
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRhenium(I) complexation–dissociation strategy for synthesising fluorine-18 labelled pyridine bidentate radiotracers†
Mitchell A. Klenner ab,
Bo Zhangc,
Gianluca Ciancaleonid,
James K. Howarda,
Helen E. Maynard-Caselya,
Jack K. Clegge,
Massimiliano Massib,
Benjamin H. Fraser‡
*a and
Giancarlo Pascali‡*af
ab,
Bo Zhangc,
Gianluca Ciancaleonid,
James K. Howarda,
Helen E. Maynard-Caselya,
Jack K. Clegge,
Massimiliano Massib,
Benjamin H. Fraser‡
*a and
Giancarlo Pascali‡*af
aHuman Health & National Deuteration Facility, Australian Nuclear Science and Technology Organisation (ANSTO), Lucas Heights, NSW 2234, Australia. E-mail: gianp@ansto.gov.au; bfr@ansto.gov.au
bSchool of Molecular and Life Sciences, Curtin University, Bentley, WA 6102, Australia
cSchool of Chemistry, Monash University, Melbourne, VIC 3800, Australia
dDipartmento di Chimica e Chimica Industriale, Universit à di Pisa, Pisa, 56127, Italy
eSchool of Chemistry and Molecular Biosciences, The University of Queensland, St. Lucia, QLD 4072, Australia
fBrain and Mind Centre, The University of Sydney, Camperdown, NSW 2050, Australia
First published on 28th February 2020
Abstract
A novel fluorine-18 method employing rhenium(I) mediation is described herein. The method was found to afford moderate to high radiochemical yields of labelled rhenium(I) complexes. Subsequent thermal dissociation of the complexes enabled the radiosynthesis of fluorine-18 labelled pyridine bidentate structures which could not be radiofluorinated hitherto. This rhenium(I) complexation–dissociation strategy was further applied to the radiosynthesis of [18F]CABS13, an Alzheimer's disease imaging agent, alongside other 2,2′-bipyridine, 1,10-phenanthroline and 8-hydroxyquinoline labelled radiotracers. Computational modelling of the reaction mechanism suggests that the efficiency of rhenium(I) activation may be attributed to both an electron withdrawal effect by the metal center and the formation of an acyl fluoride intermediate which anchors the fluoride subsequent to nucleophilic addition.
Introduction
Radiotracers are commonly used to investigate biochemical systems in living organisms and are routinely employed as nuclear medicine imaging agents to diagnose cancers and other disease pathologies.1 The discovery of new radiolabelling methods is critical to widen the portfolio of radiotracers available to diagnose currently untreated disease states. Positron emission tomography (PET) is the most sensitive of all nuclear imaging techniques and employs molecules with a specific biological role, radioactively tagged with a positron emitting isotope. Among the suite of available positron emitters, fluorine-18 is the most widely utilized radioisotope in existing PET medicines.2,3 However, fluorine-18 nucleophilic radiolabelling reactions (i.e. the main route of utilization) can be particularly challenging and low yielding due to: the limiting half-life of the radioisotope (109.7 min),4 the relatively few methods available for selective carbon–fluorine bond formation (e.g. compared to other halogens),5 the low tracer concentrations typically employed and the general requirement for anhydrous conditions.6,7 Among these reactions, pyridine structures can be yet more difficult to radiolabel with fluorine-18 and oftentimes form in very low radiochemical yields (RCYs). For example, fluorine-18 labelled acetylcholine receptor imaging agents such as 2-[18F]fluoro-3-(2(S)-azetidinylmethoxy)pyridine and 6-chloro-3-((2(S)-azetidinyl)methoxy)-5-(2-[18F]fluoropyridine-4-yl)pyridine have typically been radiosynthesis within the range of 4–10% RCY.8–12 A fluorine-18 labelled pyridine tracer designed for the PET imaging of O6-methylguanine-DNA methyltransferase (MGMT) status of tumour tissues, 2-amino-6-(2-[18F]fluoropyridine-4-ylmethoxy)-9-(octyl-β-D-glucosyl)-purine, was also synthesised in only 5% RCY.13 Given these difficulties associated with fluorine-18 labeling pyridine-containing structures, it was hypothesized that a rhenium(I) activated approach could be a powerful tool towards expanding the scope of such radiotracers.14 To test this hypothesis, we selected three common bidentate structures throughout our investigation; 1,10-phenanthroline, 2,2′-bipyridine and 8-hydroxyquinoline. In each of these studies we systematically tested the nucleophilic substitution of chloro, bromo and/or nitro leaving groups for [18F]fluoride on different positions of the molecule to establish the advantages and limitations of a thermally mediated rhenium(I) complexation–dissociation strategy. Additionally, this reaction fits within a limited scope of fluorine-18 labelling methods which work under partially aqueous conditions, inclusive of AlF,15–18 boron-dipyrromethene (BODIPY)19–21 and ethenesulfonyl fluoride (ESF) agents.22–24 This discovery led us to compare reaction conditions employing azeotropically dried and non-dried fluorine-18 sources, the latter of which typically suppresses RCY due to the decreased nucleophilicity of the [18F]fluoride in aqueous media.3Results and discussion
Each ligand precursor or non-radioactive standard was based on three core structures: 2,2′-bipyridine (BiPy), 1,10-phenanthroline (phen) or 8-hydroxyquinoline (8HQ), as shown in Fig. 1. A list of the substituted analogues studied is shown in Table 1. Rhenium(I) complexes of each of the ligand precursors and standards were prepared as facial isomers [Re(BiPy)(CO)3Cl], [Re(Phen)(CO)3Cl] or [Re(8HQ)(CO)3(NCCH3)]. Each precursor (0.08 μmol in DMSO), bearing either a chloro, bromo or nitro leaving group was reacted with azeotropically dried tetraethylammonium (TEA) [18F]fluoride (29 ± 10 MBq) under microfluidic conditions using an automated Advion synthesis module (ESI: Fig. S115, Section S.8†). The solutions were each eluted through a microreactor (15.6 μL) at a rate of 20 μL min−1 (47 s reaction time). Reaction temperatures between 50 and 190 °C were trialled for each experiment in 20 °C increments. The solutions were subsequently analysed by radioHPLC and/or radioTLC to determine the non-isolated RCYs of the radioproducts.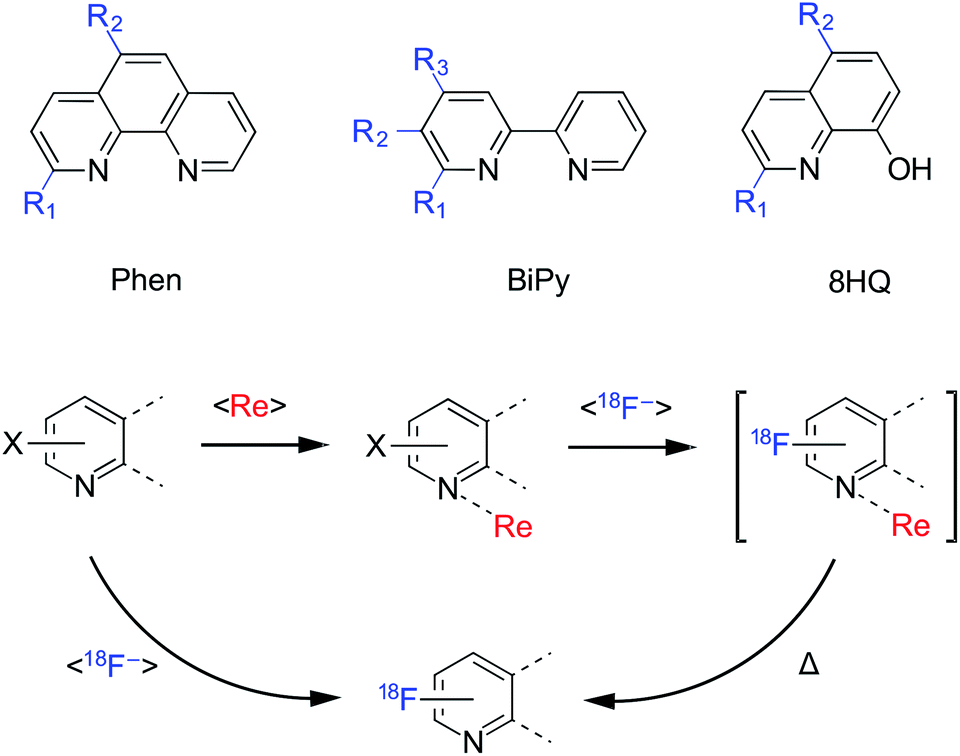 | ||
| Fig. 1 (Top) Substitution patterns for each of the BiPy, Phen and 8HQ ligands and complexes utilised as precursors or fluorinated standards. (Bottom) Model of rhenium(I) mediation. Rather than employing poor yielding direct fluorine-18 〈18F−〉 nucleophilic substitution, it was hypothesised that the pyridinyl bidentate ligands could first be complexed to a source of rhenium(I) 〈Re〉 then subsequently radiofluorinated and thermally dissociated (Δ) in a single step to afford the desired radiotracers in greater yield. | ||
| Compounda | Ligand structure | R1 | R2 | R3 |
|---|---|---|---|---|
| a [18F]L3e is also known as [18F]CABS13. | ||||
| L1a | BiPy | H | H | H |
| L1b | BiPy | Cl | H | H |
| L1c | BiPy | H | Cl | H |
| L1d | BiPy | H | H | Cl |
| L1e | BiPy | Br | H | H |
| L1f | BiPy | F | H | H |
| L1g | BiPy | H | F | H |
| L1h | BiPy | H | H | F |
| L2a | Phen | H | H | — |
| L2b | Phen | Cl | H | — |
| L2c | Phen | H | Cl | — |
| L2d | Phen | Br | H | — |
| L2e | Phen | H | NO2 | — |
| L2f | Phen | F | H | — |
| L2g | Phen | H | F | — |
| L3a | 8HQ | H | H | — |
| L3b | 8HQ | Cl | H | — |
| L3c | 8HQ | H | Cl | — |
| L3d | 8HQ | H | NO2 | — |
| L3ea | 8HQ | F | H | — |
| L3f | 8HQ | H | F | — |
Initial attempts to radiofluorinate all the BiPy ligand precursors (L1a–L1e) using standard radiofluorination conditions did not afford any of the labelled products across all trialled temperature ranges. Thus, an alternative synthetic route was hypothesised which first involved complexing the pyridine bidentate ligand to a source of [Re(CO)5Cl]. Heat supplied to the system then allowed for the rhenium(I)-mediated incorporation of fluorine-18, in concordance with a former study, whilst simultaneously facilitating expedient thermal decomplexation to liberate the radiolabelled ligand in considerable RCY. This rhenium(I) complexation–dissociation strategy is summarised in Fig. 1. Towards obtaining fluorine-18 labelled BiPy structures we first attempted the radiofluorination strategy on [Re(L1b)(CO)3Cl]. Such an approach indeed allowed for the expected formation of [18F][Re(L1f)(CO)3Cl] in a maximum 82% RCY at 130 °C. Furthermore, increasing the reaction temperature enabled such previously unreported thermal decomplexation and afforded the formerly unobtainable [18F]L1f ligand in a maximum 64% RCY at 190 °C. The two radiochromatograms showing the conversion of [18F][Re(L1f)(CO)3Cl] to [18F]L1f at 190 °C, alongside the formation of an unknown by-product, are superimposed in Fig. 2. The RCYs attained for both [18F]L1f and [18F][Re(L1f)(CO)3Cl] as a function of the reaction temperature are plotted in Fig. 3, including the reaction pathway. The trend clearly demonstrated an increase in RCY of [18F][Re(L1f)(CO)3Cl] as the temperature approached 130 °C, and subsequently decreased using higher temperatures as the ortho-substituted [18F]L1f was liberated in greater yield. Under non-azeotropically dried conditions this trend was also noticed, forming both [18F][Re(CO)3(L1f)Cl] and [18F]L1f in 29% RCY at 130 °C and 24% RCY at 190 °C, respectively (ESI: Fig. S71, Section S.4†). A similar trend was also observed for the bromo-substituted [Re(L1e)(CO)3Cl] precursor, which formed [18F][Re(L1f)(CO)3Cl] in 72% RCY peaking at 110 °C before decomplexing at higher temperatures to afford [18F]L1f in 51% RCY at 190 °C (ESI: Fig. S72, Section S.4†). On the other hand, testing the unsubstituted BiPy analogue [Re(CO)3(L1a)Cl] did not provide any radiofluorination product, save for the same radioactive by-product noticed with the previous ortho-substituted structure.
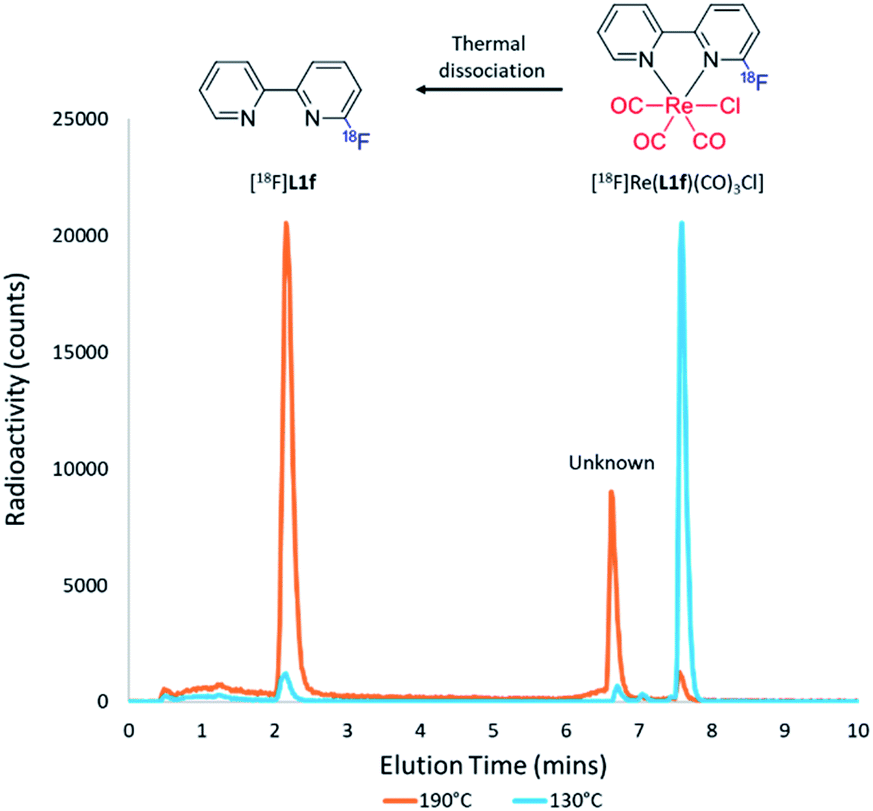 | ||
| Fig. 2 Radiochromatogram profiles for the fluorine-18 labelling of [Re(L1b)(CO)3Cl] at 130 °C (blue trace) and 190 °C (orange trace). At 130 °C [18F][Re(L1f)(CO)3Cl] (7.6 min) forms in the greatest yield, whereas at 190 °C the complex dissociates to form the otherwise unobtainable [18F]L1f ligand (2.2 min) alongside an unknown by-product (6.6 min). | ||
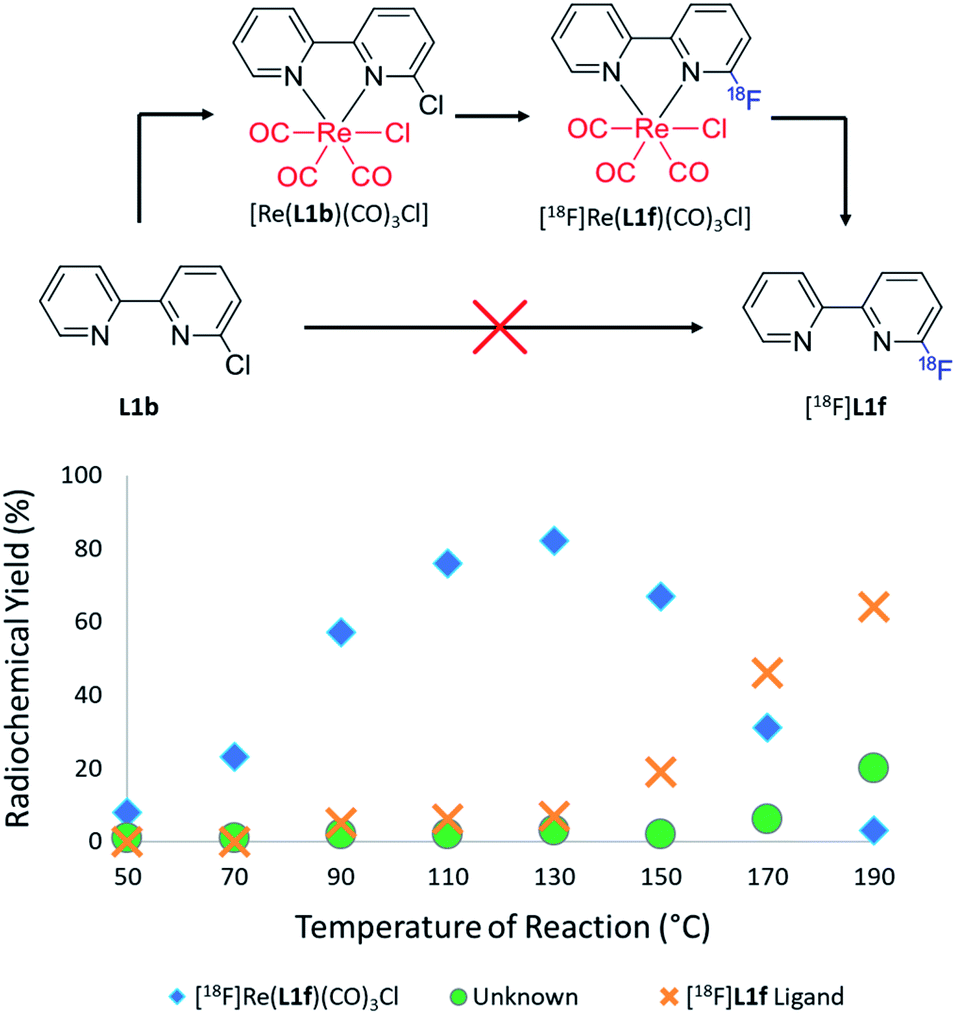 | ||
| Fig. 3 [18F]Fluoride substitution in the ortho position of [Re(L1b)(CO)3Cl] which afforded [18F][Re(L1f)(CO)3Cl] (blue diamonds). Thermal decomplexation led to the subsequent formation of [18F]L1f (orange crosses) and an unknown by-product (green circles) with the RCYs plotted as a function of reaction temperature. [18F]L1f was unable to be synthesised from L1b without first complexing to a rhenium(I) centre. | ||
Given the successful radiolabelling of the ortho-position of the pyridine ring when coordinated to the rhenium(I) center, the [Re(L1d)(CO)3Cl] precursor was subsequently synthesised to assess fluorine-18 labelling in the para-position. Such SNAr fluorinations of [Re(L1d)(CO)3Cl] afforded the radiolabelled complex in 68% RCY at 130 °C and exhibited thermal dissociation to afford the previously unsynthesisable [18F]L1h ligand in 27% RCY at 190 °C. This trend is illustrated in Fig. 4 which also exhibits the formation of a chromatographically distinct unknown by-product at higher temperatures, which formed in a maximum of 31% RCY at 190 °C. Increasing the reaction time from 47 s to 188 s resulted in the suppression of this reaction by-product, however, and the formation of [18F]L1h in 37% RCY. Despite the successful radiolabelling of the ortho and para positions of the pyridinyl moieties, [18F]fluoride substitution at the meta position of [Re(CO)3(L1c)Cl] afforded negligible yields of [18F][Re(CO3)(L1g)Cl] which were unable to be thermally dissociated to form the [18F]L1g ligand. Such results are consistent with the documented substitution efficiencies for SNAr reactions of aryl halides.
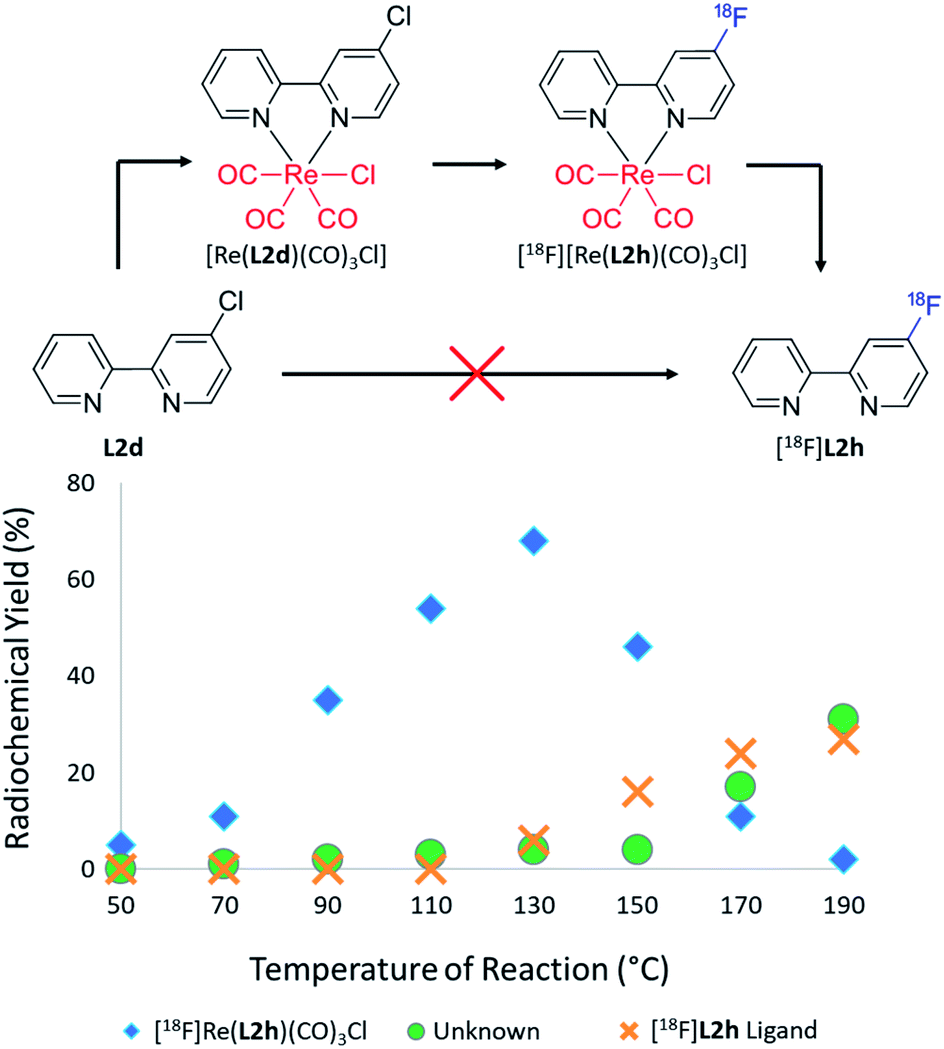 | ||
| Fig. 4 RCYs of [18F][Re(L2h)(CO)3Cl] (blue diamonds), [18F]L2h (orange crosses) and an unknown by-product (green circles) plotted as a function of the reaction temperature for the [18F]fluoride labelling of the para substituted [Re(CO)3(L2d)Cl] complex. [18F]L2h was unable to be synthesised from L2d unless mediated by rhenium(I) complexation as per the above scheme. | ||
Translation of the rhenium(I) complexation approach to Phen ligand precursors (L2a–L2e) resulted first in the formation of [18F][Re(CO)3(L2f)Cl] in 90% RCY at 90 °C from [Re(CO)3(L2b)Cl]. Minor thermal dissociation liberated [18F]L2f in 13% RCY at 170 °C (ESI: Fig. S62, Section S.4†), though this radioligand was found to form in 61% RCY without the aid of rhenium(I) complexation given the same 0.08 μmol concentration of L2b precursor and reaction conditions. Radiofluorination of the bromo substituted [Re(CO)3(L2d)Cl] precursor also mimicked this trend, forming [18F][Re(CO)3(L2f)Cl] in 69% RCY at 110 °C, though decomplexing only 8% RCY of [18F]L2f at 170 °C (ESI: Fig. S63, Section S.4†). This was not true for the fluorine-18 labelling of [Re(CO)3(L2c)Cl], however, which consisted of a chloro leaving group in the 5-position rather than the 2-position of the phenanthroline ring. In this case, [18F][Re(CO)3(L2g)Cl] still formed in 33% RCY (Section S.4, Fig. S64 of the ESI†) but did not dissociate to form the [18F]L2g ligand. Direct fluorine-18 labelling of both the L2c and L2e ligand precursors did not afford the [18F]L2g radio product either. Fluorine-18 labelling of the [Re(CO)3(L2e)Cl] precursor, however, afforded not only [18F][Re(CO)3(L2g)Cl] in 39% RCY at 110 °C but also 2% RCY of the otherwise unobtainable [18F]L2g ligand. This may likely be due to the enhanced electron withdrawal effect of the nitro group which improved yields of the fluorine-18 labelled rhenium(I) complex sufficient for the dissociation of the radioligand in detectable quantities. Non-azeotropically distilled media still allowed for the syntheses of [18F]Re(CO)3(L2f)Cl] and [18F][Re(CO)3(L2g)Cl] from [Re(CO)3(L2b)Cl] and [Re(CO)3(L2c)Cl] precursors in 59% and 2% RCY, respectively. An unknown by-product was also found to form from each of the [Re(CO)3(phen)Cl] complexes, including the non-functionalized [Re(CO)3(L2a)Cl] precursor, as was observed for the suite of [Re(CO)3(BiPy)Cl] precursor analogues. A possible explanation for the discrepancy between 2,2-bipyridine and 1,10-phenanthroline rhenium-decomplexation efficiencies could be the contribution of an approximate 20–40° twist between the two pyrindyl units of [Re(BiPy)(CO)3Cl] complexes, which result in lesser molecular rigidity and greater RCYs via dissociation.25–28 The optimum RCYs attained for each of the radiofluorinated BiPy, Phen, [Re(CO)3(BiPy)Cl] and [Re(CO)3(Phen)Cl] radiotracers, as dissociated from their respective rhenium(I) precursors at the listed temperatures, are itemised in Table 2.
| Precursor | Radiotracer | T (°C) | RCY (%) |
|---|---|---|---|
| a Reaction time of 188 s used instead of 47 s. | |||
| [18F][Re(CO)3(L1b)Cl] | [18F][Re(CO)3(L1f)Cl] | 130 | 82 |
| [18F]L1f | 190 | 64 | |
| [18F][Re(CO)3(L1d)Cl] | [18F][Re(CO)3(L1h)Cl] | 130 | 68 |
| [18F]L1ha | 190 | 37 | |
| [18F][Re(CO)3(L1e)Cl] | [18F][Re(CO)3(L1f)Cl] | 110 | 69 |
| [18F]L1f | 190 | 51 | |
| [18F][Re(CO)3(L2b)Cl] | [18F][Re(CO)3(L2f)Cl] | 90 | 91 |
| [18F]L2f | 170 | 13 | |
| [18F][Re(CO)3(L2d)Cl] | [18F][Re(CO)3(L2f)Cl] | 110 | 69 |
| [18F]L2f | 170 | 8 | |
| [18F][Re(CO)3(L2e)Cl] | [18F][Re(CO)3(L2g)Cl] | 110 | 39 |
| [18F]L2g | 130 | 2 | |
After establishing the efficiency of the rhenium(I) complexation–dissociation method for the fluorine-18 labelling of diimine ligands, there was much interest in translating the method to 8HQ structures. This interest was due to the proposed use of 8HQ derivatives as markers of Amyloid beta (Aβ) plaques characteristic of Alzheimer's disease.29–33 In particular, it was hypothesized that the rhenium(I) complexation–dissociation approach could be applied to the [18F]CABS13 ([18F]L3e) Alzheimer's disease PET diagnostic imaging agent34,35 Current radiosyntheses of [18F]CABS13 require a multi-step process of radiolabelling and deprotection of the alcohol, as shown in Fig. 5. By protecting the alcohol via a dative Re–O bond in a [Re(CO)3(L3b)(NCCH3)] precursor, as also shown in Fig. 5, we envisioned that the radiosynthesis of [18F]CABS13 could be simplified to a single step protocol. Initial fluorine-18 radiolabelling of [Re(CO)3(L3b)(NCCH3)] under the standard conditions afforded only 2% RCY of both the [18F][Re(CO)3(L3e)(NCCH3)] complex and the [18F]CABS13 radiotracer at 190 °C. Thus the precursor mass was increased ten-fold to 0.8 μmol which resulted in a remarkable increase to 18% RCY of the radiolabelled [18F][Re(CO)3(L3e)(NCCH3)] complex and 5% of the dissociated [18F]CABS13 tracer, as shown by the radiochromatogram in Fig. 5. While the RCY of [18F]CABS13 achieved via the rhenium(I) complexation–dissociation approach is still lower than the 19 ± 5% achieved by Liang, et al.,35 the synthesis is certainly still simplified by a single step and the 18% RCY of [18F][Re(CO)3(L3e)(NCCH3)] implies that more of the desired tracer could still be obtained under optimised dissociation conditions. Indeed, such results also suggest that greater RCYs from all trialled precursors herein could be attained by modifying the precursor mass. More intriguingly, the rhenium(I) complexation approach was also translated to the radiosynthesis of [18F]L3f. Experiments have demonstrated that L3f is more efficient at disaggregating Aβ plagues than CABS13 (L3e),36 however this structure was unable to be radiolabelled with fluorine-18 via traditional means. By applying the rhenium(I) complexation–dissociation strategy, it was discovered that the [18F]L3f tracer was able to be synthesised from both [Re(CO)3(L3c)(NCCH3)] and [Re(CO)3(L3d)(NCCH3)] precursors in 1% and 2% RCY, respectively. The [Re(CO)3(L3f)(NCCH3)] intermediate was also found to form in 3% and 6% RCY from the [Re(CO)3(L3c)(NCCH3)] and [Re(CO)3(L3d)(NCCH3)] precursors, respectively. Worthwhile mentioning was that the fluorine-18 labelling of such [Re(CO)3(8HQ)(NCCH3)] structures formed no unknown by-products, as had been observed in the analogous complexes of BiPy and Phen ligands. This observation was further evidenced by the radiolabelling of unsubstituted [Re(CO)3(L3a)(NCCH3)] which afforded no radioproducts at all. Such observations have led us to speculate that the unknown by-product may correlate with a Re–18F bonded species, albeit non-radioactive analogues of this nature are difficult to both synthesise and characterise for the sake of confirmation (particulary given that Re–Cl & Re–F bonds readily dissociate under ESI-MS conditions).37,38 The RCYs attained for each of the labelled 8HQ structures as a function of reaction temperature can be viewed in Section S.4 of the ESI† for both azeotropically dried and non-dried conditions.
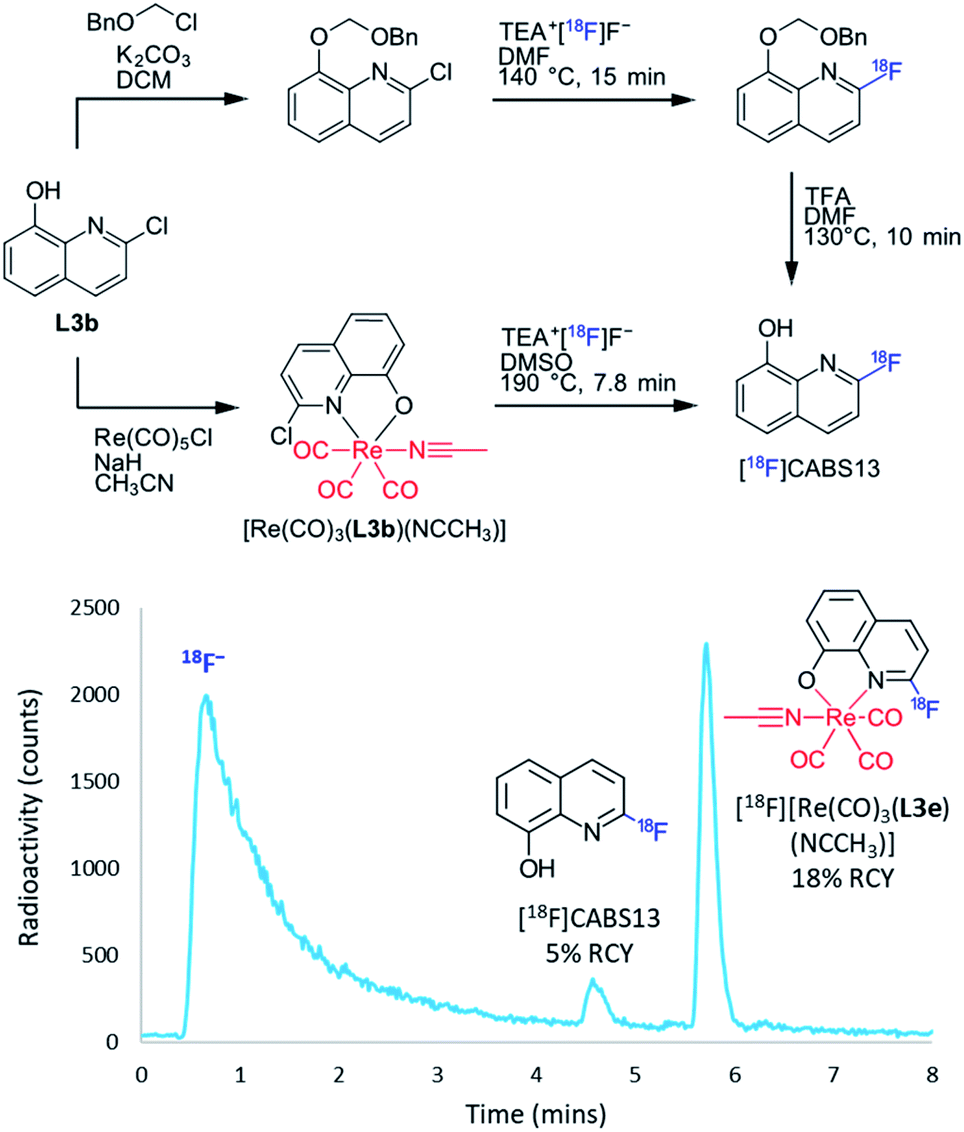 | ||
| Fig. 5 (Top) Alternative radiosynthesis of [18F]CABS13 via the rhenium(I) complexation–dissociation method circumvented multiple-step synthesis, as reported by Liang, et al.34 (Bottom) Radiochromatogram depicting the formation of [18F]CABS13 in 5% RCY from the thermal dissociation of [18F][Re(CO)3(L3e)(NCCH3)] which formed in 18% RCY. | ||
Towards understanding the mechanism by which rhenium(I) complexation–dissociation improves the fluorination of a pyridine bidentate structure, we opted to model the reaction via density functional theory (DFT) calculations at the B97-D3/def2-TZVP level of theory. The input coordinates were acquired from the XRD data of crystals of the rhenium(I) complexes grown from slow evaporation in DMSO solution. First, the coordination on the rhenium(I) centre led to a depletion of electronic density on the ligand, a phenomenon also demonstrated by the downfield chemical shifts observed in 1H-NMR spectra of the rhenium(I) complexes, as shown by the example spectra of L2b and [Re(L2b)(CO)3Cl] in Fig. 6. This depletion can be quantified by the Natural Orbitals for Chemical Valence-Charge Displacement (NOCV-CD),39 according to which the L → M σ-donation was 0.266e and the L ← M π back-donation was found to be 0.116e in the case of [Re(CO)3(L1f)Cl]. Such electronic depletion favours nucleophilic addition of the fluoride on the carbon bound to the chlorine atom. The DFT computed activation energies were 6.3 and 34 kcal mol−1 in the presence and absence of the metal fragment, respectively. Secondly, the fluoride was discovered to bond to the carbonyl in the cis position relative to the diimine ligand, forming an acyl fluoride intermediate as shown in Fig. 7. This intermediate was notably unstable, however, and quickly detached from the carbonyl anchor undergoing SNAr for the chlorine atom. The proposed fluorination mechanism, occurring between the complexation and dissociation phases, is illustrated in Fig. 8. From the experimental data it was observed that [Re(CO)3(L1f)Cl] formed in a greater fluorination yield than [Re(CO)3(L1h)Cl], whereas the [Re(CO)3(L1g)Cl] complex formed in almost no appreciable yield at all. This trend can be explained by calculating the atomic charges by Natural Population Analysis, whereupon it was shown that the carbon atoms bound to the chlorine consisted of partial charges equal to 0.29, −0.01 and 0.04e for the [Re(CO)3(L1b)Cl], [Re(CO)3(L1c)Cl] and [Re(CO)3(L1d)Cl] precursors, respectively. These values are in line with the observed reactivity, whereby a positive partial charge facilitates fluorination and a negative partial charge inhibits nucleophilic addition of the fluoride. For example, [Re(L1b)(CO)3Cl] and [Re(L2b)(CO)3Cl] were calculated to have the highest partial charges of 0.29e and 0.31e which correlated with high yields of 82% and 91% RCY for their radiolabelled analogues, respectively. Whereas [Re(CO)3(L1c)Cl], for which a negative partial charge of −0.01e was calculated, did not form the radiolabelled complex in ample quantity for detection. The partial charges are plotted in Table 3 for some select Phen and 8HQ complexes as well, alongside their associated ligands. In each case the partial charge for the carbon bound to the chlorine or nitro leaving group was found to be greater in the complex than in the ligand, save for [Re(CO)3(L2e)Cl] and L2e which differed only by 0.01e.
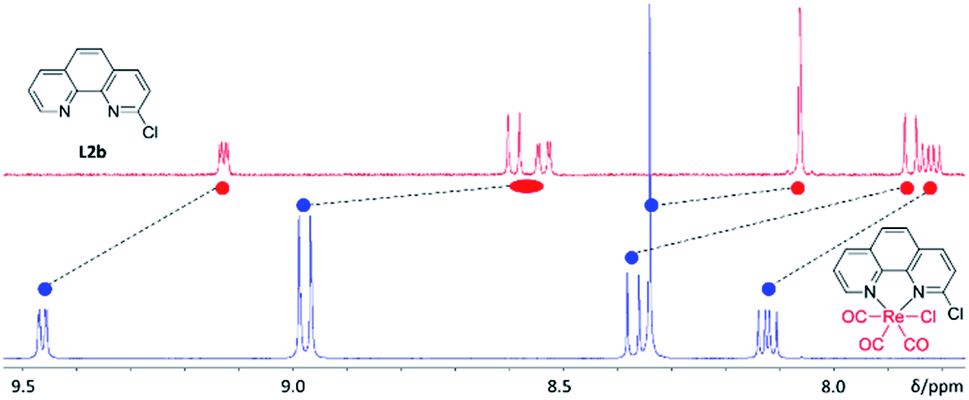 | ||
| Fig. 6 Example of the downfield chemical shifts in 1H-NMR aromatic signals observed when complexing L2b (top, red) with [Re(CO)5Cl] to form [Re(L2b)(CO)3Cl] (bottom, blue). The shifts suggest electron withdrawal from the π-aromatic system which likely facilitates nucleophilic substitution for [18F]fluoride. | ||
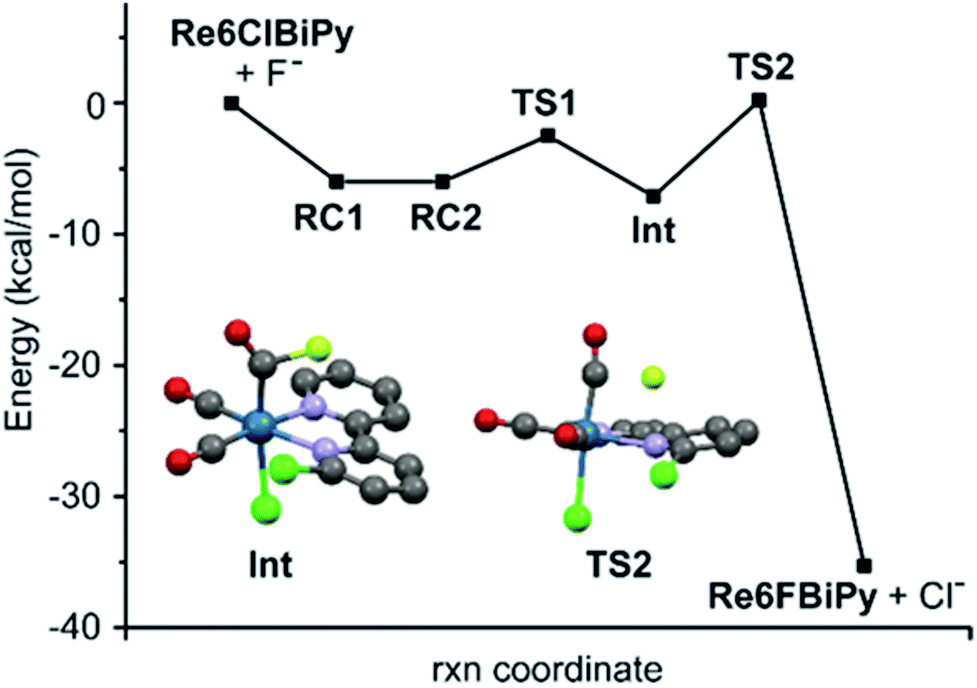 | ||
| Fig. 7 DFT modelling of the reaction mechanism forming [Re(CO)3(L1h)Cl] (Re6FBiPy) from [Re(CO)3(L1b)Cl] (Re6ClBiPy). The energies for the optimized geometries of the acyl fluoride intermediate species (Int) and the rate-determining transition states (TS) are plotted along the reaction coordinate. | ||
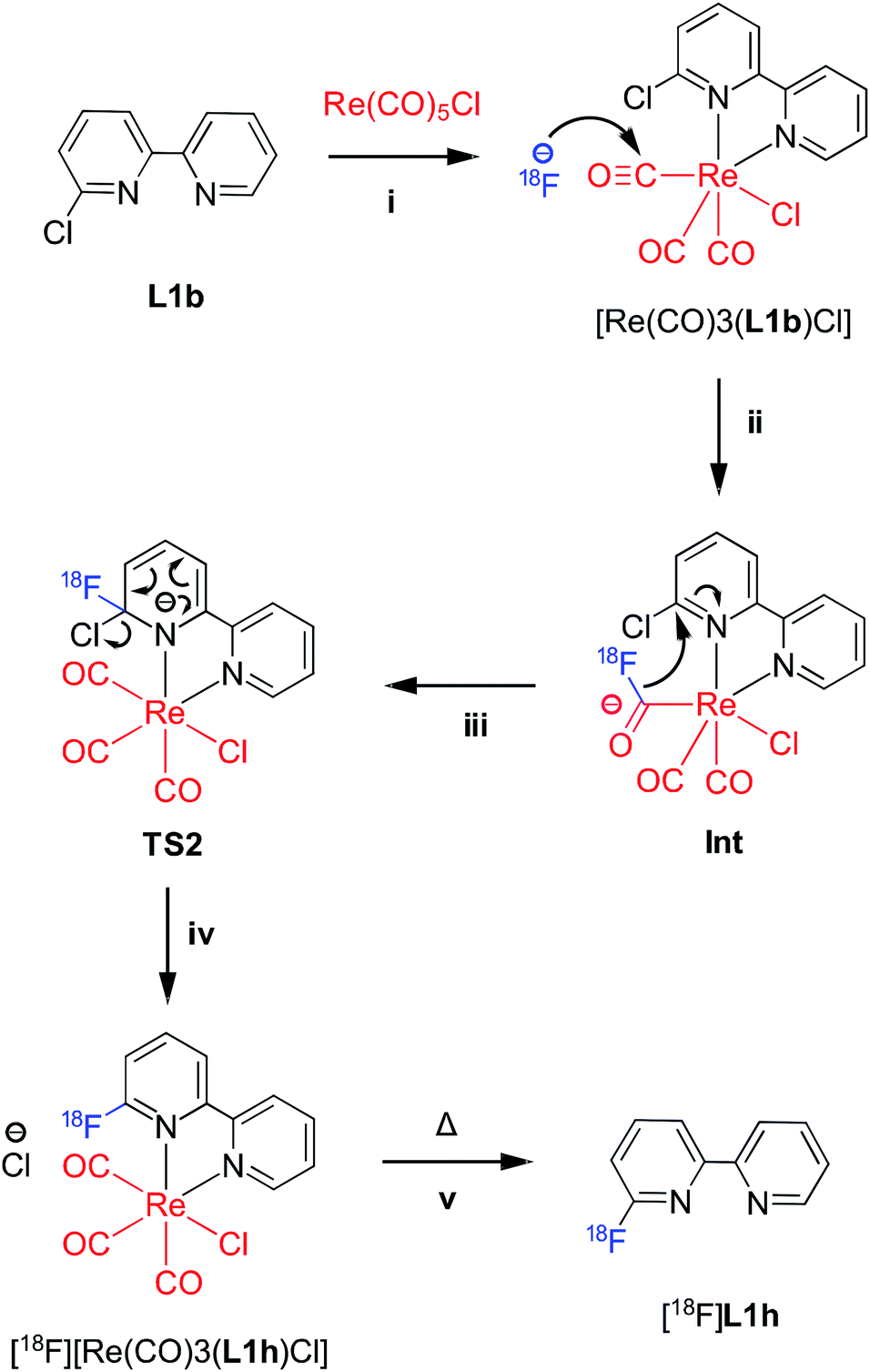 | ||
| Fig. 8 Example of the proposed rhenium-assisted radiolabelling mechanism: (i) precursor ligand L1b is complexed to a source of [Re(CO5)Cl] to form [Re(CO)3(L1b)Cl]; (ii) [18F]fluoride reacts with the cis-carbonyl of [Re(CO)3(L1b)Cl] to from an acyl fluoride intermediate species (Int); (iii) the fluorine-18 radioisotope is in proximity of the chlorine leaving group, enabling SNAr to form the second transition state (TS2); (iv) chloride is displaced from the aromatic ring, thus forming [18F][Re(CO)3(L1h)Cl]; (v) thermal decomplexation of [18F][Re(CO)3(L1h)Cl] liberates the desired [18F]L1h radiotracer. | ||
| Ligand | q (C) | Re(I) complex | q (C) | ϕ (%) |
|---|---|---|---|---|
| L1b | 0.26 | [Re(CO)3(L1b)Cl] | 0.29 | 0.5 |
| L1e | 0.17 | [Re(CO)3(L1e)Cl] | 0.18 | — |
| L1c | −0.03 | [Re(CO)3(L1c)Cl] | −0.01 | 1.0 |
| L1d | 0.03 | [Re(CO)3(L1d)Cl] | 0.04 | 0.6 |
| L2b | 0.27 | [Re(CO)3(L2b)Cl] | 0.31 | 0.3 (ref. 14) |
| L2e | 0.12 | [Re(CO)3(L2e)Cl] | 0.11 | — |
| L3d | — | [Re(CO)3(L2e)(NCCH3)] | 0.08 | — |
Finally, quantum yields of select complexes taking the form of [Re(CO)3(BiPy)Cl] were performed to determine the utility of such complexes as multimodal PET-optical probes. The quantum yields were determined to be 0.5%, 1.0% and 0.6% for the [Re(CO)3(L1b)Cl], [Re(CO)3(L1c)Cl] and [Re(CO)3(L1d)Cl] precursors in DMSO solution, respectively. These quantum yields are depicted in Table 3, alongside the [Re(CO)3(L2b)Cl] precursor for which a quantum yield of 0.3% was determined in one of our former studies.14 The fluorine substituted [Re(CO)3(L1f)Cl] was also found to have a quantum yield of 0.6% in DMSO solution. Emission spectra from these photophysical assessments can be found in Section S.6, Fig. S82–S85 of the ESI.† Transitions to the metal–ligand charge transfer (MLCT) excited state occurred in the UV region, with corresponding dπ → π* electronic transitions occurring at 380 nm for [Re(CO)3(L1b)Cl], 379 nm for [Re(CO)3(L1c)Cl] and at 369 nm for both [Re(CO)3(L1d)Cl] and [Re(CO)3(L1f)Cl] complexes, as depicted by the UV-Vis spectra in Section S.2 of the ESI.†
Conclusions
A novel rhenium(I) complexation–dissociation method for labelling with the fluorine-18 radioisotope is reported herein. The rhenium(I) centre improved fluorination via an electron withdrawal effect as suggested by the NMR data and, in accordance with our computational models, may facilitate radiofluorination via formation of an acyl fluoride intermediate with a carbonyl ancillary ligand. Through a series of microfluidic radiosyntheses we verified that moderate to high RCYs could be obtained by substituting [18F]fluoride in the rhenium(I) activated ortho and para positions of bidentate pyridine ligands, as well as in the 5-position of a Phen ligand. These radiolabelled rhenium(I) complexes are currently being further pursued for utility as PET-optical multimodal agents. Thermal decomplexation of such rhenium(I) complexes were shown to be particularly useful for synthesizing fluorine-18 labelled Bipy ligands, which were unable to be synthesised under analogous conditions without rhenium(I) activation. The formerly unobtainable [18F]L3f radio product was also attained from the rhenium(I) complexation–dissociation approach, which will enable its further investigation as an alternative 8HQ Alzheimer's disease imaging tracer to [18F]CABS13. Furthermore, the rhenium(I) complexation–dissociation strategy joins the limited suite of radiofluorinating methods which can afford radiotracers under partially aqueous conditions. Future work towards alternative decomplexation methods (e.g. photodissociation or acid dissociation), as well as attempts to fluorine-18 label monodentate pyridine ligands complexed to the rhenium(I) core, may further expand the scope of radiotracer syntheses via this rhenium(I) mediated approach.Experimental section
Synthetic protocols for rhenium complexes
Complex derivatives of [Re(CO)3(Phen)Cl] and [Re(CO)3(BiPy)Cl] were prepared by refluxing rhenium(I) pentacarbonyl chloride (300 mg, 830 μmol, 1.0 eq.) and the appropriately substituted chloro, bromo or nitro 2,2-bipyridine or 1,10-phenanthroline (1.1 eq.) in a solution of anhydrous toluene (10 mL) for 4 hours in an inert nitrogen environment, resulting in a colour change from blue or colourless to yellow or orange. The solid precipitate was isolated by vacuum filtration and washed with diethyl ether. The crude material was then dissolved in a minimum of DCM and eluted over neutral alumina Brockmann grade II using a MeOH/DCM gradient mobile phase. The eluents containing the product were subsequently combined and evaporated down under reduced pressure to afford the yellow or orange powder.Complex derivatives of [Re(CO)3(8HQ)(NCCH3)] were prepared by first refluxing rhenium(I) pentacarbonyl chloride (100 mg, 275 μmol, 1.0 eq.) in anhydrous acetonitrile (10 mL) for 4 hours in an inert nitrogen environment to synthesise the rhenium(I) diacetonitrilo tricarbonyl chloride intermediate, which was monitored by HPLC and IR spectroscopy. Simultaneously, the appropriate chloro or nitro substituted 8-hydroxyquinoline (1.0 eq.) was deprotonated with sodium hydride (60% wt/wt in mineral oil washed with n-hexanes) in anhydrous acetonitrile (2 mL) at room temperature under an inert nitrogen environment, resulting in a vibrant green solution. The green solution was then filtered (0.2 μm PTFE) and added dropwise to the solution containing rhenium. This solution was then left to stir for 12 hours at room temperature eventually forming an orange solution. The orange solution was added to a volume of aqueous triflic acid (0.1 M, 10 mL) and evaporated under reduced pressure to remove the acetonitrile, thus precipitating out a yellow or orange solid in aqueous suspension. The crude material was then isolated by vacuum filtration, dissolved in a minimum of acetonitrile and eluted over neutral alumina Brockmann grade II using an isocratic acetonitrile mobile phase. The eluents containing the product were subsequently combined and evaporated down under reduced pressure to afford the yellow or orange powder.
Further details on the syntheses of the rhenium(I) complexes and the fluorinated ligands can be found in Sections S.2 and S.3 of the ESI.† Characterisation data for the rhenium(I) complexes tricarbonylchloro(2,2′-bipyridine)rhenium(I) ([Re(CO)3(L1a)Cl]),40,41 tricarbonylchloro(1,10-phenanthroline)rhenium(I) ([Re(CO)3(L2a)Cl])42 and acetonitriletricarbonyl(8-quinolate)rhenium(I) ([Re(CO)3(L3a)NCCH3]) can be found in the existing literature.43
Characterisation data for novel complexes
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O, δ 197.3CO, δ 189.5CO, δ 157.2, δ 155.6, δ 153.2, δ 152.8, δ 142.7, δ 140.3, δ 128.0, δ 127.7, δ 125.3, δ 123.1. FTIR (ATR corr. ṽ/cm−1): ṽ 3075 (w, C–H sp2 str.), ṽ 2018 (s, A′(1) CO str.), ṽ 1882 (s, A′(2) & A′′ CO str.). UV/Vis (λ/nm, ε/L mol−1 cm−1): λ 380 (dπ → π*, ε 3375.36), λ 301 (π → π*, ε 14
O, δ 197.3CO, δ 189.5CO, δ 157.2, δ 155.6, δ 153.2, δ 152.8, δ 142.7, δ 140.3, δ 128.0, δ 127.7, δ 125.3, δ 123.1. FTIR (ATR corr. ṽ/cm−1): ṽ 3075 (w, C–H sp2 str.), ṽ 2018 (s, A′(1) CO str.), ṽ 1882 (s, A′(2) & A′′ CO str.). UV/Vis (λ/nm, ε/L mol−1 cm−1): λ 380 (dπ → π*, ε 3375.36), λ 301 (π → π*, ε 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 990.57), λ 326 (π → π*, ε 10920.28), λ 233 (π → π*, ε 19259.41). LRMS (ESI+): [M–Cl]+ m/z cald: 460.97, m/z obvs 460.95. HRMS (ESI+): [M–Cl]+ m/z cald: 460.9703, m/z obvs 460.9694 (Δ −2.0 ppm). EA (%): calc. C 31.46, H 1.42, N 5.64. Found C 31.59, H 1.17, N 5.70.O, δ 197.2CO, δ 189.6CO, δ 154.2, δ 154.1, δ 153.0, δ 150.9, δ 140.4, δ 140.2, δ 133.9, δ 128.1, δ 125.3, δ 124.8. FTIR (ATR corr. ṽ/cm−1): ṽ 3113 (w, C–H sp2 str.), ṽ 2021 (s, A′(1) CO str.), ṽ 1883 (s, A′(2) & A′′ CO str.). UV/Vis (λ/nm, ε/L mol−1 cm−1): λ 379 (dπ → π*, ε 7346.37), λ 299 (π → π*, ε 36136.21), λ 241 (π → π*, ε 42589.10). LRMS (ESI+): [M–Cl]+ m/z cald: 460.97, m/z obvs 460.95. HRMS (ESI+): [M–Cl]+ m/z cald: 460.9703, m/z obvs 460.9730 (Δ 5.9 ppm). EA (%): calc. C 31.46, H 1.42, N 5.64. Found C 31.61, H 1.14, N 5.44.O, δ 197.5CO, δ 189.7CO, δ 156.9, δ 154.3, δ 153.8, δ 153.1, δ 147.0, δ 140.3, δ 128.4, δ 127.7, δ 125.0, δ 124.7. FTIR (ATR corr. ṽ/cm−1): ṽ 3117 (w, C–H sp2 str.), ṽ 2019 (s, A′(1) CO str.), ṽ 1884 (s, A′(2) & A′′ CO str.). UV/Vis (λ/nm, ε/L mol−1 cm−1): λ 380 (dπ → π*, ε 7548.93), λ 292 (π → π*, ε 38022.44), λ 235 (π → π*, ε 48347.07). LRMS (ESI+): [M–Cl]+ m/z cald: 460.97, m/z obvs 460.95. HRMS (ESI+): [M–Cl]+ m/z cald: 460.9703, m/z obvs 460.9712 (Δ 2.0 ppm). EA (%): calc. C 31.46, H 1.42, N 5.64. Found C 31.81, H 1.20, N 5.32.O, δ 197.2CO, δ 190.0CO, δ 157.6, δ 156.0, δ 152.7, δ 146.2, δ 142.0, δ 140.2, δ 131.9, δ 127.8, δ 125.3, δ 123.3. FTIR (ATR corr. ṽ/cm−1): ṽ 3074 (w, C–H sp2 str.), ṽ 2017 (s, A′(1) CO str.), ṽ 1884 (s, A′(2) & A′′ CO str.). LRMS (ESI+): [M–Cl]+ m/z cald: 504.92, m/z obvs 505.04. EA (%): calc. C 28.87, H 1.30, N 5.18. Found C 28.90, H 1.05, N 5.21.O, δ 196.6CO (d, Jd = 16.59 Hz), δ 188.9CO, δ 164.0, δ 161.5, δ 154.4 (d, Jd = 15.92 Hz), δ 153.1, δ 145.9 (d, Jd = 10.15 Hz), δ 140.4, δ 128.2, δ 125.1, δ 121.4 (d, Jd = 2.94 Hz), δ 112.7 (d, Jd = 30.19 Hz). 19F{1H} NMR (376 MHz, DMSO-d6, δ/ppm): δ −51.75. FTIR (ATR corr. ṽ/cm−1): ṽ 3040 (w, C–H sp2 str.), ṽ 2016 (s, A′(1) CO str.), ṽ 1880 (s, A′(2) & A′′ CO str.). UV/Vis (λ/nm, ε/L mol−1 cm−1): λ 369 (dπ → π*, ε 2768.36), λ 296 (π → π*, ε 12485.88), λ 235 (π → π*, ε 15367.33). LRMS (ESI+): [M–Cl]+ m/z cald: 445.00, m/z obvs 445.10. EA (%): calc. C 32.54, H 1.47, N 5.84. Found C 32.59, H 1.16, N 5.88.O, δ 197.6CO, δ 189.9CO, δ 168.3, δ 168.3, δ 158.9 (d, Jd = 10.26 Hz), δ 156.2 (d, Jd = 9.99 Hz), δ 154.6, δ 153.1, δ 140.5, δ 128.5, δ 124.9, δ 116.0 (d, Jd = 19.7 Hz), δ 113.0 (d, Jd = 22.79 Hz). 19F{1H} NMR (376 MHz, DMSO-d6, δ/ppm): δ −94.71. FTIR (ATR corr. ṽ/cm−1): ṽ 3117 (w, C–H sp2 str.), ṽ 2018 (s, A′(1) CO str.), ṽ 1884 (s, A′(2) & A′′ CO str.). LRMS (ESI+): [M–Cl]+ m/z cald: 445.00, m/z obvs 444.92. EA (%): calc. C 32.54, H 1.47, N 5.84. Found C 32.52, H 1.33, N 5.81.O), δ 197.65 (CO), δ 189.83 (CO), δ 154.75, δ 154.11, δ 147.19, δ 146.56, δ 142.81, δ 139.92, δ 131.69, δ 129.66, δ 128.37, δ 128.09, δ 127.30, δ 127.24. FTIR (ATR corr. ṽ/cm−1): ṽ 3042 (w, C–H sp2 str.), ṽ 2021 (s, A′(1) CO str.), ṽ 1889 (s, A′(2) & A′′ CO str.). UV/Vis (CH3CN, λ/nm, ε/L mol−1 cm−1): λ 368 (Re(dπ) → π*, ε 374.65), λ 270 (π → π*, ε 2612.13), λ 218 (π → π*, ε 4703.92), λ 204 (π → π*, ε 4589.45). LRMS (ESI+): [M–Cl]+ m/z calc. 484.97, m/z obsv. 485.07. HRMS (ESI+): [M + Na]+ m/z calc. 540.9261 (C15H7N2O3FCl2ReNa+); m/z obsv. 540.9266 (Δ 0.90 ppm). EA (%): calc. C 34.62, H 1.36, N 5.38, Cl 13.63; obsv. C 34.45, H 0.95, N 5.36, Cl 13.55.O, δ 197.2CO, δ 189.6CO, δ 154.2, δ 154.1, δ 153.0, δ 150.9, δ 140.4, δ 140.2, δ 133.9, δ 128.1, δ 125.3, δ 124.8. FTIR (ATR corr. ṽ/cm−1): ṽ 3113 (w, C–H sp2 str.), ṽ 2021 (s, A′(1) CO str.), ṽ 1883 (s, A′(2) & A′′ CO str.). UV/Vis (λ/nm, ε/L mol−1 cm−1): λ 379 (dπ → π*, ε 7346.37), λ 299 (π → π*, ε 36136.21), λ 241 (π → π*, ε 42589.10). LRMS (ESI+): [M–Cl]+ m/z cald: 460.97, m/z obvs 460.95. HRMS (ESI+): [M–Cl]+ m/z cald: 460.9703, m/z obvs 460.9730 (Δ 5.9 ppm). EA (%): calc. C 31.46, H 1.42, N 5.64. Found C 31.61, H 1.14, N 5.44.O, δ 197.1CO, δ 189.8CO, δ 153.5, δ 147.8, δ 147.2, δ 146.4, δ 141.4, δ 139.4, δ 131.3, δ 130.8, δ 129.4, δ 127.8, δ 127.7, δ 126.6. FTIR (ATR corr. ṽ/cm−1): ṽ 3043.35 (w, C–H sp2 str.), ṽ 2014.27 (s, A′(1) CO str.), ṽ 1883.98 (s, A′(2) & A′′ CO str.). EA (%): calc. C 31.90, H 1.25, N 4.96. Found C 31.94, H 1.00, N 5.08.O, δ 194.4CO, δ 178.1CO, δ 151.6, δ 143.3, δ 137.1, δ 132.9, δ 130.8, δ 127.0, δ 126.9, δ 121.4, δ 114.2. FTIR (ATR corr. ṽ/cm−1): ṽ 2939 (w, C–H sp2 str.), ṽ 2918 (w, C–H sp2 str.), ṽ 2328 (w, NC str.), ṽ 2019 (s, A′(1) CO str.), ṽ 1900 (s, A′(2) & A′′ CO str.).O), δ 196.54 (d, Jd = 14 Hz, CO), δ 189.99 (CO), δ 163.77, δ 161.18, δ 153.90, δ 145.79 (d, Jd = 11 Hz), δ 145.09 (d, Jd = 2 Hz), δ 144.07 (d, Jd = 3 Hz), δ 139.48, δ 130.99, δ 128.29 (d, Jd = 1 Hz), δ 127.32 (d, Jd = 2 Hz), δ 127.09 (d, Jd = 61 Hz), δ 113.48 (d, Jd = 32 Hz). 19F NMR (376 MHz, DMSO-d6, δ/ppm): δ −36.75 (dd, Jd = 6.92, 1.92 Hz), δ −106.00 (ISTD: para-DFB). FTIR (ATR corr. ṽ/cm−1): ṽ 3077 (w, C–H sp2 str.), ṽ 2021 (s, A′(1) CO str.), ṽ 1890 (s, A′(2) & A′′ CO str.). UV/Vis (CH3CN, λ/nm, ε/L mol−1 cm−1): λ 375 (Re(dπ) → π*, ε 2720.95), λ 343 (π → π*, ε 2821.73), λ 269 (π → π*, ε 22069.94), λ 262 (π → π*, ε 22170.71), λ 215 (π → π*, ε 38798.75), λ 200 (π → π*, ε 40915.05). LRMS (ESI+): [M–Cl]+ m/z calc. 469.00, m/z obsv. 469.12. HRMS (ESI+): [M + Na]+ m/z calc. 524.9557 (C15H7N2O3FClReNa+); m/z obsv. 524.9579 (Δ 4.20 ppm). EA (%): calc. C 3575, H 1.40, N 5.56, Cl 7.04; obsv. C 35.73, H 0.98, N 5.59, Cl 6.83.O, δ 197.3CO, δ 189.6CO, δ 156.8, δ 154.5, δ 154.3, δ 152.9 (d, Jd = 2 Hz), δ 146.5 (d, Jd = 7 Hz), δ 143.6, δ 139.1 (d, Jd = 5 Hz), δ 133.0 (d, Jd = 4 Hz), δ 129.6 (d, Jd = 4 Hz), δ 126.9 (d, Jd = 3 Hz), δ 122.8 (d, Jd = 23 Hz), δ 109.3 (d, Jd = 22 Hz). 19F{1H} NMR (376 MHz, CD3CN, δ/ppm): δ −120.8.O, δ 195.3CO, δ 195.3CO, δ 170.2, δ 152.3, δ 144.3, δ 142.6, δ 131.2, δ 130.1, δ 123.8, δ 120.8, δ 117.7, δ 112.4, δ 30.3. FTIR (ATR corr. ṽ/cm−1): ṽ 2925.41 (w, C–H sp2 str.), ṽ 2303.92 (w, NC str.), ṽ 2016.96 (s, A′(1) CO str.), ṽ 1871.84 (s, A′(2) & A′′ CO str.). UV/Vis (λ/nm, ε/L mol−1 cm−1): λ 437 (dπ → π*, ε 235.06), λ 281 (π → π*, ε 2340.84), λ 237 (π → π*, ε 3104.80). EA (%): calc. C 34.32, H 1.65, N 5.72. Found C 34.89, H 1.54, N 5.60.O, δ 196.8CO, δ 195.7CO, δ 169.6, δ 151.0, δ 144.9, δ 136.5, δ 130.9, δ 128.9, δ 124.1, δ 115.4, δ 113.2. FTIR (ATR corr. ṽ/cm−1): ṽ 2959 (w, C–H sp2 str.), ṽ 2911 (w, C–H sp2 str.), ṽ 2322 (w, NC str.), ṽ 2016 (s, A′(1) CO str.), ṽ 1874 (s, A′(2) & A′′ CO str.).O, δ 194.4CO, δ 178.1CO, δ 151.6, δ 143.3, δ 137.1, δ 132.9, δ 130.8, δ 127.0, δ 126.9, δ 121.4, δ 114.2. FTIR (ATR corr. ṽ/cm−1): ṽ 2939 (w, C–H sp2 str.), ṽ 2918 (w, C–H sp2 str.), ṽ 2328 (w, NC str.), ṽ 2019 (s, A′(1) CO str.), ṽ 1900 (s, A′(2) & A′′ CO str.).O, δ 194.9CO, δ 194.2CO, δ 168.0 (d, Jd = 2 Hz), δ 161.5 (d, Jd = 256 Hz), δ 145.0 (d, Jd = 2 Hz), δ 139.7, δ 129.6 (d, Jd = 2 Hz), δ 128.6, δ 116.5, δ 111.5, δ 109.8 (d, Jd = 33 Hz). 19F{1H} NMR (376 MHz, CD3CN, δ/ppm): δ −56.94. FTIR (ATR corr. ṽ/cm−1): ṽ 2925 (w, C–H sp2 str.), ṽ 2853 (w, C–H sp2 str.), ṽ 2322 (w, NC str.), ṽ 2020 (s, A′(1) CO str.), ṽ 1875 (s, A′(2) & A′′ CO str.).O, δ 196.9CO, δ 196.0CO, δ 166.4 (d, Jd = 2 Hz), δ 151.3, δ 147.1 (d, Jd = 235 Hz), δ 143.0 (d, Jd = 5 Hz), δ 133.1 (d, Jd = 3 Hz), δ 123.3 (d, Jd = 2 Hz), δ 121.1 (d, Jd = 21 Hz), δ 144.5 (d, Jd = 20 Hz), δ 113.0 (d, Jd = 7 Hz). 19F{1H} NMR (376 MHz, CD3CN, δ/ppm): δ −144.8. FTIR (ATR corr. ṽ/cm−1): ṽ 2954 (w, C–H sp2 str.), ṽ 2909 (w, C–H sp2 str.), ṽ 2298 (w, NC str.), ṽ 2019 (s, A′(1) CO str.), ṽ 1892 (s, A′(2) & A′′ CO str.).
990.57), λ 326 (π → π*, ε 10920.28), λ 233 (π → π*, ε 19259.41). LRMS (ESI+): [M–Cl]+ m/z cald: 460.97, m/z obvs 460.95. HRMS (ESI+): [M–Cl]+ m/z cald: 460.9703, m/z obvs 460.9694 (Δ −2.0 ppm). EA (%): calc. C 31.46, H 1.42, N 5.64. Found C 31.59, H 1.17, N 5.70.O, δ 197.2CO, δ 189.6CO, δ 154.2, δ 154.1, δ 153.0, δ 150.9, δ 140.4, δ 140.2, δ 133.9, δ 128.1, δ 125.3, δ 124.8. FTIR (ATR corr. ṽ/cm−1): ṽ 3113 (w, C–H sp2 str.), ṽ 2021 (s, A′(1) CO str.), ṽ 1883 (s, A′(2) & A′′ CO str.). UV/Vis (λ/nm, ε/L mol−1 cm−1): λ 379 (dπ → π*, ε 7346.37), λ 299 (π → π*, ε 36136.21), λ 241 (π → π*, ε 42589.10). LRMS (ESI+): [M–Cl]+ m/z cald: 460.97, m/z obvs 460.95. HRMS (ESI+): [M–Cl]+ m/z cald: 460.9703, m/z obvs 460.9730 (Δ 5.9 ppm). EA (%): calc. C 31.46, H 1.42, N 5.64. Found C 31.61, H 1.14, N 5.44.O, δ 197.5CO, δ 189.7CO, δ 156.9, δ 154.3, δ 153.8, δ 153.1, δ 147.0, δ 140.3, δ 128.4, δ 127.7, δ 125.0, δ 124.7. FTIR (ATR corr. ṽ/cm−1): ṽ 3117 (w, C–H sp2 str.), ṽ 2019 (s, A′(1) CO str.), ṽ 1884 (s, A′(2) & A′′ CO str.). UV/Vis (λ/nm, ε/L mol−1 cm−1): λ 380 (dπ → π*, ε 7548.93), λ 292 (π → π*, ε 38022.44), λ 235 (π → π*, ε 48347.07). LRMS (ESI+): [M–Cl]+ m/z cald: 460.97, m/z obvs 460.95. HRMS (ESI+): [M–Cl]+ m/z cald: 460.9703, m/z obvs 460.9712 (Δ 2.0 ppm). EA (%): calc. C 31.46, H 1.42, N 5.64. Found C 31.81, H 1.20, N 5.32.O, δ 197.2CO, δ 190.0CO, δ 157.6, δ 156.0, δ 152.7, δ 146.2, δ 142.0, δ 140.2, δ 131.9, δ 127.8, δ 125.3, δ 123.3. FTIR (ATR corr. ṽ/cm−1): ṽ 3074 (w, C–H sp2 str.), ṽ 2017 (s, A′(1) CO str.), ṽ 1884 (s, A′(2) & A′′ CO str.). LRMS (ESI+): [M–Cl]+ m/z cald: 504.92, m/z obvs 505.04. EA (%): calc. C 28.87, H 1.30, N 5.18. Found C 28.90, H 1.05, N 5.21.O, δ 196.6CO (d, Jd = 16.59 Hz), δ 188.9CO, δ 164.0, δ 161.5, δ 154.4 (d, Jd = 15.92 Hz), δ 153.1, δ 145.9 (d, Jd = 10.15 Hz), δ 140.4, δ 128.2, δ 125.1, δ 121.4 (d, Jd = 2.94 Hz), δ 112.7 (d, Jd = 30.19 Hz). 19F{1H} NMR (376 MHz, DMSO-d6, δ/ppm): δ −51.75. FTIR (ATR corr. ṽ/cm−1): ṽ 3040 (w, C–H sp2 str.), ṽ 2016 (s, A′(1) CO str.), ṽ 1880 (s, A′(2) & A′′ CO str.). UV/Vis (λ/nm, ε/L mol−1 cm−1): λ 369 (dπ → π*, ε 2768.36), λ 296 (π → π*, ε 12485.88), λ 235 (π → π*, ε 15367.33). LRMS (ESI+): [M–Cl]+ m/z cald: 445.00, m/z obvs 445.10. EA (%): calc. C 32.54, H 1.47, N 5.84. Found C 32.59, H 1.16, N 5.88.O, δ 197.6CO, δ 189.9CO, δ 168.3, δ 168.3, δ 158.9 (d, Jd = 10.26 Hz), δ 156.2 (d, Jd = 9.99 Hz), δ 154.6, δ 153.1, δ 140.5, δ 128.5, δ 124.9, δ 116.0 (d, Jd = 19.7 Hz), δ 113.0 (d, Jd = 22.79 Hz). 19F{1H} NMR (376 MHz, DMSO-d6, δ/ppm): δ −94.71. FTIR (ATR corr. ṽ/cm−1): ṽ 3117 (w, C–H sp2 str.), ṽ 2018 (s, A′(1) CO str.), ṽ 1884 (s, A′(2) & A′′ CO str.). LRMS (ESI+): [M–Cl]+ m/z cald: 445.00, m/z obvs 444.92. EA (%): calc. C 32.54, H 1.47, N 5.84. Found C 32.52, H 1.33, N 5.81.O), δ 197.65 (CO), δ 189.83 (CO), δ 154.75, δ 154.11, δ 147.19, δ 146.56, δ 142.81, δ 139.92, δ 131.69, δ 129.66, δ 128.37, δ 128.09, δ 127.30, δ 127.24. FTIR (ATR corr. ṽ/cm−1): ṽ 3042 (w, C–H sp2 str.), ṽ 2021 (s, A′(1) CO str.), ṽ 1889 (s, A′(2) & A′′ CO str.). UV/Vis (CH3CN, λ/nm, ε/L mol−1 cm−1): λ 368 (Re(dπ) → π*, ε 374.65), λ 270 (π → π*, ε 2612.13), λ 218 (π → π*, ε 4703.92), λ 204 (π → π*, ε 4589.45). LRMS (ESI+): [M–Cl]+ m/z calc. 484.97, m/z obsv. 485.07. HRMS (ESI+): [M + Na]+ m/z calc. 540.9261 (C15H7N2O3FCl2ReNa+); m/z obsv. 540.9266 (Δ 0.90 ppm). EA (%): calc. C 34.62, H 1.36, N 5.38, Cl 13.63; obsv. C 34.45, H 0.95, N 5.36, Cl 13.55.O, δ 197.2CO, δ 189.6CO, δ 154.2, δ 154.1, δ 153.0, δ 150.9, δ 140.4, δ 140.2, δ 133.9, δ 128.1, δ 125.3, δ 124.8. FTIR (ATR corr. ṽ/cm−1): ṽ 3113 (w, C–H sp2 str.), ṽ 2021 (s, A′(1) CO str.), ṽ 1883 (s, A′(2) & A′′ CO str.). UV/Vis (λ/nm, ε/L mol−1 cm−1): λ 379 (dπ → π*, ε 7346.37), λ 299 (π → π*, ε 36136.21), λ 241 (π → π*, ε 42589.10). LRMS (ESI+): [M–Cl]+ m/z cald: 460.97, m/z obvs 460.95. HRMS (ESI+): [M–Cl]+ m/z cald: 460.9703, m/z obvs 460.9730 (Δ 5.9 ppm). EA (%): calc. C 31.46, H 1.42, N 5.64. Found C 31.61, H 1.14, N 5.44.O, δ 197.1CO, δ 189.8CO, δ 153.5, δ 147.8, δ 147.2, δ 146.4, δ 141.4, δ 139.4, δ 131.3, δ 130.8, δ 129.4, δ 127.8, δ 127.7, δ 126.6. FTIR (ATR corr. ṽ/cm−1): ṽ 3043.35 (w, C–H sp2 str.), ṽ 2014.27 (s, A′(1) CO str.), ṽ 1883.98 (s, A′(2) & A′′ CO str.). EA (%): calc. C 31.90, H 1.25, N 4.96. Found C 31.94, H 1.00, N 5.08.O, δ 194.4CO, δ 178.1CO, δ 151.6, δ 143.3, δ 137.1, δ 132.9, δ 130.8, δ 127.0, δ 126.9, δ 121.4, δ 114.2. FTIR (ATR corr. ṽ/cm−1): ṽ 2939 (w, C–H sp2 str.), ṽ 2918 (w, C–H sp2 str.), ṽ 2328 (w, NC str.), ṽ 2019 (s, A′(1) CO str.), ṽ 1900 (s, A′(2) & A′′ CO str.).O), δ 196.54 (d, Jd = 14 Hz, CO), δ 189.99 (CO), δ 163.77, δ 161.18, δ 153.90, δ 145.79 (d, Jd = 11 Hz), δ 145.09 (d, Jd = 2 Hz), δ 144.07 (d, Jd = 3 Hz), δ 139.48, δ 130.99, δ 128.29 (d, Jd = 1 Hz), δ 127.32 (d, Jd = 2 Hz), δ 127.09 (d, Jd = 61 Hz), δ 113.48 (d, Jd = 32 Hz). 19F NMR (376 MHz, DMSO-d6, δ/ppm): δ −36.75 (dd, Jd = 6.92, 1.92 Hz), δ −106.00 (ISTD: para-DFB). FTIR (ATR corr. ṽ/cm−1): ṽ 3077 (w, C–H sp2 str.), ṽ 2021 (s, A′(1) CO str.), ṽ 1890 (s, A′(2) & A′′ CO str.). UV/Vis (CH3CN, λ/nm, ε/L mol−1 cm−1): λ 375 (Re(dπ) → π*, ε 2720.95), λ 343 (π → π*, ε 2821.73), λ 269 (π → π*, ε 22069.94), λ 262 (π → π*, ε 22170.71), λ 215 (π → π*, ε 38798.75), λ 200 (π → π*, ε 40915.05). LRMS (ESI+): [M–Cl]+ m/z calc. 469.00, m/z obsv. 469.12. HRMS (ESI+): [M + Na]+ m/z calc. 524.9557 (C15H7N2O3FClReNa+); m/z obsv. 524.9579 (Δ 4.20 ppm). EA (%): calc. C 3575, H 1.40, N 5.56, Cl 7.04; obsv. C 35.73, H 0.98, N 5.59, Cl 6.83.O, δ 197.3CO, δ 189.6CO, δ 156.8, δ 154.5, δ 154.3, δ 152.9 (d, Jd = 2 Hz), δ 146.5 (d, Jd = 7 Hz), δ 143.6, δ 139.1 (d, Jd = 5 Hz), δ 133.0 (d, Jd = 4 Hz), δ 129.6 (d, Jd = 4 Hz), δ 126.9 (d, Jd = 3 Hz), δ 122.8 (d, Jd = 23 Hz), δ 109.3 (d, Jd = 22 Hz). 19F{1H} NMR (376 MHz, CD3CN, δ/ppm): δ −120.8.O, δ 195.3CO, δ 195.3CO, δ 170.2, δ 152.3, δ 144.3, δ 142.6, δ 131.2, δ 130.1, δ 123.8, δ 120.8, δ 117.7, δ 112.4, δ 30.3. FTIR (ATR corr. ṽ/cm−1): ṽ 2925.41 (w, C–H sp2 str.), ṽ 2303.92 (w, NC str.), ṽ 2016.96 (s, A′(1) CO str.), ṽ 1871.84 (s, A′(2) & A′′ CO str.). UV/Vis (λ/nm, ε/L mol−1 cm−1): λ 437 (dπ → π*, ε 235.06), λ 281 (π → π*, ε 2340.84), λ 237 (π → π*, ε 3104.80). EA (%): calc. C 34.32, H 1.65, N 5.72. Found C 34.89, H 1.54, N 5.60.O, δ 196.8CO, δ 195.7CO, δ 169.6, δ 151.0, δ 144.9, δ 136.5, δ 130.9, δ 128.9, δ 124.1, δ 115.4, δ 113.2. FTIR (ATR corr. ṽ/cm−1): ṽ 2959 (w, C–H sp2 str.), ṽ 2911 (w, C–H sp2 str.), ṽ 2322 (w, NC str.), ṽ 2016 (s, A′(1) CO str.), ṽ 1874 (s, A′(2) & A′′ CO str.).O, δ 194.4CO, δ 178.1CO, δ 151.6, δ 143.3, δ 137.1, δ 132.9, δ 130.8, δ 127.0, δ 126.9, δ 121.4, δ 114.2. FTIR (ATR corr. ṽ/cm−1): ṽ 2939 (w, C–H sp2 str.), ṽ 2918 (w, C–H sp2 str.), ṽ 2328 (w, NC str.), ṽ 2019 (s, A′(1) CO str.), ṽ 1900 (s, A′(2) & A′′ CO str.).O, δ 194.9CO, δ 194.2CO, δ 168.0 (d, Jd = 2 Hz), δ 161.5 (d, Jd = 256 Hz), δ 145.0 (d, Jd = 2 Hz), δ 139.7, δ 129.6 (d, Jd = 2 Hz), δ 128.6, δ 116.5, δ 111.5, δ 109.8 (d, Jd = 33 Hz). 19F{1H} NMR (376 MHz, CD3CN, δ/ppm): δ −56.94. FTIR (ATR corr. ṽ/cm−1): ṽ 2925 (w, C–H sp2 str.), ṽ 2853 (w, C–H sp2 str.), ṽ 2322 (w, NC str.), ṽ 2020 (s, A′(1) CO str.), ṽ 1875 (s, A′(2) & A′′ CO str.).O, δ 196.9CO, δ 196.0CO, δ 166.4 (d, Jd = 2 Hz), δ 151.3, δ 147.1 (d, Jd = 235 Hz), δ 143.0 (d, Jd = 5 Hz), δ 133.1 (d, Jd = 3 Hz), δ 123.3 (d, Jd = 2 Hz), δ 121.1 (d, Jd = 21 Hz), δ 144.5 (d, Jd = 20 Hz), δ 113.0 (d, Jd = 7 Hz). 19F{1H} NMR (376 MHz, CD3CN, δ/ppm): δ −144.8. FTIR (ATR corr. ṽ/cm−1): ṽ 2954 (w, C–H sp2 str.), ṽ 2909 (w, C–H sp2 str.), ṽ 2298 (w, NC str.), ṽ 2019 (s, A′(1) CO str.), ṽ 1892 (s, A′(2) & A′′ CO str.).Synthetic protocols for fluorinated ligands
Fluorinated ligands (L1h, L1f, L2f, L2g and L3e-OBn) were prepared by azeotropically drying masses of potassium fluoride (12–25 eq.) and 18-crown-6 ether (4–10 eq.) via the dropwise addition of anhydrous acetonitrile in an inert nitrogen gas environment. A mass of the chlorinated or nitrated precursor (1.0 eq.) was dissolved in anhydrous DMSO and added to the complex. The solution was then heated to between 90-250 °C and stirred for 96–120 h. The reaction mixture was then cooled and filtered through a PTFE membrane (0.2 μm). The filtrate was eluted over C18 (40 g) using an acetonitrile/TFA (0.1% in water) gradient mobile phase. The eluents containing the product were subsequently combined and lyophilised to afford a pure white, pink or orange powder.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C πAr str.), ṽ 1497 (s, CC πAr str.), ṽ 1416 (s, CC πAr str.). UV/Vis (CH3CN, λ/nm, ε/L mol−1 cm−1): λ 262 (π → π*, ε 28595.96), λ 227 (π → π*, ε 47402.32), λ 200 (π → π*, ε 16444.83). LRMS (ESI+): [M + H]+ m/z calc. 199.07, m/z obsv. 199.27. HRMS (ESI+): [M + H]+ m/z calc. 199.06660 (C12H8N2F+); m/z obsv. 199.06673 (Δ 0.64 ppm); [M–F]+ m/z calc. 179.06092 (C12H7N2+); m/z obsv. 179.06050 (Δ 2.36 ppm).
C πAr str.), ṽ 1497 (s, CC πAr str.), ṽ 1416 (s, CC πAr str.). UV/Vis (CH3CN, λ/nm, ε/L mol−1 cm−1): λ 262 (π → π*, ε 28595.96), λ 227 (π → π*, ε 47402.32), λ 200 (π → π*, ε 16444.83). LRMS (ESI+): [M + H]+ m/z calc. 199.07, m/z obsv. 199.27. HRMS (ESI+): [M + H]+ m/z calc. 199.06660 (C12H8N2F+); m/z obsv. 199.06673 (Δ 0.64 ppm); [M–F]+ m/z calc. 179.06092 (C12H7N2+); m/z obsv. 179.06050 (Δ 2.36 ppm).All other ligands were acquired through commercially available sources (Sigma Aldrich or BePharm Ltd). Further details regarding ligand syntheses, including the experimental spectra, can be found in Section S.7 of the ESI.†
Radiolabelling protocols
Aqueous [18F]fluoride was produced on an IBA Cyclone 18 Twin cyclotron via the 18O(p,n)18F nuclear reaction. The [18F]fluoride was trapped by a QMA cartridge and eluted off with tetraethylammonium bicarbonate in 10% v/v water/acetonitrile for wet conditions, and azeotropically dried and reconstituted in anhydrous DMSO for dry conditions. Fig. 9 depicts the set-up of the synthesis module used to automate the radiolabelling reactions. Microfluidic radiosyntheses were performed in Discovery Mode using a NanoTek LF Microfluidic Synthesis System (Advion) connected to a standard laptop using NanoTek software v1.4.0 GMP Lite. Microreactors were made of fused silica tubing (100 μm × 2 m, 15.6 μL) coiled tightly into a brass ring and held by a thermoresistant polymer. Radio HPLC analyses were carried out using a Shimadzu system comprised of a CBM-20 controller, LC-20AD pump, SIL-20AHT autoinjector, SPD-M20A PDA detector and a Lablogic Posi-RAM gamma detector. Analyses were performed using gradient conditions over 15 minutes, consisting of 95:5 water:acetonitrile for the first 3 minutes followed by a linear transposition to 5:95 water:acetonitrile over the remaining 12 minutes using a 2 mL min−1 flow rate. A Chromolith RP column (monolith system, Merck 50 × 4.6 mm), demonstrated to have <8% [18F]fluoride retention in low pH ranges,44 was employed as the stationary phase. Radio HPLC derived non-isolated RCYs were calculated via the integrated peak area ratio between the radio product and other radioactive species present, inclusive of any unreacted [18F]fluoride, using Laura V4.1.70 SP2 HPLC data analysis software. Further information on the radiochemical syntheses can be found in Section S.9 of the ESI.†
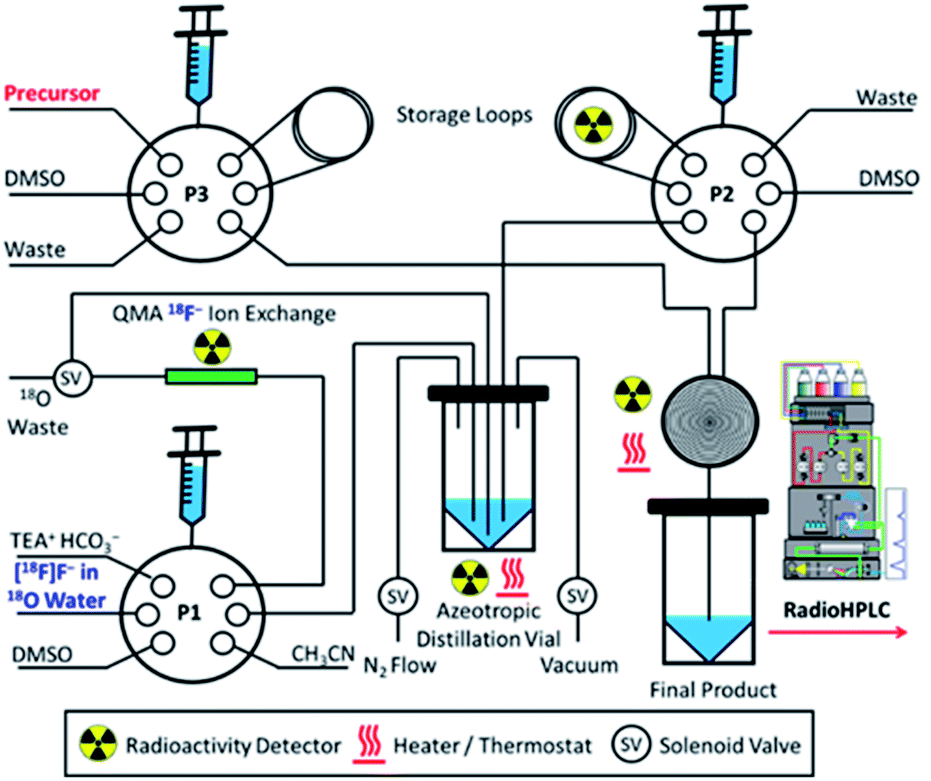 | ||
| Fig. 9 Automated synthesis module for the azeotropic drying of cyclotron generated [18F]fluoride and subsequent microfluidic set-up for the rhenium(I) complexation–dissociation reactions under varying reaction environments. | ||
Computational protocols
Geometry optimisations and bond analyses were computed using ORCA software45 at the DFT level using def2-TZVP basis sets and with the B97 density functional46 corrected using the Grimme D3 correction with Becke–Johnson damping47 to the DFT energy. The resolution of identity (RI) approximation48 and the Effective Core potential LANL2DZ49,50 were also applied. The structures were confirmed to be local energy minima except for the transition states (one imaginary frequency rather than none). Thermodynamic properties were computed at the def2-TZVP/B97-D3 level of theory and calculated at 298 K. Zero-point energy corrections were included and the DMSO solvent was accounted for through the Continuum Polarizable Medium (PCM) model.51 Natural Bond Order (NBO) analyses were performed using the NBO6 suite of programs.52 The bond analyses between the rhenium(I) and diimine ligands were performed with the combined NOCV53 and CD54 frameworks. Within the NOCV framework,39,55 the interaction between the [Re(CO)3Cl] fragment (A) and the N,N diimine ligand fragment (B) were analysed by comparing the wavefunction of the adduct AB (ψAB) with the antisymmetrized and renormalized product of the wavefunctions of the isolated fragments (ψ0). The charge rearrangement (Δρ) upon formation of the interaction between A and B fragments is defined through the following equation: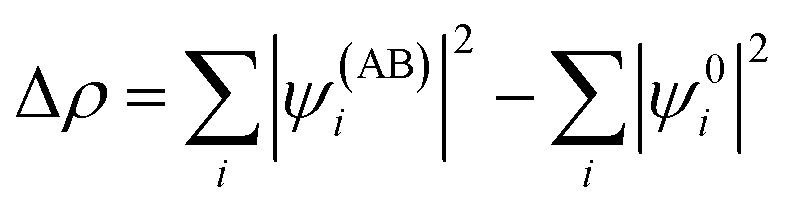
The charge displacement was separated in diagonal contributions through the valence operator (![[V with combining circumflex]](https://www.rsc.org/images/entities/i_char_0056_0302.gif) ),56–58 producing pairs of complementary orbitals (φk, φ−k) corresponding to eigenvalues with the same absolute value but opposite sign (±υk). Accordingly, Δρ can be expressed in terms of NOCV pairs via the following equation:
),56–58 producing pairs of complementary orbitals (φk, φ−k) corresponding to eigenvalues with the same absolute value but opposite sign (±υk). Accordingly, Δρ can be expressed in terms of NOCV pairs via the following equation:
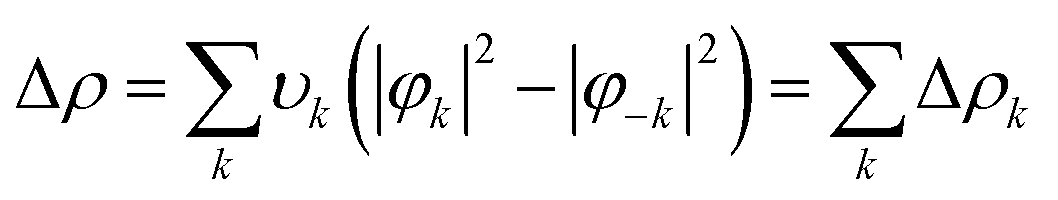
The different Δρk were then also integrated along the axis (z) aligned with the Re(I) and diimine N atom in order to quantify the fraction of electrons (Δq) involved in the φ−k → φk transition and, therefore, in the bond component, throughout the whole molecule.
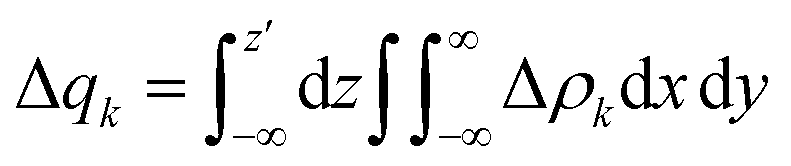
Atomic charges were calculated through the NBO. Further computational details can be found in Section S.5 of the ESI.†
Photophysical protocols
Photophysical analyses were performed using an Edinburg FLS980 photospectrometer. Light from a 450 W xenon arc lamp source was passed through excitation and emission monochromators to acquire the corresponding spectra. The quantum yields (ϕ) were calculated against a [Ru(BiPy)3]Cl2 reference solution (ϕr) via the following equation:where correction factors are applied for the integrated area of the emission spectra (I), absorbances of the solutions at the MLCT excitation wavelengths (A) and the refractive indices of the solvents (n) utilised.59 Further information on the photophysical analyses can be found in Section S.6 of the ESI.†
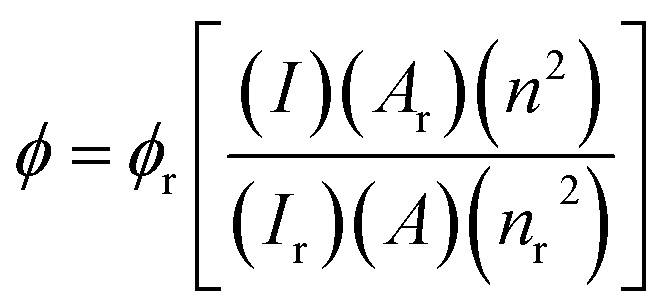
Single crystal X-ray diffraction
Data were with collected using either an Oxford Rigaku Synergy-S employing confocal mirror monochromated Mo-Kα radiation generated from a microfocus source (0.71073 Å) with ω and ψ scans at 100(2) K or at the MX1 beamline of the Australian Synchrotron with silicon double crystal monochromated radiation at 100(2) K (λ = 0.7108 Å).60 Data integration and reduction were undertaken with CrysAlisPro or XDS.61 Subsequent computations were carried out using Olex2.62 Structures were solved with ShelXT63 and refined and extended with ShelXL64 Carbon-bound hydrogen atoms were included in idealised positions and refined using a riding model. The instrument design of the Synchrotron beamline restricts data collection to rotation around a single axis which limits available completeness for low symmetry crystal systems. Ellipsoid plots for the five rhenium(I) complexes can be viewed in the ESI: Fig. S116–S120, Section S.10.† Crystallographic data is summarised below and the CIFs have been deposited at the Cambridge Crystallographic Data Centre with CCDC 1970989–1970993.[Re(L3d)(CO)3(NCCH3)]; C14H8N3O6Re (M =500.44 g mol−1): monoclinic, space group P21/c (no. 14), a = 13.0671(4) Å, b = 15.9871(5) Å, c = 7.5731(2) Å, β = 96.415(3)°, V = 1572.14(8) Å3, Z = 4, T = 100.00(10) K, μ (MoKα) = 7.764 mm−1, Dcalc = 2.123 g cm−3, 19869 reflections measured (5.096° ≤ 2θ ≤ 56.562°), 3893 unique (Rint = 0.0967, Rsigma = 0.0600) which were used in all calculations. The final R1 was 0.0390 (I > 2σ(I)) and wR2 was 0.0969 (all data).
[Re(L2b)(CO)3Cl]; C15H7Cl2N2O3Re (M =520.33 g mol−1): triclinic, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no. 2), a = 8.1930(16) Å, b = 8.6870(17) Å, c = 10.559(2) Å, α = 95.04(3)°, β = 93.13(3)°, γ = 97.76(3)°, V = 740.1(3) Å3, Z = 2, T = 100(2) K, μ (synchrotron) = 8.586 mm−1, Dcalc = 2.335 g cm−3, 13499 reflections measured (3.882° ≤ 2θ ≤ 63.35°), 3590 unique (Rint = 0.0627, Rsigma = 0.0533) which were used in all calculations. The final R1 was 0.0331 (I > 2σ(I)) and wR2 was 0.0858 (all data).
(no. 2), a = 8.1930(16) Å, b = 8.6870(17) Å, c = 10.559(2) Å, α = 95.04(3)°, β = 93.13(3)°, γ = 97.76(3)°, V = 740.1(3) Å3, Z = 2, T = 100(2) K, μ (synchrotron) = 8.586 mm−1, Dcalc = 2.335 g cm−3, 13499 reflections measured (3.882° ≤ 2θ ≤ 63.35°), 3590 unique (Rint = 0.0627, Rsigma = 0.0533) which were used in all calculations. The final R1 was 0.0331 (I > 2σ(I)) and wR2 was 0.0858 (all data).
[Re(L2c)(CO)3Cl]; C13H7Cl2N2O3Re (M =496.3159 g mol−1): triclinic, space group P (no. 2), a = 11.318(2) Å, b = 12.872(3) Å, c = 15.669(3) Å, α = 102.17(3)°, β = 99.94(3)°, γ = 102.50(3)°, V = 2120.8(8) Å3, Z = 6, T = 100(2) K, μ (synchrotron) = 8.981 mm−1, Dcalc = 2.331 g cm−3, 31430 reflections measured (3.354° ≤ 2θ ≤ 54.97°), 8464 unique (Rint = 0.0480, Rsigma = 0.0438) which were used in all calculations. The final R1 was 0.0382 (I > 2σ(I)) and wR2 was 0.0975 (all data)
[Re(L1b)(CO)3Cl]; C13H7Cl2N2O3Re (M =496.31 g mol−1): monoclinic, space group P21/c (no. 14), a = 14.655(3) Å, b = 10.996(2) Å, c = 9.0920(18) Å, β = 107.76(3)°, V = 1395.3(5) Å3, Z = 4, T = 100(2) K, μ (synchrotron) = 9.032 mm−1, Dcalc = 2.363 g cm−3, 22464 reflections measured (2.918° ≤ 2θ ≤ 56.54°), 3218 unique (Rint = 0.0448, Rsigma = 0.0259) which were used in all calculations. The final R1 was 0.0554 (I > 2σ(I)) and wR2 was 0.1411 (all data).
[Re(L1f)(CO)3Cl]; C13H7ClFN2O3Re (M =479.86 g mol−1): monoclinic, space group P21/n (no. 14), a = 7.9600(16) Å, b = 17.171(3) Å, c = 10.245(2) Å, β = 99.25(3)°, V = 1382.1(5) Å3, Z = 4, T = 100(2) K, μ (synchrotron) = 9.007 mm−1, Dcalc = 2.306 g cm−3, 19995 reflections measured (4.674° ≤ 2θ ≤ 54.968°), 2754 unique (Rint = 0.0324, Rsigma = 0.0191) which were used in all calculations. The final R1 was 0.0226 (I > 2σ(I)) and wR2 was 0.0683 (all data).
Conflicts of interest
The authors have no conflicts of interest to declare.Acknowledgements
The authors kindly acknowledge the Australian Institute for Nuclear Science and Engineering (AINSE) for the provision of a Postgraduate Research Award (PGRA). Part of this research was undertaken on the MX1 beamline of the Australian Synchrotron, Clayton, Victoria, Australia, part of ANSTO. We thank the Australian Synchrotron for travel support and their staff for assistance. Finally, the authors also acknowledge the efforts of Michael Tran and Gary Perkins for cyclotron production as part of the National Imaging Facility (NIF).References
- S. M. Ametamey, M. Honer and P. A. Schubiger, Chem. Rev., 2008, 108, 1501–1516 CrossRef CAS PubMed.
- D. van der Born, A. Pees, A. J. Poot, R. V. A. Orru, A. D. Windhorst and D. J. Vugts, Chem. Soc. Rev., 2017, 46, 4709–4773 RSC.
- O. Jacobson, D. O. Kiesewetter and X. Chen, Bioconjugate Chem., 2015, 26, 1–18 CrossRef CAS PubMed.
- A. F. Brooks, J. J. Topczewski, N. Ichiishi, M. S. Stanford and P. J. H. Scott, Chem. Sci., 2014, 5, 4545–4553 RSC.
- T. Furuya, J. E. Klein and T. Ritter, Synthesis, 2010, 11, 1804–1821 Search PubMed.
- A. J. Plamer, J. C. Clark and R. W. Goulding, Int. J. Appl. Radiat. Isot., 1977, 28, 53–65 CrossRef.
- O. Jacobson and X. Chen, Med. Chem., 2010, 10, 1048–1059 CAS.
- Y. Zhang and A. G. Horti, J. Label. Compd. Radiopharm., 2004, 47, 947–952 CrossRef CAS.
- Y. Zhang, A. W. Hall and A. G. Horti, J. Label. Compd. Radiopharm., 2004, 47, 385–392 CrossRef CAS.
- A. G. Horti, A. O. Koren, H. T. Ravert, J. L. Musachio, W. B. Mathews, E. D. London and R. F. Dannals, J. Label. Compd. Radiopharm., 1998, 41, 309–318 CrossRef CAS.
- Y. Zhang, O. A. Pavlova, S. I. Chefer, A. W. Hall, V. Kurian, L. L. Brown, A. S. Kimes, A. G. Mukhin and A. G. Horti, J. Med. Chem., 2004, 47, 2453–2465 CrossRef CAS PubMed.
- Y.-S. Ding, N. Liu, T. Wang, J. Marecek, V. Garza, I. Ojima and J. S. Fowler, Nucl. Med. Biol., 2000, 27, 381–389 CrossRef CAS PubMed.
- R. Schrirrmacher, U. Muhlhausen, B. Wangler, E. Schirrmacher, J. Reinhard, G. Nagel, B. Kaina, M. Piel, M. Wiebler and F. Rosch, Tetrahedron Lett., 2002, 43, 6301–6304 CrossRef.
- M. A. Klenner, G. Pascali, B. Zhang, T. R. Sia, L. K. Spare, A. M. Krause-Heuer, J. R. Aldrich-Wright, I. Greguric, A. J. Guastella, M. Massi and B. H. Fraser, Chem.–Eur. J., 2017, 23, 6499–6503 CrossRef CAS PubMed.
- F. Cleeran, J. Lecina, J. Bridoux, N. Devoogdt, T. Tshibangu, C. Xavier and G. Bormans, Nat. Protoc., 2018, 13, 2330–2347 CrossRef PubMed.
- P. Laverman, W. J. McBride, R. M. Sharkey, D. M. Goldenberg and O. C. Boerman, J. Labelled Compd. Radiopharm., 2014, 57, 219–223 CrossRef CAS PubMed.
- S. H. Hausner, B. Bauer and J. L. Sutcliffe, Nucl. Med. Biol., 2014, 41, 43–50 CrossRef CAS PubMed.
- N. Malik, B. Baur, G. Winter, S. N. Reske, A. J. Beer and C. Solbach, Mol. Imag. Biol., 2015, 17, 777–785 CrossRef CAS PubMed.
- Z. Li, T.-P. Lin, S. Liu, C.-W. Huang, T. W. Hudnall, F. P. Gabbi and P. S. Conti, Chem. Commun., 2011, 47, 9324–9326 RSC.
- S. Liu, D. Li, Z. Zhang, G. K. Surya-Praskash, P. S. Conti and Z. Li, Chem. Commun., 2014, 50, 7371–7373 RSC.
- H. Kim, K. Kim, S.-H. Son, J. Y. Choi, K.-H. Lee, B.-T. Kim, Y. Byun and Y. S. Choe, ACS Chem. Neurosci., 2019, 10, 1445–1451 CrossRef CAS PubMed.
- B. Zhang, G. Pascali, N. Wyatt, L. Matesic, M. A. Klenner, T. R. Sia, A. J. Guastella, M. Massi, A. J. Robinson and B. H. Fraser, J. Labelled Compd. Radiopharm., 2018, 61, 847–856 CrossRef CAS PubMed.
- B. Zhang, B. H. Fraser, M. A. Klenner, Z. Chen, S. H. Liang, M. Massi, A. J. Robinson and G. Pascali, Chem. - Eur. J., 2019, 25, 7613–7617 CrossRef CAS PubMed.
- M. A. Klenner, G. Pascali, B. Zhang, G. Ciancaleoni, M. Massi and B. H. Fraser, Aust. J. Chem., 2019, 72, 288–294 CrossRef CAS.
- L. N. Winslow, D. P. Rillema, J. H. Welch and P. Singh, Inorg. Chem., 1989, 28, 1596–1599 CrossRef CAS.
- J. R. Kirchhoff and K. Kirschbaum, Polyhedron, 1998, 17, 4033–4039 CrossRef CAS.
- A. Kochel, M. Holynska and K. Twarog, Inorg. Chem. Commun., 2012, 24, 47–49 CrossRef CAS.
- M. Zhang, P. Lu, Y. Ma and J. Shen, J. Phys. Chem. B, 2003, 107, 6535–6538 CrossRef CAS.
- M. I. Fernandez-Bachiller, C. Perez, G. C. Gonzalez-Munoz, S. Conde, M. G. Lopez, M. Villarroya, A. G. Garcia and M. I. Rodriguez-Franco, J. Med. Chem., 2010, 53, 4927–4937 CrossRef CAS PubMed.
- C. Deraeve, M. Pitie, H. Mazarguil and B. Meuiner, New J. Chem., 2007, 31, 193–195 RSC.
- C. Deraeve, C. Boldron, A. Maraval, H. Mazarguil, H. Gornitzka, L. Vendier, M. Pitie and B. Meuiner, Chem.–Eur. J., 2008, 14, 682–696 CrossRef CAS PubMed.
- F. Prati, C. Bergamini, R. Fato, O. Soukup, J. Korbecny, V. Andrisano, M. Bartolini and M. L. Bolognesi, ChemMedChem, 2016, 11, 1284–1295 CrossRef CAS PubMed.
- D. Antequera, M. Bolos, C. Spuch, C. Pascual, I. Ferrer, M. I. Fernandez-Bachiller, M. I. Rodriguez-Franco and E. Carro, Neurobiol. Dis., 2012, 46, 682–691 CrossRef CAS PubMed.
- N. Vasdev, P. Cao, E. M. van Oosten, A. A. Wilson, S. Houle, G. Hao, X. Sun, N. Slavine, M. Alhasan, P. P. Antich, F. J. Bonte and P. Kulkarni, MedChemComm, 2012, 3, 1228–1230 RSC.
- S. H. Liang, J. P. Holland, N. A. Stephenson, A. Kassenbrock, B. H. Rotstein, C. P. Diagnault, R. Lewis, L. Collier, J. M. Hooker and N. Vasdev, ACS Chem. Neurosci., 2015, 6, 535–541 CrossRef CAS PubMed.
- S. H. Liang, A. G. Southon, B. H. Fraser, A. M. Krause-Heuer, B. Zhang, T. M. Shoup, R. Lewis, I. Volitakis, Y. Han, I. Greguric, A. I. Bush and N. Vasdev, ACS Med. Chem. Lett., 2015, 6, 1025–1029 CrossRef CAS PubMed.
- E. Horn and M. R. Snow, Aust. J. Chem., 1984, 37, 35–45 CrossRef CAS.
- E. Horn and M. R. Snow, Aust. J. Chem., 1981, 34, 737–743 CrossRef CAS.
- G. Bistoni, S. Rampino, F. Tarantelli and L. Belpassi, J. Chem. Phys., 2015, 142, 084112 CrossRef PubMed.
- E. Portenkirchner, A. Apaydin, G. Aufischer, M. Havlicek, M. White, M. C. Scharber and N. S. Sariciftci, ChemPhysChem, 2014, 15, 3634–3638 CrossRef CAS PubMed.
- K. Kalyanasundaram, J. Chem. Soc., Faraday Trans. 2, 1986, 82, 2401–2415 RSC.
- M. Wrighton and D. L. Morse, J. Am. Chem. Soc., 1974, 96, 998–1003 CrossRef CAS.
- H. C. Zhao, B. Mello, B.-L. Fu, H. Chowdhury, D. J. Szalda, M.-K. Tsai, D. C. Grills and J. Rochford, Organometallics, 2013, 32, 1832–1841 CrossRef CAS.
- D. Ory, J. Van den Brande, T. de Groot, K. Serdons, M. Bex, L. Declercq, F. Cleeren, M. Ooms, K. Van Laere, A. Verbruggen and G. Bormans, J. Pharm. Biomed. Anal., 2015, 111, 209–214 CrossRef CAS PubMed.
- F. Nesse, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2017, e1327 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1997, 107, 8554–8560 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. A. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- R. A. Kendall and H. A. Fruchtl, Theor. Chim. Acta, 1997, 97, 158–163 CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- W. R. Wadt and P. J. Hay, J. Chem. Phys, 1985, 82, 284–298 CrossRef CAS.
- A. Klamt and G. Schuurmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
- E. D. Glendening, C. R. Landis and F. Weinhold, J. Comput. Chem., 2019, 40, 2234–2241 CAS.
- M. Mitoraj and A. Michalak, J. Mol. Model., 2007, 13, 347–355 CrossRef CAS PubMed.
- L. Belpassi, I. Infante, F. Tarantelli and L. Visscher, J. Am. Chem. Soc., 2008, 130, 1048–1060 CrossRef CAS PubMed.
- A. Altun, F. Nesse and G. Bistoni, J. Chem. Theory Comput., 2019, 15, 215–228 CrossRef CAS PubMed.
- R. F. Nalewajski, A. M. Koster and K. Jug, Theor. Chim. Acta, 1993, 85, 463–484 CrossRef CAS.
- R. F. Nalewajski and J. Ozek, Int. J. Quantum Chem., 1994, 51, 187–200 CrossRef CAS.
- R. F. Nalewajski, J. Mrozek and A. Michalak, Int. J. Quantum Chem., 1997, 61, 589–601 CrossRef CAS.
- P. J. Wright, S. Muzziloi, M. V. Werrett, P. Raiteri, B. W. Skelton, D. S. Silvester, S. Stagni and M. Massi, Organometallics, 2012, 31, 7566–7578 CrossRef CAS.
- T. M. McPhillips, S. E. McPhillips, H. J. Chiu, A. E. Cohen, A. M. Deacon, P. J. Ellis, E. Garman, A. Gonzalez, N. K. Sauter, R. P. Phizackerley, S. M. Soltis and P. J. Kuhn, J. Synchrotron Radiat., 2002, 9, 401–406 CrossRef CAS PubMed.
- W. J. Kabsch, J. Appl. Crystallogr., 1993, 26, 795–800 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. J. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1970989–1970993. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra00318b |
| ‡ Equal contributions for project conception: Dr G. Pascali and Dr B. H. Fraser. |
| This journal is © The Royal Society of Chemistry 2020 |