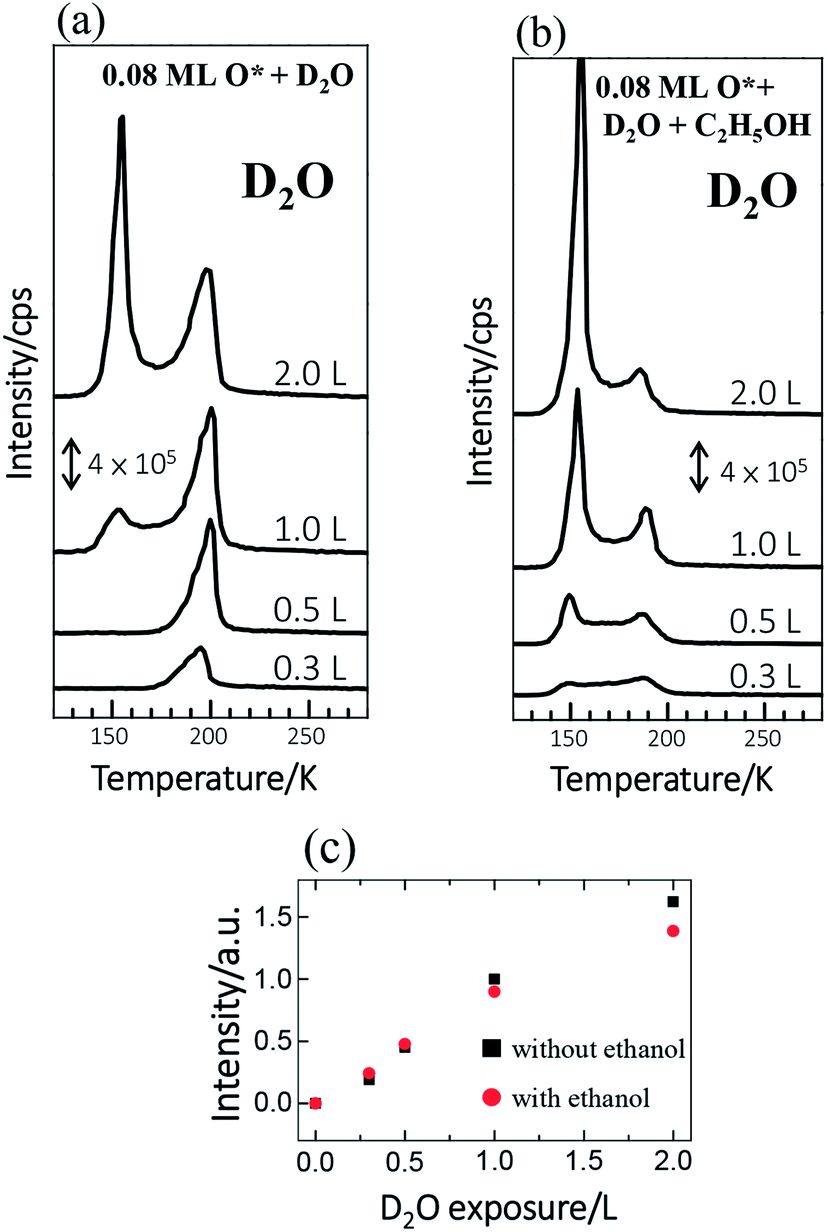 | ||
| Fig. 1 D2O TPD spectra from Rh(111)O*(0.08 ML) exposed to (a) D2O of varied amounts, as indicated, and to (b) D2O of varied amounts and subsequently 3.0 L ethanol. (c) Plots the integrated intensities of the D2O desorption features in (a) and (b) as a function of D2O exposure; black squares and red spheres denote the data from the sample without and with ethanol, respectively. | ||
The TPD spectra for the reaction products of ethanol reveal more the effect of D2O. Fig. 2a–c show the TPD spectra from 3.0 L ethanol adsorbed on Rh(111)O*(0.08 ML), Rh(111)D2O*(1.0 L)/O*(0.08 ML) and Rh(111)D2O*(2.0 L)/O*(0.08 ML). The ethanol (C2H5OH, m/z = 45 u) spectra (top lines in Fig. 2a–c) show desorption at 150 and 200 K, corresponding to multilayer and monolayer ethanol respectively. The CO (m/z = 28 u), ethylene (CH2CH2, m/z = 27 u), H2O (m/z = 18 u), methane (CH4, m/z = 16 u) and H2 (m/z = 2 u) spectra also show their desorption at various temperatures, the second to the bottom lines in Fig. 2a–c, reflecting the reforming reaction of adsorbed ethanol. Preceding studies argued that adsorbed ethanol on Rh(111) produced ethoxy readily via O–H bond scission and the ethoxy decomposed predominantly via C–Hβ bond cleavage, which led to formation of oxometallacycle intermediate (CH2CH2O*) and further decomposition producing CO, H2 and surface carbon ultimately;12,38,39 on Rh(111)O*, the decomposition probability was enhanced and the reaction pathway was largely altered to the one via C–Hα bond cleavage, which formed acetaldehyde (CH3CHO*) intermediates and promoted the production of H2 along with the side products of CH4 and H2O.11,13,39,40 The evident CH4 and H2O signals in Fig. 2a confirm the altered reaction pathway; the CH2CH2 signals implies the existence of CH2CH2O*, whose C–O bond cleavage yields CH2CH2,40 and hence that the channel via CH2CH2O* remained active. The observed desorbing species from Rh(111)D2O*/O*(0.08 ML) (Fig. 2b and c) were the same as those from Rh(111)O*(0.08 ML) (Fig. 2a) whereas their intensities differed. Both desorbing multilayer (150 K) and monolayer ethanol (200 K), which refer respectively to the ethanol on or in the D2O–ethanol mixed overlayers and that diffusing to the Rh surface, obviously decreased with increased D2O (Fig. 2b and c); the sticking coefficient for ethanol onto water overlayers was smaller than that for ethanol onto Rh(111)O*(0.08 ML) (Fig. S3†). The H2O (m/z = 18 u) spectra became highly enhanced (Fig. 2b and c) and resembled the corresponding D2O spectra (Fig. 1b), as the signals were contributed primarily from adsorbed background H2O and the cracking pattern of desorbing D2O (DO, m/z = 18 u). The CO, H2 and CH2CH2 signals altered only a little while the CH4 ones attenuated systematically with increased D2O (Fig. 2a–c). The signals of either D2 or DH were very small (Fig. S4†), indicating few D* and hence limited dissociation of D2O* into D* and OD*; the OD* was formed predominantly via the process D2O* + O* → 2OD*.
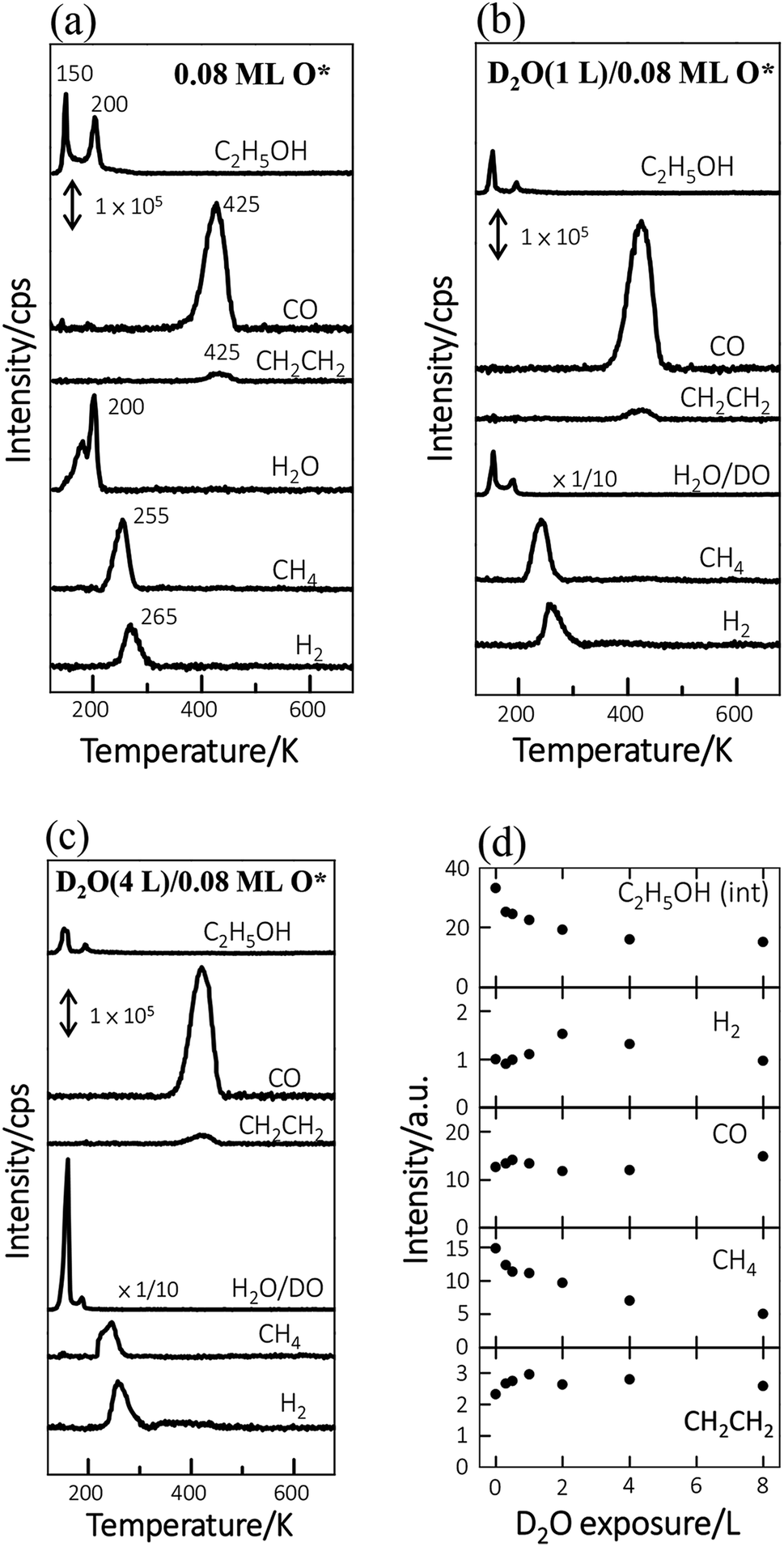 | ||
| Fig. 2 TPD spectra from 3.0 L ethanol adsorbed on (a) Rh(111)O*(0.08 ML), (b) Rh(111)D2O*(1.0 L)/O*(0.08 ML) and (c) Rh(111)D2O*(4.0 L)/O*(0.08 ML). (d) Plots the quantities of ethanol(int) and reaction products, measured with the integrated intensities of the corresponding desorption features, as a function of D2O exposure. The ethanol(int) includes those desorbing from and reacting at Rh(111) surface. | ||
Fig. 2d plots the quantities of the ethanol interacting with Rh(111)D2O*/O*(0.08 ML) (denoted as ethanol(int)) and the produced species from ethanol(int), as a function of D2O exposure. These quantities were measured with the integrated intensities of their desorption features and had taken into account their various ionization cross-sections. Ethanol(int) consisted of ethanol adsorbed directly on Rh(111) and also those which adsorbed atop D2O overlayers and migrated to the D2O–Rh(111) interface to react or desorb, so contained desorbing and decomposing ethanol at the Rh(111) surface; they were estimated according to desorbing and remaining carbon-related species.41 The ethanol(int) decreased when D2O overlayers increased; it decreased at 8.0 L D2O (corresponding about to 8.0 water overlayers) to 50% of that on Rh(111)O*(0.08 ML) (top of Fig. 2d). The decrease occurred largely because of a smaller sticking coefficient for ethanol onto D2O overlayers than that onto Rh(111)O*(0.08 ML). We note that the ethanol(int) decreased remarkably between 0.0–2.0 D2O overlayers but only a little between 2.0–8.0 D2O overlayers; increasing D2O above 2.0 overlayers blocked ineffectively the diffusion of adsorbed ethanol toward the Rh(111) surface. Additionally, total adsorbed ethanol (including both multilayer ethanol and ethanol(int)) decreased with D2O overlayers in a similar manner (Fig. S3†); the ethanol(int) made up a great proportion, about 70 ± 5%, of total adsorbed ethanol and the proportion varied insignificantly with increasing D2O overlayers. The result agrees with the above D2O TPD spectra (Fig. 1). The produced species responded with increased D2O in separate manners. The produced CH4, like ethanol(int), decreased monotonically with increased D2O; the H2 increased at D2O overlayers ≤2.0 L but decreased at higher ones; the CO varied little; the CH2CH2 increased with D2O whereas became saturated above 1.0 L. As ethanol(int) decreased with the D2O overlayers, the comparison implies that the probability of the ethanol(int) undergoing decomposition to produce CO and CH2CH2 was enhanced under the D2O overlayers.
Fig. 3 plots the ratios of the quantities of the produced species to ethanol(int) as a function of D2O exposure, to illuminate the altered probability; the red lines are drawn to guide the eyes. The ratios for all products have a similar trend below 2.0 L D2O exposure—they all increased with D2O exposure despite of varied increasing rates. Above 2.0 L D2O exposure, two separate trends are exhibited. For the first kind, the ratio was either saturated, such as H2 and CH2CH2 (first and bottom), or increased slowly, such as CO (second); the other kind, for CH4, showed a decreasing trend (third). Among these four products, CH2CH2 was exclusively contributed from the reaction route via CH2CH2O* intermediates and CH4 via CH3CHO* intermediates; the other two products, H2 and CO, were produced from both the reaction routes. The dissimilar production probabilities of these four products above 2.0 L D2O exposure are not simply concluded according to the separate reaction routes. Nevertheless, the similar increasing trend below 2.0 L D2O exposure can be understood through the formation and increased concentration of surface hydroxyl (OD*).
 | ||
| Fig. 3 Ratios of the quantities of reaction products to ethanol(int) as a function of O* coverage. These quantities were measured with the integrated intensities of the corresponding desorption features. The ethanol(int) contains those desorbing from and reacting at Rh(111) surface. The red lines are drawn to guide the eyes. | ||
Previous studies indicate that hydroxyl (OH* or OD*) can further enhance the reaction probability of ethanol on Rh surfaces pre-covered with O*, through the intermolecular hydrogen bonding between surface OH* (OD*) with ethanol or its fragments.13 The dependence on D2O coverages of the above production probabilities is strongly correlated with the quantities of OD*. To examine the correlation, we have monitored the production of OH* on Rh(111)H2O*/O*(0.08 ML) with PES spectra. No substantial difference is anticipated in the formation of OH* on Rh(111)H2O*/O*(0.08 ML) and OD* on Rh(111)D2O*/O*(0.08 ML). Fig. 4a exemplifies the O 1s PES spectra for the produced OH* as a function of H2O exposure. The bottom panel shows the O 1s line, centered about 529.6 eV, for 0.08 ML O*; upon adsorption of 0.3 L H2O at 120 K, the O 1s signals for OH* appeared about 530.5 eV (the second from the bottom), in addition to those for H2O* centered about 532.4 eV.37 The OH* was formed mainly by O* abstracting H from H2O* (H2O* + O* → 2OH*). With increased H2O coverage up to 1.0 L, both H2O* (light blue fitting curve) and OH* (blue) signals increased while O* (red) ones decreased – O* was protonated to OH*; above 1.0 L, the OH* signals became saturated despite the H2O* ones continued to grow (top). As the observed OH* signals measured the numbers of OH* on the Rh surface, the quantities of OH* increased monotonically with H2O exposure up to 1.0 L and became saturated above 1.0 L (Fig. 4b). The signal at 2.0 L was slightly attenuated by multilayer water; annealing to 160 K to remove multilayer water restored the OH* signals to about that at 1.0 L. The increased OH* corresponds well to the increased production probabilities of these four products (Fig. 3) below 2.0 L D2O exposure: the OD* (OH*) promoted the production probabilities. Above 2.0 L D2O exposure, the OD* was saturated, so the production probabilities of H2 and CH2CH2 were saturated and that of CO increased only slightly; either of them agreed with the saturation of OD* to a great extent. The saturated OD* however could not explain the declining production probability of CH4 above 2.0 L D2O exposure. Our analysis based on DFT calculations below gives a more comprehensive picture to understand the evolution of the production probabilities with co-adsorbed D2O.
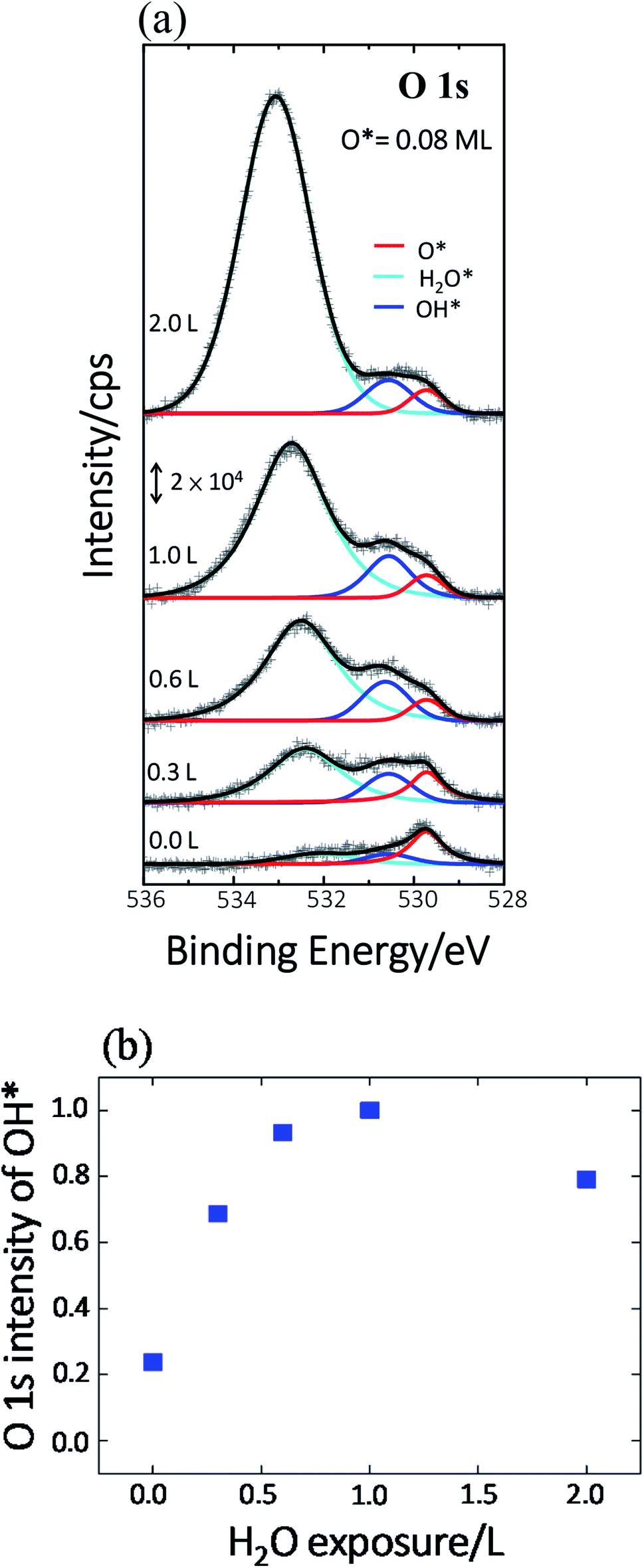 | ||
| Fig. 4 (a) O 1s spectra for Rh(111)O*(0.08 ML) (bottom) and subsequently exposed to 0.0–2.0 L H2O, as indicated, at 120 K. The black fitting curves in (a) consist of those for O* (red), OH*(blue) and H2O* (light blue). (b) Plots the quantities of OH*, measured with the integrated intensities of the fitting curve (blue) for OH*, as a function of H2O exposure. | ||
3.2 DFT computation and discussion
The experimentally observed phenomena of water and ethanol co-adsorbed on Rh(111) and Rh(111)O* surfaces are mechanistically rationalized according to the schematic plot in Fig. 5. We used H2O molecule for the computation and compared the results to the above experimental ones with D2O, because we focused on the properties associated with the electronic structures of Rh(111) surface and adsorbed water (for which H2O and D2O are identical), such as desorption energies (Edes), reaction energy (ΔE), activation energies (Ea) and electronic distributions. For water molecularly adsorbed on the surfaces, its Edes on Rh(111) surface (top panel, 0.37 eV) is slightly smaller than that on Rh(111)O* surface (middle panel, 0.48 eV), due mainly to a weak hydrogen bond between adsorbed H2O* and surface O*; the hydrogen bond is evident through the analysis of induced charge that some negative charge is induced on O* (green transparent sphere) and some positive charge on H2O* (yellow transparent sphere); the computed Bader charge for O* is −0.90|e| and those for O and H of H2O* are 1.00 and −1.92|e|, respectively. The increased Edes on Rh(111)O* contributes partly to the increased desorption temperature of first water overlayer (from 170 K to 195 K on Rh(111) surface) in the TPD experiment (Fig. 1a). | ||
| Fig. 5 Schemes of reactions of molecular water, atomic oxygen and ethanol co-adsorbed on a Rh(111) surface. The top panel shows that sole water on the Rh(111) surface has a smaller Edes. The middle one shows that water adsorbs on the Rh(111)O* surface with a greater Edes and dissociates into OH*; the OH* abstracts H from co-adsorbed ethanol with a small energetic barrier; the green and yellow transparent spheres denotes induced negative and positive charge respectively. The bottom panel shows that the decomposition of CH3CH2O* produces CH2CH2, H2, CH4 and CO. | ||
The H2O* on Rh(111)O* surface can further cleave its O–H bond and yield OH*, with energies ΔE/Ea = 0.17/0.93 eV. Upon adsorbing ethanol on the OH* covered surface, the hydrogen bond is readily formed (CH3CH2OH*…OH*, middle panel), revealed through the induced charges – positive one (yellow) on the H of CH3CH2OH* and negative one (green) on the O of OH*; the computed Bader charges for H and O of OH* are 1.00 and −1.52|e|, respectively and that for O of CH3CH2OH* is −1.63|e|. The hydrogen bond (0.5 eV) stabilizes the adsorption of ethanol and significantly lowers the energies for ethanol dissociation forming CH3CH2O* + H2O* (ΔE/Ea = −0.66/0.23 eV), compared to the dissociation without the hydrogen bond (−0.19/0.58).9,13 The yielded CH3CH2O*, with a much stronger adsorption energy (−2.47 eV), further decomposes (lower panel), while the yielded H2O* desorbs easily from the surface. As a result, the intermolecular hydrogen bond between co-adsorbed CH3CH2OH* and OH* stabilizes the ethanol adsorption and induces a low-barrier and highly exothermic proton transfer process so assists the ethanol dissociation and squeezes water out from the surface. The result explains the TPD observation in Fig. 1b that later adsorbed ethanol exchanged positons with pre-adsorbed surface water so water desorbing from the water multilayer regime increased.
The surface ethoxy (CH3CH2O*) is further dissociated on the surface (bottom panel, Fig. 5) through sequences of C–H, C–O and C–C bond cleavages and ultimately produces the products CH2CH2, H2, CH4 and CO, as observed in the TPD spectra (Fig. 2). The detailed energetics and reaction routes are plotted in Fig. S5 in the ESI;† the energetics showed trends similar to those from previous studies (Table S1†).20,21,32,33,42 The four products are formed through routes of two kinds, shown with the cyan and yellow arrows in the figure; the measured CH2CH2 and CO came from direct desorption of their surface adspecies (cyan arrows), while the measured H2 and CH4 from combinative desorption with proton (yellow ones) as their precursors were H* and  respectively. The quantities of CH2CH2 and CO correspond mostly to that of decomposing ethanol (schemes (a) and (b) in Fig. S5†); the increased ratios (production probabilities) of CH2CH2 and CO (Fig. 3) thus reflect a promoted decomposition probability of ethanol, by co-adsorbed water (or OD*). When OD* was saturated above 2.0 L D2O exposure, the decomposition probability became (or nearly) saturated so the production probabilities of CH2CH2 and CO either remained constant or increased only little.
respectively. The quantities of CH2CH2 and CO correspond mostly to that of decomposing ethanol (schemes (a) and (b) in Fig. S5†); the increased ratios (production probabilities) of CH2CH2 and CO (Fig. 3) thus reflect a promoted decomposition probability of ethanol, by co-adsorbed water (or OD*). When OD* was saturated above 2.0 L D2O exposure, the decomposition probability became (or nearly) saturated so the production probabilities of CH2CH2 and CO either remained constant or increased only little.
In contrast, the production of combinatively desorbing CH4 and H2 depends to a great extent on the fragments from decomposed ethanol as well as surface H* (schemes (c) and (d) in Fig. S5†). Surface OH* (OD*) from adsorbed water not only enhances ethanol decomposition (by abstracting H of ethanol) but also consumes surface H* to yield H2O* (HDO*), via a moderate energetic reaction (ΔE/Ea = −0.01/0.85 eV). Accordingly, the production of CH4 or H2 is balanced between the ethanol decomposition and the availability of H*. The raised production probabilities of CH4 and H2 at smaller water exposure (<2.0 L) correspond largely to a promoted ethanol decomposition, while the decreased (saturated) production probability of CH4 (H2) at a greater water exposure (≥2.0 L) to not only a saturated probability of ethanol decomposition but also a high consumption rate of H* (a high ratio of OH*(OD*) to ethanol(int)). As the consumption of H* by OH* (OD*) is completed about/below 200 K (Fig. 3), the formation of H2 and CH4 competes for the rest H*. It is noted that the formation of H2 was more competitive than that of CH4 even at a smaller water exposure (<2.0 L); with increased water exposure, the H2 production was increased at a rate much greater than that for the CH4 production (Fig. 3). The formation of CH4 was not favored because of an inhomogeneous distribution of  The channel of producing
The channel of producing 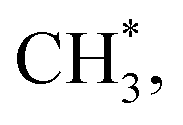 via CH3CHO* intermediates, yields less H*, so less H* is directly available to
via CH3CHO* intermediates, yields less H*, so less H* is directly available to  in contrast, both channels produce H* so H* readily finds another H* nearby to form H2. Additionally, a considerable fraction of the precursor
in contrast, both channels produce H* so H* readily finds another H* nearby to form H2. Additionally, a considerable fraction of the precursor  underwent dissociation, leading ultimately to formation of surface C*.9,13 Consequently, with limited H* at a greater water exposure, the production of H2 was sustained while that of CH4 decreased. Our DFT calculations show that an increased OH* (OD*) concentration decreases the adsorption energies of
underwent dissociation, leading ultimately to formation of surface C*.9,13 Consequently, with limited H* at a greater water exposure, the production of H2 was sustained while that of CH4 decreased. Our DFT calculations show that an increased OH* (OD*) concentration decreases the adsorption energies of  H* and OH* but in contrast, enhances the Ea for formation of CH4, H2 and H2O, which implies equally raised difficulty for their formation. The energetics varied with the OH* (OD*) concentration accounts little for the decreased production probability of CH4.
H* and OH* but in contrast, enhances the Ea for formation of CH4, H2 and H2O, which implies equally raised difficulty for their formation. The energetics varied with the OH* (OD*) concentration accounts little for the decreased production probability of CH4.
4. Conclusion
We have used TPD, PES and DFT calculations to investigate the reactions of ethanol co-adsorbed with atomic oxygen (O*) and deuterated water (D2O*) on a Rh(111) surface under UHV conditions. The results show that adsorbed ethanol penetrated readily through pre-adsorbed water overlayers to react with the Rh surface; for 2.0 L ethanol adsorbed on Rh(111)D2O*(8.0 L)/O*(0.08 ML), the ethanol(int) (which interacted with Rh surface) made up about 75% of total adsorbed ethanol (ethanol(int) + ethanol in multilayer regime), a fraction similar to that on Rh(111)O*(0.08 ML), but amounted to 50% of the ethanol(int) on Rh(111)O*(0.08 ML). The decreased ethanol(int) with water overlayers results primarily from a smaller sticking coefficient of ethanol onto the water overlayers. In the reaction aspect, the decomposition probability of ethanol(int) was remarkably enhanced, as the surface OD*, from D2O* + O* → 2OD*, abstracted readily H from ethanol(int). The production probabilities of CO, H2, CH2CH2 and CH4 were increased in proportion to the concentration of OD*, despite their increasing rates differed. Above two water overlayers, corresponding to a saturated concentration of OD*, the production probabilities of CO, H2, CH2CH2 were about saturated, whereas that of CH4 was decreased. The atypical behavior of CH4 could be additionally associated with the availability of H*. As the formation of CH4 ( + H* → CH4) competes for H* with that of H2 (H* + H* → H2) and HDO (OD* + H* → HDO), both a greater ratio OD*/ethanol(int) at a great water coverages and an inhomogeneous distribution of the precursor
+ H* → CH4) competes for H* with that of H2 (H* + H* → H2) and HDO (OD* + H* → HDO), both a greater ratio OD*/ethanol(int) at a great water coverages and an inhomogeneous distribution of the precursor  could result in the decreased production probability of CH4.
could result in the decreased production probability of CH4.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by Ministry of Science and Technology in Taiwan (MOST 107-2113-M-003-008). CPU time at Taiwan's National Center for High-performance Computing (NCHC) and Department of Applied Chemistry in Private Chinese Culture University (PCCU) was greatly appreciated. We thank G. J. Liao for his technical support.References
- R. M. Navarro, M. A. Peña and J. L. G. Fierro, Chem. Rev., 2007, 107, 3952–3991 CrossRef CAS PubMed.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed.
- L. V. Mattos, G. Jacobs, B. H. Davis and F. B. Noronha, Chem. Rev., 2012, 112, 4094–4123 CrossRef CAS PubMed.
- P. R. d. l. Piscina and N. Homs, Chem. Soc. Rev., 2008, 37, 2459–2467 RSC.
- M. Ni, D. Y. C. Leung and M. K. H. Leung, Int. J. Hydrogen Energy, 2007, 32, 3238–3247 CrossRef CAS.
- J. Kugai, S. Velu and C. Song, Catal. Lett., 2005, 101, 255–264 CrossRef CAS.
- A. M. Silva, L. O. O. Costa, A. P. M. G. Barandas, L. E. P. Borges, L. V. Mattos and F. B. Noronha, Catal. Today, 2008, 133–135, 755–761 CrossRef CAS.
- M. Li, W. Guo, R. Jiang, L. Zhao, X. Lu, H. Zhu, D. Fu and H. Shan, J. Phys. Chem. C, 2010, 114, 21493–21503 CrossRef CAS.
- J. H. Wang, C. S. Lee and M. C. Lin, J. Phys. Chem. C, 2009, 113, 6681–6688 CrossRef CAS.
- P. Y. Sheng, A. Yee, G. A. Bowmaker and H. Idriss, J. Catal., 2002, 208, 393–403 CrossRef CAS.
- B. Caglar, M. Olus Ozbek, J. W. Niemantsverdriet and C. J. Weststrate, Phys. Chem. Chem. Phys., 2016, 18(43), 30117–30127 RSC.
- B. Caglar, J. W. Niemantsverdriet and C. J. Weststrate, Phys. Chem. Chem. Phys., 2016, 18(34), 23888–23903 RSC.
- Y. Y. Hsia, Y. C. Huang, H. S. Zheng, Y. L. Lai, Y. J. Hsu, M. F. Luo and J. H. Wang, J. Phys. Chem. C, 2019, 123, 11649–11661 CrossRef CAS.
- V. Fierro, O. Akdim and C. Mirodatos, Green Chem., 2003, 5, 20–24 RSC.
- G. A. Deluga, J. R. Salge, L. D. Schmidt and X. E. Verykios, Science, 2004, 303, 993–997 CrossRef CAS PubMed.
- E. Varga, P. Pusztai, A. Oszkó, K. Baán, A. Erdőhelyi, Z. Kónya and J. Kiss, Langmuir, 2016, 32, 2761–2770 CrossRef CAS PubMed.
- M. Tóth, E. Varga, A. Oszkó, K. Baán, J. Kiss and A. Erdőhelyi, J. Mol. Catal. A: Chem., 2016, 411, 377–387 CrossRef.
- Z. Ferencz, A. Erdőhelyi, K. Baán, A. Oszkó, L. Óvári, Z. Kónya, C. Papp, H. P. Steinrück and J. Kiss, ACS Catal., 2014, 4, 1205–1218 CrossRef CAS.
- C. C. Hung, S. L. Chen, Y. K. Liao, C. H. Chen and J. H. Wang, Int. J. Hydrogen Energy, 2012, 37, 4955–4966 CrossRef CAS.
- P. Ferrin, D. Simonetti, S. Kandoi, E. Kunkes, J. A. Dumesic, J. K. Nørskov and M. Mavrikakis, J. Am. Chem. Soc., 2009, 131, 5809–5815 CrossRef CAS PubMed.
- Y. Choi and P. Liu, J. Am. Chem. Soc., 2009, 131, 13054–13061 CrossRef CAS PubMed.
- B. N. Zope, D. D. Hibbitts, M. Neurock and R. J. Davis, Science, 2010, 330, 74–78 CrossRef CAS PubMed.
- V. Stamenkovic, B. S. Mun, K. J. J. Mayrhofer, P. N. Ross, N. M. Markovic, J. Rossmeisl, J. Greeley and J. K. Norskov, Angew. Chem., Int. Ed., 2006, 45, 2897–2901 CrossRef CAS PubMed.
- I. H. Hong, T. H. Lee, G. C. Yin, D. H. Wei, J. M. Juang, T. E. Dann, R. Klauser, T. J. Chuang, C. T. Chen and K. L. Tsang, Nucl. Instrum. Methods Phys. Res., Sect. A, 2001, 467–468, 905–908 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS PubMed.
- D. M. Ceperley and B. J. Alder, Phys. Rev. Lett., 1980, 45, 566–569 CrossRef CAS.
- J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 6671–6687 CrossRef CAS PubMed.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- G. Mills, H. Jonsson and G. K. Schenter, Surf. Sci., 1995, 324, 305–337 CrossRef CAS.
- C. Michel, F. Auneau, F.-o. Delbecq and P. Sautet, ACS Catal., 2011, 1, 1430–1440 CrossRef CAS.
- D. Loffreda, C. Michel, F. Delbecq and P. Sautet, J. Catal., 2013, 308, 374–385 CrossRef CAS.
- A. A. Gokhale, J. A. Dumesic and M. Mavrikakis, J. Am. Chem. Soc., 2008, 130, 1402–1414 CrossRef CAS PubMed.
- X. Gonze, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 55, 10337 CrossRef CAS.
- G. Kresse, J. Furthmüller and J. Hafner, Europhys. Lett., 1995, 32, 729–734 CrossRef CAS.
- A. Shavorskiy, T. Eralp, E. Ataman, C. Isvoranu, J. Schnadt, J. N. Andersen and G. Held, J. Chem. Phys., 2009, 131, 214707 CrossRef CAS PubMed.
- E. Vesselli, A. Baraldi, G. Comelli, S. Lizzit and R. Rosei, ChemPhysChem, 2004, 5, 1133–1140 CrossRef CAS PubMed.
- E. Vesselli, G. Comelli, R. Rosei, S. Freni, F. Frusteri and S. Cavallaro, Appl. Catal., A, 2005, 281, 139–147 CrossRef CAS.
- C. Y. Syu and J. H. Wang, ChemCatChem, 2013, 5, 3164–3174 CrossRef CAS.
- The number of decomposing ethanol molecules are calculated with the carbon atoms of desorbing products and multiplying a factor to include the remaining carbon which is observed from the C 1s signals of photoelectron spectra.
- J. E. Sutton and D. G. Vlachos, Ind. Eng. Chem. Res., 2015, 54, 4213–4225 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra02015j |
| This journal is © The Royal Society of Chemistry 2020 |