
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Colorectal cancer stem cells: a review of targeted drug delivery by gold nanoparticles
Manali Haniti Mohd-Zahid
 a,
Rohimah Mohamud
b,
Che Azurahanim Che Abdullah
cd,
JitKang Lim
e,
Halima Alem
f,
Wan Nurhidayah Wan Hanaffib and
Iskandar Z. A.
*a
a,
Rohimah Mohamud
b,
Che Azurahanim Che Abdullah
cd,
JitKang Lim
e,
Halima Alem
f,
Wan Nurhidayah Wan Hanaffib and
Iskandar Z. A.
*a
aDepartment of Chemical Pathology, School of Medical Sciences, Universiti Sains Malaysia, 16150 Kubang Kerian, Kelantan, Malaysia. E-mail: iskandarza@usm.my
bDepartment of Immunology, School of Medical Sciences, Universiti Sains Malaysia, 16150 Kubang Kerian, Kelantan, Malaysia
cDepartment of Physics, Faculty of Science, Universiti Putra Malaysia, 43400 UPM Serdang, Selangor, Malaysia
dMaterial Synthesis and Characterization Laboratory, Institute of Advanced Technology, Universiti Putra Malaysia, 43400 UPM Serdang, Selangor, Malaysia
eSchool of Chemical Engineering, Universiti Sains Malaysia, 14300 Nibong Tebal, Penang, Malaysia
fInstitut Jean Lamour (IJL, UMR 7198), Université de Lorraine, CNRS, Campus Artem 2 allée André Guinier – BP 50840, F-54011 Nancy Cedex, France
First published on 9th January 2020
Abstract
Colorectal cancer (CRC) is one of the most common cancers with equal probability of affecting both men and women worldwide. Recently, the newly emerged theory of cancer stem cells has associated CRC stem cells (CRCSCs) with the high rates of recurrence and poor prognosis. Thus, targeting CRCSCs instead of CRC may resolve cancer relapse. Among the chemotherapeutic drugs, the antimetabolite 5-fluorouracil (5-FU) has shown high efficiency in CRC treatment. However, due to several limitations, the usefulness of 5-FU has been restricted. The application of gold nanoparticles (AuNPs) in drug delivery systems is reported to enhance the effectiveness of anticancer drugs due to their biostability, non-toxicity and feasibility for surface modification. Furthermore, the overexpression of biomolecular surface markers in CRCSCs may elevate the specific targeting by AuNPs, and hence, reduce the non-specific binding, which could lead to systemic side effects. This review briefly presents the proposed therapeutic potential of AuNPs loaded with 5-FU, conjugated with specific antibodies targeting CRCSCs, which could be valuable to improve some limitations in current CRC management.
 Manali Haniti Mohd-Zahid | Manali Haniti Mohd-Zahid received her BSc degree in Biotechnology from International Islamic University of Malaysia (IIUM) in 2014 and obtained her MSc degree in Pharmacy from Universiti Kebangsaan Malaysia (UKM) in 2018. She is currently a PhD degree candidate in the Department of Chemical Pathology, School of Medical Sciences at Universiti Sains Malaysia (USM). Her current research interests include targeted drug delivery systems using colloidal gold nanoparticles in colorectal cancer treatment. |
 Rohimah Mohamud | Rohimah Mohamud received her MSc and PhD degree in Vaccinology and Immunology from Universiti Sains Malaysia (USM) and Monash University, Australia in 2007 and 2014 respectively. Her expertise include the identification and functional studies of Regulatory T cells and dendritic cells, allergic airway inflammation mouse models, cancer immunology, and development of rBCG for TB vaccines. She is currently a University Lecturer and Principal Investigator at USM. Her current research is focused on nanoparticles that mediate immune response in asthma and cancer research both for human and animal models. |
 Che Azurahanim Che Abdullah | Che Azurahanim Che Abdullah received her PhD degree in Biophysics from University of Surrey in 2012, after a BSc in Medical Physics from Universiti Sains Malaysia (USM) in 2007. She is currently an Associate Professor in the Department of Physics, Faculty of Science at Universiti Putra Malaysia (UPM). She is an expert in nanomaterials and nanodelivery. Her current research interests include nanotheranostics. |
 JitKang Lim | JitKang Lim received his PhD degree in Chemical Engineering from Carnegie Mellon University working with Prof. Robert D. Tilton and Prof. Sara A. Majetich (Physics) and is currently a Professor of Chemical Engineering at Universiti Sains Malaysia (USM). Since October 2009, he has also held a courtesy appointment as the Visiting Research Professor of Physics at Carnegie Mellon University (CMU) in the United States. His current research interests focus on both colloidal behaviors and engineering applications of magnetic nanoparticles. |
 Halima Alem | Halima Alem is an Associate Professor at Lorraine University in the Department of Chemical Engineering. She has a strong background and more than a decade of experience in the design and elaboration of surfaces and nanomaterials for applications in healthcare and process engineering. She is an Associate Editor of the journal Nanomaterials and Nanotechnology and a member of two other editorial boards. She has co-authored more than 40 peer reviewed publications and participated in more than 70 national and international conferences, some of them as invited speakers. In October 2019, she was awarded by an Institut Universitaire de France delegation as a Junior member. |
 Iskandar Zulkarnain Alias | Iskandar Zulkarnain Alias received his PhD degree in Chemical Pathology from School of Medical Sciences at Universiti Sains Malaysia (USM) in 2006, after a Master of Community Health Sciences from Universiti Kebangsaan Malaysia (UKM) in 1999. He is currently a Senior Lecturer in the Department of Chemical Pathology at USM. His research interests include tumor marker studies in breast cancer, apoptosis and survivin expression in breast cancer and cell culture and antibody production. His current research is focused on gold nanoparticles and drug delivery systems in colorectal cancer stem cells. |
1. Introduction
Colorectal cancer (CRC) is among the most common cancers and accounts for nearly 9% of all cancers in the world1,2 with high mortality rate due to advanced stage diagnosis and high rates of recurrence.3 Studies have underlined the cause of recurrence and metastases as due to the presence of cancer stem cells (CSCs), a small subpopulation of cancer cells which are able to self-renew, differentiate and sustain tumor growth.2,4,5 Colorectal cancer stem cells (CRCSCs) are characterized by the overexpression of CD133, a five-transmembrane glycoprotein.6,7Currently, common cytotoxic chemotherapies used for the treatment of CRC are 5-fluorouracil (5-FU), oxaliplatin and cisplatin. Chemotherapeutic drugs kill cancer cells by interfering with their cell growth and division mechanisms.8 A few studies both on cellular and animal models have proved that treatment with these chemotherapeutic drugs, either alone or in combination with other chemotherapies, is effective in treating CRC.9–11 Among these chemotherapeutic agents, 5-FU is one of the most prescribed anti-tumor drugs for CRC therapy.12,13 Owing to its analogous structure to uracil, 5-FU can be incorporated into RNA and DNA and interferes with nucleoside metabolism. However, 5-FU-based chemotherapy also interferes with rapidly dividing healthy cells due to its lack of site specificity, and hence, causes common side effects such as hair loss, nausea and vomiting.14,15 In addition, rapid clearance from blood circulation and poor distribution limit the therapeutic action of 5-FU.16
Drug delivery systems in cancer therapeutics provide an alternative to minimize the limitations of conventional cancer chemotherapy by improving specific drug targeting, prolonging the circulation time and controlling drug release.17,18 In addition, targeted drug delivery increases bioavailability to maintain the drug concentration upon arriving at the cancer site.19 Within the past few decades, nanoparticles have received great interest among the CRC research community as one of the effective drug delivery systems. Some nanoparticles targeting CRC have been studied previously, including solid lipid nanoparticles,20,21 heparin-polyethyleneimine nanoparticles,22 silica nanoparticles23 and liposome-based nanoparticles.24,25 However, according to a literature survey (2005–2015) by Wilhelm et al., only 0.7% of the administered nanoparticle dose reaches the solid tumor, which eventually hinders the effective use of nanoparticles as drug delivery agents.26
Here, we summarize a proposed system of targeted drug delivery consisting of 5-FU loaded gold nanoparticles (AuNPs) conjugated with CD133 antibody to target CRCSCs, supported by previous studies (Table 1).
| System | Advantages | Reference |
|---|---|---|
| AuNPs as drug carrier | Facile synthesis | 27 |
| Biocompatible | ||
| Multi-functionalization | ||
| Tunable surface | ||
| mPEG-surface coating | Reduce protein adsorption on AuNPs | 28 and 29 |
| Avoid macrophage uptake | ||
| 5-FU | Antimetabolite | 30 |
| Inhibit DNA synthesis | ||
| Anti-CD133 monoclonal antibody conjugate | Overexpression of CD133 has been associated with the relapse, metastasis and chemotherapy resistant | 7 and 31 |
| Interfere on specific proteins overexpressed CD133 involved in tumorigenesis (targeted cancer therapy) | ||
| CRCSCs targeting | Targeting CRCSCs rather than tumor bulk could be effective in reducing risk of relapse and metastasis | 32 |
Previously, the specific targeting of the CSCs in CRC by CD133 antibody has been studied using methoxy poly(ethylene glycol)–poly(ε-caprolactone) (mPEG–PCL) nanoparticles loaded with anticancer drug 7-ethyl-10-hydroxy-camptothecin (SN-38).33 The in vitro and in vivo studies showed that CD133-targeting nanoparticles have increased cytotoxicity and inhibited tumor growth in HCT116 cells and mouse xenograft models, respectively, compared with the none antibody-targeted nanoparticles. In this case, the therapeutic effects on CRC have been enhanced by the CD133 antibody conjugates because the chemotherapeutic agent SN-38 is efficiently guided to the overexpressed CD133 markers at the tumor site.
Fig. 1 shows the proposed schematic mechanism of 5-FU loaded AuNPs conjugated with CD133 antibody to target CRCSCs. In drug delivery systems, nonionic polymer-surface modification of AuNPs by methoxy polyethylene glycol (mPEG) thiol is commonly used to improve blood residence time, reduce reticulo-endothelial system (RES) uptake and avoid non-specific targeting.28 The AuNP complexes will be internalized by CRCSCs through passive (neovasculature) and active targeting (anti-CD133 ligand-receptor docking) followed by fusion with lysosomes. Owing to the changes of some pathological events in tumor cells, such as pH and concentration of intracellular glutathione (GSH), AuNPs can be engineered to be activated by these endogenous stimuli in order to release the payloads at the tumor site and hence enhance the therapeutic effects and avoid systemic side effects to healthy cells. Regularly, the extracellular pH of solid tumor cells is acidic (5.5 to 6.5) compared to the physiological condition34 due to the anaerobic glycolysis-produced lactic acid in hypoxia35 and substantial H+ generation because of higher metabolic activity.36 Besides the acidic tumor environment, 5-FU will be released on exposure to lysosomal enzymes followed by interfering with DNA synthesis and RNA processing and functioning to cause tumor cell death.
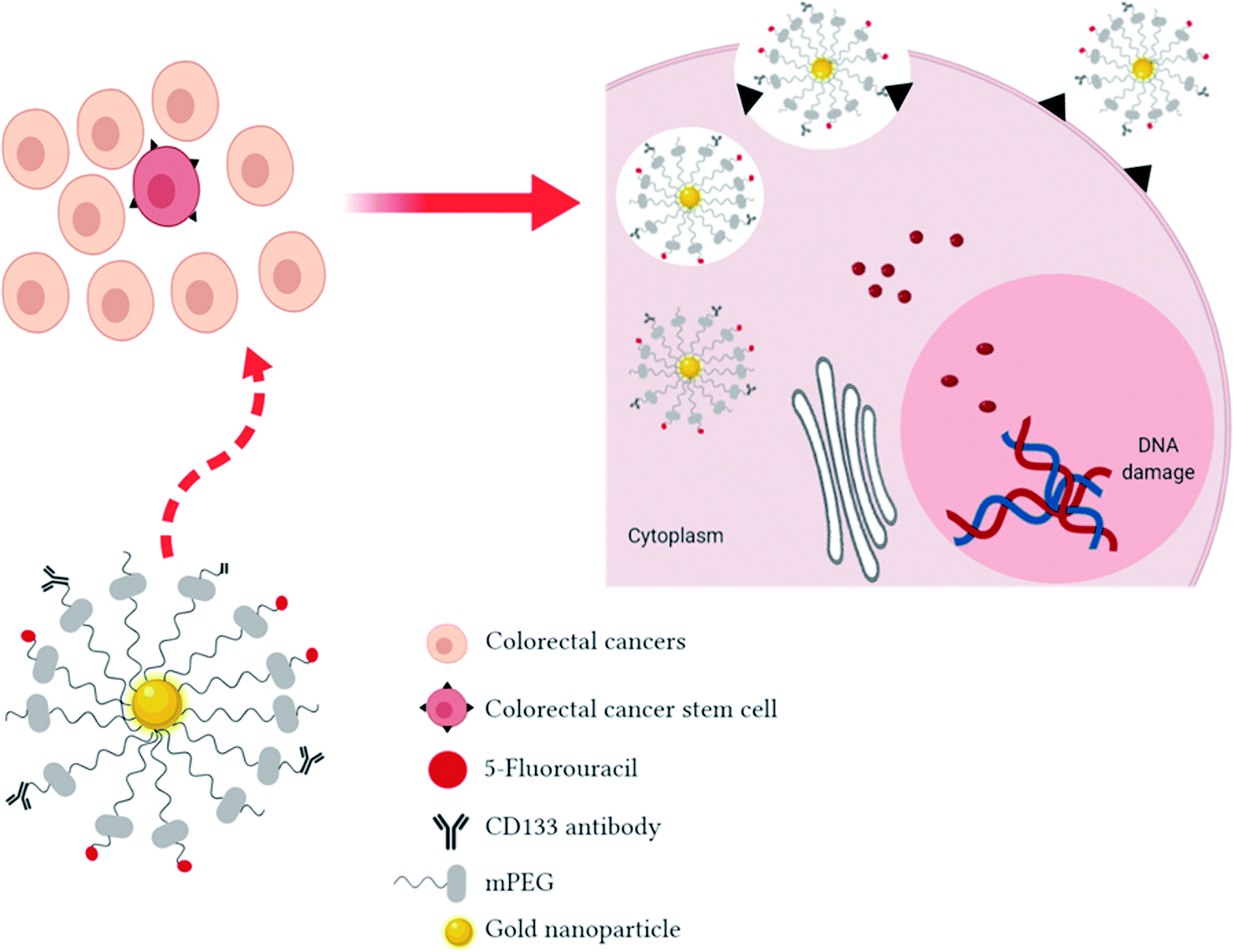 | ||
| Fig. 1 The proposed schematic mechanism of 5-fluorouracil (5-FU) loaded gold nanoparticles (AuNPs) conjugated with CD133 antibodies to target colorectal cancer stem cells (CRCSCs) in a targeted drug delivery system. Methoxy polyethylene glycol (mPEG)-stabilized AuNPs loaded with 5-FU and conjugated with CD133 antibody will target the CRCSCs instead of the bulk of colorectal cancers due to the overexpression of CD133 antigen on the surface of CRCSCs. High affinity binding of the CD133 antibody ligand with the targeted cells will increase the delivery efficiency and hence protect healthy cells. The AuNPs with loaded 5-FU will be internalized by cells via endocytosis. The acidic environment in the tumor may trigger the cleave of 5-FU from the AuNP complex inside cell endosomes to enhance cell toxicity by interfering with DNA synthesis. | ||
2. Colorectal cancer stem cells
According to the cancer stem cell (CSC) theory, a small subpopulation of cells share embryonic stem cell characteristics,2 giving rise to metastatic cancer cells and responsible for the high rate of recurrence.37 Indeed, CSCs share the same major signaling pathways with embryonic stem cells, including the Wnt, Notch, transforming growth factor beta (TGF-β), and Hedgehog signaling pathways.38 Nevertheless, quiescent CSCs express stemness genes, have the capability to self-renew and differentiate into other non-stem cancer cells and resist traditional chemotherapy and radiotherapy.39 Another crucial feature of CSCs is possessing tumorigenic potential, which is measured by the ability of the cells to initiate xenograft tumors upon transplantation.38Thus, targeting CSCs instead of the tumor bulk population is a therapeutic goal in CRC management, particularly in preventing cancer relapse. The isolation and identification of CSCs from CRC further elucidates several putative stem cell markers. The identification of CRCSC markers will be beneficial in identifying the disease progression as well as the risk of recurrence among patients.40 Table 2 shows several putative stem cell biomarkers of CRC and their prognostic value as previously reported.
| CRCSC Markers | Molecular weight | Prognostic significance | Tumorigenicity | References |
|---|---|---|---|---|
| CD133 | 120 kDa | • Highly correlated with low survival in CRC patients | • Subcutaneous injection of a CD133+, but not CD133− subpopulation of CRC into immunodeficient mice established a tumor | 41–45 |
| • Higher expression is associated with resistance of CRC to 5-FU based chemotherapy | ||||
| CD44 | 81 kDa | • Associated with lower patient survival | • CD44+ cells from a patient's tumor initiated a xenograft tumor in vivo and form a sphere in vitro | 43, 46 and 47 |
| • Not associated with prognosis | ||||
| CD166 | 100–105 kDa | • The expression correlated with a shortened period of survival | • CD166+/CD44+ or CD166+/ESA+ from human CRC cells can induce tumorigenesis in immunodeficient mice | 43, 48 and 49 |
| • No significant correlation with poor survival | ||||
| CD24 | 35–45 kDa | • No significant correlation with patient low survival | • Tumor spheroid cells capable of inducing tumors upon xenotransplantation | 4 and 50 |
| CD26 | 110 kDa | • Associated with enhanced invasiveness and chemoresistance | • Isolated CD26+, but not CD26− cells of CRC, developed distant metastasis when injected into the mouse cecal wall | 51 |
| Epithelial cell adhesion molecule (EpCAM) | 30–40 kDa | • Inversely correlated between EpCAM glycoprotein expression and tumor staging | • Injection of 200 to 500 EpCAMhigh/CD44+ cells in immunodeficient mice was sufficient to give rise to a tumor | 4 and 52 |
It is interesting to note that several discrepancies of prognosis values and tumorigenic potential have been reported among the listed CRCSC biomarkers. Despite the above listed inconsistencies, there is strong evidence from numerous studies suggesting that CD133 is a notable marker to identify CRCSCs.6,7,53 CD133, also known as prominin-1, is a 97 kDa pentaspan transmembrane glycoprotein54,55 and has been reported to be mostly localized in membrane protrusions.56 However, due to glycosylation, CD133 yields a 120 kDa protein.55 It was first discovered in 1997 as a hematopoietic stem cell (HSC) marker, which is associated with stem cell maintenance.55
Recently, CD133 has been widely studied as a putative biological marker for CRCSCs to predict tumor progression, prognosis value and chemoradiotherapy resistance.7 The enhanced CD133 expression in CRCSCs suggests that CD133 is a suitable biomarker target for the treatment of CRC. Fang et al. demonstrated that CD133+ cells isolated from primary CRC gave rise to spheroid cultures, which have the ability to self-renew and maintain CD133 expression in serum-free media.57 This finding supports the notion that CRCSCs that are CD133+ are able to initiate tumors, which may increase CRC occurrence.
It was also suggested that CRCSCs escaped conventional chemotherapy targeting rapidly dividing cells due to their quiescent nature, thereby inhibiting the complete eradication of CRC.58 In addition, the presence of overexpressed multidrug resistance protein 1 (MDR-1), detoxifying enzymes and DNA repair proteins in CSCs increased chemoresistance in malignant tumors.59,60 A study by Ong et al. reported that CD133+ CRC cells were more resistant towards 5-FU-based chemotherapy.44 Therefore, CD133-targeted therapeutic strategies in CRC might be valuable to eliminate CSC, which is associated with a high risk of relapse.
3. 5-Fluorouracil
5-FU is an antimetabolite and was discovered in 1957 to display tumor inhibitory activities by Heiderberger et al.61 Currently, it is used as a first line chemotherapy agent and is widely used as an antineoplastic drug.12,62 5-FU is a natural pyrimidine uracil analog with a fluorine atom inserted into the C-5 position in place of hydrogen (Fig. 2).63 Besides CRC, 5-FU is also commonly used to treat other solid malignancies arising from breast, stomach, pancreatic and head and neck cancers.64,65 | ||
| Fig. 2 Chemical structure of uracil and 5-fluorouracil. | ||
The regulation of 5-FU metabolism involves thymidylate synthase (TS), dihydropyrimidine dehydrogenase (DPD) and orotate phosphoribosyl transferase (OPRT) enzymes.66 It has been reported that only 1–3% of the original dose of 5-FU contributes to the cytotoxicity in rapidly proliferating tumor and normal cells via anabolic actions.67 Meanwhile, about 80–85% of 5-FU is subjected to biotransformation in the liver and catabolized into inactive metabolites, fluorinated β-alanine, by DPD67,68 and 15–20% is eliminated in urine.68 Thus, the development of drug delivery agents for 5-FU will enhance the 5-FU cytotoxicity to the targeted tumor cells. As an analog of uracil, 5-FU readily crosses the cell membrane using the same facilitated transport mechanism as uracil and is incorporated into nucleic acids, which eventually contributes to the cytotoxicity.30,69 5-FU and uracil are in continuous competition with each other due to their identical metabolic pathways (Fig. 3). Instead of uridine triphosphate (UTP), the active metabolite of 5-FU, fluorouridine triphosphate (FUTP), is incorporated into ribonucleic acid (RNA) and interferes with its processing and functioning.67 Fluorodeoxyuridine triphosphate (FdUTP) is another 5-FU metabolite that can be incorporated into deoxyribonucleic acid (DNA) instead of deoxythymidine triphosphate (dTTP). In addition, instead of normal pyrimidines, deoxyuridine monophosphate (dUMP), the metabolite of 5-FU, fluorodeoxyuridine monophosphate (FdUMP), forms a covalent ternary complex with TS and 5,10-methylene tetrahydrofolate (CH2THF) and inhibits deoxythymidine monophosphate (dTMP) production, which eventually interferes with DNA synthesis.62,67 In general, 5-FU causes cancer cell death by interfering with DNA synthesis and RNA processing and function.
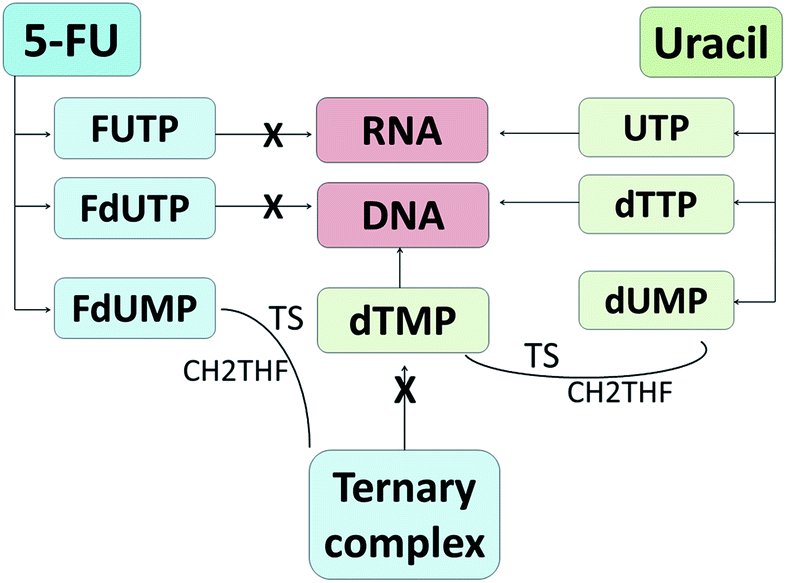 | ||
| Fig. 3 Metabolism routes of 5-fluorouracil and uracil. 5-FU metabolites inhibit DNA synthesis and RNA processing and function, which eventually results in cell death. | ||
Despite the effectiveness of 5-FU as a chemotherapeutic drug, one of its limitations is short plasma half-life (10–20 min) caused by fast degradation.63 In addition, due to the non-selectivity and poor bioavailability of 5-FU to tumor sites, a higher dose is required in order to reach the effective drug concentration. Nonetheless, high doses of chemotherapeutic agents lead to elevated systemic side effects and increased incidence of drug resistance, thereby limiting the usefulness of 5-FU in CRC chemo-treatment.
4. Considerations on using nanoparticles in drug delivery
The application of nanoparticles in drug delivery systems has enhanced the conventional anticancer treatments, particularly in terms of bioavailability, specific targeting, and reducing adverse side effects. However, nanoparticle clearance from the circulation by the reticuloendothelial system (RES), including the mononuclear phagocytic system (MNS), is one of the major concerns in drug delivery systems. The MNS consists of the liver, spleen, lymph nodes, bone marrow, skin and macrophages.26 Meanwhile, scavenger receptors on Kupffer cells are responsible for recognizing, engulfing and eliminating the opsonized nanoparticles in the liver.26,70 Thus, several requirements should be incorporated in designing nanoparticles to evade rapid clearance and enhance effective delivery of chemotherapeutic agents. Generally, the mechanisms of nanoparticles in drug delivery are affected by the physicochemical properties of the nanoparticles, such as size, surface chemistry and particle shape. These characterizations may impact nanoparticle-cell interactions, particularly in cellular and tissue uptake.First and foremost, it is apparent that nanoparticles offer an ideal system for chemotherapeutic drug delivery due to their tunable sizes. In medical applications, nanoparticles are materials with preferential dimensions of 1–100 nm.71 In addition, to avoid recognition by the RES, nano-sized particles also contribute to passive targeting into the tumor zone. The idea of nanoparticles as promising carriers for anti-tumor drugs was owing to the study by Matsumura and Maeda, which suggested that nanoparticles could accumulate in tumors.72 The formation of neovasculature in tumors increases the permeability of the vascular endothelial layer, resulting in leaks and susceptibility for the passage of molecules and/or nanosystems with sizes in the range of hundreds of nm.73,74 It is reported that the range of size of the vascular pore cut-off exhibited by most solid tumors is between 380 nm and 780 nm.75 Thus, nano-sized particles within this size range can easily pass through the cancer vascular pores. This phenomenon is known as the enhanced permeability and retention (EPR) effect (Fig. 4); it enables passive targeting of nanoparticles carrying drugs and is reported to enhance drug efficiency by up to 2.3-fold compared with when loaded with the free drugs.76
 | ||
| Fig. 4 The enhanced permeability and retention (EPR) effect, which allows passive targeting of nanoparticles. The neovasculature phenomenon in the tumor causes disordered vascular endothelial layers and permits the passage of nanoparticles. | ||
Secondly, hydrophobic and highly surface charged nanoparticles are highly recognized by the RES and removed from the circulation.77 Thus, various current nanoparticle-based drug delivery systems undergo surface modifications by grafting polymer coatings such as polyethylene glycol (PEG), poly(N-vinyl-2-pyrrolidone) (PVP) and dextran in order to avoid agglomeration and systemic clearance by macrophages, thereby increasing the bioavailability of chemotherapeutic agents.78–80 Hydrophilic PEG shielded the presence of nanoparticles to avoid opsonization by passive diffusion across the lipophilic cell membrane and hence increase the nanoparticle availability in systemic circulation.79 For instance, doxorubicin-loaded PEGylated liposomes have shown significant efficacy in breast cancer treatment.81 In addition, the stability of the formulated nanoparticles in the delivery system influences the cellular uptake. Monodispersity of nanoparticles in suspension is required to avoid agglomeration, which increases the rate of clearance. Basically, more positively or negatively charged nanoparticles prevent agglomeration in suspension due to their larger degree of repulsion. However, since many plasma membrane surfaces are negatively charged, nanoparticles with cationic surfactants display high affinity for cellular uptake via endocytosis during nanoparticle-cell adhesion.82
Lastly, nanoparticle shape is another crucial factor that can affect the cellular uptake efficiency and actions of nanoparticle delivery. In a study conducted by Huang et al., mesoporous silica nanoparticles (MSNs) of long-rod shape showed an increased number of internalized particles compared with short-rod shaped and sphere-shaped MSNs.83 However, when compared to spherical shape MSNs, the long-rod shaped MSNs showed higher cytotoxicity than the short-rod shaped and sphere-shaped particles. Another study found that the sharpness of the nanoparticles influenced the intracellular translocation and excretion of nanoparticles.84 In that study, it was reported that the sharp-shaped nanodiamonds translocated from the endosome to the cytoplasm quickly, with difficult cellular excretion compared with the round-shaped nanodiamonds, suggesting that the sharp-shaped nanoparticles ruptured the endosomal membrane, thereby reduced the excretion rate, and remained in the cytoplasm for an extended time. These studies suggest the interplay between cell interactions and nanoparticle design as one of the important aspects in developing drug delivery systems.
In addition, it is important to note that in cellular systems, particles enter the cells via the endocytosis route, which consists of phagocytosis or pinocytosis as summarized in Fig. 5. Large particles are taken up by phagocytosis/micropinocytosis, while nanoparticles with sizes lower than 200 nm are engulfed by micropinocytosis, either by a clathrin-dependent, caveolae-dependent or clathrin/caveolae-independent route.78,85
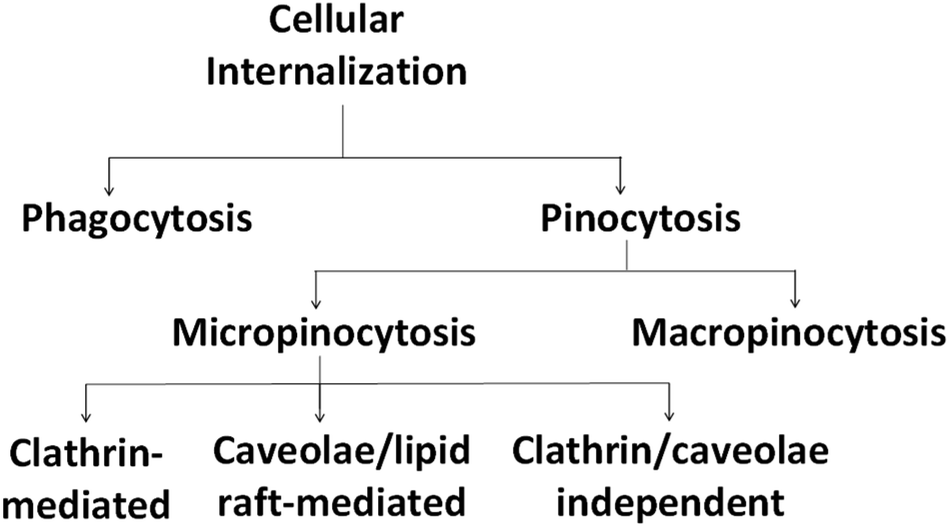 | ||
| Fig. 5 Different pathways for cellular internalization of nanoparticles. | ||
The clathrin-mediated pathway transfers the nanoparticles into lysosomes and a portion of the nanoparticles are recycled back to the extracellular space. Meanwhile, nanoparticles that follow the caveolae pathway are transported to caveosomes to be either translocated to the endoplasmic reticulum/Golgi body or enter the endosomal pathway.86 Engineered physicochemical properties as well as elemental compositions of the nanoparticles are crucial, since they are responsible for the fate of the nanoparticles generally in blood circulation and hence determine the effectiveness of the carried chemotherapeutic agents.87 Table 3 summarizes the common physicochemical properties of nanoparticles and the pathways for their internalization as described by Kou et al.78
| Physicochemical properties | Cellular internalization | |
|---|---|---|
| Size | 60–80 nm | Caveolae-mediated endocytosis which could avoid lysosomes |
| ∼100 nm | Clathrin-mediated endocytosis | |
| Up to 5 μm | Engulfed by macropinocytosis | |
| Larger particle | Opsonized and engulfed by phagocytosis | |
| Surface charge | Cationic | Have strong electrostatic interaction with the anionic cell membrane surface leading to rapid entry |
| Neutral | May interact with the cells with the aid of hydrophobic and hydrogen bond interactions | |
| Anionic | May interact with the cationic protein membrane and create repulsive interactions with the negatively charged cell surface leading to endocytosis | |
| Surface hydrophobicity | Hydrophobic | High affinity for the cell membrane |
| Hydrophilic | Suppresses interaction between the nanoparticles and lipid bilayer of cells | |
Apart from the physicochemical properties of the particles, it is also important to note that the initial materials for nanoparticles should be non-toxic materials or biocompatible to avoid toxic effects.88
5. The applications of gold nanoparticles in drug delivery systems
Gold (Au) has been applied in the biomedical field for many years. It has been used in bioimaging89 as well as in the treatment of arthritis/inflammation since the beginning of the 20th century.90 Since gold salts show anti-inflammatory activities, they have been incorporated into disease-modifying anti-rheumatic drugs (DMARDs), one of the drug classes to treat rheumatic arthritis.91 However, gold salts as DMARDs have been discontinued and replaced by others, probably due to the affinity of gold for DNA and hence its interference in cell function.80 The toxic effects of AuNPs have been supported by a study conducted by Qiu et al. which reported that acute and chronic exposure to AuNPs interrupts gene expression.92 To date, no gold-based nanomedicines have been approved by the US Food and Drug Administration (FDA) yet.80Besides as a direct therapeutic agent, AuNPs have been intensively studied in other biomedical applications, including in plasmonic photothermal therapy, photodynamic therapy and targeted drug delivery.93 To date, various studies have proven the overwhelming potential of AuNPs with some in clinical trial phases. A recent phase II clinical study into the photothermal effect (AuroLase Therapy with identification number: NCT02680535) utilized the unique optical tunability of AuNPs that can convert light into heat to thermally kill prostate cancer.94 A near-infrared laser that did not destroy healthy tissues was specifically designed to excite the accumulated AuNPs in cancerous cells and hence, the removal of the prostate tumor was precise enough without significant damage to surrounding healthy tissues.
One of the advantages of AuNPs compared to other metal nanoparticles is that the gold core is relatively inert, and so it is considered to be biocompatible and non-toxic.95 Previously, it has been reported that ionic AuNPs show obvious toxicity at 25 mM.96 Nevertheless, another study found that plain AuNPs did not show any cytotoxicity to human cells up to 250 mM.97 Thus, it is suggested that AuNPs themselves are non-toxic, but then toxicity could be derived from their functionalization, such as with surface coatings. In spite of this, AuNPs have shown promising potential to deliver chemotherapeutic drugs targeting cancer cells either via passive targeting, active targeting or both. As previously described, passive targeting is due to the neovasculature formed by tumor cells. Meanwhile, the active targeting approach is via the surface functionalization of AuNPs using immobilized ligands such as proteins, antibodies or small molecules for specific cell targeting on the targeted surface of the membrane.80 Thus, targeted cancer therapies can be beneficial for the controlled delivery of chemotherapeutic agents stemming from the fact that cancer cells overexpress specific antigens.98 Owing to the expression of specific antigens, drug carriers are often conjugated with specific antibodies to bind with the particularly expressed antigen, which will be discussed later.
The incorporation of a biocompatible coating seems to enhance the usefulness of AuNPs in drug delivery systems. A recent clinical trial on an AuNP-based product, Aurimune (CYT-6091), recombinant human tumor necrosis factor (rhTNF) bound to colloidal gold via a PEG-linker, showed that AuNP-loaded rhTNF was three times as effective in treating advanced stage cancer patients compared to the native rhTNF without introducing any significant toxic effects.99 This finding supports the notion that AuNPs with some modifications could be a promising targeted drug carrier in mediating conventional chemotherapeutic agents to eradicate tumors.
Undoubtedly, in order to justify further development, in vitro studies using human cell line models are very valuable for initial screening prior to clinical validation and translation. However, preclinical in vitro studies are beneficial to study specific tumor biology and treatment responses only. Thus, the in vitro effects may not fully represent the effects in medical application, since more complex interactions exist among the diverse organs and physiological systems in the human body. In spite of this, there are some AuNP studies that have reached clinical trials as previously discussed, and this could be a promising area for researchers to further explore their applications, particularly in drug delivery systems for cancer management.
Table 4 shows the application of functionalized-AuNPs in drug delivery systems targeting various types of cancers that have been reported in vivo and in vitro.
| Drugs loaded | Surface coatings/ligands | Treatments | Key findings | Reference |
|---|---|---|---|---|
| 5-FU | Thiols thioglycolic acid (TGA) and glutathione (GSH) | Colon cancer tissue samples were obtained from patients and incubated with TGA/GSH–AuNPs–5-FU or 5-FU alone | TGA/GSH–AuNPs–5-FU exhibited 2-fold higher anticancer effect compared with free 5-FU | 100 |
| Doxorubicin (DOX) | PEG | Carcinoma cells were intraperitoneally injected into female Balb/c mice and treated with DOX | PEG coatings showed high drug accumulation compared to passive targeting without PEG | 101 |
| TGF-β1 antibody and methotrexate | Folic acid | MDA-MB-231 breast cancer cell line | Folate conjugation enhanced the cellular uptake and TGF-β1 antibody on the AuNPs reduced extracellular TGF-β1 of cancer cells by 30% | 102 |
| Mitoxantrone (MTX) | Methoxy-polyethylene-glycol (mPEG) | Combination of photodynamic therapy (PDT) and chemotherapy on human melanoma (DFW) and breast cancer (MCF7) cell lines | MTX–mPEG–AuNP complex improved the efficacy of PDT with a light emitting diode | 103 |
| Doxorubicin (DOX) | Oligonucleotides (ONT) | DOX–ONT–AuNP complex treated SW480 CRC cell line and xenograft mice which were subcutaneously inoculated by the transfected SW480 cells | DOX–ONT–AuNP complex reduced cell viability and inhibited tumor growth in CRC xenograft mice | 104 |
6. Design of gold nanoparticles for cellular uptake
As previously discussed, physicochemical properties in terms of size, shape and surface properties play a major role in determining the competency of AuNPs as an optimal drug carrier, particularly in order to overcome certain biological barriers, such as macrophage clearance. For drug delivery purposes, the interaction between membrane receptors and nanoparticles is one of the most important aspects that regulates the rate of cellular uptake (endocytosis) and hence increases the accumulation of drug-loaded nanoparticles at the tumor site. In this regard, the nanoparticle size plays an important role to avoid early clearance by the MPS organs. Several previous studies have reported the rate of cellular uptake and AuNP accumulation among different sizes of AuNPs (Table 5). In AuNP chemical synthesis using the Turkevish method, the size of the synthesized AuNPs can be easily adjusted by varying the ratio of gold salt and citrate.105 Thus, tunability of the AuNP size during chemical synthesis can maximize the efficient delivery of therapeutic drugs to targeted cells.| Size of AuNPs (nm) | Cell lines or animals | Findings | References |
|---|---|---|---|
| 14, 30, 50, 74, 100 | HeLa cells | 50 nm AuNPs showed the maximum uptake by HeLa cells, which was 1294 nanoparticles per hour, and the maximum time for cellular uptake was 6 hours | 106 |
| 3, 10, 25, 50 | Hep-2 cells | A toxicity study of plain AuNPs on Hep-2 cells revealed that smaller AuNPs could enter the nucleus thereby damaging the cellular and nuclear membranes and it was suggested that the cytotoxicity of AuNPs is highly size-dependent (3 > 10 > 25 > 50 nm) | 107 |
| 1.4 | HeLa cells | AuNPs with the size of 1.4 nm were reported to cause necrosis due to the oxidative stress and disruption of membrane integrity | 108 |
| 1.4, 18, 80 | Pregnant rats | Smaller size of AuNPs was associated with teratogenic effects | 109 |
| 2, 6, 15 | MCF-7 cells and female Balb/c nude mice | Higher uptake of smaller AuNPs; 2 and 6 nm AuNPs distributed evenly throughout the cytoplasm and nucleus, and 15 nm AuNPs were only found in the cytoplasm and formed agglomerates | 110 |
| No significant toxic effects of AuNPs on the liver, spleen, kidneys, lungs, and heart | |||
| 2.4, 5.5, 8.2, 16, 38, 89 | COS-1 cells | Intracellular uptake and localization of AuNPs: | 111 |
| 2.4 nm: primarily in the nucleus | |||
| 5.5 and 8.2 nm: partially in the cytoplasm | |||
| 16 nm and above: no cellular uptake and located at the cellular periphery | |||
| 2, 10, 25, 40, 50, 70, 80, 100 | SK-BR-3 cells | 40 and 50 nm AuNPs showed the greatest effect on membrane receptor internalization and enhanced apoptotic activities | 112 |
| 4, 12, 17 | HeLa cells | Uptake forces via raft-mediated endocytosis increased with the size of AuNPs | 113 |
| 13, 45, 70, 110 | CL1-0 and HeLa cells | 45 nm AuNPs showed the highest uptake rate and entered cells via endocytosis | 114 |
| 30, 50, 90 | PC-3 cells | 30 and 50 nm plain AuNPs were highly taken up by the cells compared to the larger particles | 115 |
| PEGylation on the AuNP surface reduced the cellular uptake by reducing the interactions of the anionic plain AuNPs with the negatively charged domains on the cell membrane surface | |||
| 13, 45 | CF-31 cells | The major pathway for 45 nm AuNPs was clathrin-mediated endocytosis while for 13 nm AuNPs it was phagocytosis | 116 |
| 16, 26, 40, 58 | RAW264.7 and HepG2 cells | Negatively charged 40 nm AuNPs showed the highest uptake in both cells while positively charged AuNPs had no certain tendency | 117 |
| 20, 30, 50, 80 | CHO-K1 cells | Less internalization with increasing size; 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 AuNPs/cell for 20 nm AuNPs, and 50 AuNPs/cell for 80 nm AuNPs 800 AuNPs/cell for 20 nm AuNPs, and 50 AuNPs/cell for 80 nm AuNPs |
118 |
In biomedical applications, it is important to take note that the size of AuNPs should not be too small to avoid the interactions with biological macromolecules leading to cytotoxicity, but not too large to reduce the chance of macrophage clearance. Based on previous studies, it was highly suggested that the size of endocytosis-susceptible guest particles should be around 40–50 nm.106,112,114 This is in agreement with the 100 nm size limit of the plasma membrane to form endocytic vesicles.119 In addition, AuNPs with a size around 40 to 60 nm were reported to have optimal values of membrane bending rigidity and ligand–receptor binding interaction for endocytosis.114 However, it is another important consideration to note that the hydrodynamic diameter of AuNPs will increase upon coating or surface modification and hence affect the rate of cellular uptake. Thus, it is crucial to consistently monitor the increase of AuNP size before and after modifications to avoid clearance due to larger size.
In addition to size, the shape of AuNPs is another crucial physical factor to affect endocytic uptake by cells. The particle shape can enhance the process of cellular membrane wrapping during endocytosis.120 It was reported that spherical AuNPs increased the cellular uptake when compared to rod-shaped AuNPs106,115 probably due to the elongated particles requiring longer time for membrane wrapping.121 Despite the fact there were some previous studies reported that rod-shaped AuNPs enhanced the therapeutic outcomes in cancer drug delivery, including prolonged circulation times122 and enhanced drug loading,123 it is also important to note that the synthesis of gold nanorods requires the use of a growth-directing surfactant (cetyltrimethylammonium bromide, CTAB). Free CTAB without nanorods was shown to induce cytotoxicity in the human colon cancer HT-29 cell line.124 However, it was suggested that a polyacrylic acid (PAA) coating was able to reduce the exposure of the cells to the CTAB and hence attenuated the CTAB-capped gold nanorod toxicity. Thus, instead of focusing on the rate of cellular uptake alone, the toxic effects of drug carriers on the cellular system need to be prioritized as well.
In a separate study by Xie et al., mPEGylation of triangular AuNPs resulted in the highest cellular uptake by RAW264.7 cells, followed by rods and star-like structures.85 The highest uptake of the triangular-like shape was highly associated with the cytoskeleton arrangement and mediated by the dynamin-dependent endocytosis pathway. These studies indicate that the geometry of AuNPs is an additional feature that can influence the biological interactions, particularly in cellular uptake. However, more repetitive studies need to be conducted to gain a better understanding of the cellular uptake and toxicity implications of drug carriers with different shapes on the cellular system.
7. Effect of surface properties of AuNPs on cellular uptake
In addition to size and shape, the interactions of nanoparticles with lipid bilayer cell membranes mainly rely upon the chemical functionalities coated on the nanoparticle surfaces. Generally, modification of the AuNP surface is an important feature that influences the effective use of the particles for drug delivery systems. According to Pissuwan et al., surface modifications for AuNPs are needed in drug delivery systems to prolong residence of the AuNP conjugates in circulation, avoid RES clearance, ensure effective attachment of the desired targeting or therapeutic molecules, improve AuNP stability by preventing agglomeration and finally, neutralize the possible cytotoxicity caused by stabilizing surfactants during AuNP synthesis.125 For instance, the surface of AuNPs is coated by either neutral charged groups such as PEG or zwitterionic ligands, or charged functional groups that are anionic or cationic to avoid rapid clearance via non-specific uptake by the RES and provide active nanoparticle interactions with cells, respectively.126 Moreover, a previous preclinical trial of CYT-6091 showed that PEGylated AuNPs carrying rhTNF can escape immunogenicity by avoiding RES phagocytic clearance, thereby allowing the nanotherapeutic to be circulated longer in the blood circulation.99 Another study using gold nanorods reported that surface-modified gold nanorods with PEG showed no agglomeration due to the nearly complete neutral charge on the nanorod surface, as measured by zeta-potential, while increasing the circulation of the AuNP conjugates in in vivo systems.127An example of drug delivery using charged functional groups is provided by Hauck et al.128 In this study, gold nanorods of 18 × 40 nm were coated with various layer-by-layer polyelectrolyte (PE) coatings to produce positively and negatively charged nanorods by electrostatic interactions. Nanorods coated with negatively charged poly(4-styrenesulfonic acid) (PSS) followed by a layer of positively charged poly(diallyldimethylammonium chloride) (PDADMAC) exhibited a higher cellular uptake via passive targeting compared to the negatively charged PSS-coated nanorods. Electrostatic interactions between the negatively charged cellular membrane and positively charged nanoparticles may explain this higher uptake.
8. Active targeting or tumors by antibody conjugates
To date, no commercial antibody-conjugated drug-loaded nanoparticles have been applied in cancer therapy. However, antibody-conjugated drugs, such as Mylortag®, are commercially available on the market to treat acute myeloid leukaemia.75 The application of antibodies in cancer therapy has received great attention owing to the expression of specific antigens by cancerous cells. Thus, the development of antibody conjugates as the active targeting ligands in nanoparticle-targeted drug delivery will improve the specificity of chemotherapeutic drug delivery without inducing significant damage to healthy cells.Antibody grafting via covalent linkages at the surface of AuNPs is preferable compared to physical adsorption to reduce competitive displacement of the antibodies in the blood.75 To demonstrate the efficacy of functionalized antibodies in drug-loaded nanoparticles targeting tumors, Kou et al. synthesized cationic SM5-1 single chain antibody (scFv) and polylysine (SMFv-polylys)-coated poly(lactide-co-glycolide) (PLGA) nanoparticles loaded with paclitaxel to induce cytotoxicity in human hepatocellular carcinoma cell lines (Ch-hep-3).129 It was found that the paclitaxel-loaded PLGA in the presence of scFv antibody showed higher cell death compared to the paclitaxel-loaded PLGA only. In a recent AuNP study, conjugation of EpCAM or TARP antibodies with paclitaxel-loaded AuNPs via an N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC) and N-hydroxysuccinimide (NHS) (EDC/NHS) coupling reaction showed significant T47D breast cancer cell reduction compared to the paclitaxel-loaded AuNPs without antibody conjugates.130 Both studies support the notion that antibody conjugation provides specific drug delivery in order to enhance the chemotherapeutic action at the targeted tumor site, and hence could be further developed to hinder interactions with healthy cells.
9. Concluding remarks
The treatment of CRC has emerged as a topic of interest, since CRC displays high rates of recurrence as well as poor prognosis. The discovery of subpopulations in CRC known as stem cells had led to advanced studies targeting CRCSCs, owing to the stem cell theory that they are able to self-renew and sustain tumor growth. The traditional CRC chemotherapeutic agent, 5-FU, targeting RNA and DNA synthesis and function causes cytotoxicity by interfering with nucleoside metabolism. However, due to the short half-life, non-selectivity and poor biodistribution, the usefulness of 5-FU has been limited. Thus, there is an urgent need to construct an effective targeted drug delivery system to maximize the efficacy of chemotherapeutic agents. Recently, AuNPs have been shown to resolve the limitations of traditional chemotherapy via drug delivery systems. The noble AuNPs have various properties that enable them to be designed and functionalized to act as an effective drug carrier, such as biostability, non-toxicity and feasibility for surface modification. Owing to the overexpression of CD133 in CRCSCs, the specific targeting of AuNPs loaded with 5-FU towards CRCSCs can be enhanced via bioconjugation of anti-CD133 to the nanoparticles. Therefore, it will be possible for AuNPs to reach and release 5-FU at the targeted site (CRCSCs), thus reducing the interactions with the healthy cells and hence eliminating the systemic side effects previously reported for naked 5-FU without nanodrug carriers.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors would like to acknowledge Universiti Sains Malaysia under Research University Individual (RUI Grant No. 1001/PPSP/8012296) for the financial support.References
- M. Ghoncheh, M. Mohammadian, A. Mohammadian-Hafshejani and H. Salehiniya, Ann. Glob. Health, 2016, 82, 726–737 CrossRef.
- M. J. Munro, S. K. Wickremesekera, L. Peng, S. T. Tan and T. Itinteang, J. Clin. Pathol., 2018, 71, 110–116 CrossRef CAS PubMed.
- J. W. Huh, Y. A. Park, K. Y. Lee, S. A. Kim and S. K. Sohn, Yonsei Med. J., 2009, 50, 704–708 CrossRef.
- C. Fanali, D. Lucchetti, M. Farina, M. Corbi, V. Cufino, A. Cittadini and A. Sgambato, World J. Gastroenterol., 2014, 20, 923–942 CrossRef PubMed.
- C. J. Li, X. Zhang and G. W. Fan, J. Cancer Res. Ther., 2014, 10, 233–239 CrossRef.
- S. Chen, X. Song, Z. Chen, X. Li, M. Li, H. Liu and J. Li, PLoS One, 2013, 8, e56380 CrossRef CAS.
- F. Ren, W. Q. Sheng and X. Du, World J. Gastroenterol., 2013, 19, 2603–2611 CrossRef CAS.
- B. Viswanath, S. Kim and K. Lee, Int. J. Nanomed., 2016, 11, 2491–2504 CAS.
- Y. Seium, R. Stupp, T. Ruhstaller, P. Gervaz, G. Mentha, M. Philippe, A. Allal, C. Trembleau, J. Bauer, R. Morant and A. D. Roth, Ann. Oncol., 2005, 16, 762–766 CrossRef CAS PubMed.
- R. Shiragami, S. Murata, C. Kosugi, T. Tezuka, M. Yamazaki, A. Hirano, Y. Yoshimura, M. Suzuki, K. Shuto and K. Koda, Int. J. Oncol., 2013, 43, 431–438 CrossRef CAS PubMed.
- M. M. Saber, A. M. Al-Mahallawi, N. N. Nassar, B. Stork and S. A. Shouman, BMC Cancer, 2018, 18, 822 CrossRef PubMed.
- H. Wei, D. Qing, C. De-Ying, X. Bai and F. Li-Fang, Int. J. Pharm., 2008, 348, 35–45 CrossRef PubMed.
- O. Abdel-Rahman, Clin. Colorectal Cancer, 2019, 18, 58–63 CrossRef PubMed.
- S. Tummala, M. N. S. Kumar and A. Prakash, Saudi Pharm. J., 2015, 23, 308–314 CrossRef PubMed.
- B. A. Cisterna, N. Kamay, W. Choi, A. Tavakkoli, O. C. Farokhzad and C. Vilos, Nanomedicine, 2016, 11, 2443–2456 CrossRef CAS.
- K. Liu, Z. Q. Wang, S. J. Wang, P. Liu, Y. H. Qin, Y. Ma, X. C. Li and Z. J. Huo, Int. J. Nanomed., 2015, 10, 6445–6454 CAS.
- A. K. Mitra, V. Agrahari, A. Mandal, K. Cholkar, C. Natarajan, S. Shah, J. Joseph, H. M. Trinh, R. Vaishya, X. Yang, Y. Hao, V. Khurana and D. Pal, J. Controlled Release, 2015, 219, 248–268 CrossRef CAS PubMed.
- R. V. Kalaydina, K. Bajwa, B. Qorri, A. Decarlo and M. R. Szewczuk, Int. J. Nanomed., 2018, 13, 4727–4745 CrossRef CAS.
- R. R. Rasha, Am. J. Adv. Drug Delivery, 2016, 4, 55–57 Search PubMed.
- A. E. Yassin, M. K. Anwer, H. A. Mowafy, I. M. El-Bagory, M. A. Bayomi and I. A. Alsarra, Int. J. Med. Sci., 2010, 7, 398–408 CrossRef CAS PubMed.
- S. P. Chandran, K. P. Nachinmuthu, S. B. Natarajan, M. G. Inamdar and M. S. B. M. Shahimi, Curr. Cancer Ther. Rev., 2018, 14, 75–87 CrossRef CAS.
- L. Zhang, X. Gao, K. Men, B. Wang, S. Zhang, J. Qiu, M. Huang, M. Gou, N. Huang, Z. Qian, X. Zhao and Y. Wei, Int. J. Nanomed., 2011, 6, 2419–2427 CAS.
- J. P. Tiernan, N. Ingram, G. Marston, S. L. Perry, J. V. Rushworth, P. L. Coletta, P. A. Millner, D. G. Jayne and T. A. Hughes, Nanomedicine, 2015, 10, 1223–1231 CrossRef CAS.
- K. Ogawara, K. Un, K. Tanaka, K. Higaki and T. Kimura, J. Controlled Release, 2009, 133, 4–10 CrossRef CAS.
- A. Hardiansyah, L. Y. Huang, M. C. Yang, T. Y. Liu, S. C. Tsai, C. Y. Yang, C. Y. Kuo, T. Y. Chan, H. M. Zou, W. N. Lian and C. H. Lin, Nanoscale Res. Lett., 2014, 9, 497 CrossRef PubMed.
- S. Wilhelm, A. J. Tavares, Q. Dai, S. Ohta, J. Audet, H. F. Dvorak and W. C. W. Chan, Nat. Rev. Mater., 2016, 1, 16014 CrossRef CAS.
- B. Duncan, C. Kim and V. M. Rotello, J. Controlled Release, 2010, 148, 122–127 CrossRef CAS PubMed.
- S. S. Banerjee, N. Aher, R. Patil and J. Khandare, J. Drug Delivery, 2012, 2012, 103973 Search PubMed.
- T. A. Larson, P. P. Joshi and K. Sokolov, ACS Nano, 2012, 6, 9182–9190 CrossRef CAS.
- D. B. Longley, D. P. Harkin and P. G. Johnston, Nature, 2003, 3, 330–338 CAS.
- T. A. Baudino, Curr. Drug Discovery Technol., 2015, 12, 3–20 CrossRef CAS PubMed.
- X. Yu, S. S. Kanwar, B. B. Patel, J. Nautiyal, F. H. Sarkar and A. P. N. Majumdar, Transl. Oncol., 2009, 2, 321–328 CrossRef PubMed.
- S. T. Ning, S. Y. Lee, M. F. Wei, C. L. Peng, S. Y. F. Lin, M. H. Tsai, P. C. Lee, Y. H. Shih, C. Y. Lin, T. Y. Luo and M. J. Shieh, ACS Appl. Mater. Interfaces, 2016, 8, 17793–17804 CrossRef CAS PubMed.
- J. J. Casciari, S. V. Sotirchos and R. M. Sutherland, J. Cell. Physiol., 1992, 151, 386–394 CrossRef CAS PubMed.
- Y. Kato, S. Ozawa, C. Miyamoto, Y. Maehata, A. Suzuki, T. Maeda and Y. Baba, Cancer Cell Int., 2013, 13, 1–8 CrossRef PubMed.
- M. Carbone, J. King Saud Univ., Sci., 2017, 29, 284–290 CrossRef.
- C. L. Zhang, T. Huang, B. L. Wu, W. X. He and D. Liu, Oncotarget, 2017, 8, 75756–75766 Search PubMed.
- D. Abetov, Z. Mustapova, T. Saliev and D. Bulanin, Tumor Biol., 2015, 36, 1339–1353 CrossRef CAS PubMed.
- J. Xiao, J. Mu, T. Liu and H. Xu, J. Med. Discovery, 2017, 2, jmd17003 Search PubMed.
- R. C. Langan, J. E. Mullinax, M. T. Raiji, T. Upham, T. Summers, A. Stojadinovic and I. Avital, J. Cancer, 2013, 4, 241–250 CrossRef.
- C. A. O'Brien, A. Pollett, S. Gallinger and J. E. Dick, Nature, 2007, 445, 106–110 CrossRef PubMed.
- L. Ricci-Vitiani, D. G. Lombardi, E. Pilozzi, M. Biffoni, M. Todaro, C. Peschle and R. De Maria, Nature, 2007, 445, 111–115 CrossRef CAS PubMed.
- D. Horst, L. Kriegl, J. Engel, T. Kirchner and A. Jung, Cancer Invest., 2009, 27, 844–850 CrossRef PubMed.
- C. W. Ong, L. G. Kim, H. H. Kong, L. Y. Low, B. Iacopetta, R. Soong and M. Salto-Tellez, Mod. Pathol., 2010, 23, 450–457 CrossRef CAS PubMed.
- A. V. Paschall, D. Yang, C. Lu, P. S. Redd, J. H. Choi, C. M. Heaton, J. R. Lee, A. Nayak-Kapoor and K. Liu, Oncotarget, 2016, 7, 78698–78712 CrossRef PubMed.
- K. M. Ropponen, M. J. Eskelinen, P. K. Lipponen, E. Alhava and V. M. Kosma, Scand. J. Gastroenterol., 1998, 33, 301–309 CrossRef CAS PubMed.
- L. Du, H. Wang, L. He, J. Zhang, B. Ni, X. Wang, H. Jin, N. Cahuzac, M. Mehrpour, Y. Lu and Q. Chen, Clin. Cancer Res., 2008, 14, 6751–6760 CrossRef CAS PubMed.
- W. Weichert, T. Knosel, J. Bellach, M. Dietel and G. Kristiansen, J. Clin. Pathol., 2004, 57, 1160–1164 CrossRef CAS PubMed.
- P. Dalerba, S. J. Dylla, I. K. Park, R. Liu, X. Wang, R. W. Cho, T. Hoey, A. Gurney, E. H. Huang, D. M. Simeone, A. A. Shelton, G. Parmiani, C. Castelli and M. F. Clarke, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 10158–10163 CrossRef CAS PubMed.
- J. Papailiou, K. J. Bramis, M. Gazouli and G. Theodoropoulos, Int. J. Colorectal Dis., 2011, 26, 1–11 CrossRef.
- R. Pang, W. L. Law, A. C. Chu, J. T. Poon, C. S. Lam, A. K. Chow, L. Ng, L. W. H. Cheung, X. R. Lan, H. Y. Lan, V. P. Y. Tan, T. C. Yau, R. T. Poon and B. C. Y. Wong, Cell Stem Cell, 2010, 6, 603–615 CrossRef CAS.
- M. Mokhtari and Z. Zakerzade, Adv. Biomed. Res., 2017, 6, 56 Search PubMed.
- B. B. Wang, Z. J. Li, F. F. Zhang, H. T. Hou, J. K. Yu and F. Li, Histol. Histopathol., 2016, 31, 299–306 CAS.
- I. S. Hong, G. B. Jang, H. Y. Lee and J. S. Nam, Int. J. Nanomed., 2015, 10, 251–260 CAS.
- P. M. Glumac and A. M. LeBeau, Clin. Transl. Med., 2018, 7, 18 CrossRef PubMed.
- D. Corbeil, K. Roper, C. A. Fargeas, A. Joester and W. B. Huttner, Traffic, 2001, 2, 82–91 CrossRef CAS.
- D. D. Fang, Y. J. Kim, C. N. Lee, S. Aggarwal, K. McKinnon, D. Mesmer, J. Norton, C. E. Birse, T. He, S. M. Ruben and P. A. Moore, Br. J. Cancer, 2010, 102, 1265–1275 CrossRef CAS.
- B. M. Boman and E. Huang, J. Clin. Oncol., 2008, 26, 2828–2838 CrossRef PubMed.
- J. N. Rich and S. Bao, Cell Stem Cell, 2007, 1, 353–355 CrossRef CAS PubMed.
- R. Gangemi, L. Paleari, A. M. Orengo, A. Cesario, L. Chessa, S. Ferrini and P. Russo, Curr. Med. Chem., 2009, 16, 1688–1703 CrossRef CAS.
- C. Heidelberger, N. K. Chaudhuri, P. Danneberg, D. Mooren and L. Griesbach, Nature, 1957, 179, 663–666 CrossRef CAS.
- C. Focaccetti, A. Bruno, E. Magnani, D. Bartolini, E. Principi, K. Dallaglio, E. O. Bucci, G. Finzi, F. Sessa, D. M. Noonan and A. Albini, PLoS One, 2015, 10, e0115686 CrossRef PubMed.
- S. Handali, E. Moghimipour, M. Rezaei, Z. Ramezani, M. Kouchak, M. Amini, K. A. Angali, S. Saremy and F. A. Dorkoosh, Biomed. Pharmacother., 2018, 108, 1259–1273 CrossRef CAS.
- N. Zhang, Y. Yin, S. J. Xu and W. S. Chen, Molecules, 2008, 13, 1551–1569 CrossRef CAS.
- K. Bracht, A. M. Nicholls, Y. Liu and W. F. Bodmer, Br. J. Cancer, 2010, 103, 340–346 CrossRef CAS.
- M. Ishikawa, T. Miyauchi and Y. Kashiwagi, BMC Cancer, 2008, 8, 188 CrossRef PubMed.
- M. W. Saif, K. N. Syrigos and N. A. Katirtzoglou, Expert Opin. Invest. Drugs, 2009, 18, 335–348 CrossRef CAS PubMed.
- F. Casale, R. Canaparo, L. Serpe, E. Muntoni, C. D. Pepa, M. Costa, L. Mairone, G. P. Zara, G. Fornari and M. Eandi, Pharmacol. Res., 2004, 50, 173–179 CrossRef CAS.
- R. M. Wohlhueter, R. S. McIvor and G. W. Peter, J. Cell. Physiol., 1980, 104, 309–319 CrossRef CAS PubMed.
- K. Sztandera, M. Gorzkiewicz and B. Klajnert-Maculewicz, Mol. Pharm., 2018, 16, 1–23 Search PubMed.
- A. K. Biswas, M. R. Islam, Z. S. Choudhury, A. Mostafa and M. F. Kadir, Adv. Nat. Sci.: Nanosci. Nanotechnol., 2014, 5, 043001 Search PubMed.
- Y. Matsumura and H. Maeda, Cancer Res., 1986, 46, 6387–6392 CAS.
- Y. Nakamura, A. Mochida, P. L. Choyke and H. Kobayashi, Bioconjugate Chem., 2016, 27, 2225–2238 CrossRef CAS.
- R. Ngoune, A. Peters, D. V. Elverfeldt, K. Winkler and G. Pütz, J. Controlled Release, 2016, 238, 58–70 CrossRef CAS.
- M. Arruebo, M. Valladares and Á. González-Fernández, J. Nanomater., 2009, 2009, 439389 Search PubMed.
- H. Maeda, H. Nakamura and J. Fang, Adv. Drug Delivery Rev., 2013, 65, 71–79 CrossRef CAS PubMed.
- H. Kobayashi, R. Watanabe and P. L. Choyke, Theranostics, 2014, 4, 81–89 CrossRef CAS PubMed.
- L. Kou, J. Sun, Y. Zhai and Z. He, Asian J. Pharm. Sci., 2013, 8, 1–10 CrossRef CAS.
- J. R. Nicol, D. Dixon and J. A. Coulter, Nanomedicine, 2015, 10, 1315–1326 CrossRef CAS PubMed.
- D. Bobo, K. J. Robinson, J. Islam, K. J. Thurecht and S. R. Corrie, Pharm. Res., 2016, 33, 2373–2387 CrossRef CAS.
- J. W. Park, Breast Cancer Res., 2002, 4, 95–99 CrossRef CAS.
- D. L. Cooper, C. M. Conder and S. Harirforoosh, Expert Opin. Drug Delivery, 2014, 11, 1661–1680 CrossRef CAS PubMed.
- X. Huang, X. Teng, D. Chen, F. Tang and J. He, Biomaterials, 2010, 31, 438–448 CrossRef CAS PubMed.
- Z. Chu, S. Zhang, B. Zhang, C. Zhang, C. Y. Fang, I. Rehor, P. Cigler, H. C. Chang, G. Lin, R. Liu and Q. Li, Sci. Rep., 2014, 4, 4495 CrossRef PubMed.
- X. Xie, J. Liao, X. Shao, Q. Li and Y. Lin, Nature, 2017, 7, 3827 Search PubMed.
- N. Hao, L. Li, Q. Zhang, X. Huang, X. Meng, Y. Zhang, D. Chen, F. Tang and L. Li, Microporous Mesoporous Mater., 2012, 162, 14–23 CrossRef CAS.
- R. Mohamud, S. D. Xiang, C. Selomulya, J. M. Rolland, R. E. O'Hehir, C. L. Hardy and M. Plebanski, Drug Metab. Rev., 2014, 46, 176–190 CrossRef CAS PubMed.
- J. H. Adair, M. P. Parette, E. I. Altınoğlu and M. Kester, ACS Nano, 2010, 4, 4967–4970 CrossRef CAS PubMed.
- E. Hutter and D. Maysinger, Microsc. Res. Tech., 2011, 74, 592–604 CrossRef CAS PubMed.
- M. F. H. Carneiro and F. J. Barbosa, J. Toxicol. Environ. Health, 2016, 19, 129–148 CrossRef PubMed.
- S. Ramiro, C. Gaujoux-Viala, J. L. Nam, J. S. Smolen, M. Buch, L. Gossec, D. Van Der Heijde, K. Winthrop and R. Landewé, Ann. Rheum. Dis., 2014, 73, 529–535 CrossRef CAS PubMed.
- T. A. Qiu, J. S. Bozich, S. E. Lohse, A. M. Vartanian, L. M. Jacob, B. M. Meyer, I. L. Gunsolus, N. J. Niemuth, C. J. Murphy, C. L. Haynes and R. D. Klaper, Environ. Sci.: Nano, 2015, 2, 615–629 RSC.
- L. Dykman and N. Khlebtsov, Chem. Soc. Rev., 2012, 41, 2256–2282 RSC.
- A. Laukka, UTHealth News, https://www.uth.edu/news/story.htm, accessed 27 March 2019 Search PubMed.
- S. Verma, P. Utreja, M. Rahman and L. Kumar, Curr. Nanomed., 2018, 8, 1–18 Search PubMed.
- C. M. Goodman, C. D. McCusker, T. Yilmaz and V. M. Rotello, Bioconjugate Chem., 2004, 15, 897–900 CrossRef CAS PubMed.
- E. E. Connor, J. Mwamuka, A. Gole, C. J. Murphy and M. D. Wyatt, Small, 2005, 1, 325–327 CrossRef CAS PubMed.
- J. Wang, X. Hu and D. Xiang, Drug Delivery, 2018, 25, 1319–1327 CrossRef CAS PubMed.
- S. K. Libutti, G. F. Paciotti, A. A. Bymes, H. R. Alexander, W. E. Gannon, M. Walker, G. D. Seidel, N. Yuldasheva and N. Tamarkin, Clin. Cancer Res., 2010, 16, 6139–6149 CrossRef CAS PubMed.
- M. A. Safwat, G. M. Soliman, D. Sayed and M. A. Attia, Int. J. Pharm., 2016, 513, 648–658 CrossRef CAS PubMed.
- N. Elbialy, M. M. Fathy and W. M. Khalil, Int. J. Pharm., 2015, 490, 190–199 CrossRef CAS PubMed.
- N. Rizk, N. Christoforou and S. Lee, Nanotechnology, 2016, 27, 185704 CrossRef PubMed.
- A. Imanparast, M. Bakhshizadeh, R. Salek and A. Sazgarnia, Photodiagn. Photodyn. Ther., 2018, 23, 295–305 CrossRef CAS PubMed.
- C. S. Lee, H. Kim, J. Yu, S. H. Yu, S. Ban, S. Oh, D. Jeong, J. Im, M. J. Baek and T. H. Kim, Eur. J. Med. Chem., 2017, 142, 416–423 CrossRef CAS PubMed.
- E. C. Dreaden, L. A. Austin, M. A. Mackey and M. A. El-Sayed, Ther. Delivery, 2012, 3, 457–478 CrossRef CAS PubMed.
- B. D. Chithrani, A. A. Ghazani and W. C. W. Chan, Nano Lett., 2006, 6, 662–668 CrossRef CAS PubMed.
- C. Boyoglu, Q. He, G. Willing, S. Boyoglu-Barnum, V. A. Dennis, S. Pillai and S. R. Singh, ISRN Nanotechnol., 2013, 2013, 123838 Search PubMed.
- Y. Pan, A. Leifert, D. Ruau, S. Neuss, J. Bornemann, G. Schmid, W. Brandau, U. Simon and W. Jahnen-Dechent, Small, 2009, 5, 2067–2076 CrossRef CAS PubMed.
- M. Semmler-Behnke, J. Lipka, A. Wenk, S. Hirn, M. Schäffler, F. Tian, G. Schmid, G. Oberdörster and W. G. Kreyling, Part. Fibre Toxicol., 2014, 11, 1–12 CrossRef PubMed.
- K. Huang, H. Ma, J. Liu, S. Huo, A. Kumar, T. Wei, X. Zhang, S. Jin, Y. Gan, P. C. Wang, S. He, X. Zhang and X. J. Liang, ACS Nano, 2012, 6, 4483–4493 CrossRef CAS PubMed.
- E. Oh, J. B. Delehanty, K. E. Sapsford, K. Susumu, R. Goswami, J. B. Blanco-Canosa, P. E. Dawson, J. Granek, M. Shoff, Q. Zhang, P. L. Goering, A. Huston and I. L. Medintz, ACS Nano, 2011, 5, 6434–6448 CrossRef CAS PubMed.
- W. Jiang, B. Y. S. Kim, J. T. Rutka and W. C. W. Chan, Nat. Nanotechnol., 2008, 3, 145–150 CrossRef CAS PubMed.
- Y. Shan, S. Ma, L. Nie, X. Shang, X. Hao, Z. Tang and H. Wang, Chem. Commun., 2011, 47, 8091–8093 RSC.
- S. H. Wang, C. W. Lee, A. Chiou and P. K. Wei, J. Nanobiotechnol., 2010, 8, 33 CrossRef CAS PubMed.
- Arnida, A. Malugin and H. Ghandehari, J. Appl. Toxicol., 2010, 30, 212–217 CAS.
- T. Mironava, M. Hadjiargyrou, M. Simon, V. Jurukovski and M. H. Rafailovich, Nanotoxicology, 2010, 4, 120–137 CrossRef CAS.
- X. Liu, N. Huang, H. Li, Q. Jin and J. Ji, Langmuir, 2013, 29, 9138–9148 CrossRef CAS PubMed.
- A. Elbakry, E. C. Wurster, A. Zaky, R. Liebl, E. Schindler, P. Bauer-Kreisel, T. Blunk, R. Rachel, A. Goepferich and M. Breunig, Small, 2012, 8, 3847–3856 CrossRef CAS PubMed.
- F. Osaki, T. Kanamori, S. Sando, T. Sera and Y. Aoyama, J. Am. Chem. Soc., 2004, 126, 6520–6521 CrossRef CAS PubMed.
- N. Kamaly, Z. Xiao, P. M. Valencia, A. F. Radovic-Moreno and O. C. Farokhzad, Chem. Soc. Rev., 2013, 41, 2971–3010 RSC.
- V. Voliani, G. Signore, R. Nifosí, F. Ricci, S. Luin and F. Beltram, Recent Pat. Nanotechnol., 2012, 2, 34–44 CAS.
- Arnida, M. M. Janát-Amsbury, A. Ray, C. M. Peterson and H. Ghandehari, Eur. J. Pharm. Biopharm., 2011, 77, 417–423 CrossRef CAS PubMed.
- S. Pandey, R. Shah, A. Mewada, M. Thakur, G. Oza and M. Sharon, J. Mater. Sci.: Mater. Med., 2013, 24, 1671–1681 CrossRef CAS PubMed.
- A. M. Alkilany, P. K. Nagaria, C. R. Hexel, T. J. Shaw, C. J. Murphy and M. D. Wyatt, Small, 2009, 5, 701–708 CrossRef CAS PubMed.
- D. Pissuwan, T. Niidome and M. B. Cortie, J. Controlled Release, 2011, 149, 65–71 CrossRef CAS PubMed.
- A. Verma and F. Stellacci, Small, 2010, 6, 12–21 CrossRef CAS.
- T. Niidome, M. Yamagata, Y. Okamoto, Y. Akiyama, H. Takahashi, T. Kawano, Y. Katayama and Y. Niidome, J. Controlled Release, 2006, 114, 343–347 CrossRef CAS PubMed.
- T. S. Hauck, A. A. Ghazani and W. C. W. Chan, Small, 2008, 4, 153–159 CrossRef CAS PubMed.
- G. Kou, J. Gao, H. Wang, H. Chen, B. Li, D. Zhang, S. Wang, S. Hou, W. Qian, J. Dai, Y. Zhong and Y. Guo, J. Biochem. Mol. Biol., 2007, 40, 731–739 CAS.
- Z. Alhalili, J. Shapter, D. Figueroa and B. Sanderson, Drug Delivery Lett., 2018, 8, 217–225 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |