
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Imparting CO2 reduction selectivity to ZnGa2O4 photocatalysts by crystallization from hetero nano assembly of amorphous-like metal hydroxides†
Masanori Takemoto a,
Yasuaki Tokudome*a,
Soichi Kikkawab,
Kentaro Teramura*b,
Tsunehiro Tanakab,
Kenji Okadaa,
Hidenobu Murataa,
Atsushi Nakahiraa and
Masahide Takahashia
a,
Yasuaki Tokudome*a,
Soichi Kikkawab,
Kentaro Teramura*b,
Tsunehiro Tanakab,
Kenji Okadaa,
Hidenobu Murataa,
Atsushi Nakahiraa and
Masahide Takahashia
aDepartment of Materials Science, Osaka Prefecture University, 1-1, Gakuencyo, Naka-ku, Sakai, Osaka 599-8531, Japan. E-mail: tokudome@mtr.osakafu-u.ac.jp
bDepartment of Molecular Engineering, Kyoto University, Kyotodaigaku Katsura, Nishikyo-ku, Kyoto 615-8510, Japan
First published on 25th February 2020
Abstract
Imparting an enhanced CO2 reduction selectivity to ZnGa2O4 photocatalysts has been demonstrated by controlled crystallization from interdispersed nanoparticles of zinc and gallium hydroxides. The hydroxide precursor in which Zn(II) and Ga(III) are homogeneously interdispersed was prepared through an epoxide-driven sol–gel reaction. ZnGa2O4 obtained by a heat-treatment exhibits a higher surface basicity and an enhanced affinity for CO2 molecules than previously-reported standard ZnGa2O4. The enhanced affinity for CO2 molecules of the resultant ZnGa2O4 leads to highly-selective CO evolution in CO2 photo-reduction with H2O reductants. The present scheme is promising to achieve desirable surface chemistry on metal oxide photocatalysts.
Introduction
Heterogeneous catalysis for environmental-friendly production of resources has attracted remarkable attention.1 Especially, complex oxides, with high chemical/thermal stabilities and capability of their use in aqueous media, have been studied for various catalytic reactions such as CO2 reduction,2–4 water-splitting,5,6 biodiesel synthesis.7 Among many approaches, such as using co-catalysts,8–11 co-solvents,12 nanostructuration13–16 and Z-scheme photosynthesis,17,18 surface modification of catalysts is especially a promising and versatile pathway to explore attracting catalytic activities, because the adsorption of reactants on a solid surface is a key reaction step in heterogeneous catalysis.19Recently, we have reported the synthesis of metal hydroxide nanoparticle (NP) with a size of single nanometer by epoxide-mediated alkalization.20,21 This synthesis pathway allows us to obtain metal hydroxide NPs as a stable suspension at a high yield without impurities.22 Moreover, a unique and tailorable surface property has been demonstrated by the introduction of a large hetero-interface by interdispersing two different hydroxide NPs in a homogenous manner.23 For example, hetero nanointerfaces constructed between cobalt and nickel hydroxide NPs (∼2 nm) exhibit lower charge transfer resistance and improved electrochemical properties thanks to highly homogenous interdispersion of hydroxide NPs. Our motivation in the present work is to develop a straightforward route to access a “complex oxide” photocatalyst with a desirable surface chemistry of stoichiometric composition, through a low-temperature thermal treatment on this class of metal hydroxides with hetero nanostructures.
We here choose ZnGa2O4 as a representative system in order to demonstrate the present concept for yielding metal oxide photocatalysts with an enhanced catalytic activity originated from a high surface basicity. ZnGa2O4 has been reported as a promising oxide to photocatalytically reduce CO2 into CO with H2O as an electron donor by loading of Ag co-catalyst.24 On the other hand, there still remains a challenge that competitive H2 evolution as a result of the reduction of H2O solvent inevitably occurs on bare ZnGa2O4 catalyst (without co-catalyst), resulting in a low reaction selectivity toward CO2 reduction (<∼30%).24 A previous report on Zn-modified Ga2O3 photocatalyst suggests that Zn–O–Ga linkages on the surface of catalysts play an important role to inhibit H2 production in photocatalytic conversion of CO2 with H2O.4 The homogenous interdispersion of precursory metal hydroxide NPs is expected to allow for the formation of such kind of surface.
Herein, we demonstrate that a nanocomposite of precursory metal hydroxide NPs with hetero nanointerfaces can be used to prepare ZnGa2O4 photocatalyst stoichiometrically-containing Zn(II) and Ga(III) on the oxide surface, leading to a highly-enhanced selectivity toward photocatalytic CO2 reduction. The precursory nanocomposite is prepared under a highly supersaturated condition induced by epoxide-mediated alkalization.25 ZnGa2O4 is obtained from the precursory nanocomposite by heat-treatment ranging from 500 °C to 900 °C. Surface analyses confirm that ZnGa2O4 prepared in the present scheme exhibits a higher base strength and a higher affinity for CO2 compared to those of standard ZnGa2O4 obtained by a solid phase reaction from a mixture of metal oxide precursors. XPS measurement has revealed that ZnGa2O4 prepared through the present scheme has a surface of stoichiometric composition that is not the case for standard ZnGa2O4 catalysts prepared at 850 °C. As a result, the present ZnGa2O4 heat-treated at 700 °C (without co-catalyst) exhibits a reaction selectivity of 48.0% toward CO evolution, more than double to that of standard ZnGa2O4 (20.3%). Also, Ag co-catalyst, that is required to maximize the reaction selectivity toward CO evolution, can be decreased into 5% of the previous reports, thanks to the high affinity for CO2 adsorption of the resultant ZnGa2O4 surface. Based on these insights obtained in this representative ZnGa2O4 system, synthesis of complex oxide catalysts through the crystallization of interdispersed hydroxides NPs is expected to be applied to design a wide range of complex oxide catalysts with an intended surface.
Experimental section
Materials
Zn(NO3)2·6H2O (99.0%), Ga(NO3)3·nH2O (99.9%), ZnO (99.0%), Ga2O3 (99.99%), NH3 aqueous solution (25 wt%), ethanol (99.5%), methanol (99.8%), bromothymol blue, phenolphthalein, 2,4-dinitroaniline, NaHCO3 (99.5–100.3%), 0.1 M of AgNO3 aqueous solution and propylene oxide (PO, >99%) were used as received. PO was purchased from Sigma-Aldrich Corp. All other reagents were purchased from FUJIFILM Wako Pure Chemical Industries. Ultra-pure water of resistivity of 18.2 MΩ cm was used in all experiments.Preparation of ZnGa2O4
Zn(NO3)2·6H2O (0.740 g; 2.49 mmol) and Ga(NO3)3·nH2O (1.99 g; 4.98 mmol, n is set as 8 for the calculation.) were dissolved in a mixture of ultra-pure water (6.00 mL; 333 mmol) and ethanol (6.00 mL; 165 mmol). At 25 °C, PO (8.10 mL; 112 mmol) was added to the mixture solution and the solution was stirred for 30 s to form a homogeneous sol. Then, the sol kept in a closed container was placed under a static condition at 25 °C to form a wet gel. The wet gel was aged for 24 h at 25 °C. After aging process, the wet gel was dried at 40 °C to obtain a precursory xerogel. The xerogel was calcined at various temperatures ranging from 500 °C to 900 °C for 12 h to yield ZnGa2O4. ZnGa2O4 was also prepared by a solid state reaction. The details are described in ESI.†Photocatalytic conversion of CO2 with H2O reductant in liquid phase
Photocatalytic conversion of CO2, including the step of Ag-loading on ZnGa2O4, was carried out in a flow system using an inner-irradiation-type reaction vessel at 35 °C. Photocatalyst (0.5 g) was dispersed in ultra-pure water (1.0 L) containing 0.1 M of NaHCO3. Ag co-catalyst was loaded on ZnGa2O4 by photo-deposition method as follows; (1) AgNO3 was added at a concentration ranging from 2.34 × 10−6 M to 46.8 × 10−6 M. (2) Ar gas was bubbled into the suspension at a flow rate of 80 mL min−1 to purge air for 1 h. (3) The flow rate was changed to 30 mL min−1 and the suspension was illuminated with a 400 W high-pressure mercury lamp for 2 h. These steps for Ag-loading were skipped when photocatalytic reaction over the bare ZnGa2O4 photocatalyst was examined. Then, CO2 gas was bubbled into the suspension at a flow rate of 60 mL min−1 for 1 h in order to purge Ar or air. The suspension was illuminated for 5 h with a 400 W high-pressure mercury lamp with a quartz filter equipped with a water cooling system, with a CO2 flow at a rate of 30 mL min−1. The outlet of the reactor was connected to a six-way valve with a sampling loop. Evolved gaseous products of H2, O2, and CO together with CO2 gas were collected and analyzed by thermal conductivity detector-gas chromatography (TCD-GC) using a GC-8A chromatograph (Shimadzu Corp.) with a Molecular Sieve 5A column and Ar as a carrier gas, and by flame ionization detector-gas chromatography (FID-GC) with a methanizer (Shimadzu Corp.), a Shin Carbon ST column, and N2 as a carrier gas. In an isotopic experiment with 13CO2, the formation of 13CO was detected by mass spectroscopy by using a quadrupole-type mass spectrometer (BEL Japan, BEL Mass).Characterization
Crystal structures of samples were identified by powder X-ray diffraction (PXRD; MultiFlex, Rigaku, Japan). X-ray photoelectron spectroscopy (XPS) spectra were recorded by using X-ray photoelectron spectrometer (ESCA-3400, Shimadzu Corp.) under Mg Kα radiation. X-ray absorption fine structure (XAFS) spectra of Zn K-edge and Ga K-edge were collected in the transmission mode at room temperature on BL11 at the SAGA Light Source (Saga, Japan). A silicon (111) double-crystal monochrometor was used to obtain the incident X-ray beam. The intensity of the incident and transmitted X-ray was monitored by an ionization chamber. N2 adsorption–desorption isotherms at −196 °C were obtained on a volumetric gas adsorption apparatus (BELSORP-mini II, Bel Japan Inc., Japan). Prior to the measurements, sample powders were pretreated at 200 °C under a vacuum condition overnight. Specific surface area of ZnGa2O4 was estimated by the Brunauer–Emmett–Teller (BET) method. Morphologies of Zn–Ga AMH and ZnGa2O4 catalysts were observed by a field emission scanning electron microscope (FE-SEM; S-4800, Hitachi, Japan, with a thin Pt coating), transmission electron microscopes (TEM; JEM-2000FX, JEOL, Japan and Hitachi 7650, Hitachi, Japan) and a field emission transmission electron microscope (FE-TEM; JEM-2100F, JEOL, Japan) equipped with EDS. The UV-Vis diffusion reflectance spectra of ZnGa2O4 samples were measured by using UV-Visible spectrometer (V-670, JASCO, Japan) equipped with an integrating sphere. The energy gap (Eg) was estimated with the Davis–Mott method.26 Base strength of bare ZnGa2O4 catalysts was estimated by Hammett indicators; bromothymol Blue (pKa = 7.2), phenolphthalein (pKa = 9.8) and 2,4-dinitroaniline (pKa = 15.0) were used as coloring agents, and methanol was used as a solvent. When the color change of the indicator is observed, the base strength (H_) of the tested solid sample is determined to be higher than the pKa value of the indicator employed.27,28 The affinity between CO2 and the surface of ZnGa2O4 catalysts was assessed by thermal programmed desorption (TPD: BELCAT-B, Bel Japan Inc., Japan) of CO2. He gas was flowed as a carrier gas and ZnGa2O4 was degassed at 200 °C for 1 h. Then, CO2 was introduced and allowed to adsorb on ZnGa2O4 at 30 °C for 30 min. After the adsorption of CO2, the sample was heated from 30 °C to 800 °C at a heating rate of 5 °C min−1. Desorbed CO2 was monitored by using a quadrupole-type mass spectrometer (BEL Mass, Bel Japan Inc., Japan).Results and discussion
Synthesis of ZnGa2O4 through calcination of a gel composed of interdispersed metal hydroxide NPs with low crystallinities
A precursory xerogel for the synthesis of ZnGa2O4 was obtained through epoxide-mediated alkalization from an aqueous solution of Zn(NO3)2·6H2O and Ga(NO3)3·nH2O (Fig. 1(a)). Two small peaks at 2θ = 9.9° and 19.8° and the broad diffraction pattern were observed in PXRD pattern of precursory xerogel (Fig. 1(b)). Two small peaks are assigned to 003 and 006 diffraction peaks of Zn–Ga layered double hydroxide (LDH) crystal,29 whose Zn2+/Ga3+ ratio was estimated as 1.5 from TEM-EDS mapping shown in Fig. S1.† By Bragg's law using the diffraction peak at 2θ = 9.9°, the interlayer separation is calculated to be 0.89 nm, which corresponds to an interlayer space of LDH accommodating nitrate anions.30,31 The broad diffraction on the PXRD pattern of the precursory xerogel is in good agreement with that for low-crystalline Ga(OH)3 NPs with a size of 4.4 nm in diameter (SBET = 349 m2 g−1). The obtained gel is a nanocomposite of poorly-crystallized (amorphous-like) metal hydroxides containing a small amount of Zn–Ga LDH. The present epoxide-mediated alkalization allows us to yield a hydroxide gel corresponding with starting chemical composition because most of Zn2+ and Ga3+ species (96.3% of initial metals) are consumed during the reaction in a homogeneous manner (Fig. S2†). Scanning electron microscope (SEM) observation reveals that LDH platelets with a lateral size of 2 μm are embedded in the gel matrix composed of NPs (<10 nm in diameter) (Fig. 1(c–e)). Zn and Ga are homogeneously distributed in the precursory gel (Fig. S1(c)†), which confirms that both amorphous-like Zn(OH)2 and Ga(OH)3 NPs interdispersedly assembles to form the precursory gel (Fig. 1(e)). The present synthesis condition allows us to achieve a high degree of supersaturation and thereby the formation of abundant NPs to form a homogenous gel throughout the reaction solution. Hereafter, the present precursory composite gel is called as “Zn–Ga AMH” (Zn–Ga Amorphous Metal Hydroxide). | ||
| Fig. 1 (a) Photograph of precursory wet gel obtained from an aqueous solution of Zn(NO3)2·6H2O and Ga(NO3)3·nH2O under a highly supersaturated condition induced by epoxide-mediated alkalization. (b) PXRD patterns of precursory xerogel and low-crystalline Ga(OH)3 NPs obtained from an aqueous solution of Ga(NO3)3·nH2O (without Zn(NO3)2·6H2O). (c–e) SEM images of precursory xerogel at different magnifications. | ||
Fig. 2 shows powder X-ray diffraction (PXRD) patterns obtained from Zn–Ga AMH by the calcination at various temperatures ranging from 500 °C to 900 °C. Their diffraction patterns are ascribed to the typical pattern of ZnGa2O4 (ICDD PDF 00-038-1240). Interestingly, the synthesis of ZnGa2O4 as a single phase is possible even when the calcination temperature is as low as 500 °C by using the present Zn–Ga AMH as a precursor. Morphological observation by SEM (Fig. S3†) reveals that the size of ZnGa2O4 crystals obtained from Zn–Ga AMH by the calcination at 500 °C is >10 nm and becomes larger with increasing calcination temperature due to crystal growth. The achievement of low-temperature synthesis of ZnGa2O4 as a single phase is mainly due to highly-homogeneous dispersion of low-crystalline Zn(OH)2 and Ga(OH)3 NPs in Zn–Ga AMH as discussed in the former paragraph. As is well-known, cations are homogeneously distributed in hydroxide layers of Zn–Ga LDH in atomic scale,32,33 suggesting that the contaminated small amount of Zn–Ga LDH also contributes to the low-temperature synthesis of ZnGa2O4. ZnGa2O4 samples obtained as a single phase from Zn–Ga AMH are hereinafter named as “AMH-derived ZnGa2O4-X” (X is calcination temperature).
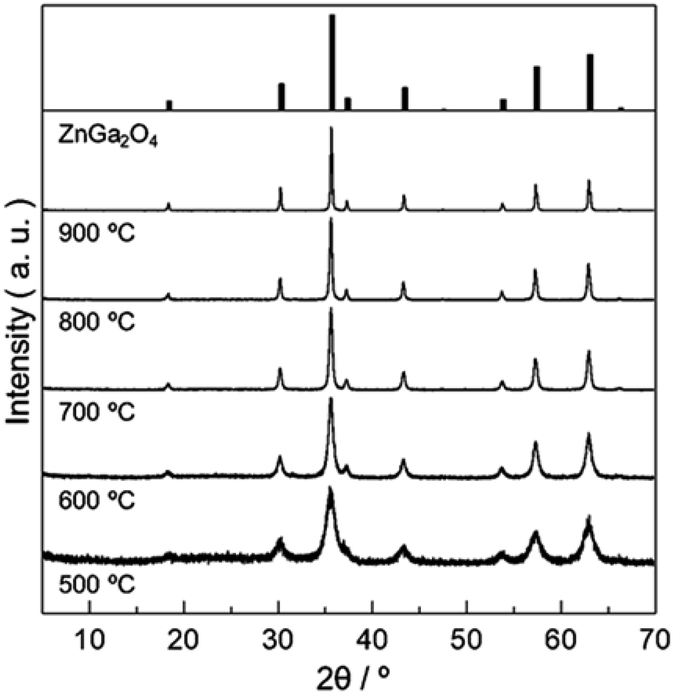 | ||
| Fig. 2 PXRD patterns of ZnGa2O4 obtained from Zn–Ga AMH by calcination in air at various temperatures for 12 h. | ||
Surface basicity and affinity for CO2 of ZnGa2O4
The surface basicity of AMH-derived ZnGa2O4 was investigated in comparison with previously-reported one. Hereafter, ZnGa2O4 prepared at 850 °C through a solid phase reaction (Fig. S4†), whose catalytic activity is reportedly highest in the report24 is called as “standard ZnGa2O4”. We found that ZnGa2O4 obtained from Zn–Ga AMH exhibits a unique surface nature different from standard ZnGa2O4. Fig. 3(a) shows AMH-derived ZnGa2O4-700 and standard ZnGa2O4 immersed in a methanolic solution of Hammett indicator (phenolphthalein). AMH-derived ZnGa2O4-700 changes its color from white to red upon the immersion in the indicator solution, whilst the change of color is not observed for standard ZnGa2O4, indicating that base strength of AMH-derived ZnGa2O4-700 in methanol is stronger than that of standard ZnGa2O4. Base strength, crystallite size, and specific surface area of ZnGa2O4 samples are summarized in Table S1.† Base strength of AMH-derived ZnGa2O4 decreases by the heat treatment above at 700 °C. Nonetheless, the base strength of AMH-derived ZnGa2O4-900 is found to be still higher than that of standard ZnGa2O4 prepared at a relatively lower calcination temperature of 850 °C. The difference in coloring behavior cannot be explained by difference in surface area, but difference in surface nature; AMH-derived ZnGa2O4-900 (SBET = 0.53 m2 g−1) exhibits a stronger surface basicity than standard ZnGa2O4 (SBET = 1.3 m2 g−1). The epoxide-mediated alkalization employed in the present study clearly contributes to yield ZnGa2O4 with the high basic strength.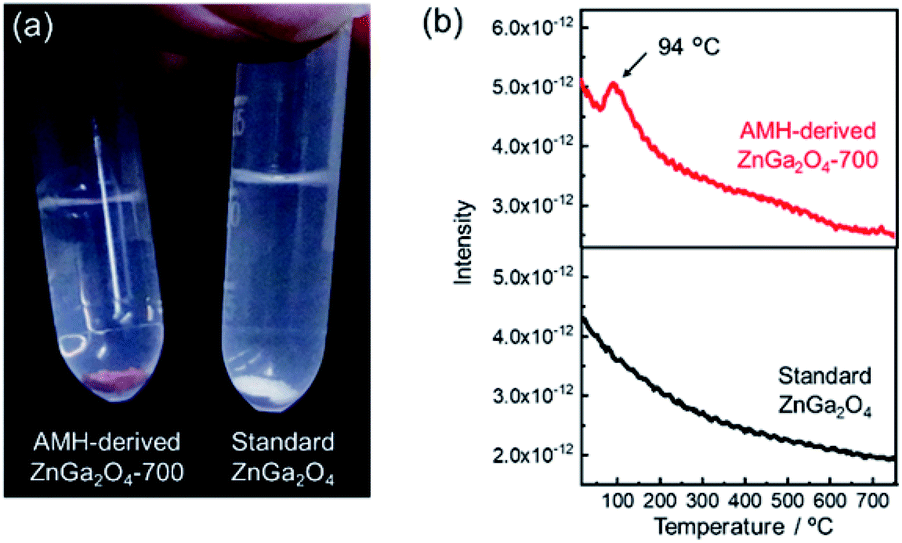 | ||
| Fig. 3 (a) Photograph of AMH-derived ZnGa2O4-700 and standard ZnGa2O4 immersed in a methanolic solution of Hammett indicator (phenolphthalein). (b) CO2-TPD curves of AMH-derived ZnGa2O4-700 and standard ZnGa2O4 (sample mass: 0.5 g). | ||
CO2 temperature programmed desorption (CO2-TPD) curves of AMH-derived ZnGa2O4-700 and standard ZnGa2O4 are plotted in Fig. 3(b). The peak at 94 °C, originated from desorption of CO2, is observed only for the TPD curve of AMH-derived ZnGa2O4-700, suggesting that the surface of AMH-derived ZnGa2O4 has a higher affinity and a stronger adsorptive nature for CO2 than standard ZnGa2O4. The peak at 94 °C could be detected even when the sample mass of AMH-derived ZnGa2O4-700 was decreased to 1/5 in order to compensate the difference in SBET between AMH-derived ZnGa2O4-700 (SBET = 5.0 m2 g−1) and standard ZnGa2O4 (SBET = 1.3 m2 g−1). The result reveals that the difference in SBET is not the cause of the different CO2 adsorption capacity of the two samples. The peak in low-temperature range is also observed for TPD curves of AMH-derived ZnGa2O4-500 and AMH-derived ZnGa2O4-600 (not shown). From the above all results, we can conclude that single phase ZnGa2O4 with a high base strength and a high affinity for CO2 can be obtained by the calcination of Zn–Ga AMH.
Structure analysis of ZnGa2O4 catalysts were performed by XAFS and XPS. The difference of surface structure between AMH-derived ZnGa2O4 and standard ZnGa2O4 was investigated by XPS. XAFS measurement was employed as a technique to understand local environments of the atoms for bulk samples. Fig. 4 shows Zn K-edge and Ga K-edge EXAFS and Fourier transforms (FT) of EXAFS spectra of AMH-derived ZnGa2O4-700 and standard ZnGa2O4. The same characteristic distance is observed in the first neighbor peaks (Zn–O shells in Zn K-edge EXAFS and Ga–O shells in Ga K-edge EXAFS) and the second neighbor peaks (Zn–Ga and Zn–Zn shells in Zn K-edge EXAFS, and Ga–Ga and Ga–Zn shells in Ga K-edge EXAFS). These results support that the average local environments of Zn and Ga atoms are comparable in AMH-derived ZnGa2O4-700 and standard ZnGa2O4.
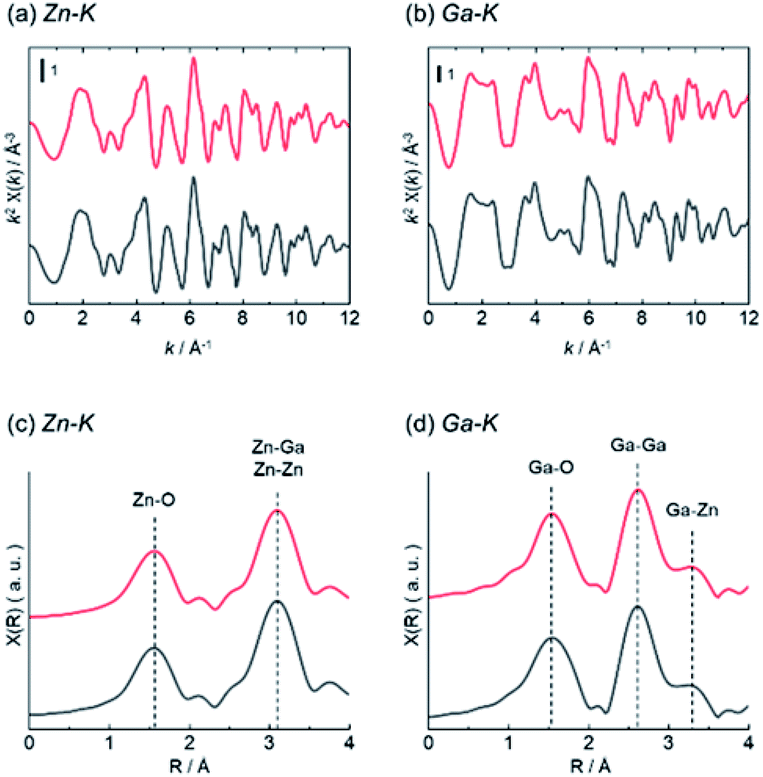 | ||
| Fig. 4 (a) Zn K-edge and (b) Ga K-edge EXAFS (c and d) Fourier transforms of EXAFS spectra of AMH-derived ZnGa2O4-700 (red line) and standard ZnGa2O4 (black line). | ||
XPS analysis was employed to compare the chemical composition for AMH-derived ZnGa2O4-700 and standard ZnGa2O4, where the sampling depth is estimated to be ca. 3.0 nm. The XPS peaks of Zn 3d, Ga 3d, Zn 3p, Ga 3p, Zn 3s, Ga 3s, O 1s, Zn 2p and Ga 2p are observed in wide-scan XPS spectra of both ZnGa2O4 catalysts (Fig. S5†). Narrow spectra and estimated molar ratio values of Zn/Ga of two ZnGa2O4 catalysts are summarized in Fig. 5. The peak positions of Zn 2p and Ga 2p in narrow scan XPS spectra are comparable for two ZnGa2O4 catalysts. It should be highlighted that there is a clear difference in Zn/Ga ratio between two ZnGa2O4 photocatalysts. Preferential segregation of Zn on the surface (Zn/Ga = 0.7) is confirmed for standard ZnGa2O4. A higher temperature would be required to obtain ZnGa2O4 with the surface of stoichiometric composition in this solid state reaction, whereas the calcination at a higher temperature promotes the sintering of complex oxide particles, leading to decrease of accessible sites by reactants on complex oxide catalyst. On the other hand, the stoichiometric Zn/Ga molar ratio (Zn/Ga = 0.5) on the surface is confirmed for AMH-derived ZnGa2O4-700, thanks to the highly-homogeneous distribution of hydroxide NPs in Zn–Ga AMH precursor. The results of XPS analyses suggest that a larger amount of Zn–O–Ga linkages is formed in the case of AMH-derived ZnGa2O4-700 compared to standard ZnGa2O4. Although the further study is required to elucidate exact adsorption sites on the surface for CO2, such as hydroxyl groups, and oxygen in Zn–O–Ga linkages, it can be concluded that AMH-derived ZnGa2O4-700 with the stoichiometric surface exhibits the higher affinity for CO2 molecules because of the introduction of the stronger Lewis basic sites. The use of interdispersed amorphous Zn and Ga metal hydroxide NPs is a key to yield complex oxide with the surface of stoichiometric composition even by a low-temperature synthesis.
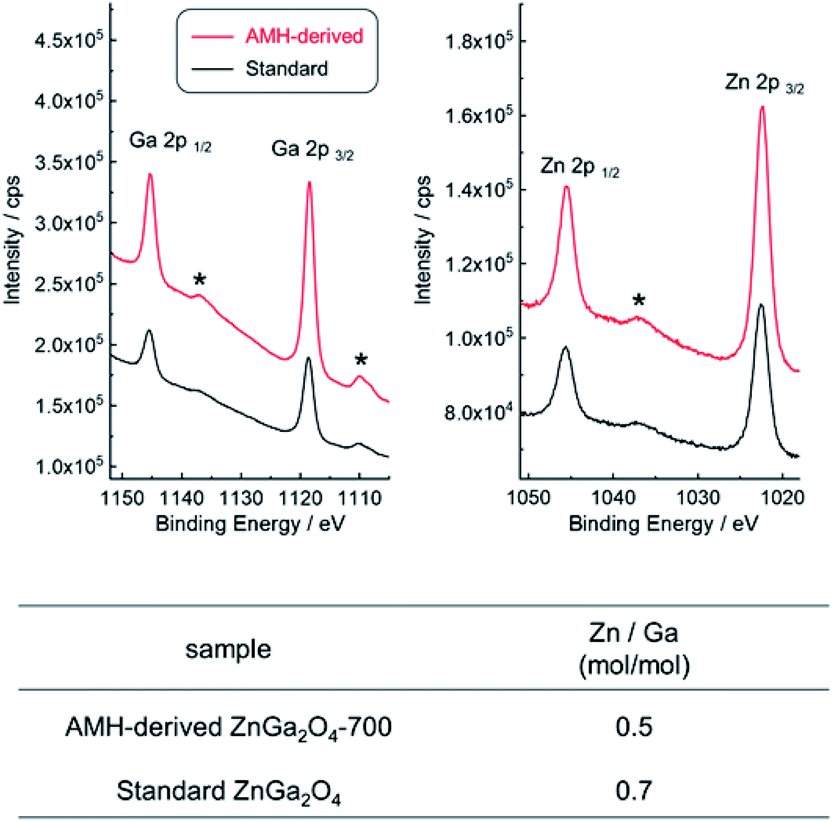 | ||
| Fig. 5 Narrow scan XPS spectra of AMH-derived ZnGa2O4-700 (red line) and standard ZnGa2O4 (black line) and the values of molar ratio of Zn/Ga. Surface chemical composition of ZnGa2O4 photocatalysts analyzed by XPS measurement *: satellite peaks. | ||
Photocatalytic CO2 reduction over bare ZnGa2O4 (without co-catalyst)
Photocatalytic properties of AMH-derived ZnGa2O4 catalysts were systematically investigated under the experimental condition optimized in a literature.24 AMH-derived ZnGa2O4-700 and standard ZnGa2O4 exhibit the best catalytic activities in terms of CO evolution and reaction selectivity after the calcination temperature of 700 and 850 °C,24 respectively (Fig. S6†). These two best catalysts for respective systems were closely examined in the following section.The isotopic experiment was also carried out using 13CO2 as a gaseous reactant. Q-Mass spectra in m/z = 28 and m/z = 29 were collected for the photocatalytic conversion of 13CO2 with H2O over AMH-derived ZnGa2O4-700 (Fig. 6). The peak derived from 13CO is observed on Q-Mass spectrum of m/z = 29. It has been evidenced that AMH-derived ZnGa2O4-700 photocatalytically reduces introduced 13CO2 into 13CO. Fig. S7† shows time course of gas evolutions for photocatalytic conversion of CO2 with water over AMH-derived ZnGa2O4-700. The linear increase of evolved gases indicates that the present ZnGa2O4 catalyst possesses the stability in the photocatalytic reaction in aqueous solution. Furthermore, any impurities are not observed on the XRD pattern of the spent catalyst, which also supports the stability of AMH-derived ZnGa2O4-700 (Fig. S8†). Rates of gas evolution (eqn (1)–(3)) and reaction selectivity toward CO evolution (eqn (4)) over bare ZnGa2O4 catalysts (without co-catalysts) are represented in Fig. 7. Both ZnGa2O4 catalysts work as photocatalysts for CO2 reduction along with evolving gaseous products by reduction and oxidation in a stoichiometric ratio, (H2 + CO)/O2 = 2.0. This further supports that the formation of CO over both ZnGa2O4 catalysts is derived from not a decomposition of contaminated organic compounds but photocatalytic conversion of introduced CO2 molecules.34
 | (1) |
| CO2 + 2H+ + 2e− → CO + H2O | (2) |
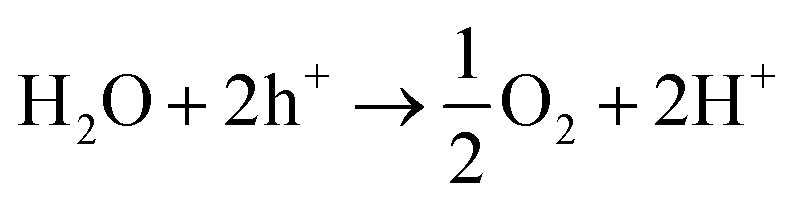 | (3) |
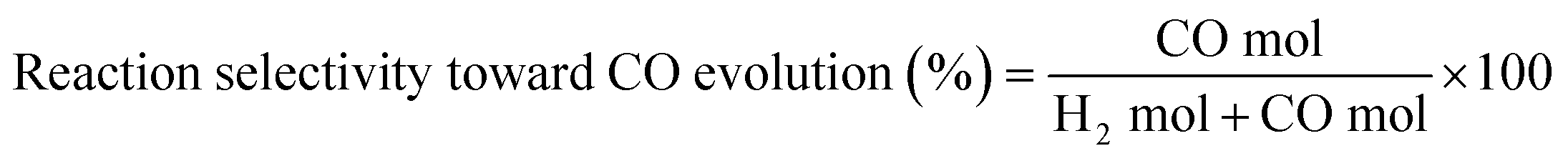 | (4) |
 | ||
| Fig. 6 Mass spectra of (a) m/z = 28 and (b) m/z = 29 in the photocatalytic conversion of 13CO2 by H2O over bare AMH-derived ZnGa2O4-700 photocatalyst. | ||
 | ||
| Fig. 7 Rates of H2 (grey), O2 (sky blue) and CO (red) evolution and reaction selectivity toward CO evolution (◆) over bare AMH-derived ZnGa2O4-700 and bare standard ZnGa2O4. | ||
There is no difference between two photocatalysts in the formation rates of oxidation product (O2) and sum of reduction products (H2 + CO). The rates of gas evolution do not degrade during photocatalytic reaction for 5 h for both photocatalysts. It is interesting to note that the clear difference is observed in the reaction selectivity toward the CO evolution between bare AMH-derived ZnGa2O4-700 (48.0%) and bare standard ZnGa2O4 (20.3%). Energy gap (Eg) estimated by UV-Vis spectroscopy, crystallite size, and specific surface area of ZnGa2O4 catalysts are summarized in Fig. S9 and Table S2.† There can be seen no relationship between these parameters and reaction selectivity toward CO evolution. More clearly, a high reaction selectivity toward CO evolution with a stoichiometric ratio, (H2 + CO)/O2 = 2.0, is observed even in the case of AMH-derived ZnGa2O4-900 (47.3%) whose Eg and SBET are comparable to and smaller than standard ZnGa2O4, respectively. As discussed in the previous section, CO2 adsorption capacity per unit surface area of catalysts is larger for AMH-derived ZnGa2O4-700. The high reaction selectivity toward CO evolution over AMH-derived ZnGa2O4-700 can be reasonably explained by the high affinity for CO2 of AMH-derived ZnGa2O4-700.
Photocatalytic CO2 reduction over Ag-loaded ZnGa2O4
It has been well-known that metal co-catalysts, including Ag NP, are capable to reduce the recombination between excited electrons and holes.35,36 Fig. 8(a) shows the rates of gas evolution for H2, O2 and CO, and the reaction selectivity toward CO evolution over AMH-derived ZnGa2O4-700 with Ag co-catalyst at various loadings. The rate of H2 evolution decreases with increasing the loaded amount of Ag co-catalyst. This trend is corresponding to those demonstrated in previous reports.24,37,38 Ag was reported to work as a preferential active sites for CO2 reduction and thereby inhibits H2 evolution with increasing coverage with Ag, resulting in enhanced reaction selectivity toward CO evolution. The maximum activity for CO evolution (32.3 μmol h−1) could be achieved at 0.05 wt% of Ag co-catalyst with a drastically improved reaction selectivity toward CO evolution (75.9%). Compared to a previous ZnGa2O4 catalyst with an optimized Ag loading of 1 wt%,24 95% of Ag can be cut down in the present case, thanks to the high affinity for CO2 of AMH-derived ZnGa2O4-700. Fig. 8(b) shows a TEM image of AMH-derived ZnGa2O4-700 at Ag loading of 0.1 wt%. The surface of AMH-derived ZnGa2O4-700 is partially covered by dispersedly-loaded Ag NPs. Fig. 8(c) represents a schematic illustration explaining the relationship between surface properties and photocatalytic activity toward CO2 reduction. The surface of AMH-derived ZnGa2O4-700 with a homogeneous distribution of Zn and Ga works as an efficient CO2 absorber and concentrate CO2 to be reduced. The preferential adsorption of CO2 on AMH-derived ZnGa2O4-700, evidenced by CO2-TPD measurement (Fig. 3(b)), allows for the photocatalytic reduction of CO2 at catalytic active sites, vicinity of Ag co-catalysts, in a more selective manner even at a low Ag-loading.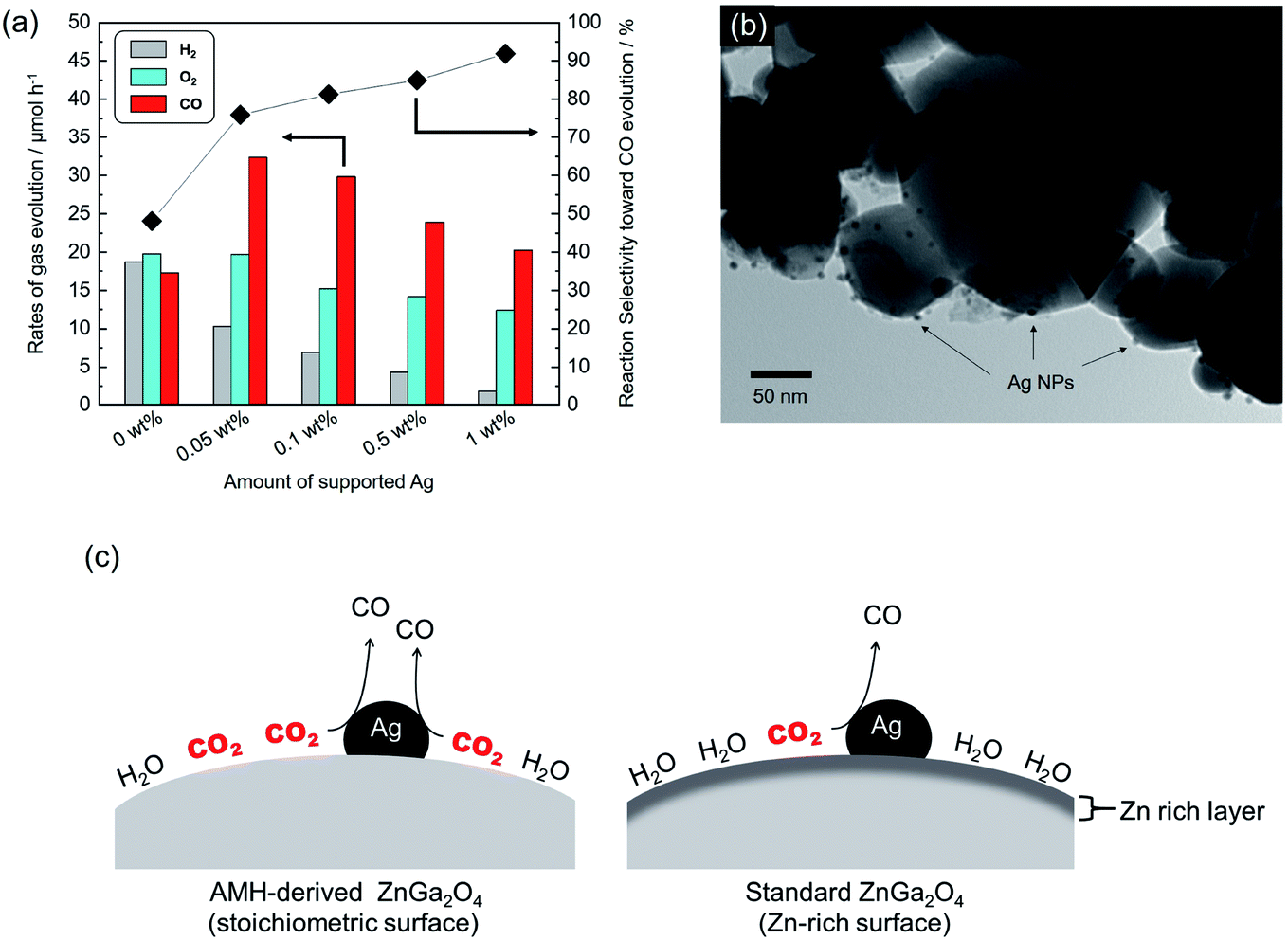 | ||
| Fig. 8 (a) Rates of gas evolution for H2 (grey), O2 (sky blue) and CO (red) and reaction selectivity toward CO evolution over Ag-loaded AMH-derived ZnGa2O4-700 at different Ag loadings. (b) TEM image of Ag-loaded AMH-derived ZnGa2O4-700 with 0.1 wt% of Ag co-catalyst. (c) Schematic illustration of photocatalytic CO2 reduction over AMH-derived ZnGa2O4 (left) and standard ZnGa2O4 (right) with Ag loading. | ||
In summary, the present work has demonstrated the formation of desirable surfaces on complex metal oxides by using precursors of interdispersed nanometric hydroxides. ZnGa2O4 was chosen as a representative system to demonstrate the present concept for yielding metal oxide photocatalysts with an enhanced catalytic activity originated from a high surface basicity. The scheme of using NPs as precursors is further expected to be available for designing a wide range of metal oxide catalysts with nano/macrotextures by integrating with the nano building block approach.20,22,39
Conclusions
The metal hydroxides precursor (Zn–Ga AMH) with a large hetero-interfaces of Zn(OH)2 and Ga(OH)3 NPs was prepared through an epoxide mediated alkalization. ZnGa2O4 catalysts as a single phase were synthesized using the Zn–Ga AMH precursor. Surface analysis with Hammett indicators reveals that AMH-derived ZnGa2O4 catalysts exhibit the stronger base strength even after the calcination at high temperature, ≥700 °C, compared to ZnGa2O4 prepared through a solid phase reaction. CO2–TPD measurements revealed that the peak at 94 °C, originated from desorption of CO2, was observed only for the TPD curve of AMH-derived ZnGa2O4-700, suggesting that the surface of AMH-derived ZnGa2O4 has a higher affinity and a stronger adsorptive nature for CO2 than standard ZnGa2O4. Structural analysis by XPS reveals that AMH-derived ZnGa2O4-700 has a surface of the stoichiometric composition, resulting in a higher affinity for CO2 than standard ZnGa2O4. The bare AMH-derived ZnGa2O4-700 with a high affinity for CO2 exhibits the highest reaction selectivity toward the CO2 reduction (48.0%) among previously-reported co-catalysts-free metal oxide catalysts for the photocatalytic conversion of CO2 with liquid H2O. Additionally, the loading amount of Ag co-catalyst decreased by 95% due to the higher affinity for CO2. Based on these insights obtained in this representative ZnGa2O4 system, the present scheme using interdispersed hydroxide nanoparticles will pave the way to general method to tune the surface properties of metal oxide catalysts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The present work is partially supported by the JSPS KAKENHI, Grant-in-Aid for Scientific Research on Innovative Areas, Grant for Basic Science Research Projects from The Sumitomo Foundation. The experiments using synchrotron radiation were performed at the beamlines BL11 of the SAGA Light Source (Proposal No. 1904019T/BL11). Y. T. thanks Dr Vanessa Prevot, CNRS, France and Prof. Claude Forano, Université Clermont Auvergne, France, for their helpful suggestions. The JSPS-CNRS bilateral program is also acknowledged for the encouragement of the discussion on the present study. We thank Dr Naoki Tarutani, Hosei University, Japan for FE-TEM observation.Notes and references
- Y. Liu, G. Zhao, D. Wang and Y. Li, Natl. Sci. Rev., 2015, 2, 150 CrossRef CAS.
- K. Li, B. Peng and T. Peng, ACS Catal., 2016, 6, 7485 CrossRef CAS.
- J. L. White, M. F. Baruch, J. E. Pander III, Y. Hu, I. C. Fortmeyer, J. E. Park, T. Zhang, K. Liao, J. Gu, Y. Yan, T. W. Shaw, E. Abelev and A. B. Bocarsly, Chem. Rev., 2015, 115, 12888 CrossRef CAS PubMed.
- Z. Wang, K. Teramura, Z. Huang, S. Hosokawa, Y. Sakata and T. Tanaka, Catal. Sci. Technol., 2016, 6, 1025 RSC.
- I. Papadas, J. A. Christodoulides, G. Kioseoglou and G. S. Armatas, J. Mater. Chem. A, 2015, 3, 1587 RSC.
- K. Maeda and K. Domen, J. Phys. Chem. Lett., 2010, 1, 2655 CrossRef CAS.
- E. Santacesaria, G. M. Vicente, M. Di Serio and R. Tesser, Catal. Today, 2012, 195, 2 CrossRef CAS.
- K. Iizuka, T. Wato, Y. Miseki, K. Saito and A. Kudo, J. Am. Chem. Soc., 2011, 133, 20863 CrossRef CAS PubMed.
- Y. Sakata, Y. Matsuda, T. Yanagida, K. Hirata, H. Imamura and K. Teramura, Catal. Lett., 2008, 125, 22 CrossRef.
- Y. Wei, X. Wu, Y. Zhao, L. Wang, Z. Zhao, X. Huang, J. Liu and J. Li, Appl. Catal., B, 2018, 236, 445 CrossRef CAS.
- Y. Zhao, Y. Wei, X. Wu, H. Zheng, Z. Zhao, J. Liu and J. Li, Appl. Catal., B, 2018, 226, 360 CrossRef CAS.
- Z. B. Todorović, O. S. Stamenković, I. S. Stamenković, J. M. Avramović, A. V. Veličković, I. B. Banković-Ilić and V. B. Veljković, Fuel, 2013, 107, 493 CrossRef.
- Q. Liu, D. Wu, Y. Zhou, H. Su, R. Wang, C. Zhang, S. Yan, M. Xiao and Z. Zou, ACS Appl. Mater. Interfaces, 2014, 6, 2356 CrossRef CAS PubMed.
- S. C. Yan, S. X. Ouyang, J. Gao, M. Yang, J. Y. Feng, X. X. Fan, L. J. Wan, Z. S. Li, J. H. Ye, Y. Zhou and Z. G. Zou, Angew. Chem., Int. Ed., 2010, 49, 6400 CrossRef CAS PubMed.
- M. Zhao, Y. Gu, P. Chen, Z. Xin, H. Zhu, B. Wang, K. Zhu, S. Yan and Z. Zou, J. Mater. Chem. A, 2019, 7, 9316 RSC.
- X. Wu, C. Wang, Y. Wei, J. Xiong, Y. Zhao, Z. Zhao, J. Liu and J. Li, J. Catal., 2019, 377, 309 CrossRef CAS.
- R. Abe, J. Photochem. Photobiol., C, 2010, 11, 179 CrossRef CAS.
- C. Wang, Y. Zhao, H. Xu, Y. Li, Y. Wei, J. Liu and Z. Zhao, Appl. Catal., B, 2020, 263, 118314 CrossRef CAS.
- J. Zhu and X. Lin, J. Colloid Interface Sci., 1997, 196, 110 CrossRef PubMed.
- Y. Tokudome, T. Morimoto, N. Tarutani, P. D. Vaz, C. D. Nunes, V. Prevot, G. B. Stenning and M. Takahashi, ACS Nano, 2016, 10, 5550 CrossRef CAS PubMed.
- Y. Tokudome, M. Fukui, S. Iguchi, Y. Hasegawa, K. Teramura, T. Tanaka, M. Takemoto, R. Katsura and M. Takahashi, J. Mater. Chem. A, 2018, 6, 9684 RSC.
- N. Tarutani, Y. Tokudome, M. Jobbágy, F. A. Viva, G. J. A. A. Soler-Illia and T. Masahide, Chem. Mater., 2016, 28, 5606 CrossRef CAS.
- N. Tarutani, Y. Tokudome, M. Jobbágy, G. J. A. A. Soler-Illia and M. Takahashi, J. Mater. Chem. A, 2019, 7, 25290 RSC.
- W. Zheng, T. Kentaro, H. Saburo and T. Tsunehiro, J. Mater. Chem. A, 2015, 3, 11313 RSC.
- E. G. Alexander, M. T. Thomas, H. S. Joe Jr, F. P. John, W. H. Lawrence and L. S. Randall, Chem. Mater., 2001, 13, 999 CrossRef.
- E. A. Davis and N. F. Mott, Philos. Mag., 1970, 22, 903 CrossRef CAS.
- J. L. Shumaker, C. Crofcheck, S. A. Tackett, E. Santillan-Jimenez and M. Crocker, Catal. Lett., 2007, 115, 56 CrossRef CAS.
- W. Xie and H. Li, J. Mol. Catal. A: Chem., 2006, 255, 1 CrossRef CAS.
- G. S. Thomas and P. V. Kamath, Solid State Sci., 2006, 8, 1181 CrossRef CAS.
- H.-Y. Zeng, X. Deng, Y.-J. Wang and K.-B. Liao, AIChE J., 2009, 55, 1229 CrossRef CAS.
- G. A. Caravaggio, C. Detellier and Z. Wronski, J. Mater. Chem., 2001, 11, 912 RSC.
- M. Vucelic, W. Jones and G. D. Moggridge, Clays Clay Miner., 1997, 45, 803 CrossRef CAS.
- S. Cadars, G. r. Layrac, C. Gérardin, M. l. Deschamps, J. R. Yates, D. Tichit and D. Massiot, Chem. Mater., 2011, 23, 2821 CrossRef CAS.
- K. Teramura and T. Tanaka, Phys. Chem. Chem. Phys., 2018, 20, 8423 RSC.
- J. Yang, D. Wang, H. Han and C. Li, Acc. Chem. Res., 2013, 46, 1900 CrossRef CAS PubMed.
- Y. Liu, S. Yang, S. Zhang, H. Wang, H. Yu, Y. Cao and F. Peng, Int. J. Hydrogen Energy, 2018, 43, 5551 CrossRef CAS.
- K. Teramura, H. Tatsumi, Z. Wang, S. Hosokawa and T. Tanaka, Bull. Chem. Soc. Jpn., 2015, 88, 431 CrossRef CAS.
- Z. Wang, K. Teramura, S. Hosokawa and T. Tanaka, Appl. Catal., B, 2015, 163, 241 CrossRef CAS.
- N. Tarutani, Y. Tokudome, M. Jobbágy, G. J. A. A. Soler-Illia, Q. Tang, M. Müller and M. Takahashi, Chem. Mater., 2018, 31, 322 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra00710b |
| This journal is © The Royal Society of Chemistry 2020 |