
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnraveling how the Gly526Ser mutation arrests prostaglandin formation from arachidonic acid catalyzed by cyclooxygenase-2: a combined molecular dynamics and QM/MM study†
Adrián Suñer-Rubio a,
Anna Cebrián-Pratsa,
Àngels González-Lafontab and
José M. Lluch*ab
a,
Anna Cebrián-Pratsa,
Àngels González-Lafontab and
José M. Lluch*ab
aDepartament de Química, Universitat Autònoma de Barcelona, 08193 Bellaterra, Barcelona, Spain. E-mail: JoseMaria.Lluch@uab.cat
bInstitut de Biotecnologia i de Biomedicina (IBB), Universitat Autònoma de Barcelona, 08193 Bellaterra, Barcelona, Spain
First published on 3rd January 2020
Abstract
Cyclooxygenases (COXs) are the enzymes responsible for the biosynthesis of prostaglandins, eicosanoids that play a major role in many physiological processes. Particularly, prostaglandins are known to trigger inflammation, and COX-2, the enzyme isoform associated with this inflammatory response, catalyzes the cyclooxidation of arachidonic acid, leading to prostaglandin G2. For this reason, COX-2 has been a very important pharmacological target for several decades now. The catalytic mechanism of COX-2, a so-called all-radical mechanism, consists of six chemical steps. One of the most intriguing aspects of this mechanism is how COX-2 manages to control the regio- and stereospecificity of the products formed at each step. Mutagenesis experiments have previously been performed in an attempt to find those hot-spot residues that make such control possible. In this context, it is worth mentioning that in experiments with the Gly526Ser COX-2 mutant, prostaglandins were not detected. In this paper, we have combined molecular dynamics simulations and quantum mechanics/molecular mechanics calculations to analyze how the COX-2 catalytic mechanism is modified in the Gly526Ser mutant. Therefore, this study provides new insights into the COX-2 catalytic function.
1 Introduction
Lipid mediators1 are bioactive lipids involved in many physiological processes, and are generated through specific biosynthetic pathways. They bind to their corresponding G protein-coupled receptors and transmit signals to target cells. The synthesis of many lipid mediators begins with the oxygenation of polyunsaturated fatty acids by some enzymes.2 One of the most well known and important cases is the dioxygenation of arachidonic acid (AA, 20:4 n-6) by the enzyme cyclooxygenase-2 (COX-2), which leads to the formation of the eicosanoid prostaglandins. Prostaglandins control many pathological events, such as inflammation, fever, pain, cardiovascular diseases, and cancer.1AA is released from membrane glycerophospholipids. COX-2 is a membrane-associated homodimeric bifunctional hemoprotein.3 Only one monomer acts as a catalyst at a given time.4 The determination of the corresponding crystal structure3 has shown that in the murine cyclooxygenase active site of the catalytic monomer the AA carboxylate lies near the side chains of Arg120 and Tyr355, whereas the ω-end is positioned in the hydrophobic groove above Ser530, exhibiting an “L-shaped” binding conformation. A tyrosyl radical is generated as a consequence of one-electron transfer from Tyr385 to the oxy-ferryl protoporphyrin IX radical cation formed in the peroxidase active site after activation by an alkyl hydroperoxide of its heme group. The C13 carbon atom of AA is positioned 2.95 Å below the phenolic oxygen of Tyr385. The nascent tyrosyl radical starts the cyclooxygenase reaction by abstraction of the C13 pro-S hydrogen atom when AA occupies the cyclooxygenase active site.5,6 Afterwards, addition of two oxygen molecules converts AA to prostaglandin G2 (PGG2). Later, PGG2 is released to the peroxidase active site, where its 15-hydroperoxyl group is reduced to give prostaglandin H2 (PGH2).
The biosynthesis of PGG2 in the cyclooxygenase active site is achieved by a six-step all-radical mechanism2,7–13 (see Scheme 1) derived from proposals by Hamberg and Samuelsson. It successively consists of: a C13 pro-S hydrogen abstraction from AA by the tyrosyl radical 385, forming a planar delocalized C11–C15 pentadienyl radical (step 1); an antarafacial molecular oxygen addition at C11 to give an 11R peroxyl radical (step 2); a 9,11-cyclization leading to a C8-radical cyclic endoperoxide (step 3); an 8,12-cyclization yielding a bicyclo endoperoxide and a C13–C15 allyl radical (step 4); a second antarafacial molecular oxygen addition at the 15S position (step 5); and a final back transfer of hydrogen from Tyr385 to the peroxyl radical at C15 (step 6), leading to the formation of PGG2.
 | ||
| Scheme 1 The all-radical mechanism for the formation of PGG2 from arachidonic acid catalyzed by wild-type COX-2. | ||
One of the most amazing features of the six-step all-radical mechanism is the extremely efficient way how COX-2 manages to control the stereochemistry and regiochemistry of the reactions it catalyzes. AA is a very long substrate (it contains 20 carbon atoms). It is simultaneously a flexible molecule (owing to its multiple single bonds) and a rigid molecule (owing to its four C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds). The role of many active site residues of COX-2 is to make this control possible. A number of mutagenesis experiments have been performed to better understand the way they act.14–16 Particularly interesting is the case of the Gly526Ser mutant human COX-2.16 This mutation does not appear to interfere with the first three reaction steps. However, no formation of prostaglandins was detected, 11R-hydroperoxyeicosatetraenoic acid (11R-HPETE) becoming the main product. Brash and co-workers16 proposed that the 8,12-cyclization in the Gly526Ser mutant is sterically hindered by misalignment of the substrate carbon chains. So, instead of prostaglandins they found novel products, which were identified as 8,9-11,12-diepoxy-13R- (or 15R)-hydroperoxy derivatives of arachidonic acid.
C double bonds). The role of many active site residues of COX-2 is to make this control possible. A number of mutagenesis experiments have been performed to better understand the way they act.14–16 Particularly interesting is the case of the Gly526Ser mutant human COX-2.16 This mutation does not appear to interfere with the first three reaction steps. However, no formation of prostaglandins was detected, 11R-hydroperoxyeicosatetraenoic acid (11R-HPETE) becoming the main product. Brash and co-workers16 proposed that the 8,12-cyclization in the Gly526Ser mutant is sterically hindered by misalignment of the substrate carbon chains. So, instead of prostaglandins they found novel products, which were identified as 8,9-11,12-diepoxy-13R- (or 15R)-hydroperoxy derivatives of arachidonic acid.
The aim of this paper is to provide a detailed molecular view of the way how an apparently little significant mutation is able to fully arrest the PGG2 biosynthesis, instead yielding diepoxy products. Thus, we have combined molecular dynamics (MD) simulations with quantum mechanics/molecular mechanics (QM/MM) calculations to analyze how the Gly526Ser mutant of COX-2 alters the six-step all-radical mechanism shown in Scheme 1 that holds for wild-type COX-2, so contributing to the understanding of the very important catalytic function of COX-2.
2 Computational methods
2.1 System setup
In a previous paper we studied13 the mechanism that governs the biosynthesis of PGG2 catalyzed by wild-type COX-2. That system was built from the murine COX-2/AA complex (PDB code 3HS5)3, replacing the Co3+ complexed to the protoporphyrin IX group by Fe3+. The heme prosthetic group was tethered to the axial histidine (His388). Only the monomer B (the one where AA adopts an “L-shaped” productive binding configuration, with the C13 carbon atom of AA near Tyr385) was modeled in the cyclooxygenase active site. After a 100 ns MD simulation a pair of snapshots was selected, and we showed that both were able to follow the all-radical mechanism displayed in Scheme 1, eventually yielding PGG2. In this paper, we have taken one of those snapshots (snapshot 1), and we have replaced the Gly526 residue by a serine using the CHIMERA17 program. All the amino acid residues of the protein but His388 and Tyr385 were described using the AMBER ff14SB force field.18 All water molecules were deleted and the system was solvated again by means of the AMBER18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 19 tleap program. A pH of 7 was assumed for the titratable residues. Five Na+ ions were added at the protein surface to neutralize the total charge of the system. An almost cubic box (100.2 Å × 113.4 Å × 92.5 Å) of TIP3P water molecules20 was constructed, with a minimum cutoff distance of 15 Å between the atoms of the enzyme and the edge of the periodic box. All the water molecules closer than 2.2 Å to any atom of the enzyme or AA were removed. In all, the system contained about 80000 atoms.
19 tleap program. A pH of 7 was assumed for the titratable residues. Five Na+ ions were added at the protein surface to neutralize the total charge of the system. An almost cubic box (100.2 Å × 113.4 Å × 92.5 Å) of TIP3P water molecules20 was constructed, with a minimum cutoff distance of 15 Å between the atoms of the enzyme and the edge of the periodic box. All the water molecules closer than 2.2 Å to any atom of the enzyme or AA were removed. In all, the system contained about 80000 atoms.
2.2 Parameterization
First of all, the parameters of the heme group were taken from a recent study.21 However, the corresponding partial charges of the ferric penta-coordinated high spin group (Fe3+) were calculated using the Restrained Electrostatic Potential (RESP)22 method at the HF/6-31G(d,p) level of theory. In addition, the parameters of the histidine coordinated to the metal were described by the general AMBER force field (GAFF)23 using the residuegen and parmchk modules of AMBER14.24 Secondly, the AA substrate, the delocalized C11–C15 pentadienyl radical of AA, and its delocalized C13–C15 allyl radical parameters were also taken from a recent study.25 Finally, the parameterization of the tyrosyl radical was performed by calculating first the optimized structure of this residue using the B3LYP functional26,27 and the 6-31G(d,p)28,29 basis set for all atoms, followed by an RESP calculation of the partial atomic charges at the same level of theory using Gaussian09.30 At this point, as the residue is not a standard tyrosine, the parameters of its bonds, angles, dihedrals and van der Waals terms should be re-calculated. Therefore, the force constants associated with each bond and angle were fitted using a harmonic function to reproduce the B3LYP/6-31G(d,p) potential energy profiles generated for the corresponding stretching and bending vibrations. The dihedral parameters were not calculated as they do not present significant differences with the standard tyrosine residue. The van der Waals parameters were taken from the GAFF force field of AMBER14, assigning the oxygen radical as an ether or ester oxygen.2.3 Molecular dynamics simulations
Starting from the structure resulting from the setup, we carried out three successive molecular dynamics simulations using the same protocol before the production stage. For the first MD simulation the monomer B of Gly526Ser COX-2, with AA and the radical Tyr385 within the cyclooxygenase active site, was optimized following three successive molecular mechanics (MM) minimizations using the steepest descent and conjugate gradient methods. This was done to relax the system. Firstly, we applied harmonic restraints on the enzyme, the heme prosthetic group and AA, keeping the solvation water molecules free. In the second minimization, only the heme prosthetic group and His388 were restrained. In the last minimization only the protein side chains were restrained, the rest of the system remaining free. Then, we carried out the MD simulation employing periodic boundary conditions (PBCs), with a cutoff of 10 Å for all Lennard-Jones and electrostatic interactions and the particle-mesh Ewald method31 to treat long-range electrostatic effects. The system was gradually heated from 0 to 300 K, with an increment of 30 K, during 10 steps of 20 ps each, imposing a harmonic restraint (5 kcal mol−1 Å−2) to the heme prosthetic group, under the canonic (NVT) ensemble using Langevin dynamics.32 Next, a simulation was performed under the isothermic–isobaric (NPT) ensemble to ensure that the box did not deform, and to achieve an adequate density. Thus, once at 300 K, we carried out four steps of 10 ps each, at 1 atm pressure, with a weak restraint on the residue side chains, using an isotropic weak-coupling algorithm and the Berendsen barostat,33 and then one more step of 160 ps with the same restraint up to a density of around 1 g cm−3. Finally, an equilibration stage of 10 ns followed by a production period of 100 ns were calculated at 300 K under the NVT ensemble without any restraints apart from employing the SHAKE algorithm34 to constrain all the covalent bonds containing hydrogen.The other two MD simulations will be explained below. All the MD simulations were performed using the AMBER18 GPU (CUDA) version of the PMEMD package.35,36
2.4 QM/MM calculations
We used quantum mechanics/molecular mechanics (QM/MM) calculations for the energy optimization of the structures. Each QM/MM system consisted of nearly 10500 atoms, including all the residues of the monomer B of Gly526Ser COX-2 (with the radical Tyr385), the complete AA, and the 500 water molecules closest to C10 of AA. The active region, where all atoms moved freely during the optimization processes, involved around 2100 atoms whereas the rest of the system was kept frozen. This region was defined by all residues and water molecules within a 15 Å radius sphere centering on C10, including the complete AA and the heme prosthetic group.
The QM/MM calculations were carried out with the modular package ChemShell,37,38 using Gaussian09 for the density functional theory (DFT) calculations of the quantum mechanics (QM) part. The calculations of the MM part were carried out with the DL_POLY module39 in Chemshell, employing the AMBER force field.18 The QM region (being the Fe atom in the MM part because it did not belong to the cyclooxygenase active site) was described by the B3LYP functional with the 6-31G(d,p) basis set for all atoms, with a zero total charge and doublet multiplicity in all reaction steps. This QM region presented different sizes depending on the particular step studied in the catalytic mechanism (see Fig. S1†). The polarizing effect of the enzyme over the QM part was included by the electrostatic embedding scheme,40 and the QM/MM boundary was treated using hydrogen link atoms with the charge-shift model.41 Finally, no cutoffs were introduced for the nonbonding MM and QM/MM interactions.
To determine the potential energy profiles, a series of optimizations were carried out imposing harmonic restraints on the reaction coordinate, with an increment of 0.2 Å at each step. The energy minimizations were done with the limited-memory Broyden–Fletcher–Goldfarb–Shanno (L-BFGS) algorithm.42 A combination of the partitioned rational function optimizer (P-RFO)39 and the L-BFGS methods was used to locate transition state structures. All these algorithms are included in the Hybrid Delocalized Internal Coordinate Optimiser (HDLCopt)39 module of Chemshell. All the pictures of molecules were generated with the VMD43 and CHIMERA17 programs.
3 Results and discussion
3.1 Molecular dynamics simulations
As explained above, the first MD simulation of the Gly526Ser COX-2/AA Michaelis complex was carried out over 100 ns starting from the structure resulting from the setup. The evolution of the distances between the C13 pro-S hydrogen or the C13 carbon atom and the hydrogen-acceptor oxygen of the Tyr385 radical is displayed in Fig. 1 (for greater clarity, see the separated evolution of both distances in Fig. S2 and S3†). The distributions of both distances practically match throughout the simulation. The distances oscillate between about 5 Å and 7 Å most of the time for the first 50 ns, but, after a sudden jump, both remain between about 7 Å and 9 Å for the last 50 ns. | ||
| Fig. 1 Evolution of the C13 pro-S hydrogen–O(Tyr385) (red line) and C13–O(Tyr385) (blue line) distances over 100 ns of the first MD simulation of the Gly526Ser COX-2/AA Michaelis complex. | ||
A typical structure corresponding to the second half of the MD simulation is pictured in Fig. 2. It can be seen that AA is forced to undergo a major repositioning as a consequence of the Gly526Ser mutation, moving from the “L-shaped” binding conformation that it adopts in the wild-type COX-23 to a conformation where the chains corresponding to the AA ω-end and the AA carboxylate-end become roughly parallel. This motion causes the C13 pro-S hydrogen to be too far from the hydrogen-acceptor oxygen of the Tyr385 radical for the abstraction of the first step of the all-radical mechanism to occur.
 | ||
| Fig. 2 A typical structure corresponding to the second half of the first MD simulation of the Gly526Ser COX-2/AA Michaelis complex. | ||
This rather unexpected behavior of AA inside the Gly526Ser mutant could not explain the experimental results by Brash and co-workers.16 Thus, we checked if the problem lay in the fact that we generated the setup of the mutant keeping the substrate AA within it. That is, we had not thus far allowed a free relaxation of the mutant enzyme in the absence of AA. Then, we eliminated AA from the setup (see Section 2.1), we repeated the protocol previous to the production stage as described in Section 2.3, and we ran a 20 ns MD simulation of the Gly526Ser COX-2 mutated enzyme. During these 20 ns, Ser526 rotates in order that the hydrogen of its OH group forms a hydrogen bond with the oxygen atom of the backbone of Met522 (this hydrogen bond is not possible with Gly526 in wild-type COX-2). This position is mostly maintained during this MD simulation (see Fig. S4†). As a consequence, the two hydrogen atoms of the Cβ of Ser530 point towards the hydrophobic groove where the AA ω-end will finally lie.
Next, we re-introduced AA into the cavity of the mutant. To this aim, we eliminated the water molecules from the setup (see Section 2.1) and from the last structure of the previous 20 ns MD simulation, and we overlaid the enzyme residues of both structures. Then, we re-solvated the system, as explained in Section 2.1, and we repeated the protocol described in Section 2.3 to run another 100 ns MD simulation of the Gly526Ser COX-2/AA Michaelis complex. The evolution of the distances between the C13 pro-S hydrogen or the C13 carbon atom and the hydrogen-acceptor oxygen of the Tyr385 radical (see Fig. 3) shows that most of the structures are now ready for the abstraction in the first step of the all-radical mechanism, especially for the first 90 ns, when the distances C13 pro-S hydrogen–O(Tyr385) and C13–O(Tyr385) are 3.4 ± 0.8 Å and 3.8 ± 0.8 Å, respectively.
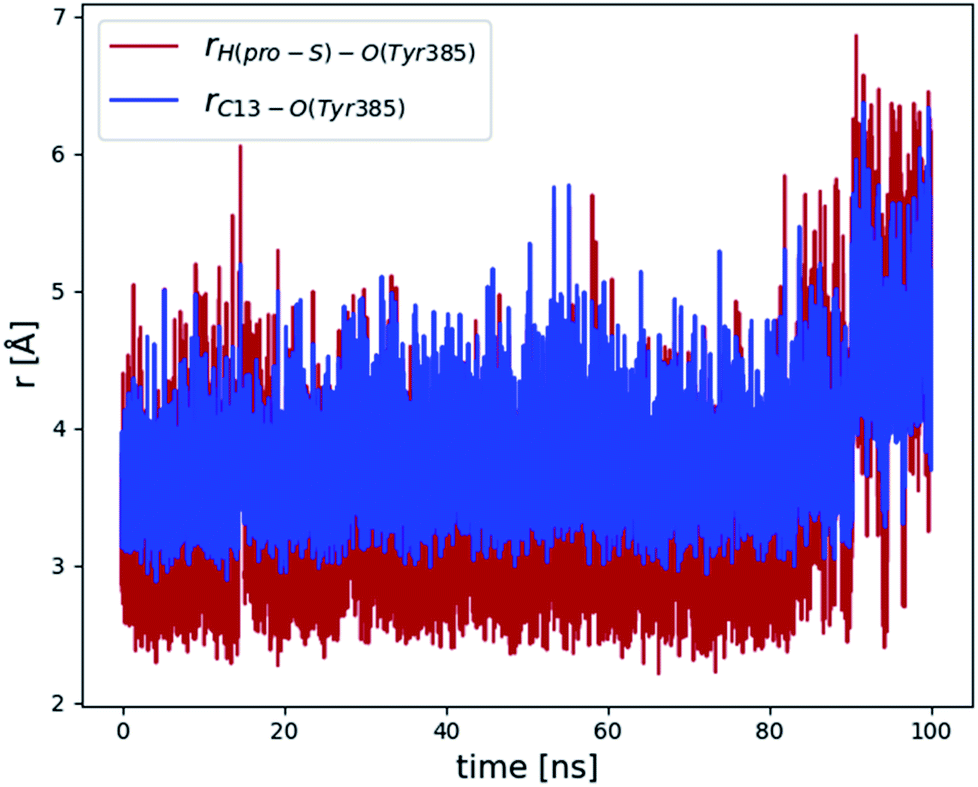 | ||
| Fig. 3 Evolution of the C13 pro-S hydrogen–O(Tyr385) (red line) and C13–O(Tyr385) (blue line) distances over 100 ns of the last MD simulation of the Gly526Ser COX-2/AA Michaelis complex. | ||
In order to choose a snapshot representative of the ensemble of structures generated along this last MD simulation of the Gly526Ser COX-2/AA Michaelis complex, we picked one structure each 10 ps and filtered the structures by imposing the conditions r(C13 pro-S H–O(Tyr385)) ≤ 3 Å and r(C13 pro-S H–O(Tyr385)) < r(C13–O(Tyr385)). So, we obtained 3049 snapshots, which we clustered using a root mean square deviation (RMSD) of 1.3 Å for the heavy atoms of AA. The three most populated clusters included 1737, 1000 and 305 structures, respectively. Next, we determined the average position of the heavy atoms of AA over all the structures belonging to the most populated cluster, and we found the snapshot of that cluster with the smallest AA heavy atoms RMSD with respect to that average position. This snapshot, the so-called centroid snapshot for the most populated cluster, is pictured in Fig. 4. This structure has no similarities with the one shown in Fig. 2 in the case of the unrelaxed mutant enzyme. Now the “L-shaped” productive binding configuration of AA, as observed in the above-mentioned COX-2 crystal structure,3 has been recovered, with the AA ω-end extending along the hydrophobic groove above Ser530, and the C13 carbon atom and the C13 pro-S hydrogen atom of AA located below and near the phenolic oxygen of Tyr385. A comparison between the structure corresponding to snapshot 1 extracted from the MD simulation of the COX-2/AA Michaelis complex (see ref. 13) and the centroid snapshot of the most populated cluster of the last MD simulation of the Gly526Ser COX-2/AA Michaelis complex (Fig. 4) is pictured in Fig. S5.† In addition, the distances between side chain carbon atoms of selected residues of that snapshot 1 (COX-2/AA) or that centroid snapshot (Gly526Ser COX-2/AA) and selected carbon atoms of AA bound in the corresponding active site are shown in Table S1.†
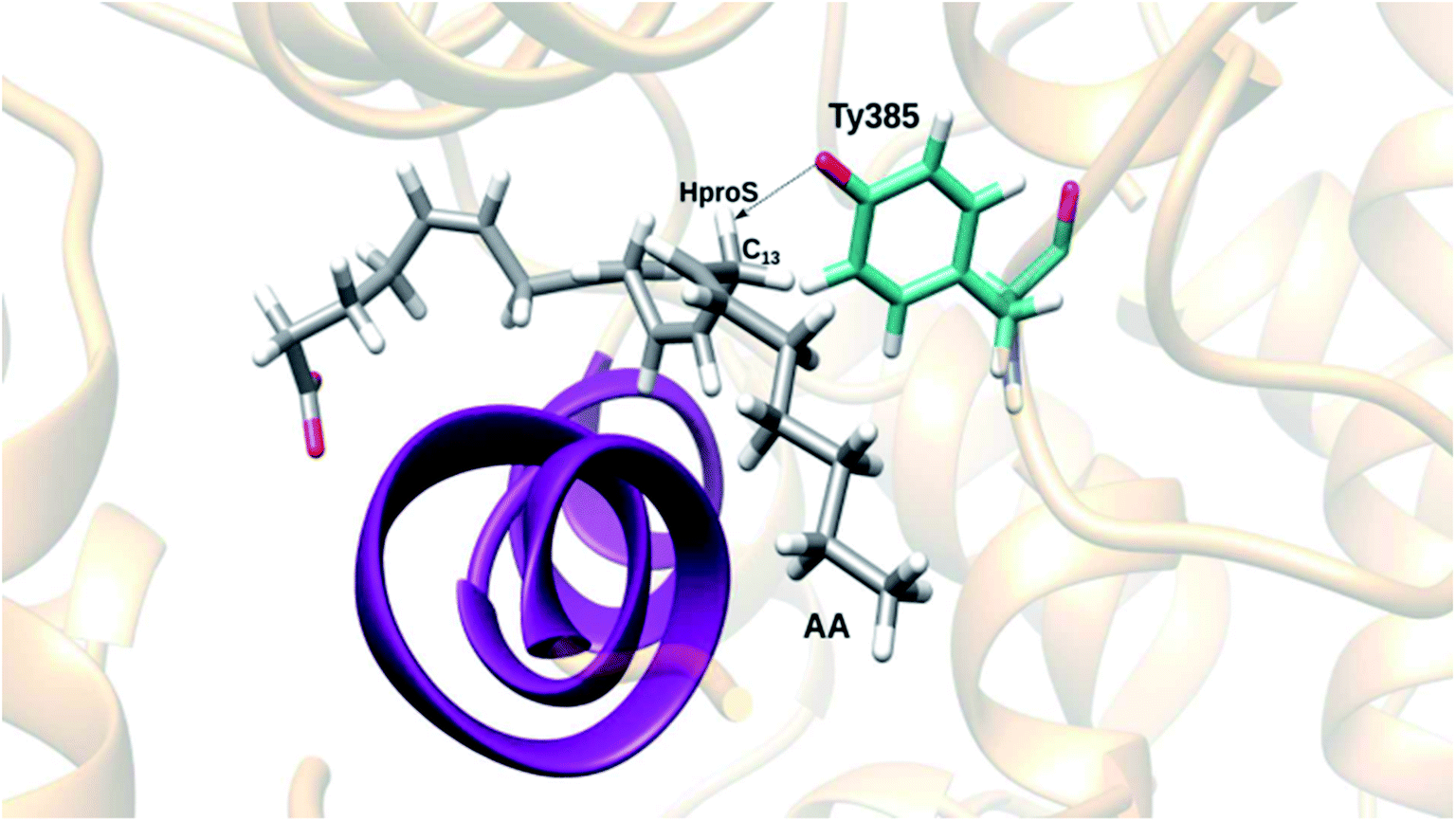 | ||
| Fig. 4 A centroid snapshot of the most populated cluster, chosen as a snapshot representative of the ensemble of structures generated during the last MD simulation of the Gly526Ser COX-2/AA Michaelis complex. | ||
The centroids of the second and third most populated clusters were also determined. They are compared with the centroid of the most populated cluster in Fig. S6.† An “L-shaped” binding configuration of AA is obtained in the three cases.
In order to check if the 100 ns MD simulation shown in Fig. 3 is actually representative of the configurational space visited by an equilibrated Gly526Ser COX-2/AA Michaelis complex, we carried out two additional MD simulations. In the first, we lengthened this MD simulation for an additional 150 ns (see Fig. S7†). In the second, we selected at random one of the structures of the 20 ns MD simulation of the Gly526Ser COX-2 mutated enzyme, and after re-introducing AA into the cavity of the mutant as explained above, we generated a 100 ns MD simulation of the Gly526Ser COX-2/AA Michaelis complex (see Fig. S8†). This new MD simulation is equivalent to the one shown in Fig. 3, but starting from a different structure and different initial velocities. Comparison of Fig. 3 and S7, S8† confirms that, except for some local deviations, the Gly526Ser COX-2/AA Michaelis complex populates a region of the configurational space where most of the structures are ready for the C13 pro-S hydrogen abstraction by O(Tyr385) in the first step of the all-radical mechanism.
3.2 QM/MM calculations
Now we used QM/MM calculations to determine if the Gly526Ser mutant of COX-2 is capable of driving the centroid snapshot of AA throughout the all-radical mechanism. Firstly, we built the potential energy profile of the C13 pro-S hydrogen abstraction by the tyrosyl radical (step 1) as a function of the reaction coordinate. In this case, the reaction coordinate was taken as the difference between the distances corresponding to the breaking bond (C13-pro-S hydrogen) and the forming bond (pro-S hydrogen–O(Tyr385)). The energy profile was calculated both forward and backward to avoid hysteresis (see Fig. 5). Once the reactants, transition state structure, and products had been located, the potential energy barrier turned out to be 24.8 kcal mol−1. The values of the distances corresponding to the three atoms that directly participate in the breaking/forming bonds are given in Table S2† for each structure. As expected for a hydrogen transfer, the hydrogen donor and acceptor atoms approach each other to make the hydrogen transfer easier. After the transfer, both atoms move away. The transition state structure appears in the region with the closest distance (see Fig. S9†). | ||
| Fig. 5 Forward (blue line) and backward (ochre line) potential energy profiles corresponding to C13 pro-S hydrogen abstraction by the tyrosyl radical. The circles indicate the locations of the stationary points. | ||
The first step produces a planar delocalized C11–C15 pentadienyl radical AA. In the second step the addition of the oxygen molecule to C11 can be antarafacial (with the face of the C11–C15 pentadienyl system opposite to the tyrosyl radical), giving an 11R stereochemistry, or suprafacial (on the same side as the tyrosyl radical), leading to an 11S stereochemistry. We chose different possible starting positions of the oxygen molecule for the attack to C11, as we had already done in previous work.13 That C11 atom was chosen as the origin of coordinates, and the oxygen molecules, when possible, were placed around it along the x, y and z Cartesian axes, along the bisector axes contained in the xy, xz and yz planes, or near them. In all, 53 initial oxygen molecules were set close to C11, at distances of 2.5, 3, and 3.5 Å, corresponding to 9, 18, and 26 O2 molecules, respectively. Fifty-three QM/MM single point energy calculations were carried out for each one. Then, the structures with the highest energies were discarded and the most stable structures were optimized. The optimized structures were taken as starting points to build the potential energy profiles for the oxygen addition to C11, the distance O (the one closest to C11)–C11 being the reaction coordinate. Only six different reaction paths leading to the peroxyl radical in C11 were obtained. In Fig. 6 we display these reactions paths (labeled I to VI) according to the structure (that is, the initial optimized position of the oxygen molecule) from which the reaction path starts. The corresponding potential energy barriers and geometrical information are given in Table 1. The suprafacial and antarafacial oxygen additions lead to peroxides with R and S configurations at C11, respectively. This scenario is the opposite to the case of wild-type COX-2, where the suprafacial and antarafacial oxygen attacks produce the S and R configurations at C11, respectively.13 On the other hand, the potential energy barriers (7.1 to 11.2 kcal mol−1) turn out to be small in comparison with that for hydrogen abstraction (24.8 kcal mol−1).
 | ||
| Fig. 6 Potential energy profiles corresponding to the addition of the oxygen molecule to C11. The first point of reaction path I sets the origin of potential energies. O1 stands for the oxygen atom of the oxygen molecule that is closest to C11. | ||
| Reaction path | Initial distance O–C11 (Å) | ΔE (kcal mol−1) | O2 attack | Stereochemistry at C11 |
|---|---|---|---|---|
| I | 3.18 | 7.7 | Suprafacial | R |
| II | 3.43 | 8.4 | Suprafacial | R |
| III | 3.12 | 8.6 | Suprafacial | R |
| IV | 4.04 | 11.2 | Suprafacial | R |
| V | 3.70 | 7.3 | Antarafacial | S |
| VI | 3.84 | 7.1 | Antarafacial | S |
The third step consists of the 9,11-cyclization, leading to a C8-radical cyclic endoperoxide. In Fig. 7 we have plotted the potential energy profiles obtained as a function of the reaction coordinate O–C9 for the six reaction paths. Some of the profiles involve two potential energy maxima. In these cases, in the first stage the free oxygen of the oxygen molecule added to C11 has to rotate around the C11–O bond in order to approach C9 (first maximum) and then the 9,11-cyclization occurs (second maximum). The potential energy barriers corresponding to the higher energy transition state structures for each reaction path are shown in Table 2, along with stereochemical information. The four paths corresponding to an R configuration at C11 involve barriers 2–3 kcal mol−1 lower than the two paths with an S configuration at C11. Most interestingly, we have obtained (9S, 11R) and (9R, 11S) cyclic endoperoxides, but not (9R, 11R). This fact will have important consequences for the following mechanistic step.
 | ||
| Fig. 7 Potential energy profiles corresponding to the 9,11-cyclization leading to a C8-radical cyclic endoperoxide. O2 stands for the oxygen atom of the oxygen molecule that is free and can attack C9. The potential energy of the initial structure of reaction path I is taken as the origin of energies. | ||
| Reaction path | ΔE (kcal mol−1) | Stereochemistry at C11 | Stereochemistry at C9 |
|---|---|---|---|
| I | 8.0 | R | S |
| II | 7.3 | R | S |
| III | 7.7 | R | S |
| IV | 8.1 | R | S |
| V | 9.9 | S | R |
| VI | 10.7 | S | R |
For the fourth step we chose the distance C8–C12 to define the reaction coordinate for the 8,12-cyclization. When that distance was shortened the O–O bond of the cyclic endoperoxide was broken in all cases. Instead of the formation of the cyclopentane ring, a hydrogen transfer from C11 to C8 followed by the breakage of the O–O bond occurred, leading to the formation of a ketone group in C11 and a radical centered at the other oxygen atom (the one bonded to C9). The corresponding potential energy profiles are displayed in Fig. 8. This process turns out to be very exothermic, and the hydrogen transfer and the O–O cleavage can take place in a concerted way or in two stages, depending on the reaction path. The potential energy barriers range from 15.5 kcal mol−1 to 25 kcal mol−1.
 | ||
| Fig. 8 Potential energy profiles as a function of the C8–C12 distance taken as the reaction coordinate. The potential energy of the initial structure of reaction path I is taken as the origin of energies. | ||
Our calculations confirm the experimental finding by Brash and co-workers,16 who found that the 8,12-cyclization does not occur in the Gly526Ser mutant COX-2. We know that the cyclopentane ring closure only occurs with a (9R, 11R) configuration,13,16 but this is just the stereochemical configuration that cannot be reached after the third mechanistic step in the mutant, as mentioned above. The reason for that can be understood by analyzing Fig. 9, where, for the sake of example, we have pictured the cyclic endoperoxide corresponding to reaction path II as a function of the C8–C12 distance at a point before the hydrogen transfer and the O–O cleavage (Fig. 9A), and at a point after that (Fig. 9B). As commented in Section 3.1, in the Gly526Ser mutant COX-2 the hydrogen of the Ser526 OH group forms a hydrogen bond (see Fig. S5†) with the oxygen atom of the Met522 backbone (a hydrogen bond that cannot exist with Gly526 in wild-type COX-2). As a consequence, the two hydrogen atoms of the Cβ Ser530 point towards the hydrophobic groove where the AA ω-end lies. This position makes the helix D containing residues from 521 to 535 quite rigid and forces Ser530 to adopt the position shown in Fig. 9A (both Ser residues belong to that helix). As a result, the hydrogen at C9 cannot be at the same side as the hydrogen at C11 (this is equivalent to saying that the (9R, 11R) configuration cannot be reached), and the 8,12-cyclization cannot happen. In Fig. 9B the hydrogen transfer from C11 to C8 and the breakage of the O–O bond have already taken place.
 | ||
| Fig. 9 Cyclic endoperoxide corresponding to reaction path II as a function of the C8–C12 distance at a point before the hydrogen transfer and the O–O cleavage (A), and at a point after that (B). Distances are given in Å. | ||
At this point it is clear that the Gly526Ser mutation hinders the formation of prostaglandins because the 8,12-cyclization does not occur, and the cyclic endoperoxide breaks. Nevertheless, this breakage does not experimentally take place in the way described above according to Fig. 8 and 9B. In effect, as already found by Brash and co-workers,16 the homolytic cleavage of the O–O bond actually leads to the formation of 8,9-11,12-diepoxy derivatives of the arachidonic acid. Taking the distance O (bonded to C9)–C8 as the reaction coordinate, the 8,9- and 11,12-epoxides are formed in a concerted way for reaction paths I and II (see Fig. 10A), but just the 8,9-epoxide for reaction paths III–VI (see also Fig. 10A). The 11,12-epoxide for these last reaction paths is formed next when the distance O (bonded to C11)–C12 is chosen to define the reaction coordinate (see Fig. 10B). Anyway, the product contains an allyl radical delocalized from C13 to C15. In all cases the potential energy barriers for the formation of the epoxides are clearly lower than the ones corresponding to the above-discussed O–O cleavage involving a hydrogen transfer from C11 to C8, so explaining the experimental formation of the 8,9-11,12-diepoxy derivatives.
 | ||
| Fig. 10 Potential energy profiles corresponding to the formation of (A) the 8,9- and 11,12-epoxides in a concerted way for reaction paths I and II, but just the 8,9-epoxide for reaction paths III–VI, as a function of the O2 (bonded to C9)–C8 distance taken as the reaction coordinate; and (B) the 11,12-epoxide for reaction paths III to VI, as a function of the O1 (bonded to C11)–C12 distance taken as the reaction coordinate. | ||
Both epoxides are stabilized by interactions with several residues like Ser526. The 11,12-epoxide is especially stabilized by formation of a hydrogen bond with Tyr385, which, in turn, is hydrogen-bonded to Tyr348 (see Fig. 11). As a matter of fact, the two early maxima that appear for the formation of the 11,12-epoxide in the case of reaction paths III and IV (Fig. 10B) just correspond to the previous reorganization of Tyr385 in order to be able to interact with the epoxide to be formed. It is interesting to note that, conversely, Tyr385 does not interact with either of the two oxygen atoms of the O–O bridge after the 8,12-cyclization to give a bicyclo endoperoxide which occurs in wild-type COX-2 (see Fig. S10,† derived from the calculations in our previous work13 for the sake of comparison).
 | ||
| Fig. 11 Structure of the 8,9- and 11,12-epoxides corresponding to (A) reaction path I; and (B) reaction path III. Distances are given in Å. | ||
4. Conclusions
The biosynthesis of prostaglandins from arachidonic acid catalyzed by cyclooxygenase-2 is the result of a complex mechanism where every residue of the cyclooxygenase active site plays its role to achieve exceptional stereochemical and regiochemical control of the outcome. Unraveling these roles is not an easy task. Particularly remarkable is the case of the Gly526 residue. Brash and co-workers16 have shown that the Gly526Ser mutant of human COX-2 is unable to produce the 8,12-cyclization required for the formation of prostaglandin. Instead, they found novel products that were identified as 8,9-11,12-diepoxy-13R- (or 15R)-hydroperoxy derivatives of arachidonic acid. In this work we have combined molecular dynamics simulations and QM/MM calculations to shed light on the molecular details of the effects of such a mutation.Our results show that inside the Gly526Ser COX-2 binding pocket, AA adopts the same “L-shaped” productive binding configuration as the one observed in the COX-2 crystal structure,3 with the AA ω-end extending along the hydrophobic groove above Ser530, and the C13 carbon atom and the C13 pro-S hydrogen atom of AA located below and near the phenolic oxygen of Tyr385. Then, the first three reaction steps of the all-radical mechanism7,13 can take place, leading to C8-radical cyclic endoperoxides, in both wild-type COX-2 and Gly526Ser COX-2. The difference between the two cases lies in the fact that only COX-2, but not the mutant, is able to produce cyclic endoperoxides with a (9R, 11R) configuration, the stereochemistry that is required to make the 8,12-cyclization possible. This is a consequence of the fact that in the Gly526Ser mutant COX-2, the hydrogen of the Ser526 OH group forms a hydrogen bond with the oxygen atom of the Met522 backbone (a hydrogen bond that cannot exist with Gly526 in wild-type COX-2). Then, the two hydrogen atoms of Cβ Ser530 point towards the hydrophobic groove where the AA ω-end is located, in a position that hinders the hydrogen at C9 from being at the same side as the hydrogen at C11 (this is equivalent to saying that the (9R, 11R) configuration cannot be reached). Consequently, the 8,12-cyclization towards the formation of prostaglandin cannot take place in Gly526Ser COX-2. In fact, all attempts to force the shortening of the C8–C12 distance provoke hydrogen transfer from C11 to C8 followed by the breakage of the O–O bond of the cyclic endoperoxide. Anyway, the homolytic cleavage of the O–O bond leading to the formation of 8,9-11,12-diepoxy derivatives of the arachidonic acid turns out to occur faster, thus explaining the experimental findings by Brash and co-workers.16 We hope that this work can contribute to better molecular comprehension of the catalytic mechanism of COX-2, one of the main enzymes responsible for inflammation processes in humans.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Spanish “Ministerio de Ciencia, Innovación y Universidades” (project CTQ2017-83745-P).References
- M. Murakami, Exp. Anim., 2011, 60, 7–20 CrossRef CAS PubMed.
- C. Schneider, D. A. Pratt, N. A. Porter and A. R. Brash, Chem. Biol., 2007, 14, 473–488 CrossRef CAS PubMed.
- A. J. Vecchio, D. M. Simmons and M. G. Malkowski, J. Biol. Chem., 2010, 285, 22152–22163 CrossRef CAS PubMed.
- C. Yuan, C. J. Rieke, G. Rimon, B. A. Wingerd and W. L. Smith, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 6142–6147 CrossRef CAS PubMed.
- W. L. Smith, D. L. DeWitt and R. M. Garavito, Annu. Rev. Biochem., 2000, 69, 145–182 CrossRef CAS PubMed.
- B. J. Orlando, P. P. Borbat, E. R. Georgieva, J. H. Freed and M. G. Malkowski, Biochemistry, 2015, 54, 7309–7312 CrossRef CAS PubMed.
- M. Hamberg and B. Samuelsson, J. Biol. Chem., 1967, 242, 5336–5343 CAS.
- K. Marnett and L. J. Maddipati, ed. M. Grisham, J. Everse and K. Everse, Prostaglandin H Synthase, CRC Press, Florida, Unites States, Boca Raton, 1991, vol. 1, pp. 293–334 Search PubMed.
- W. L. Smith and L. J. Marnett, Biochim. Biophys. Acta, 1991, 1083, 1–17 CrossRef CAS.
- C. Schneider, W. E. Boeglin, S. Lai, J. K. Cha and A. R. Brash, Anal. Biochem., 2000, 284, 125–135 CrossRef CAS PubMed.
- L. J. Marnett, Curr. Opin. Chem. Biol., 2000, 4, 545–552 CrossRef CAS PubMed.
- P. J. Silva, P. A. Fernandes and M. J. Ramos, Theor. Chem. Acc., 2003, 110, 345–351 Search PubMed.
- A. Cebrián-Prats, À. González-Lafont and J. M. Lluch, ACS Omega, 2019, 4, 2063–2074 Search PubMed.
- S. W. Rowlinson, B. C. Crews, D. C. Goodwin, C. Schneider, J. K. Gierse and L. J. Marnett, J. Biol. Chem., 2000, 275, 6586–6591 CrossRef CAS PubMed.
- C. Schneider, W. E. Boeglin, J. J. Prusakiewicz, S. W. Rowlinson, L. J. Marnett, N. Samel and A. R. Brash, J. Biol. Chem., 2002, 277, 478–485 CrossRef CAS PubMed.
- C. Schneider, W. E. Boeglin and A. R. Brash, J. Biol. Chem., 2004, 279, 4404–4414 CrossRef CAS PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
- J. A. Maier, C. Martinez, K. Kasavajhala, L. Wickstrom, K. E. Hauser and C. Simmerling, J. Chem. Theory Comput., 2015, 11, 3696–3713 CrossRef CAS PubMed.
- D. A. Case, I. Y. Ben-Shalom, S. R. Brozell, D. S. Cerutti, T. E. Cheatham III, V. W. D. Cruzeiro, T. A. Darden, R. E. Duke, D. Ghoreishi, M. K. Gilson, H. Gohlke, A. W. Goetz, D. Greene, R. Harris, N. Homeyer, S. Izadi, A. Kovalenko, T. Kurtzman, T. S. Lee, S. LeGrand, P. Li, C. Lin, J. Liu, T. Luchko, R. Luo, D. J. Mermelstein, K. M. Merz, Y. Miao, G. Monard, C. Nguyen, H. Nguyen, I. Omelyan, A. Onufriev, F. Pan, R. Qi, D. R. Roe, A. Roitberg, C. Sagui, S. Schott-Verdugo, J. Shen, C. L. Simmerling, J. Smith, R. Salomon-Ferrer, J. Swails, R. C. Walker, J. Wang, H. Wei, R. M. Wolf, X. Wu, L. Xiao, D. M. York and P. A. Kollman, AMBER 2018, University of California, San Francisco, 2018 Search PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- K. Shahrokh, A. Orendt, G. S. Yost and T. E. Cheatham, J. Comput. Chem., 2012, 33, 119–133 CrossRef CAS PubMed.
- C. I. Bayly, P. Cieplak, W. Cornell and P. A. Kollman, J. Phys. Chem., 1993, 97, 10269–10280 CrossRef CAS.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- D. A. Case, V. Babin, J. T. Berryman, R. M. Betz, Q. Cai, D. S. Cerutti, T. E. Cheatham III, T. A. Darden, R. E. Duke, H. Gohlke, A. W. Goetz, S. Gusarov, N. Homeyer, P. Janowski, J. Kaus, I. Kolossváry, A. Kovalenko, T. S. Lee, S. LeGrand, T. Luchko, R. Luo, B. Madej, K. M. Merz, F. Paesani, D. R. Roe, A. Roitberg, C. Sagui, R. Salomon-Ferrer, G. Seabra, C. L. Simmerling, W. Smith, J. Swails, R. C. Walker, J. Wang, R. M. Wolf, X. Wu and P. A. Kollman, AMBER 14, University of California, San Francisco, 2014 Search PubMed.
- P. Tosco, J. Am. Chem. Soc., 2013, 135, 10404–10410 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. DeFrees and J. A. Pople, J. Chem. Phys., 1982, 77, 3654–3665 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Farkas, J. B. Foresman, J. V Ortiz, J. Cioslowski, D. J. Fox, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Had and D. J. Fox, Gaussian, Inc., Wallingford CT, 2009.
- M. Deserno and C. Holm, J. Chem. Phys., 1998, 109, 7678–7693 CrossRef CAS.
- H. Lei, N. A. Baker and X. Li, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 14183–14188 CrossRef CAS PubMed.
- H. J. C. Berendsen, J. P. M. Postma, W. F. Van Gunsteren, A. Dinola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
- J. P. Ryckaert, G. Ciccotti and H. J. C. Berendsen, J. Comput. Phys., 1977, 23, 327–341 CrossRef CAS.
- S. Le Grand, A. W. Götz and R. C. Walker, Comput. Phys. Commun., 2013, 184, 374–380 CrossRef CAS.
- R. Salomon-Ferrer, A. W. Götz, D. Poole, S. Le Grand and R. C. Walker, J. Chem. Theory Comput., 2013, 9, 3878–3888 CrossRef CAS PubMed.
- S. Metz, J. Kästner, A. A. Sokol, T. W. Keal and P. Sherwood, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 101–110 CAS.
- P. Sherwood, A. H. de Vries, M. F. Guest, G. Schreckenbach, C. R. A. Catlow, S. A. French, A. A. Sokol, S. T. Bromley, W. Thiel, A. J. Turner, S. Billeter, F. Terstegen, S. Thiel, J. Kendrick, S. C. Rogers, J. Casci, M. Watson, F. King, E. Karlsen, M. Sjøvoll, A. Fahmi, A. Schäfer and C. Lennartz, J. Mol. Struct.: THEOCHEM, 2003, 632, 1–28 CrossRef CAS.
- W. Smith and T. R. R. Forester, J. Mol. Graphics, 1996, 14, 136–141 CrossRef CAS PubMed.
- D. Bakowies and W. Thiel, J. Phys. Chem., 1996, 100, 10580–10594 CrossRef CAS.
- H. M. Senn and W. Thiel, Angew. Chem., Int. Ed., 2009, 48, 1198–1229 CrossRef CAS PubMed.
- D. C. Liu and J. Nocedal, Math. Program., 1989, 45, 503–528 CrossRef.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The QM region of each particular reaction step; evolution of the C13 pro-S hydrogen–O(Tyr385) distance during 100 ns of the first MD simulation of the Michaelis complex Gly526Ser COX-2/AA; evolution of the C13–O(Tyr385) distance during 100 ns of the first MD simulation of the Michaelis complex Gly526Ser COX-2/AA; distance between the hydrogen of the Ser526 OH group and the oxygen atom of the backbone of Met522 during 10 ns (equilibration) + 20 ns (production) MD simulation of the Gly526Ser COX-2 mutated enzyme without AA; comparison between the structure corresponding to snapshot 1 extracted from the MD simulation of the COX-2/AA Michaelis complex and the centroid snapshot of the most populated cluster of the last MD simulation of the Gly526Ser COX-2/AA Michaelis complex; comparison between the centroid of the most populated clusters; evolution of the C13 pro-S hydrogen–O(Tyr385) and C13–O(Tyr385) distances during 250 ns of the last MD simulation of the Gly526Ser COX-2/AA Michaelis complex; evolution of the C13 pro-S hydrogen–O(Tyr385) and C13–O(Tyr385) distances during a new 100 ns MD simulation of the Gly526Ser COX-2/AA Michaelis complex; distances between side chain carbon atoms of selected residues of snapshot 1 extracted from the MD simulation of the COX-2/AA Michaelis complex or the centroid snapshot of the most populated cluster of the last MD simulation of the Gly526Ser COX-2/AA Michaelis complex, and arachidonic acid bound in the corresponding active site active site; distances corresponding to the three atoms that directly participate in the breaking/forming of bonds for the reactant, transition state structure and product corresponding to the C13 pro-S hydrogen abstraction by the tyrosyl radical; evolution of the key distances during C13 pro-S hydrogen abstraction by the tyrosyl radical; structure obtained after the 8,12-cyclization to give a bicyclo endoperoxide which occurs in wild-type COX-2. See DOI: 10.1039/c9ra08860a |
| This journal is © The Royal Society of Chemistry 2020 |