
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStereodivergent access to all four stereoisomers of chiral tetrahydrobenzo[f][1,4]oxazepines, through highly diastereoselective multicomponent Ugi–Joullié reaction†
Alessandro Pinnaa,
Andrea Basso a,
Chiara Lambruschinia,
Lisa Monia,
Renata Rivab,
Valeria Roccaa and
Luca Banfi*a
a,
Chiara Lambruschinia,
Lisa Monia,
Renata Rivab,
Valeria Roccaa and
Luca Banfi*a
aDepartment of Chemistry and Industrial Chemistry, Università di Genova, Italy. E-mail: banfi@chimica.unige.it
bDepartment of Pharmacy, Università di Genova, Italy
First published on 3rd January 2020
Abstract
Starting from easily accessible chiral enantiopure 1,2-amino alcohols and salicylaldehydes, a concise route to cyclic imines has been developed. These chiral cyclic imines undergo a highly diastereoselective Ugi–Joullié reaction to give trans tetrahydrobenzo[f][1,4]oxazepines with the introduction of up to 4 diversity inputs. The cis isomer may also be attained, thanks to a thermodynamically controlled base catalysed epimerization. Free secondary amines have been obtained using an unprecedented “removable” carboxylic acid.
Introduction
Multicomponent reactions (MCRs) are processes where three or more substrates are combined in a single step, to give a product that contains all the essential parts of the used components.1 Thanks to their high step economy and to the possibility to introduce several diversity inputs in a single step, they are considered a very powerful tool for the construction of libraries of drug-like entities.2–4 While the use of MCRs in medicinal chemistry is well assessed, their application in other fields is less pronounced. Recently, MCRs were found to be successful in the synthesis of functionalized fluorophores.5–7 On the other hand, only a few papers deal with the use of MCRs for the combinatorial assembly of chiral organocatalysts.8–10 For this latter goal, the control of stereochemistry is clearly essential.Isocyanide-based processes (e.g. the Ugi and Passerini reactions) are among the most popular MCRs. However, they suffer from a typical lack of diastereoselection when chiral inputs are employed. This limits their utility, because of the generation of diastereomeric mixtures.
Recently, some examples of diastereoselective Passerini reactions on chiral enantiopure aldehydes or ketones have been reported,11–16 but the related Ugi reaction is more problematic, not only because of the low diastereoselectivity typically achieved, but also for the easy racemization of aldehydes with α-stereogenic centres.17
Better results have been accomplished with the so called Ugi–Joullié reaction.18,19 This is a 3-component variant of the Ugi reaction that employs preformed cyclic imines. When these cyclic imines are chiral and enantiopure, racemization at the α-position is less likely, although yet possible.20 Furthermore, thanks to a higher rigidity, several examples of highly diastereoselective Ugi–Joullié reactions have been reported. Most of the previous studies involve 5-membered (pyrrolines, thiazolines and so on),9,21–31 or 6-membered cyclic imines (e.g. tetrahydropyridines).32–38 The use of compounds with other ring size is much less investigated. Only recently an Ugi–Joullié reaction of a 3-membered azirine was reported.39 In our group we are particularly interested in applying MCRs for the synthesis of benzo-fused 7-membered nitrogen heterocycles,40 whose interest in medicinal chemistry is steadily growing.41 Towards this goal, we have already studied the diastereoselective Ugi–Joullié reaction of some 7-membered chiral enantiopure cyclic imines.20,42
We thought that enantiopure dihydrobenzoxazepines of general formula 3 could be very useful intermediates for the diastereoselective synthesis of a variety of tetrahydrobenzoxazepines 4 or 5, through multicomponent processes. For example, the Ugi–Joullié reaction would give peptide-like compounds 4. Removal of the acyl group from 4 will then afford secondary amines 5. Secondary amines 5 might also be directly obtained by exploiting other MCRs, such as the Betti, or the Ugi-tetrazole reactions. Compounds 4 and 5, containing a typical privileged structure, may be interesting for library production, whereas chiral enantiopure amines 5 may be investigated as potential organocatalysts (Scheme 1).
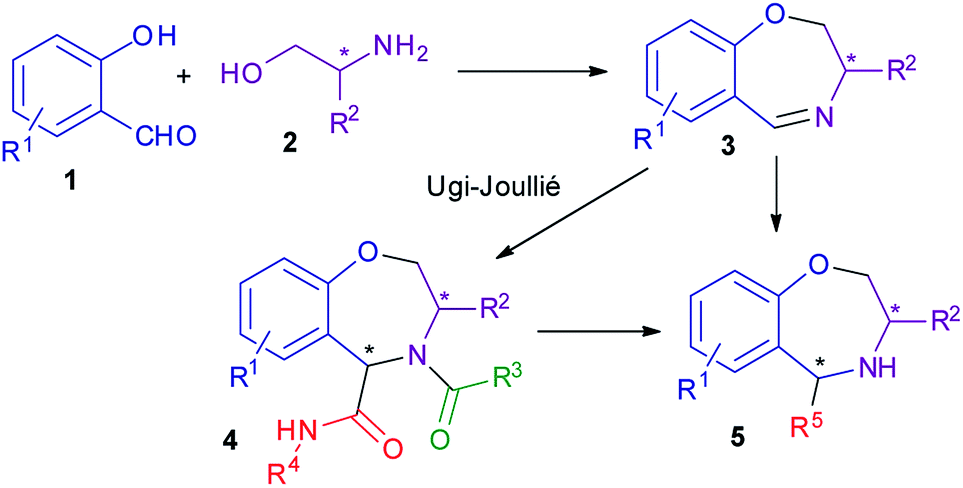 | ||
| Scheme 1 General strategy. | ||
In order to make the whole methodology highly practical, it was essential to develop a fast and diversity-oriented synthesis of chiral imines 3 in enantiopure form. Our plan was to employ amino alcohols 2, in turn derived from widely available α-amino acids.
In this paper we report the successful accomplishment of a quite short synthesis of imines 3, and their diastereodivergent conversion into both diastereomers of 4, through an Ugi–Joullié reaction. Since α-amino acids are available in both enantiomeric forms, this means the possibility to get all four stereoisomers of 4. Furthermore, the efficient obtainment of secondary amines 5 is demonstrated as well.
Results and discussion
The key step for the synthesis of cyclic imines 3 is the Mitsunobu coupling of salicylaldehydes 1 with Boc-protected aminoalcohols 7. All employed alcohols 7 are known,43–45 and they can be easily synthesized by reduction of Boc-protected α-amino acids, in turn commercially available in both enantiomeric forms with a wide variety of R2 substituents. Alternatively, some of them (7d, 7e) have been obtained by protection of the commercially available enantiopure amino alcohols (see ESI†).We have previously shown42 that protection of the aldehyde group was essential. Thus, salicylaldehydes 1 were quantitatively converted into the acetals 6. They were not isolated but, after a very simple work-up, directly coupled with amino alcohols 7a–e. This step proved to be very critical and was optimized using 1a and 7a. Working on Boc-protected 1-amino-2-alkanols, we had previously selected di-tert-butylazodicarboxylate (TBAD) and PPh3 as the best reagents. However, in the present case, the yield was rather poor (25%) and the known aziridine 9a46 was obtained as a major product. Shifting to other typical azodicarboxylates such as DEAD or DIAD, the yield remained unsatisfactory, due to the formation of substantial amounts of 9a. This result was not completely unexpected, taking into account some literature precedents on Mitsunobu reactions of phenols with this type of Boc-amino alcohols, where yields are often low and strongly depend on the nature of the starting phenol.47–49 After some investigation (see ESI for details†), we found out that the formation of 9a could be nearly completely suppressed by using a special azodicarboxylate, namely 1,1′-(azodicarbonyl)dipiperidine (ADDP).50 Its use led to a 78% yield of the desired product 8a. With other amino alcohols the yields were somehow lower (see Table 1), but still satisfactory. Chromatographic separation from aziridines 9 were quite easy, whereas the remaining excess of phenol 6 was conveniently removed by basic extraction (Scheme 2).
 | ||
| Scheme 2 Synthetic sequence from salicyladehydes 1 to Ugi adducts 10–11 (or ent-10–ent-11). | ||
Ethers 8 were then converted very easily into the cyclic imines 3, by acid promoted removal of both protecting group. The crude imines were subjected, without any intermediate purification, to the Ugi–Joullié reaction with a variety of isocyanides and carboxylic acids. It is worth noting that the whole sequence from 1 to the Ugi adducts 10–11 required just a single intermediate purification, at the level of ethers 8. This straightforward route to enantiopure cyclic imines 3 allows the introduction of two diversity inputs and, due to the wide availability of both (S) and (R) α-amino acids, gives an access to both enantiomeric series.
As for the Ugi–Joullié reaction, we tried different reaction conditions, as detailed in the ESI,† eventually finding that the best conditions, both in terms of isolated yield and of diastereoselectivity, involved the use of methanol as solvent with no additive at room temperature.
As shown in Scheme 3, the diastereoselectivities were in all cases excellent. The yields were moderate to good, but it should be stressed that they refer to the overall yield of two steps, including also formation of the cyclic imines 3 by removal of the protecting groups. In some instances, the Ugi–Joullié reactions were incomplete, and thus a higher yield could probably be obtained by using a higher excess of isocyanide or by longer reaction times.
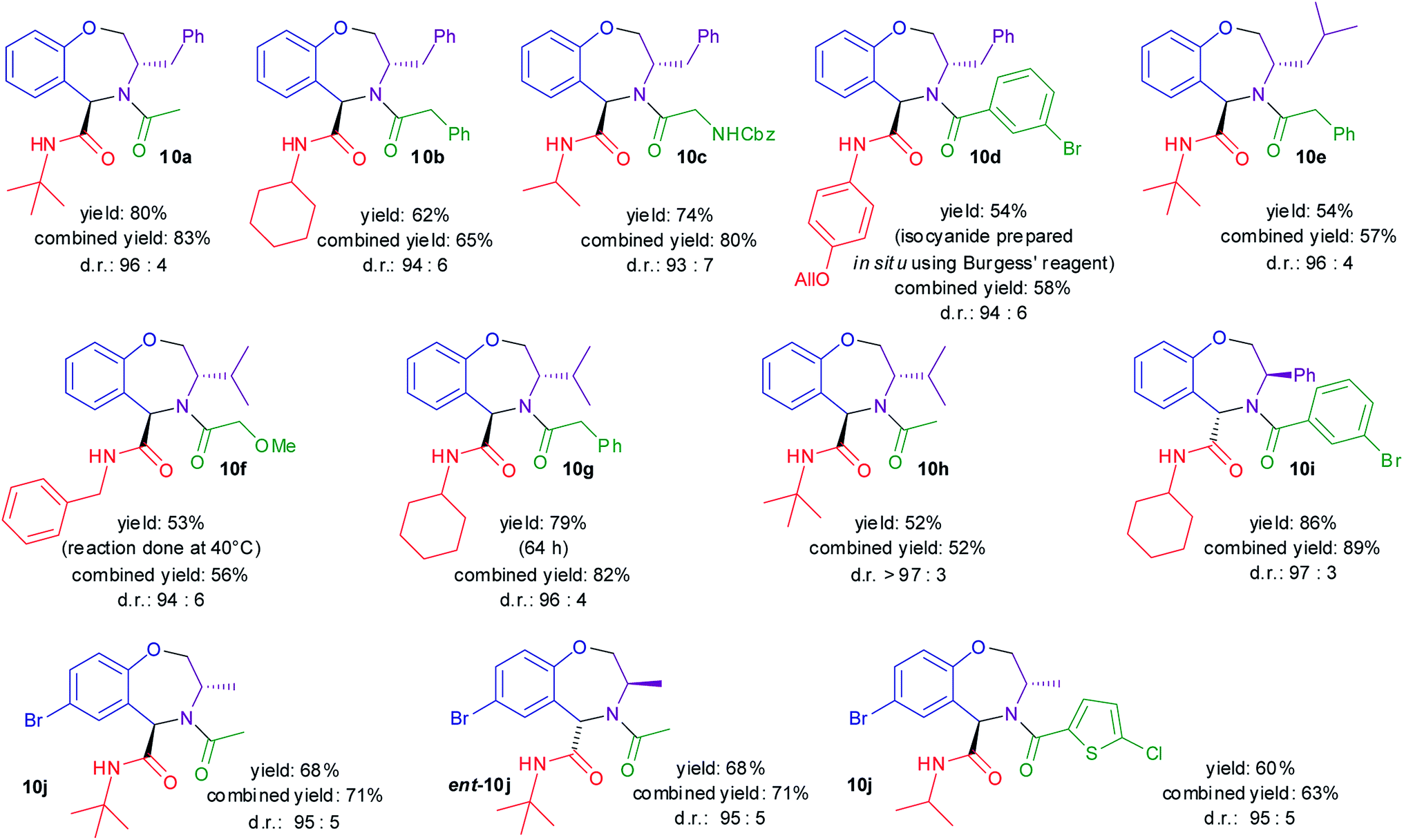 | ||
| Scheme 3 Scope of the methodology. Ugi–Joullié reaction were carried out at rt for 48 h, unless otherwise noted. All reactions were carried out on 200–400 μmol of starting 8. Yield = isolated yield of 10 from 8. Combined yield = isolated or calculated yield of 10 + 11 from 8. D.r. were determined by HPLC on the crude product. | ||
The results reported in Scheme 3 show that it is possible to synthesize enantiopure trans tetrahydrobenzo[f][1,4]oxazepines 10, introducing up to four diversity inputs: the salicylaldehyde 1, the amino alcohol 7, the isocyanide, and the carboxylic acid. The methodology is operationally simple (just two chromatographies) and highly diastereoselective. Finally, both enantiomeric series are accessible, thanks to the easy availability of 1,2-amino alcohols or α-amino acids in either enantiomeric form. Tetrahydrobenzoxazepines 10 may be useful in the medicinal chemistry realm, since several N-acylated tetrahydrobenzo[f][1,4]oxazepines have been reported to be endowed with very interesting pharmacological properties.51–55 On the other hand, the 5-substituted compounds are largely unexplored from this point of view, although some stimulating examples can be found in the literature.56–58 The possibility to control stereochemistry in their synthesis will facilitate the exploration of their chemical space.
However, apart from medicinal chemistry applications, we also wanted to use this chemistry for the combinatorial synthesis of chiral enantiopure secondary amines, to be used as organocatalysts. A possibility to obtain amines like 13 or 14 would be to apply a “truncated” Ugi on cyclic imines 3. In the literature we could find only few examples of “truncated” Ugi reaction of aldehydes, primary amines and isocyanides,59–61 employing either Lewis or Brønsted acids (or enzymes). However, to our knowledge, no example of “truncated” Ugi applied to cyclic imines was reported.
We tried various Lewis acids (e.g. ZnBr2 or ZnCl2),62 obtaining in all cases poor results in terms of yields. Similar unsatisfactory outcomes were obtained using boric acid63 or sulphinic acid.64
Thus, we decided to follow a different approach, performing a normal Ugi–Joullié reaction, but using an easily removable carboxylic acid. While there are many examples of “convertible” isocyanides, the use of removable carboxylic acids in the Ugi or Ugi–Joullié reaction is less explored. An acid component sometimes used for this scope is 4-pentenoic acid, that can be removed by treatment with iodine.28,65
However, as removable carboxylic acid, we selected known compound 12 (Scheme 4), that was prepared by a modification of the reported procedures (see ESI†).66,67 The Ugi–Joullié reaction using this acid proceeded as usual, affording adduct 10l with high d.r.
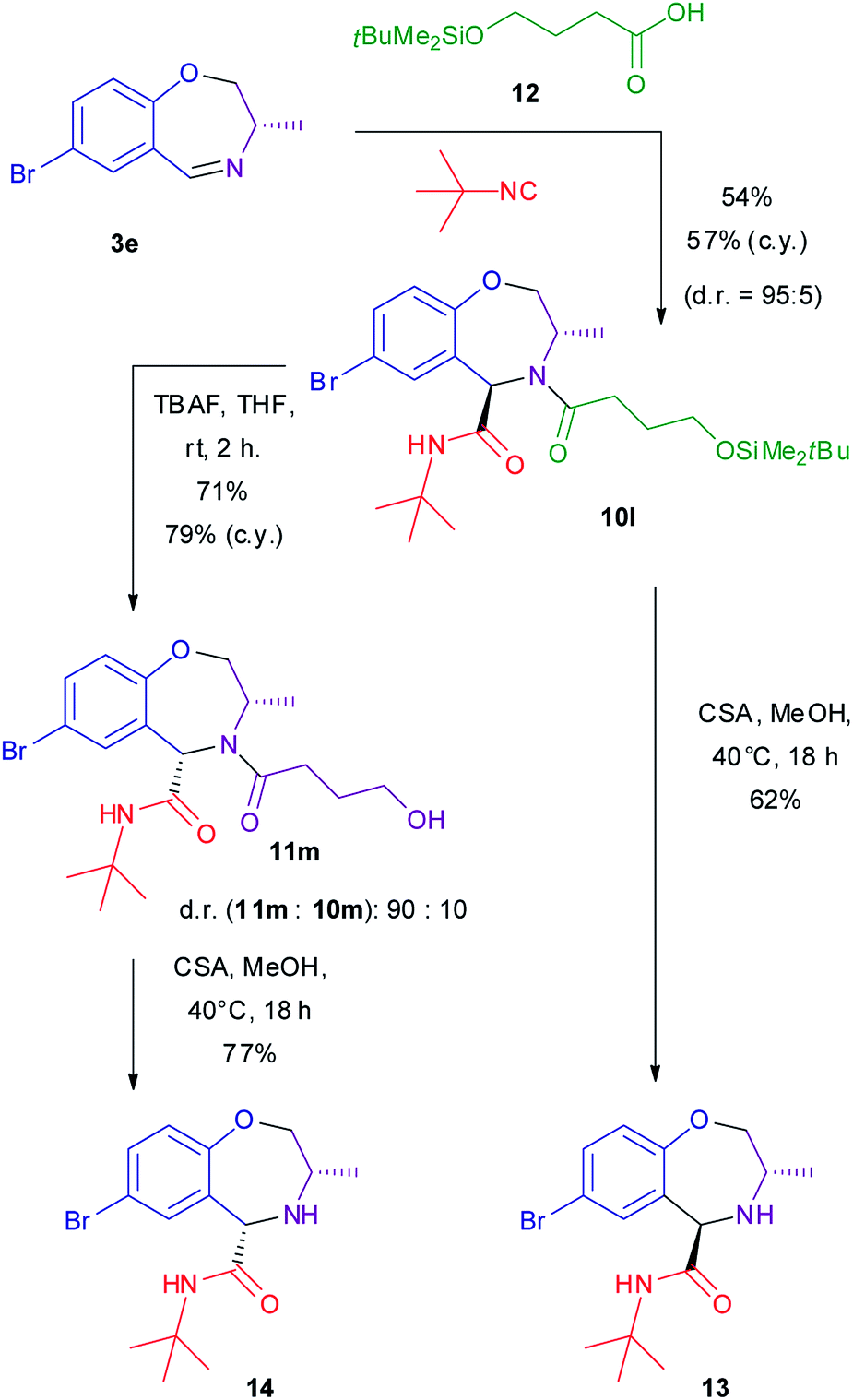 | ||
| Scheme 4 Synthesis of diastereomeric amines 13 and 14. c.y. = combined yield of both diastereomers. CSA = camphorsulphonic acid. TBAF = tetrabutylammonium fluoride. | ||
Our initial plan was to use tetrabutylammonium fluoride to promote desilylation of the alcohol and to catalyse the subsequent intramolecular acyl transfer to give γ-butyrolactone and the free secondary amine 13. With our surprise, not only the reaction stopped at the level of free alcohols 10m–11m, TBAF being unable to promote intramolecular acyl transfer, but a nearly complete epimerization took place, converting trans 10m into cis epimer 11m (d.r. = 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10). Alcohol 11m can be isolated in diastereomerically pure form by chromatography.
:10). Alcohol 11m can be isolated in diastereomerically pure form by chromatography.
On the other hand, by treatment of 10l with an acid catalyst in methanol, the desilylation reaction takes place quickly without any epimerization at rt. Then, by raising the temperature to 40 °C intramolecular acyl transfer leads to the release of γ-butyrolactone and to the formation of diastereomerically pure amine 13. The same conditions convert cis alcohol 11m to diastereomerically pure amine 14. Thus, thanks to the TBAF promoted epimerization, it is possible to stereodivergently convert trans 10 into both trans and cis amines 13 and 14, depending on reaction conditions. Since both enantiomers of amino alcohols 7 are available, this means that all four stereoisomers of these secondary amines are easily accessible. This fact is quite useful in view of investigation of these secondary amines as organocatalysts: we can explore both decoration diversity (up to 3 diversity inputs) and stereochemical diversity. It is worth noting that amines 13 and 14 are completely stable against epimerization under basic conditions (TBAF or KOH), indicating that only N-acylated compounds are prone to epimerization.
In order to get some additional information on the epimerization mechanism, we submitted 10j (obtained from imine (S)-3e) to basic conditions (KOH in MeOH) (Scheme 5). Again, epimerization took place, giving a cis:trans ratio of 68:32. Analysis by HPLC on chiral stationary phase of the two crudes, showed that both 10j and 11j were enantiomerically pure, and different from ent-10j and ent-11j obtained from Ugi–Joullié reaction of imine (R)-3e. This fact clearly demonstrates that epimerization occurs at carbon 5 and not at carbon 3, otherwise 10j would have been converted into ent-11j.
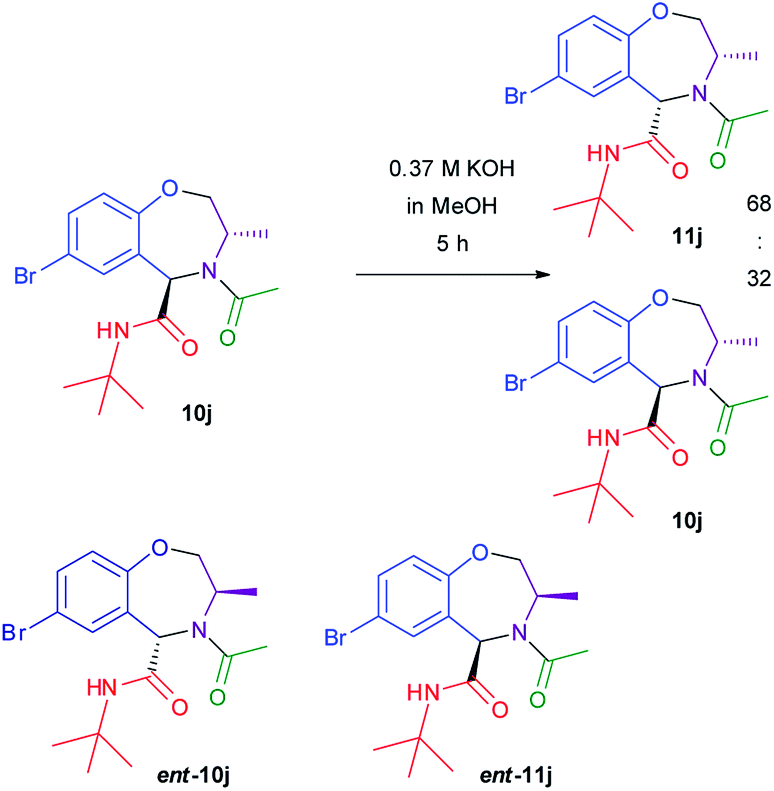 | ||
| Scheme 5 Epimerization of 10j and ent-10j. | ||
Treatment of isolated pure cis compound 11j under the same conditions again affords a 68:32 cis:trans ratio, proving that this is the thermodynamic ratio. Finally, also in the case of compound 10a epimerization takes place under these conditions, affording a cis:trans ratio = 68:32.
These experiments demonstrate that, for 10a–11a, 10j–11j, or 10m–11m (and probably also for all the other Ugi–Joullié products here described) the “kinetic” product is not the most stable one. Furthermore, the thermodynamic preference for the cis isomer is stronger for 10m–11m than for 10a–11a or 10j–11j. This suggest some influence of the free hydroxy group.
The relative configuration was established, at the level of amines 13 and 14, by a very clear NOE effect between H-5 and H-3, which is present only in the cis compound. Although the conformational equilibrium of these benzoxazepines is rather complex (see a discussion in the ESI†), no conformation of the trans compound would be able to experience this NOE.
We could not find in the literature conformational studies on these systems, apart from our own paper on products similar to 10–11, but with the substituent bound at carbon 2, instead of at carbon 3.42 The ESI† contains a discussion on these complex conformational equilibria, applied to 10j–11j. The most stable conformations are either half-chair or twist, where the substituent at C-5 occupies an axial position. Among them, the best one is a twist conformation of the cis stereoisomer 11j where the methyl group is in equatorial position. This result agrees with the thermodynamic preference for the cis product.
The conformation of imine 3e is a sort of half-chair, where the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond is coplanar with the benzene ring. Among the two possible half-chairs, the one with the methyl in pseudo-equatorial position seems more stable. In any case the face opposite to the substituent is less encumbered, leading to a kinetic preference for the trans compound.
N bond is coplanar with the benzene ring. Among the two possible half-chairs, the one with the methyl in pseudo-equatorial position seems more stable. In any case the face opposite to the substituent is less encumbered, leading to a kinetic preference for the trans compound.
Conclusions
In conclusion we have developed a concise and diversity-oriented methodology to obtain both N-acylated, and N-free tetrahydrobenzo[f][1,4]oxazepines with a high control of configuration of the two stereogenic centres. The enantiomeric purity is granted by the starting materials (1,2-amino alcohols of α-amino acids), which are easily accessible from the chiral pool. The methodology is based on a highly diastereoselective Ugi–Joullié reaction that strongly favours the trans products. However, also the thermodynamically favoured cis isomers can be obtained thanks to a base mediated epimerization. These seven-membered nitrogen heterocycles can find applications in medicinal chemistry, because tetrahydro-1,4-benzoxazepines are typical privileged structures. On the other hand, secondary amines such as 13, 14 or their analogues may be explored as asymmetric organocatalysts. An easy combinatorial synthesis of such potential catalysts, exploring both decoration and stereochemical diversity, is indeed possible. Studied directed towards this goal are in progress.Experimental section
Typical procedure for the Mitsunobu reaction: (S)-tert-butyl (1-(2-(dimethoxymethyl)phenoxy)-3-phenylpropan-2-yl)carbamate 8a
A solution of salicylaldehyde (0.770 mL, 901 mg, 7.38 mmol) in dry MeOH (8.1 mL) was treated with trimethyl orthoformate (3.50 mL, 32.0 mmol) and Amberlyst 15 (100 mg). The resulting mixture was stirred at room temperature for 20 h, then diluted with CH2Cl2 and filtered. The filtrate was treated with solid NaHCO3 (25 mg) and filtered again. The filtrate was evaporated, and the residue was taken up in CH2Cl2/toluene, and again evaporated to azeotropically remove all methanol. The crude acetal 6a was obtained as a yellow oil (1.380 g). This acetal (301.7 mg, theoretical 1.61 mmol, 1.2 equiv.) was dissolved in dry THF (2.60 mL), cooled to 0 °C, and treated sequentially with Boc-amino alcohol 7a (330.5 mg, 1.31 mmol, 1 equiv.), PPh3 (171.8 mg, 0.655 mmol, 0.5 equiv.) and 1,1′-(azodicarbonyl)dipiperidine (ADDP) (165.5 mg, 0.656 mmol, 0.5 equiv.). After stirring at 0 °C for 60 min, further PPh3 (171.8 mg, 0.655 mmol) and ADDP (165.5 mg, 0.656 mmol) were added. Finally, after 60 minutes, an identical amount of both reagents was added. The mixture was further stirred overnight at rt and evaporated to dryness. The crude was taken up in Et2O and washed with 1 M NaOH, to remove excess of 6a. The organic phase was washed with aqueous saturated NH4Cl, evaporated and purified by chromatography (PE/AcOEt 11:1) to give pure 8a as an oil (411.4 mg, 78% from 7a). For characterization data, see ESI.†
Typical procedure for the Ugi–Joullié reaction: (3S,5R)-4-acetyl-3-benzyl-N-(tert-butyl)-2,3,4,5-tetrahydrobenzo[f][1,4]oxazepine-5-carboxamide 10a
Acetal 8a (312.4 mg, 778 μmol) was dissolved in CH2Cl2 (3.0 mL) and treated with 37% aqueous HCl (640 μL, 7.73 mmol). The mixture was stirred at rt for 5 h. Then it was diluted with CH2Cl2, and cautiously treated with 5% aqueous Na2CO3 (25 mL). After checking that pH > 9, the two phases were separated, washed with brine, and evaporated to dryness. The resulting crude imine 3a (oil) was taken up in dry methanol (3.90 mL), and treated with acetic acid (53 μL, 927 μmol), and tert-butyl isocyanide (106 μL, 937 μmol). The mixture was stirred at rt for 45 h and evaporated to dryness. It was taken up in AcOEt, washed with saturated aqueous NaHCO3 (to remove excess carboxylic acid), evaporated, and chromatographed (PE/AcOEt 2:1) to give pure 10a (237 mg, 80%). The ratio 10a:11a was determined by HPLC on the crude product and resulted = 95:5. For HPLC conditions and characterization data, see ESI.†
(3S,5R)-7-Bromo-N-(tert-butyl)-3-methyl-2,3,4,5-tetrahydrobenzo[f][1,4]oxazepine-5-carboxamide 13
Compound 10l (75.2 mg, 139 μmol) was dissolved in a 1.0 M solution of camphorsulfonic acid in dry MeOH (0.60 mL), and stirred overnight at 40 °C. Then it was diluted with AcOEt and washed with a 1:1 mixture of saturated aqueous NaHCO3 and brine. The final pH of aqueous phase was 8. The phases were separated, and the organic one evaporated to dryness and chromatographed (PE/AcOEt 2:1 + 1% EtOH → PE/AcOEt 1:1 + 1% EtOH) to give pure 13 as a white solid (29 mg, 62%). Rf = 0.55 (PE/AcOEt 2:1). [α]D = +28.6 (c 0.6, CHCl3). Mp: 154.0–156.2 °C. 1H NMR (300 MHz, CDCl3, 20 °C): δ = 7.34 (1H, dd, J = 8.4, 2.4 Hz), 7.29 (1H, d, J = 2.4 Hz), 6.92 (1H, d, J = 8.4 Hz), 5.96 (1H, broad s, NH), 4.40 (1H, s, CHCO), 4.12 (1H, dd, J = 11.6, 2.0 Hz, CHHO), 3.50–3.31 (2H, m, CHHO, CHN), 1.32 (9H, s, C(CH3)3), 1.07 (3H, d, J = 6.3 Hz, CH3CH). 13C NMR (75 MHz, CDCl3, 25 °C): δ = 169.3 (CO), 157.7, 133.7, 116.2 (quat.), 133.6, 132.3, 123.4 (aromatic CH), 78.4 (CH2O), 64.3 (CHCO), 51.5 (CNH), 50.8 (CHN), 28.7 (C(CH3)3), 17.8 (CH3CH). IR (ATR): νmax 3320, 3295, 2995, 2970, 2930, 2869, 1745, 1673, 1520, 1502, 1483, 1452, 1391, 1363, 1345, 1298, 1268, 1243, 1222, 1194, 1166, 1114, 1080, 1045, 1016, 985, 941, 928, 907, 874, 819, 786, 761, 726, 661 cm−1. HRMS (ESI+): found 341.0870 [calcd for C15H22BrN2O2+ (M + H)+ 341.0865].
(3S,5S)-7-Bromo-N-(tert-butyl)-3-methyl-2,3,4,5-tetrahydrobenzo[f][1,4]oxazepine-5-carboxamide 14
Compound 10l (103.7 mg, 191 μmol) was dissolved in dry THF (1.22 mL) and treated with a 1 M solution of tetrabutylammonium fluoride (TBAF) in THF (230 μL, 230 μmol). The mixture was stirred at rt for 2 h. Then it was diluted with AcOEt and washed with a 1:1 mixture of brine and water. The organic phases were evaporated. HPLC analysis of this crude product indicated a d.r. of 90:10 (see ESI for details†). This crude product was chromatographed (PE/AcOEt 1:2 + 1% EtOH) to give pure cis compound 11m as a colorless oil (58 mg, 71%). The overall yield, calculated from d.r., was 79%. This intermediate was dissolved in a 1.0 M solution of camphorsulfonic acid in dry MeOH (0.63 mL), and stirred overnight at 40 °C. Then it was diluted with AcOEt and washed with a 1:1 mixture of saturated aqueous NaHCO3 and brine. The final pH of aqueous phase was 8. The phases were separated, and the organic one evaporated to dryness and chromatographed (PE/AcOEt 2:1 + 1% EtOH → PE/AcOEt 1:1 + 1% EtOH) to give pure 14 as a white solid (36 mg, 77%). Rf = 0.55 (PE/AcOEt 2:1). [α]D = −57.0 (c 1.0, CHCl3). Mp: 107.6–110.0 °C. 1H NMR (300 MHz, CDCl3, 20 °C): δ = 7.39 (1H, d, J = 2.4 Hz), 7.30 (1H, dd, J = 8.4, 2.4 Hz), 7.10 (1H, broad s, NH), 6.90 (1H, d, J = 8.4 Hz), 4.53 (1H, s, CHCO), 4.24 (1H, d, J = 9.0 Hz, CHHO), 3.32–3.18 (2H, m, CHHO, CHN), 1.43 (9H, s, C(CH3)3), 1.03 (3H, d, J = 5.6 Hz, CH3CH). 13C NMR (75 MHz, CDCl3, 25 °C): δ = 169.6 (CO), 158.0, 137.0, 116.8 (quat.), 131.6, 130.4, 123.3 (aromatic CH), 78.6 (CH2O), 61.4 (CHCO), 54.5 (CHNH), 51.1 (CNH), 28.8 (C(CH3)3), 17.7 (CH3CH). IR (ATR): νmax 3338, 3286, 2965, 2931, 2875, 1661, 1513, 1477, 1457, 1392, 1365, 1316, 1300, 1286, 1264, 1245, 1223, 1164, 1144, 1104, 1085, 1047, 1009, 994, 939, 915, 903, 884, 874, 852, 829, 823, 805, 774, 751, 730, 675, 623 cm−1. HRMS (ESI+): found 341.0866 [calcd for C15H22BrN2O2+ (M + H)+ 341.0865].
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Mr Francesco Bellina for his collaboration to this work, Dr Giuliana Ottonello and Dr Andrea Armirotti for HRMS.Notes and references
- Science of Synthesis: Multicomponent Reactions 1 and 2, ed. T. J. J. Müller, Thieme, Stuttgart, 2014 Search PubMed.
- P. Slobbe, E. Ruijter and R. V. A. Orru, RSC Med. Chem., 2012, 3, 1189–1218 RSC.
- E. Ruijter and R. Orru, Drug Discov. Today Technol., 2018, 29, 1–2 CrossRef PubMed.
- B. Biersack, K. Ahmed, S. Padhye and R. Schobert, Expert Opin. Drug Discov., 2018, 13, 39–49 CrossRef CAS PubMed.
- F. de Moliner, N. Kielland, R. Lavilla and M. Vendrell, Angew. Chem., Int. Ed., 2017, 56, 3758–3769 CrossRef CAS PubMed.
- V. Maffeis, L. Moni, D. Di Stefano, S. Giordani and R. Riva, Front. Chem. Sci. Eng., 2019 DOI:10.1007/s11705-019-1833-0.
- L. Moni, C. F. Gers-Panther, M. Anselmo, T. J. J. Muller and R. Riva, Chem.–Eur. J., 2016, 22, 2020–2031 CrossRef CAS PubMed.
- A. F. de la Torre, G. S. Scatena, O. Valdes, D. G. Rivera and M. W. Paixao, Beilstein J. Org. Chem., 2019, 15, 1210–1216 CrossRef CAS PubMed.
- A. Znabet, E. Ruijter, F. J. J. de Kanter, V. Kohler, M. Helliwell, N. J. Turner and R. V. A. Orru, Angew. Chem., Int. Ed., 2010, 49, 5289–5292 CrossRef CAS PubMed.
- O. I. Shmatova and V. G. Nenajdenko, J. Org. Chem., 2013, 78, 9214–9222 CrossRef CAS PubMed.
- R. C. Cioc, V. Estevez, D. J. van der Niet, C. M. L. Vande Velde, N. G. Turrini, M. Hall, K. Faber, E. Ruijter and R. V. A. Orru, Eur. J. Org. Chem., 2017, 1262–1271 CrossRef CAS PubMed.
- L. Moni, L. Banfi, A. Basso, A. Bozzano, M. Spallarossa, L. Wessjohann and R. Riva, Molecules, 2016, 21, 1153 CrossRef PubMed.
- K. Vlahovicek-Kahlina, M. Vazdar, A. Jakas, V. Smrecki and I. Jeric, J. Org. Chem., 2018, 83, 13146–13156 CrossRef CAS PubMed.
- M. Zimuwandeyi, F. Kola, A. Lemmerer, D. Brady, A. L. Rousseau and M. L. Bode, Tetrahedron, 2018, 74, 2925–2941 CrossRef CAS.
- P. R. Krishna, G. Dayaker, D. V. Ramana and R. Kunde, Helv. Chim. Acta, 2014, 97, 1076–1087 CrossRef.
- L. Moni, L. Banfi, A. Basso, E. Martino and R. Riva, Org. Lett., 2016, 18, 1638–1641 CrossRef CAS PubMed.
- L. Moni, L. Banfi, A. Basso, L. Carcone, M. Rasparini and R. Riva, J. Org. Chem., 2015, 80, 3411–3428 CrossRef CAS PubMed.
- S. Gazzotti, G. Rainoldi and A. Silvani, Expert Opin. Drug Discov., 2019, 14, 639–652 CrossRef CAS PubMed.
- A. Katsuyama, A. Matsuda and S. Ichikawa, Org. Lett., 2016, 18, 2552–2555 CrossRef CAS PubMed.
- L. Moni, L. Banfi, A. Basso, A. Galatini, M. Spallarossa and R. Riva, J. Org. Chem., 2014, 79, 339–351 CrossRef CAS PubMed.
- A. Katsuyama, F. Yakushiji and S. Ichikawa, J. Org. Chem., 2018, 83, 7085–7101 CrossRef CAS PubMed.
- G. Rainoldi, F. Begnini, M. de Munnik, L. Lo Presti, C. M. L. Vande Velde, R. Orru, G. Lesma, E. Ruijter and A. Silvani, ACS Comb. Sci., 2018, 20, 98–105 CrossRef CAS PubMed.
- E. Speich, L. Banfi, L. Moni, R. Riva, V. Rocca and A. Basso, Chem. Heterocycl. Comp., 2018, 54, 329–333 CrossRef CAS.
- V. Cerulli, L. Banfi, A. Basso, V. Rocca and R. Riva, Org. Biomol. Chem., 2012, 10, 1255–1274 RSC.
- D. Hayashi, N. Tsukioka, Y. Inoue, Y. Matsubayashi, T. Iizuka, K. Higuchi, Y. Ikegami and T. Kawasaki, Bioorg. Med. Chem., 2015, 23, 2010–2023 CrossRef CAS PubMed.
- P. Szczesniak, E. Maziarz, S. Stecko and B. Furman, J. Org. Chem., 2015, 80, 3621–3633 CrossRef CAS PubMed.
- E. R. van Rijssel, T. P. M. Goumans, G. Lodder, H. S. Overkleeft, G. A. van der Marel and J. D. C. Codee, Org. Lett., 2013, 15, 3026–3029 CrossRef CAS PubMed.
- T. Wennekes, K. M. Bonger, K. Vogel, R. J. B. H. N. van den Berg, A. Strijland, W. E. Donker-Koopman, J. M. F. G. Aerts, G. A. van der Marel and H. S. Overkleeft, Eur. J. Org. Chem., 2012, 6420–6454 CrossRef CAS.
- A. Znabet, M. M. Polak, E. Janssen, F. J. J. de Kanter, N. J. Turner, R. V. A. Orru and E. Ruijter, Chem. Commun., 2010, 46, 7918–7920 RSC.
- L. Banfi, A. Basso, G. Guanti and R. Riva, Tetrahedron Lett., 2004, 45, 6637–6640 CrossRef CAS.
- M. M. Bowers, P. Carroll and M. M. Joullié, J. Chem. Soc., Perkin Trans. 1, 1989, 857–865 RSC.
- J. Hoogenboom, M. Lutz, H. Zuilhof and T. Wennekes, J. Org. Chem., 2016, 81, 8826–8836 CrossRef CAS PubMed.
- A. L. Chandgude, D. Narducci, K. Kurpiewska, J. Kalinowska-Tluscik and A. Domling, RSC Adv., 2017, 7, 49995–49998 RSC.
- T. Ramanivas, G. Gayatri, D. Priyanka, V. L. Nayak, K. K. Singarapu and A. K. Srivastava, RSC Adv., 2015, 5, 73373–73380 RSC.
- K. Katayama, T. Okamura, T. Sunadome, K. Nakagawa, H. Takeda, M. Shiro, A. Matsuda and S. Ichikawa, J. Org. Chem., 2014, 79, 2580–2590 CrossRef CAS PubMed.
- C. S. Azad and A. K. Saxena, Org. Chem. Front., 2015, 2, 665–669 RSC.
- D. Zhu, L. Xia, L. Pan, S. Li, R. Chen, Y. Mou and X. Chen, J. Org. Chem., 2012, 77, 1386–1395 CrossRef CAS PubMed.
- G. van der Heijden, T. B. van Schaik, V. Mouarrawis, M. J. M. de Wit, C. M. L. V. Velde, E. Ruijter and R. V. A. Orru, Eur. J. Org. Chem., 2019, 5313–5325 CrossRef CAS.
- A. Angyal, A. Demjen, E. Weber, A. K. Kovacs, J. Wolfling, L. G. Puskas and I. Kanizsai, J. Org. Chem., 2018, 83, 3570–3581 CrossRef CAS PubMed.
- L. Banfi, A. Basso, C. Lambruschini, L. Moni and R. Riva, Chem. Heterocycl. Comp., 2017, 53, 382–408 CrossRef CAS.
- O. Bakulina, M. Chizhova, D. Dar'in and M. Krasavin, Eur. J. Org. Chem., 2018, 2018, 362–371 CrossRef CAS.
- L. Banfi, A. Bagno, A. Basso, C. De Santis, R. Riva and F. Rastrelli, Eur. J. Org. Chem., 2013, 2013, 5064–5075 CrossRef CAS.
- S. K. Reddy Guduru, S. Chamakuri, I. O. Raji, K. R. MacKenzie, C. Santini and D. W. Young, J. Org. Chem., 2018, 83, 11777–11793 CrossRef CAS PubMed.
- K. Jedrzejczak, P. Hrynczyszyn, M. Szczesio, J. Artym, T. Jastrzabek, M. Kocieba, M. Glowka, K. Huben, I. Kochanowska, M. Zimecki, J. Zabrocki, S. Jankowski and B. Kolesinska, Bioorg. Med. Chem., 2017, 25, 4265–4276 CrossRef CAS PubMed.
- A. Del Vecchio, F. Caille, A. Chevalier, O. Loreau, K. Horkka, C. Halldin, M. Schou, N. Camus, P. Kessler, B. Kuhnast, F. Taran and D. Audisio, Angew. Chem., Int. Ed., 2018, 57, 9744–9748 CrossRef CAS PubMed.
- A. L. Braga, D. S. Ludtke, M. W. Paixao, E. E. Alberto, H. A. Stefani and L. Juliano, Eur. J. Org. Chem., 2005, 4260–4264 CrossRef CAS.
- T. Fujimoto, J. Kunitomo, Y. Tomata, K. Nishiyama, M. Nakashima, M. Hirozane, S. Yoshikubo, K. Hirai and S. Marui, Bioorg. Med. Chem. Lett., 2011, 21, 6414–6416 CrossRef CAS PubMed.
- D. Z. Yang, P. Wang, J. Z. Liu, H. L. Xing, Y. Liu, W. C. Xie and G. S. Zhao, Bioorg. Med. Chem., 2014, 22, 366–373 CrossRef CAS PubMed.
- K. Sagi, T. Nakagawa, M. Yamanashi, S. Makino, M. Takahashi, M. Takayanagi, K. Takenaka, N. Suzuki, S. Oono, N. Kataoka, K. Ishikawa, S. Shima, Y. Fukuda, T. Kayahara, S. Takehana, Y. Shima, K. Tashiro, H. Yamamoto, R. Yoshimoto, S. Iwata, T. Tsuji, K. Sakurai and M. Shoji, J. Med. Chem., 2003, 46, 1845–1857 CrossRef CAS PubMed.
- D. Obrecht, P. Ermert, S. Oumouch, A. Piettre, J.-F. Gosalbes and M. Thommen, World Intellectual Property Organization Pat., WO 2013/139697 A1, 2013.
- S. Naganathan, D. L. Andersen, N. G. Andersen, S. Lau, A. Lohse and M. D. Sorensen, Org. Process Res. Dev., 2015, 19, 721–734 CrossRef CAS.
- A. Ebrahimi, K. Sevinc, G. G. Sevinc, A. P. Cribbs, M. Philpott, F. Uyulur, T. Morova, J. E. Dunford, S. Goklemez, S. Ari, U. Oppermann and T. T. Onder, Nat. Chem. Biol., 2019, 15, 519 CrossRef CAS PubMed.
- T. A. Popp, C. Tallant, C. Rogers, O. Fedorov, P. E. Brennan, S. Muller, S. Knapp and F. Bracher, J. Med. Chem., 2016, 59, 8889–8912 CrossRef CAS PubMed.
- C. S. Takeuchi, B. G. Kim, C. M. Blazey, S. Ma, H. W. B. Johnson, N. K. Anand, A. Arcalas, T. G. Baik, C. A. Buhr, J. Cannoy, S. Epshteyn, A. Joshi, K. Lara, M. S. Lee, L. C. Wang, J. W. Leahy, J. M. Nuss, N. Aay, R. Aoyama, P. Foster, J. Lee, I. Lehoux, N. Munagala, A. Plonowski, S. Rajan, J. Woolfrey, K. Yamaguchi, P. Lamb and N. Miller, J. Med. Chem., 2013, 56, 2218–2234 CrossRef CAS PubMed.
- J. E. Wilson, R. Kurukulasuriya, C. Sinz, M. Lombardo, K. Bender, D. Parker, E. C. Sherer, M. Costa, K. Dingley, X. F. Li, S. Mitelman, S. Tong, R. Bugianesi, A. Ehrhardt, B. Priest, K. Ratliff, F. Ujjainwalla, R. Nargund, W. K. Hagmann and S. Edmondson, Bioorg. Med. Chem. Lett., 2016, 26, 2947–2951 CrossRef CAS PubMed.
- M. Kono, T. Oda, M. Tawada, T. Imada, Y. Banno, N. Taya, T. Kawamoto, H. Tokuhara, Y. Tomata, N. Ishii, A. Ochida, Y. Fukase, T. Yukawa, S. Fukumoto, H. Watanabe, K. Uga, A. Shibata, H. Nakagawa, M. Shirasaki, Y. Fujitani, M. Yamasaki, J. Shirai and S. Yamamoto, Bioorg. Med. Chem., 2018, 26, 470–482 CrossRef CAS PubMed.
- S. J. Taylor, E. H. Demont, J. Gray, N. Deeks, A. Patel, D. Nguyen, M. Taylor, S. Hood, R. J. Watson, R. A. Bit, F. McClure, H. Ashall and J. Witherington, J. Med. Chem., 2015, 58, 8236–8256 CrossRef CAS PubMed.
- L. Toth, Y. Fu, H. Y. Zhang, A. Mandi, K. E. Kover, T. Z. Illyes, A. Kiss-Szikszai, B. Balogh, T. Kurtan, S. Antus and P. Matyus, Beilstein J. Org. Chem., 2014, 10, 2594–2602 CrossRef PubMed.
- S. Klossowski, B. Wiraszka, S. Berlozecki and R. Ostaszewski, Org. Lett., 2013, 15, 566–569 CrossRef CAS PubMed.
- Q. Wang, D. X. Wang, M. X. Wang and J. P. Zhu, Acc. Chem. Res., 2018, 51, 1290–1300 CrossRef CAS PubMed.
- M. Milen, A. Dancso, T. Foldesi and B. Volk, Tetrahedron, 2017, 73, 70–77 CrossRef CAS.
- A. Shaabani, S. Keshipour, S. Shaabani and M. Mahyari, Tetrahedron Lett., 2012, 53, 1641–1644 CrossRef CAS.
- A. Kumar, D. Saxena and M. K. Gupta, RSC Adv., 2013, 3, 4610–4612 RSC.
- B. Saha, B. Frett, Y. Wang and H.-y. Li, Tetrahedron Lett., 2013, 54, 2340–2343 CrossRef CAS.
- R. Madsen, C. Roberts and B. Fraser-Reid, J. Org. Chem., 1995, 60, 7920–7926 CrossRef CAS.
- D. Nakashima and H. Yamamoto, J. Am. Chem. Soc., 2006, 128, 9626–9627 CrossRef CAS PubMed.
- P. Renton, L. Shen, J. Eckert, G. M. Lee, D. Gala, G. Chen, B. Pramanik and D. Schumacher, Org. Process Res. Dev., 2002, 6, 36–41 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra10689h |
| This journal is © The Royal Society of Chemistry 2020 |