
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of oxygen functionalized carbon nanotubes and their application for selective catalytic reduction of NOx with NH3†
Bora Yea,
Sun-I Kima,
Minwoo Leeb,
Mohammadamin Ezazic,
Hong-Dae Kim a,
Gibum Kwon*c and
Duck Hyun Lee*a
a,
Gibum Kwon*c and
Duck Hyun Lee*a
aGreen Materials & Processes R&D Group, Korea Institute of Industrial Technology, Ulsan 44413, Republic of Korea. E-mail: dulee@kitech.re.kr
bSmall & Medium Class Vessel Convergence Technology Team, Korea Marine Equipment Research Institute, Gunsan 54001, Republic of Korea
cDepartment of Mechanical Engineering, University of Kansas, Lawrence, Kansas 66045, USA. E-mail: gbkwon@ku.edu
First published on 28th April 2020
Abstract
Oxygen functionalized carbon nanotubes synthesized by surface acid treatment were used to improve the dispersion properties of active materials for catalysis. Carbon nanotubes have gained attention as a support for active materials due to their high specific surface areas (400–700 m2 g−1) and chemical stability. However, the lack of surface functionality causes poor dispersion of active materials on carbon nanotube supports. In this study, oxygen functional groups were prepared on the surface of carbon nanotubes as anchoring sites for decoration with catalytic nanoparticles. The oxygen functional groups were prepared through a chemical acid treatment using sulfuric acid and nitric acid, and the amount of functional groups was controlled by the reaction time. Vanadium, tungsten, and titanium oxides as catalytic materials were dispersed using an impregnation method on the synthesized carbon nanotube surfaces. Due to the high density of oxygen functional groups, the catalytic nanoparticles were well dispersed and reduced in size on the surface of the carbon nanotube supports. The selective catalytic reduction catalyst with the oxygen functionalized carbon nanotube support exhibited enhanced NOx removal efficiency of over 90% at 350–380 °C which is the general operating temperature range of catalysis in power plants.
1. Introduction
Various carbon materials (graphene, graphene oxide, reduced graphene oxide, carbon nanotubes (CNTs), and activated carbon) have been used as support materials in various fields such as catalysis,1 fuel cells,2 and water purification3 due to their high specific surface areas and chemical stability.4,5 When carbon materials are applied as supports, they prevent aggregation of the catalyst material and increase the catalytic active sites, which result in enhanced catalytic activities at the temperatures below 200 °C.1,4 Although graphene oxide can improve the dispersion of active materials on the surface because of the abundant oxygen functional groups, they also accelerate the thermal decomposition at the temperatures above 150 °C.6 Reduced graphene oxide is relatively stable at high temperatures compared with graphene oxide because the surface oxygen functional groups have been reduced. However, reduced graphene oxide is prone to oxidation under long exposures to high-temperature oxygen atmospheres.7 Therefore, it is necessary to design thermally stable carbon materials to utilize them as SCR catalyst supports that can function at high temperatures (>350 °C).CNTs have cylindrical structures of rolled-up sheets comprising a single layer of hybridized carbon atoms. Various studies have been conducted on CNTs because of their high specific surface areas, mechanical strength, and electrochemical characteristics.2,8 However, CNTs are primarily composed of carbon bonds with very small amounts of functional groups on the surface, which hinders the dispersion of catalysts due to insufficient anchoring sites. Therefore, doping of oxygen-, sulfur-, and nitrogen-containing groups has been used to functionalize CNT surfaces and improve their electrical and dispersion properties.8–10
Nitrogen oxides (NOx) are emitted from both stationary sources (power plants, cement, chemical plants, steel mills, etc.) and mobile sources (large vessels, automobiles) and exist in various forms such as NO, and NO2.11,12 NOx have been identified as the main source of fine particulate matter (PM2.5), and various studies have been conducted to reduce their emissions from the sources.13,14 Selective catalytic reduction (SCR) of NOx with NH3 is a representative NOx removal method that can reduce NOx emissions by 80–100%. In recent years, many researches have been conducted to improve the catalytic activity in an extended temperature range of SCR catalyst by adding the various catalysts components such as Fe, Ce, Mn and Cu.14 It was reported by Han et al. that the SCR catalyst having good sulfur-poisoning resistance was prepared by adding Fe2O3 to a support.15–17 The typical vanadium-based catalysts have high NOx removal efficiencies (>90%) at temperatures between 300 and 400 °C, even at small loads (<2 wt%), and MoO3 or WO3 can be added as a promoter to enhance the thermal, structural stability and the SCR performance of V-based catalysts.18–20 However, vanadium decompose NH3 at high temperatures,21 and it causes harmful effects to the human body.
Here, we synthesized oxygen functionalized CNTs (O-CNTs), and applied them in V–W/TiO2 SCR catalysts. An acid treatment process using sulfuric acid and nitric acid was conducted to functionalize the CNT surfaces with abundant oxygen functional groups, and the amount of functional groups was adjusted by controlling the acid treatment time. The vanadium, tungsten, and titanium oxide catalyst materials were well dispersed on the synthesized O-CNTs because the surface functional groups which acted as anchoring sites of catalyst materials, and they inhibited the aggregation of active materials and reduced the size of catalysts nanoparticles. As a result, the synthesized catalysts have enhanced catalytic acid sites and high specific surface area, and the NOx removal efficiency can be enhanced even with the less amounts of active materials.22
2. Experimental
2.1. Preparation of oxygen functionalized CNTs
Oxygen functionalized CNTs were synthesized by chemical oxidation using sulfuric acid and nitric acid as oxidants. Commercial CNTs (0.3 g, JEIO, diameter 7–9 nm, length 10–50 μm, aligned bundle type) were added to a 4 M sulfuric acid (H2SO4, Daejung Chemicals, 95%) and 4 M nitric acid (HNO3, Daejung Chemicals, 70%) solution, followed by vigorous stirring for 0–6 h to control the amount of oxygen functional groups. Next, the mixtures were washed and filtered several times with deionized (DI) water using a vacuum filtration system until the solution pH reached 7–8. Finally, the O-CNTs were obtained in the form of a precipitate.2.2. Synthesis of O-CNT-supported SCR catalyst
The O-CNT-supported SCR catalyst was prepared by the impregnation method. The O-CNTs and TiO2 particles were dispersed in respective DI water. Next, 1 wt% ammonium metavanadate (AMV) (NH4VO3, Sigma-Aldrich, 99.99%) and oxalic acid (HO2CCO2H, Sigma-Aldrich, 99.0%) (molar ratio = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) were dissolved in DI water, and 6 wt% ammonium metatungstate (AMT) ((NH4)6H2W12O40·xH2O, Sigma-Aldrich, 99.99%) was dissolved in DI water. The prepared TiO2, AMV, and AMT solutions were slowly added to the first prepared CNT dispersion solution to synthesize the O-CNT-supported SCR catalyst (VW/O-CNT/Ti), and the mixture was stirred for 7 h at 60 °C. The mixtures were then dried by rotary evaporation at 10.0 mbar and 40 °C, and the dried powder was heat-treated at 500 °C for 2 h under a nitrogen atmosphere to synthesize the O-CNT-supported SCR catalyst (VW/O-CNT/Ti). For comparison, SCR catalysts without CNTs and with pristine, non-functionalized CNTs were prepared under the same conditions and named as VW/Ti and VW/CNT/Ti, respectively.
:3) were dissolved in DI water, and 6 wt% ammonium metatungstate (AMT) ((NH4)6H2W12O40·xH2O, Sigma-Aldrich, 99.99%) was dissolved in DI water. The prepared TiO2, AMV, and AMT solutions were slowly added to the first prepared CNT dispersion solution to synthesize the O-CNT-supported SCR catalyst (VW/O-CNT/Ti), and the mixture was stirred for 7 h at 60 °C. The mixtures were then dried by rotary evaporation at 10.0 mbar and 40 °C, and the dried powder was heat-treated at 500 °C for 2 h under a nitrogen atmosphere to synthesize the O-CNT-supported SCR catalyst (VW/O-CNT/Ti). For comparison, SCR catalysts without CNTs and with pristine, non-functionalized CNTs were prepared under the same conditions and named as VW/Ti and VW/CNT/Ti, respectively.
2.3. Catalyst characterization
X-ray photoelectron spectroscopy (XPS) (Thermo Fisher, K-Alpha) was carried out with an Al Kα source to observe the contents of carbon and oxygen bonds in the O-CNTs according to acid treatment time. Fourier-transform infrared spectroscopy (FT-IR) (Varian, 670 FTIR) was performed in the wavelength range from 500 to 4000 cm−1, and Raman spectroscopy (WITec, alpha300s) was performed at an excitation wavelength of 532 nm to determine the intensity changes for the D and G bands from the CNT surfaces depending on the acid treatment time. The morphologies of the dispersed active materials on the CNT supports and synthesized catalysts were obtained by transmission electron microscopy (TEM) (JEOL, JEM-2100F) and field emission scanning electron microscopy (FE-SEM) (Hitachi, SU8020) at an accelerating voltage of 15 kV. The specific surface areas and pore properties were calculated by the Brunauer–Emmett–Teller (BET) equation and Barrett–Joyner–Halenda (BJH) method using N2 physisorption (Micrometrics, ASAP 2020). Before the analysis, all of the catalysts were degassed at 150 °C for 4 h to remove water and gas adsorbed on the catalyst surface. The structural phases and crystallite sizes were confirmed by X-ray diffraction (XRD, ULTIMA 4) using a Cu Kα radiation source (λ = 0.15406 nm) at 2θ ranging from 10° to 90°.2.4. Catalytic activity test
The NOx removal efficiencies of the synthesized SCR catalysts were measured in a fixed-bed reactor, where the sample was placed on a stainless-steel tube with an inner diameter of 9.5 mm and analyzed using a powder catalyst. The analysis temperature varied from 250 to 400 °C, and the analytical conditions were as follows: 500 ppm NOx, 5% O2, and 500 ppm NH3 (NH3/NOx = 1.0) with N2 balance under a total flow of 500 sccm. The flow rate for each gas was controlled by a mass flow controller. The feed gases were mixed and preheated to 420 °C at the front of the reactor, and the lines were heated to 200 °C to prevent the precipitation of ammonium sulfate or ammonium nitrate salts from the high-reactivity gas. The sample conditions were set at grams, and the weight hourly space velocity (WHSV) was set at 300000 ml (g−1 h−1). After analyzing the NOx concentration at the reactor inlet and outlet through a chemi-luminescent detection analyzer (CLD), the de-NOx efficiency was calculated according to the following equation.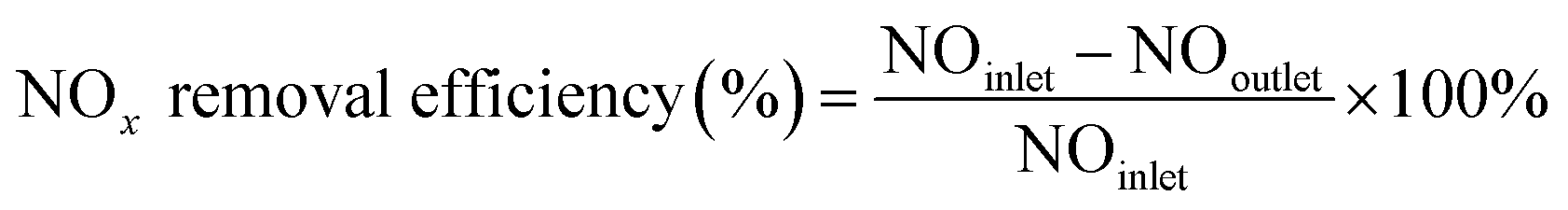
3. Results and discussion
3.1. Effect of acid treatment time on CNTs
Fig. 1 illustrates the synthesis of the O-CNTs by acid treatment and V–W–Ti doping of the O-CNTs as an SCR catalyst using the impregnation method. As mentioned in the experimental section, oxygen groups were formed on the surface of commercial CNTs using a sulfuric acid and nitric acid solution. Among the various synthesis methods for O-CNTs, chemical functionalization is advantageous in that it is simple, inexpensive, and easy to adjust in terms of the amount of functional groups. It is important to form an appropriate amount of oxygen groups on the surface of the CNT supports to improve the catalytic properties. A suitable amount of generated functional groups can aid in dispersing the metal oxide nanoparticles onto the surface and prevent the particles from aggregating. However, excess amounts of oxygen functional groups on the surface could easily decompose at high temperatures, which would decrease the thermal stability of the CNTs and cause the structure to collapse during reactions. Therefore, the synthesis conditions were controlled by changing the acid treatment time from 1 to 6 h to determine the condition for optimum oxygen functionalization and thus the best NOx removal efficiency.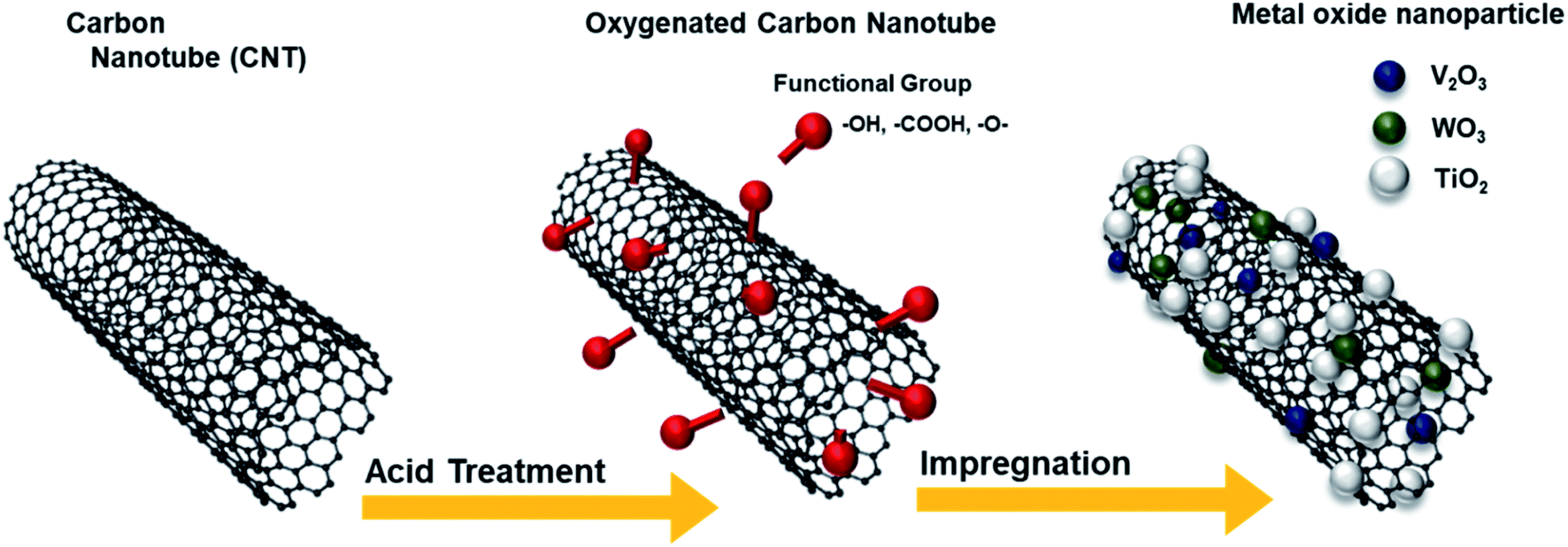 | ||
| Fig. 1 Schematic diagram of the synthesis of O-CNTs and V–W–Ti-doped CNT SCR catalysts. | ||
The SEM images in Fig. 2a and b show the morphology of the commercial pristine CNTs used in this study at low magnification. The CNTs had an aligned bundle-type appearance with diameters of 8 to 10 nm, lengths of 35 to 45 μm, and bundle sizes of approximately 1.5 to 4 μm. The surface properties of the initial CNTs including specific surface area and pore properties were analyzed using the BET method and BHJ equation, and the results are shown in Fig. 2c and d. The N2 adsorption–desorption isotherms represent typical type IV adsorption isotherms of mesopores according to the six representative adsorption isotherms defined by the International Union of Pure and Applied Chemistry (IUPAC). The hysteresis loops in the isotherms exhibit type H3 behavior at a relative pressure range of 0.7–1.0, and the pore distribution determined by the BJH formula indicates mainly micro-pores (<2 nm) and meso-pores ranging in size from more than 2 nm to less than 50 nm.4 The pristine CNTs had a high specific surface area of 441 m2 g−1 that, along with the mesoporous properties, makes them an advantageous support for active materials to increase the catalytic reaction sites.23 However, on the SCR catalyst synthesized by impregnating vanadium oxide and tungsten oxide onto the commercial pristine CNTs without functional groups, the active materials formed large (tens of nanometers) secondary particles as shown in Fig. S1.† Therefore, there was a limit to how evenly the particles could be distributed onto the thin nanotubes. In order to yield smaller catalyst particles, it is necessary to use a carbon material with a sufficient amount of functional groups attached to the surface.
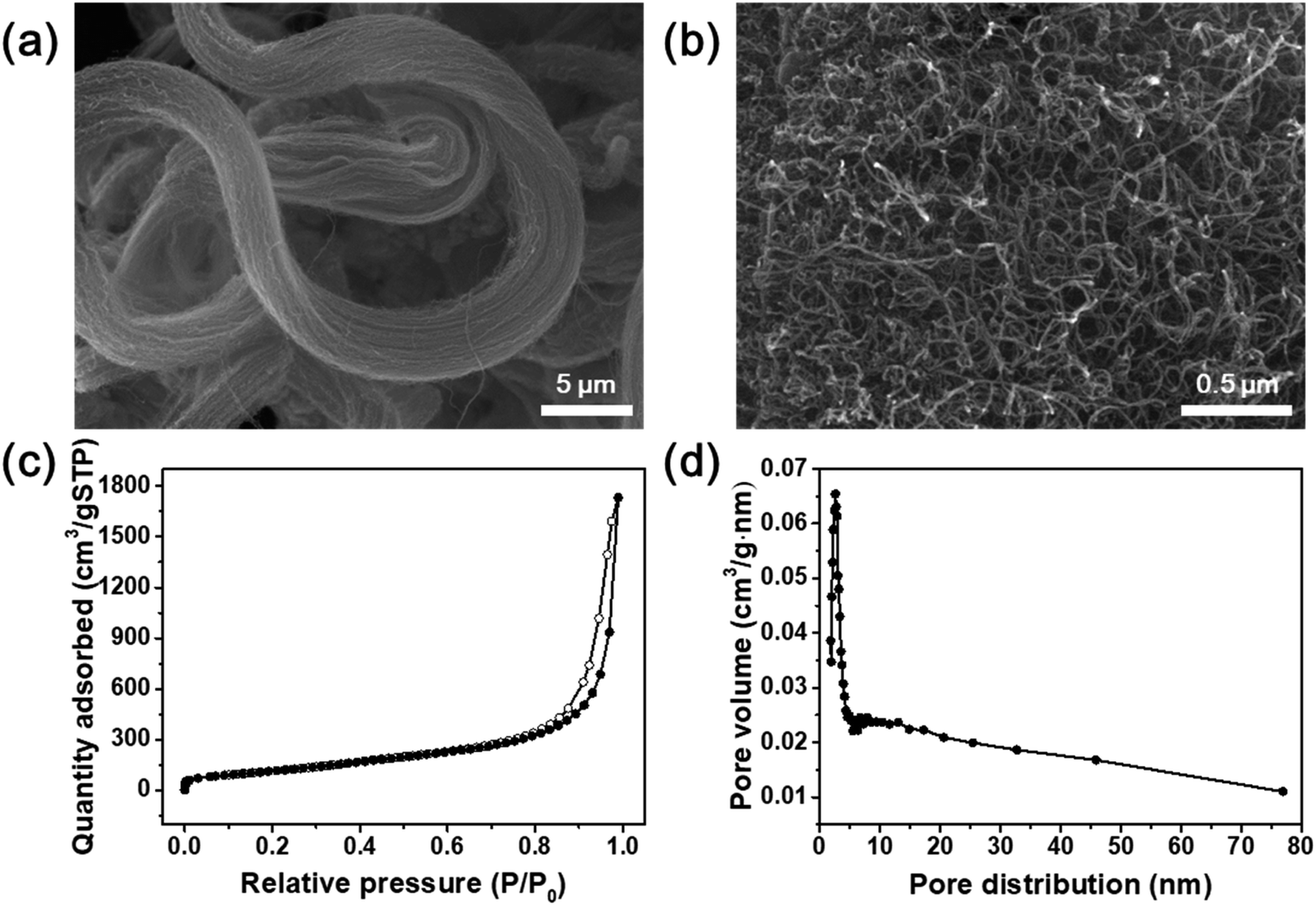 | ||
| Fig. 2 (a and b) SEM images of commercial CNTs. (c) Nitrogen adsorption–desorption isotherms and (d) pore distribution of commercial CNTs. | ||
XPS analysis was conducted to confirm the oxygen concentration and binding structures on the CNT surfaces depending on acid treatment time. Specific oxygen and carbon contents and ratios of each bond type are shown in detail in Table 1. Fig. S2† shows that the pristine CNTs without any acid treatment showed no oxygen peak on the surface in the XPS survey results. On the other hand, the oxygen content gradually increased with acid treatment time, and the highest intensity was observed after acid treatment for 3 h (O/C = 67.81). According to the many literatures related to the chemical acid treatment of pristine CNT using the mixture of HNO3 and H2SO4, it serves to remove the amorphous carbon and impurities present on the surface of the CNT. And in particular, HNO3 plays a role in forming oxygen function groups such as carboxyl groups by expanding the end cap of the pristine CNT surface and forming defects in the wall.24–26 Therefore, the O 1s peak is increased through the chemical treatment with the mixture of HNO3 and H2SO4, and in can be seen that the amount of oxygen functional groups formed on the CNT surface increased with the acid treatment time. Fig. 3a and b shows the C 1s and O 1s peaks and the proportion of each bond by peak deconvolution. The separated C 1s features contain C–C and C–O peaks at 284.3 and 285.6 eV, respectively, which were the same for all CNT samples.27 As acid treatment time increased, the intensity of the C–C bond peak gradually increased compared with the C–O bond peak. This can be inferred as the formation of surface carbon-to-carbon bonds as the CNT structure collapses due to the harsh strong acid synthesis conditions.28 In Fig. 3b, the formation of oxygen functional groups on the CNT surfaces was more clearly confirmed by the changes in integrated peak areas. The features corresponding to oxygen, C–O, C–OH, and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O appeared at 533.2, 531.6, and 530.1 eV, respectively, but were not present in the pristine CNT spectrum. Based on these results, the acid treatment produced chemically functional oxygen groups on the surface of the CNTs.29 The amount of functional groups gradually increased with acid treatment time, then decreased after 3 h, which agrees with the results shown in Fig. S2.† Particularly, the sample treated in acid for 3 h had the highest ratio of C–OH bonds, followed by C–O and CO bonds.30 These oxygen functional groups act as chemically attachable sites to help disperse the catalyst particles without aggregation and prevent the growth of catalytic particle size.2,31 As the acid treatment time increased beyond 3 h, the oxygen content decreased, and the overall XPS peak intensity decreased. These results were also clearly supported by the results of the FT-IR analysis. As shown in Fig. S2b,† the CO, CC, and C–O bond peaks (carbonyl and carboxyl groups) appeared respectively at 1565, 1384, and 1158 cm−1, and the oxygen-related peaks gradually increased in intensity with acid treatment time because the oxygen functional groups were formed by the acid treatment process.25 Additionally, the peak appearing at 3440 cm−1 is the O–H stretch, which is closely related to water molecules adsorbed on the surface of the CNTs.25,30 Particularly, the CC bond peak had the highest intensity after acid treatment for 6 h due to the coupling of carbons on the CNT surface caused by structural collapse as described above.28
O appeared at 533.2, 531.6, and 530.1 eV, respectively, but were not present in the pristine CNT spectrum. Based on these results, the acid treatment produced chemically functional oxygen groups on the surface of the CNTs.29 The amount of functional groups gradually increased with acid treatment time, then decreased after 3 h, which agrees with the results shown in Fig. S2.† Particularly, the sample treated in acid for 3 h had the highest ratio of C–OH bonds, followed by C–O and CO bonds.30 These oxygen functional groups act as chemically attachable sites to help disperse the catalyst particles without aggregation and prevent the growth of catalytic particle size.2,31 As the acid treatment time increased beyond 3 h, the oxygen content decreased, and the overall XPS peak intensity decreased. These results were also clearly supported by the results of the FT-IR analysis. As shown in Fig. S2b,† the CO, CC, and C–O bond peaks (carbonyl and carboxyl groups) appeared respectively at 1565, 1384, and 1158 cm−1, and the oxygen-related peaks gradually increased in intensity with acid treatment time because the oxygen functional groups were formed by the acid treatment process.25 Additionally, the peak appearing at 3440 cm−1 is the O–H stretch, which is closely related to water molecules adsorbed on the surface of the CNTs.25,30 Particularly, the CC bond peak had the highest intensity after acid treatment for 6 h due to the coupling of carbons on the CNT surface caused by structural collapse as described above.28
| Acid treatment time | C (%) | O (%) | C 1s scan (%) | O 1s scan (%) | |||
|---|---|---|---|---|---|---|---|
| C–C | C–O–C | C–OH/C–O–C | C–O | CO |
|||
| 0 h | 98.0 | 2.0 | 54.0 | 46.0 | 93.4 | 0.2 | 6.5 |
| 1 h | 93.6 | 6.1 | 54.8 | 45.2 | 76.7 | 21.5 | 1.7 |
| 2 h | 93.7 | 6.3 | 60.2 | 39.8 | 72.8 | 22.1 | 5.0 |
| 3 h | 87.6 | 13.4 | 57.7 | 42.3 | 83.1 | 5.9 | 10.9 |
| 4 h | 90.2 | 9.8 | 61.8 | 38.2 | 73.0 | 6.1 | 20.9 |
| 5 h | 92.5 | 7.6 | 59.0 | 41.0 | 68.2 | 8.5 | 23.3 |
| 6 h | 91.7 | 8.3 | 59.0 | 41.0 | 62.1 | 5.3 | 32.6 |
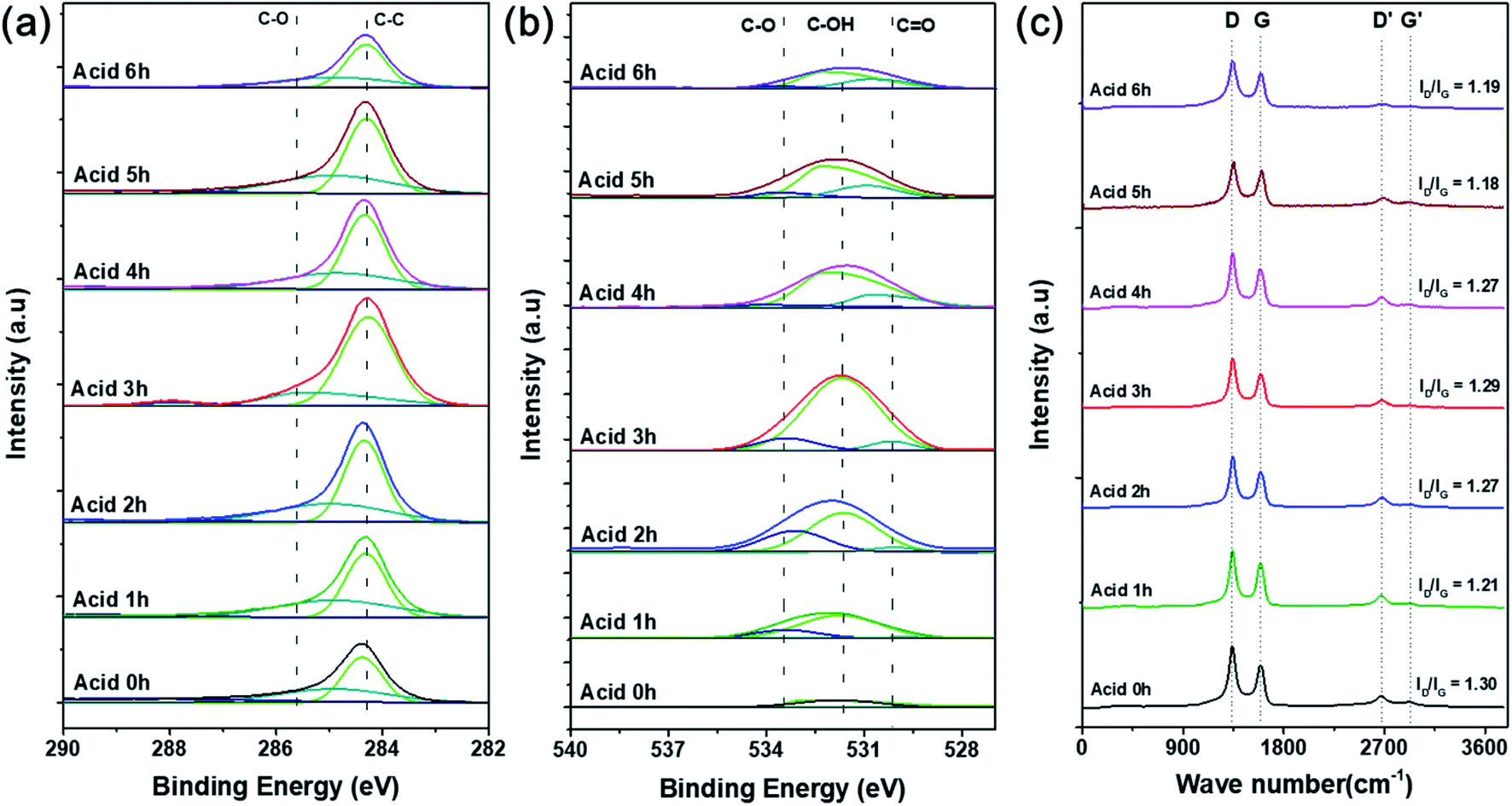 | ||
| Fig. 3 Deconvoluted (a) C 1s and (b) O 1s XPS curves and (c) Raman spectra of O-CNTs prepared with various acid treatment times. | ||
To confirm the surface architecture of the CNTs, Raman spectroscopy was performed with an excitation wavelength of 532 nm. Generally, carbonaceous materials have two main peaks: the G band at 1580 cm−1 and D band at 1350 cm−1. The G band corresponds to well-ordered graphite crystallites comprising sp2 carbon bonds that appear in all graphites, and the D band corresponds to surface structural defects or amorphous carbon in a disordered CNT structure.32 Furthermore, the crystallinity or disorder of the carbon material can be confirmed by the intensity ratio of the D and G bands (ID/IG). However, Fig. 3c indicates that the ID/IG ratios of the pristine and acid-treated CNTs were 1.30 and 1.18–1.29, respectively. During the acid treatment, oxygen functional groups are attached to the surface of the CNTs and impart defects onto the sidewalls, and the treatment simultaneously removes any amorphous carbon present on the commercial CNT surfaces.26 Therefore, the acid treated CNTs showed lower ratio of ID/IG because the surface amorphous carbon or impurities were removed.33,34 As the acid treatment time increases from 1 to 3 h, the intensity of the D band slightly increased because the chemical modification increased the oxygen functional groups, and the disordered structure were formed.33,35 However, when the acid treatment time increased to more than 3 h, the ratio of ID/IG was slightly decreased again because the oxygen functional groups were removed, which agreed well with the results of the XPS (Fig. 3).
Table 2 summarizes the specific surface areas and pore characteristics of the pristine CNTs and O-CNTs prepared with various acid treatment times. The specific surface area of CNTs depends on the number of walls, inner diameter, length, degree of surface functionalization, and presence of impurities such as metals and amorphous carbon.26 Compared with the pristine CNTs, the specific surface area of the O-CNTs increased at first with acid treatment time (Table 2). Because the acid treatment opens the end of the nanotubes, forming sidewall defects and removes amorphous carbon present on the surface of the commercially available CNTs, the specific surface area increased by 20–40 m2 g−1.36 However, the sample treated for 3 h had a low specific surface area because the acid-treated functional groups blocked the pores of the CNTs, thereby reducing the pore size and volume.37 In addition, the O-CNTs treated for 3 h led the aggregation of CNTs due to the presence of abundant oxygen functional groups, and it caused the reduction of specific surface area. Therefore, the pore volume of the pristine CNTs decreased from 2.61 to 1.05 cm3 g−1 with the increased acid treatment time, and the pore size decreased from 24.27 to 10.38 nm. However, the specific surface area increased again with the acid treatment time more than 4 h because of the formation of defects and removal of the agglomerated structure which caused by the surface etching through acid treatment.35 Functionalized CNTs can also cause de-bundling and increase the specific surface area as they disturb the π–π interactions on the surface.36
| Acid treatment time | 0 h | 1 h | 2 h | 3 h | 4 h | 5 h | 6 h |
| SBET (m2 g−1) | 441.57 | 482.50 | 467.72 | 361.55 | 500.98 | 470.84 | 423.59 |
| Pore volume (cm3 g−1) | 2.61 | 1.96 | 1.91 | 1.05 | 1.93 | 1.82 | 1.70 |
| Pore size (nm) | 24.27 | 17.05 | 15.10 | 10.38 | 14.60 | 14.67 | 14.84 |
Based on the changes in surface binding structure, oxygen content, and specific surface area with acid treatment time, the O-CNTs treated for 3 h have the highest oxygen content and a large amount of surface functional groups to attach more catalytically active materials as a support.
3.2. Characteristics of O-CNT-supported SCR catalyst
The particle size and morphology of the SCR catalysts with and without O-CNT support were confirmed by TEM analysis as shown in Fig. 4. Fig. 4a and b show the synthesized VW/Ti catalyst without O-CNT support. The catalyst particles were agglomerated and formed 30–50 nm secondary particles. On the other hand, with the O-CNTs as a support for the SCR catalyst, the nano-sized active materials (V, W) were evenly dispersed on the surface of the O-CNTs with tube diameters of approximately 10 nm (Fig. 4c and d). As shown in the Fig. S1a and b,† when the oxygen functional groups were not formed through additional acid treatment of the CNTs, it was found that the V and W particles were hardly dispersed and CNT have clean surface without any impregnation. Similar trends were observed in the VW/O-CNT/Ti images (Fig. 4e and f) indicating that the aggregation of nano-particles was significantly inhibited when O-CNT support was used as compared to Fig. S1c and d.† This is because the O-CNTs have oxygen functional groups on the surface after the acid treatment process for 3 h, which serve as anchoring sites for particle attachment.38,39 Commercial TiO2 powders for SCR catalysts generally have a primary particle size of several tens of nanometers, and the particles form secondary particles upon aggregating with each other. However, after impregnation of the catalyst components onto the O-CNTs, the vanadium, tungsten, and titanium oxide particles were dispersed across the tube surface without particle aggregation and with a high binding energy.40 Compared with the VW/Ti catalyst, which agglomerated into secondary particles 50 nm in size, the TiO2 particles were less than 20 nm in size when prepared on the O-CNTs. | ||
| Fig. 4 TEM images of synthesized (a and b) VW/Ti catalyst, (c and d) V–W impregnated O-CNT nanocomposite, and (e and f) VW/O-CNT/Ti catalyst. | ||
XRD analysis was performed to confirm the structural changes of the catalysts with the addition of the O-CNT support. Fig. 5a shows the XRD patterns of the two synthesized SCR catalysts, which indicated a distinct anatase TiO2 phase (JCPDS PDF 21) with characteristic peaks at 2θ = 25.3° (101), 37.8° (112), 48.0° (200), 53.9° (105), and 55.0° (211).41 Aside from the TiO2 peaks, no V2O5 or WO3 peaks were identified because the amount of active materials (less than 10 wt%) was small and the dispersed oxides were mainly amorphous in phase.20,42 Only slight intensity and FWHM changes were found between the XRD patterns of the SCR catalysts with and without O-CNT support. The particle diameter of the TiO2 crystallites were estimated using the Scherrer equation (Dβ = Kλ/βcosθ).39 The average TiO2 crystallite sizes in VW/Ti and VW/O-CNT/Ti were calculated as 18.8 and 16.2 nm, respectively.43
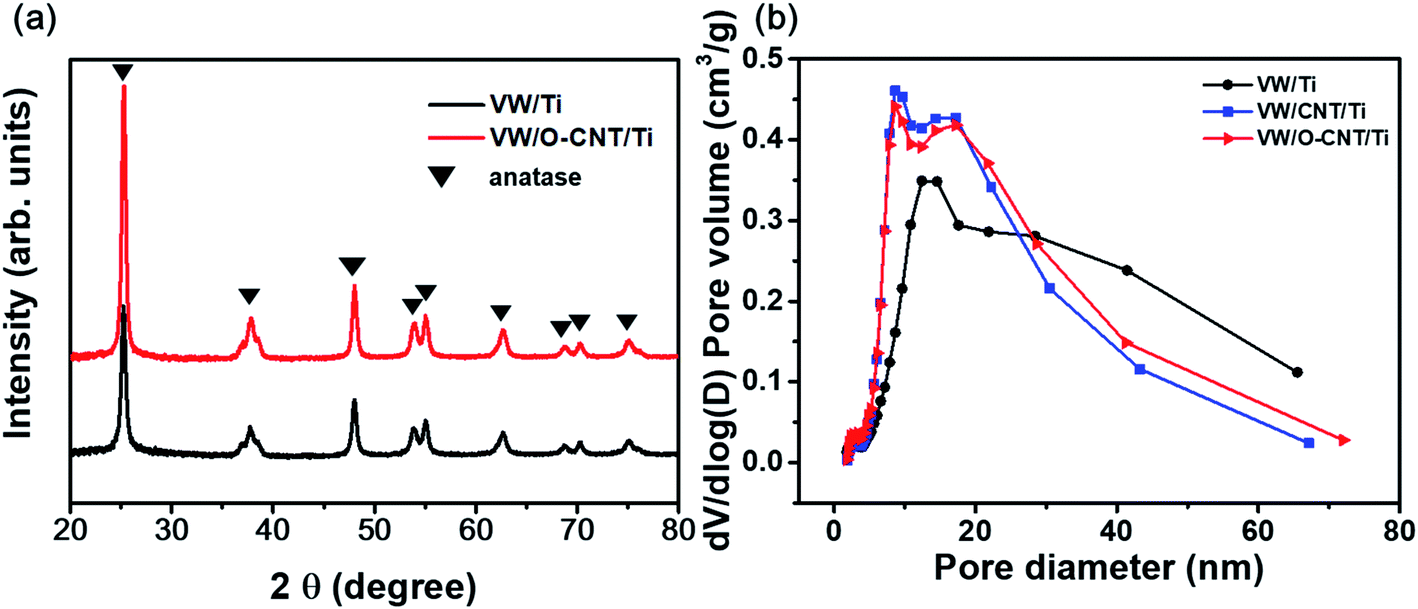 | ||
| Fig. 5 (a) XRD patterns and (b) pore size distributions of the synthesized SCR catalysts. | ||
Fig. 5b shows the pore size distributions of the SCR catalysts with and without O-CNTs calculated from the N2 desorption isotherms using the BJH equation. The three synthesized catalysts (VW/Ti, VW/CNT/Ti, and VW/O-CNT/Ti samples) had the same mesoporous structures with pore sizes of 2 to 50 nm, although smaller, finer pores were mainly formed in the O-CNT-supported catalyst. If micro-pores or meso-pores develop inside the solid catalyst, they could lead to high specific surface areas. As listed in Table 3, the VW/Ti catalyst exhibited a specific surface area of 81.90 m2 g−1, whereas that of the O-CNT-supported catalyst was 105.86 m2 g−1, which represents a 20% increase.
| Sample | SBET (m2 g−1) | Pore volume (cm3 g−1) | Average pore size (nm) | Nanoparticle size (nm) |
|---|---|---|---|---|
| VW/Ti | 81.90 | 0.34 | 15.02 | 73.26 |
| VW/CNT/Ti | 98.16 | 0.34 | 12.50 | 61.13 |
| VW/O-CNT/Ti | 105.86 | 0.34 | 12.19 | 56.68 |
In the case of VW/CNT/Ti samples without any surface treatment of CNTs, the specific surface area was increased as 17 m2 g−1 compared to the VW/Ti catalyst. This is because CNTs with high specific surface area properties were added to the catalyst, as shown in the Table 2, rather than due to the enhancement of dispersion and acidity characteristics. Because the catalysts are involved in surface reactions, the nano-sized active materials must be well dispersed on the catalyst surface to yield effective catalytic properties.1,4 Therefore, the O-CNT-supported catalyst is expected to exhibit high NOx removal efficiencies because of its high specific surface area and well-dispersed catalyst particles on the surface.
3.3. Catalytic performance
The catalytic performance of the synthesized SCR catalysts for NOx reduction in the presence of NH3 as a reducing agent was evaluated between 250 and 400 °C, and the results are shown in Fig. 6. The NOx removal efficiencies of the VW/Ti, pristine CNT-supported VW/Ti (VW/CNT/Ti), and O-CNT-supported VW/Ti (VW/O-CNT/Ti) catalysts were measured in a fixed-bed reactor. All the catalysts showed NOx removal efficiencies below 85% at low temperatures (<300 °C). However, at 350 °C, the O-CNT-supported catalyst afforded the highest efficiency (∼90%) compared with those of VW/Ti (88%) and VW/CNT/Ti (76%). Although only 1 wt% vanadium oxide was added to the O-CNT-supported catalyst as the main active material, the resulting catalyst had a high NOx removal efficiency close to 90% at temperatures between 350 and 400 °C. In contrast, the pristine CNT-supported catalyst had a lower efficiency than the other two catalysts across the entire temperature range. This is because when a large amount of active materials was impregnated onto the CNT surface without functional groups, the CNTs could not effectively function as a support to increase the catalytic active sites (Fig. S1†). Therefore, when commercial CNTs is added to disperse the active materials, it can be seen that even if the specific surface area characteristics are high, but the NOx removal efficiency is low because the characteristics of dispersing catalytically active materials are poor. When a small amount of vanadium oxide was added to the catalyst (less than 1 wt%), a superior catalytic activity occurred only for the high-temperature region.44 However, Fig. 6 indicates that the O-CNT-supported catalyst yielded a higher NOx removal efficiency compared with the other catalysts (>85% at 300 °C). In addition, at temperatures above 400 °C, the nano-dispersion of WO3 used as a promoter maintained the thermal and structural stability and activity required for the SCR catalyst (Fig. S3†). This suggests that when CNTs are used as a support, in addition to the high specific surface area affecting the dispersion of catalytic particles, the catalyst efficiency is also increased by the presence of sufficient surface functional groups that effectively disperse the nanoparticles without aggregation.45,46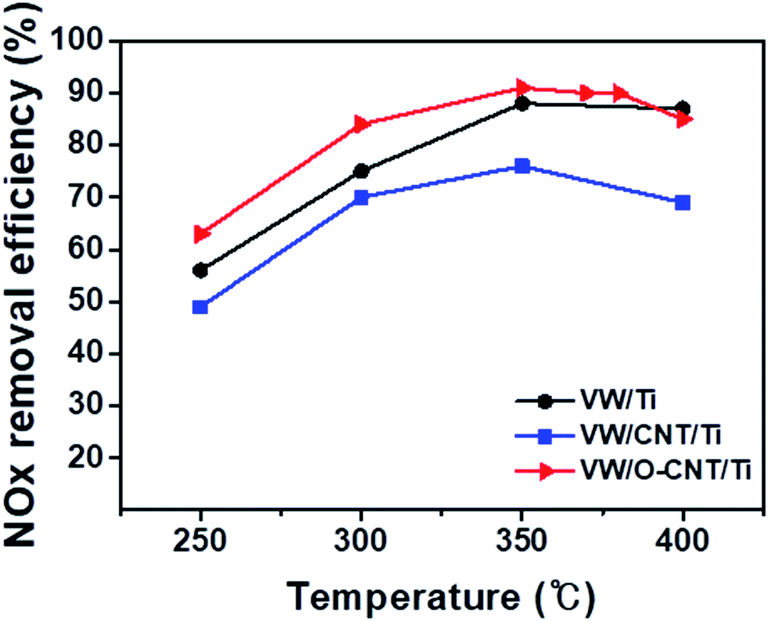 | ||
| Fig. 6 NOx removal efficiencies* of synthesized VW/Ti, VW/CNT/Ti, and VW/O-CNT/Ti catalysts as a function of temperature from 250 to 400 °C. *Catalytic reaction conditions: [NOx] = 500 ppm, [NH3] = 500 ppm, [O2] = 5%, balance gas N2, total flow 500 sccm, and GHSV = 300000 ml gcat−1 h−1. | ||
4. Conclusions
In summary, we have presented an SCR catalyst supported by CNTs with added surface oxygen functional groups. A chemical acid treatment was performed using an aqueous nitric acid and sulfuric acid solution to attach oxygen functional groups to the surface of the CNTs, and the SCR catalyst was synthesized by impregnating vanadium, tungsten, and titanium oxides onto the functional groups of the synthesized O-CNTs. The inhibition of agglomeration and the nano-dispersion of active materials on the O-CNT supports were confirmed by TEM analysis, and the specific surface area and pore characteristics of the catalysts were also improved by the addition of the O-CNT support. Based on the results, the catalysts with O-CNTs exhibited higher NOx removal efficiencies (90% between 350 and 400 °C) than the synthesized VW/Ti and pristine CNT-supported catalyst over the entire temperature range of 250–400 °C. The results reported here indicate that applying CNTs with surface oxygen functional groups as supports can improve the catalytic activity by suppressing the aggregation and enhancing the dispersion of catalytically active particles. Therefore, this method could be applied to various catalyst fields in addition to SCR.Conflicts of interest
There are no conflicts to declare.Acknowledgements
DHL acknowledge the financial support from the Korea Institute of Industrial Technology (KITECH), [grant number EE190053, JE200010] and the Ministry of Trade, Industry and Energy (MOTIE), [grant number 20005721]. GBK acknowledge the financial support from the US Department of Interior-Bureau Reclamation [grant number R17AC00132] and the University of Kansas [grant number 2234508-099-MESUPPRV].References
- B. Ye, M. Lee, B. Jeong, J. Kim, D. H. Lee, J. M. Baik and H. D. Kim, Catal. Today, 2019, 328, 300–306 CrossRef CAS
.
- B. C. Cha, S. Jun, B. Jeong, M. Ezazi, G. Kwon, D. Kim and D. H. Lee, J. Power Sources, 2018, 401, 296–302 CrossRef CAS
- S. Y. Lee, D. H. Kim, S. C. Choi, D. J. Lee, J. Y. Choi and H. D. Kim, Microporous Mesoporous Mater., 2014, 194, 46–51 CrossRef CAS
- B. Ye, B. Jeong, M. Lee, H. D. Kim and J. M. Baik, Adv. Mater. Technol., 2019, 4, 1–7 Search PubMed
- A. Gonçalves, J. Silvestre-Albero, E. V. Ramos-Fernández, J. C. Serrano-Ruiz, J. J. M. Órfão, A. Sepúlveda-Escribano and M. F. R. Pereira, Appl. Catal., B, 2012, 113–114, 308–317 CrossRef
- X. Huang, G. Zhao and X. Wang, RSC Adv., 2015, 5, 49973–49978 RSC
- M. E. Assal, M. R. Shaik, M. Kuniyil, M. Khan, A. Al-Warthan, M. R. H. Siddiqui, S. M. A. Khan, W. Tremel, M. N. Tahir and S. F. Adil, RSC Adv., 2017, 7, 55336–55349 RSC
- S. K. Park, Q. Mahmood and H. S. Park, Nanoscale, 2013, 5, 12304–12309 RSC
- W. J. Lee, D. H. Lee, T. H. Han, S. H. Lee, H. S. Moon, J. A. Lee and S. O. Kim, Chem. Commun., 2011, 47, 535–537 RSC
- W. J. Lee, J. M. Lee, S. T. Kochuveedu, T. H. Han, H. Y. Jeong, M. Park, J. M. Yun, J. Kwon, K. No, D. H. Kim and S. O. Kim, ACS Nano, 2012, 6, 935–943 CrossRef CAS PubMed
- M. D. Amiridis and J. P. Solar, Ind. Eng. Chem. Res., 1996, 35, 978–981 CrossRef CAS
- G. Qi, R. T. Yang and R. Chang, Appl. Catal., B, 2004, 51, 93–106 CrossRef CAS
- K. Baker and P. Scheff, Atmos. Environ., 2007, 41, 6185–6195 CrossRef CAS
- L. Han, S. Cai, M. Gao, J. Y. Hasegawa, P. Wang, J. Zhang, L. Shi and D. Zhang, Chem. Rev., 2019, 119, 10916–10976 CrossRef CAS PubMed
- L. Han, M. Gao, C. Feng, L. Shi and D. Zhang, Environ. Sci. Technol., 2019, 53, 5946–5956 CrossRef CAS PubMed
- L. Kang, L. Han, J. He, H. Li, T. Yan, G. Chen, J. Zhang, L. Shi and D. Zhang, Environ. Sci. Technol., 2019, 53, 938–945 CrossRef CAS PubMed
- L. Han, M. Gao, J. Y. Hasegawa, S. Li, Y. Shen, H. Li, L. Shi and D. Zhang, Environ. Sci. Technol., 2019, 53, 6462–6473 CrossRef CAS PubMed
- X. Wang, X. Li, Q. Zhao, W. Sun, M. Tade and S. Liu, Chem. Eng. J., 2016, 288, 216–222 CrossRef CAS
- C. J. G. van der
Grift, A. F. Woldhuis and O. L. Maaskant, Catal. Today, 1996, 27, 23–27 CrossRef
- H. K. Matralis, C. Papadopoulou, C. Kordulis, A. Aguilar Elguezabal and V. Cortes Corberan, Appl. Catal., A, 1995, 126, 365–380 CrossRef CAS
- J. Z. Luyao Zong, F. Dong, G. Zhang, W. Han and Z. Tang, Catal. Surv. Asia, 2017, 103–113 CrossRef
- Y. Shao, G. Yin, Y. Gao and P. Shi, J. Electrochem. Soc., 2006, 153, 1093–1097 CrossRef
- M. Casanova, K. Schermanz, J. Llorca and A. Trovarelli, Catal. Today, 2012, 184, 227–236 CrossRef CAS
- F. Avilés, J. V. Cauich-Rodríguez, L. Moo-Tah, A. May-Pat and R. Vargas-Coronado, Carbon, 2009, 47, 2970–2975 CrossRef
- S. Sahebian, S. M. Zebarjad, J. vahdati Khaki and A. Lazzeri, J. Nanostruct. Chem., 2015, 5, 287–293 CrossRef CAS
- K. A. Wepasnick, B. A. Smith, K. E. Schrote, H. K. Wilson, S. R. Diegelmann and D. H. Fairbrother, Carbon, 2011, 49, 24–36 CrossRef CAS
- X. C. Li, F. S. She, D. Shen, C. P. Liu, L. H. Chen, Y. Li, Z. Deng, Z. H. Chen and H. E. Wang, RSC Adv., 2018, 8, 28625–28631 RSC
- P. Van Trinh, N. N. Anh, N. T. Tam, N. T. Hong, P. N. Hong, P. N. Minh and B. H. Thang, RSC Adv., 2017, 7, 49937–49946 RSC
- J. Yue, K. Zhang, H. Yu, L. Yu, T. Hou, X. Chen, H. Ge, T. Hayat, A. Alsaedi and S. Wang, J. Mater. Sci., 2019, 54, 6140–6150 CrossRef CAS
- M. Wongaree, S. Chiarakorn and S. Chuangchote, J. Nanomater., 2015, 2015, 1–10 CrossRef
- K. A. Wepasnick, B. A. Smith, J. L. Bitter and D. Howard Fairbrother, Anal. Bioanal. Chem., 2010, 396, 1003–1014 CrossRef CAS PubMed
- Y. Qiao, R. Han, Y. Pang, Z. Lu, J. Zhao, X. Cheng, H. Zhang, Z. Yang, S. Yang and Y. Liu, J. Mater. Chem. A, 2019, 7, 11400–11407 RSC
- R. Yudianti, H. Onggo, Y. Saito, T. Iwata and J. Azuma, Open Mater. Sci. J., 2011, 5, 242–247 CrossRef CAS
- V. Datsyuk, M. Kalyva, K. Papagelis, J. Parthenios, D. Tasis, A. Siokou, I. Kallitsis and C. Galiotis, Carbon, 2008, 46, 833–840 CrossRef CAS
- H. Liu, J. Wang, J. Wang and S. Cui, Materials, 2018, 11, 2442 CrossRef CAS PubMed
- R. L. H. M. E. Birch, T. A. Ruda-Eberenz, M. Chai and R. Andrews, Ann. Occup. Hyg., 2013, 57, 1148–1166 Search PubMed
- A. H. Norzilah, A. Fakhru’L-Razi, T. S. Y. Choong and A. L. Chuah, J. Nanomater., 2011, 2011 Search PubMed
- K. Hareesh, R. P. Joshi, D. V. Sunitha, V. N. Bhoraskar and S. D. Dhole, Appl. Surf. Sci., 2016, 389, 1050–1055 CrossRef
- Y. Xiong, C. Tang, X. Yao, L. Zhang, L. Li, X. Wang, Y. Deng, F. Gao and L. Dong, Appl. Catal., A, 2015, 495, 206–216 CrossRef CAS
- M. Park, B. H. Kim, S. Kim, D. S. Han, G. Kim and K. R. Lee, Carbon, 2011, 49, 811–818 CrossRef CAS
- M. Lee, B. Ye, B. Jeong, H. Chun, D. H. Lee, S. Park, H. Lee and H.-D. Kim, Korean J. Chem. Eng., 2018, 35, 1988–1993 CrossRef CAS
- C. Fang, D. Zhang, L. Shi, R. Gao, H. Li, L. Ye and J. Zhang, Catal. Sci. Technol., 2013, 3, 803–811 RSC
- A. L. Castro, M. R. Nunes, A. P. Carvalho, F. M. Costa and M. H. Florêncio, Solid State Sci., 2008, 10, 602–606 CrossRef CAS
- B. Jeong, B. Ye, E. S. Kim and H. D. Kim, Catal. Commun., 2017, 93, 15–19 CrossRef CAS
- M. jin Lee, D. H. Kim, M. Lee, B. Ye, B. Jeong, D. H. Lee, H. D. Kim and H. Lee, Environ. Sci. Pollut. Res., 2019, 26, 36107–36116 CrossRef PubMed
- J. Y. Choi, B. Jeong, E.-S. Kim, H.-Y. Chun, D.-W. Shin, D.-H. Kim and H.-D. Kim, J. Nanosci. Nanotechnol., 2015, 15, 9083–9087 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra01665a |
| This journal is © The Royal Society of Chemistry 2020 |