
An aminotetracyanocyclopentadienide system: light-induced formation of a thermally stable cyclopentadienyl radical†
Patrick R.
Nimax
a,
Florian
Zoller
a,
Tobias
Blockhaus
a,
Teresa
Küblböck
a,
Dina
Fattakhova-Rohlfing
b and
Karlheinz
Sünkel
 *a
*a
aDepartment Chemistry, Ludwig-Maximilians-University Munich, Butenandtstr. 9-11, 81377 Munich, Germany. E-mail: suenk@cup.uni-muenchen.de
bElectrochemical Storage Department, Institut für Energie- und Klimaforschung: Werkstoffsynthese und Herstellungsverfahren (IEK-1), Forschungszentrum Jülich GmbH Wilhelm-Johnen-Straße, 52425 Juelich, Germany
First published on 18th November 2019
Abstract
Crystals of the aminotetracyanocyclopentadienyl radical were obtained from the reaction of CaCl2 with Ag[C5(CN)4(NH2)] and recrystallization in MeOH, performed in sunlight. The radical was characterized by X-ray diffraction, EPR and UV Vis spectroscopy as well as by cyclovoltammetry and DFT calculations.
Introduction
More or less stable cyclopentadienyl radicals have been known for nearly 100 years.1 In 1925 the first synthesis of the [C5Ph5] radical (Chart 1, A, R = H) was reported,2 which was later characterized by X-ray crystallography,3 and EPR and UV-Vis-spectroscopy.4 Closely related [C5Ar5] radicals, for example A (see Chart 1, R = Me), were described in 2002, and were shown to be thermally stable, but sensitive to O2 or H2O.5 The stability of these radicals was attributed to steric reasons, and thus it was not so surprising that three other sterically very encumbered radicals, B–D in Chart 1,6–8 could be isolated and fully characterized. There were also several cyclopentadienyl radicals reported that were not sterically hindered, but contained electron-withdrawing substituents: the intermediate existence of radicals E and F was postulated from the cyclovoltammetric data of the corresponding anions, but neither the radicals themselves nor any of their decomposition products could be isolated.9,10 Irradiation of the σ-dimers [C5R5]2 (R = Me,11 COOMe,12) within an EPR spectrometer showed the formation of the corresponding radicals G and H, but these could not be isolated. The pentahalocyclopentadienyl radicals [C5X5] (X = F,13 Cl,14 and Br15) could be isolated in frozen matrices and characterized by EPR spectroscopy, but their inherent instability prevented further characterization. Chart 1 shows the structures of these known cyclopentadienyl radicals.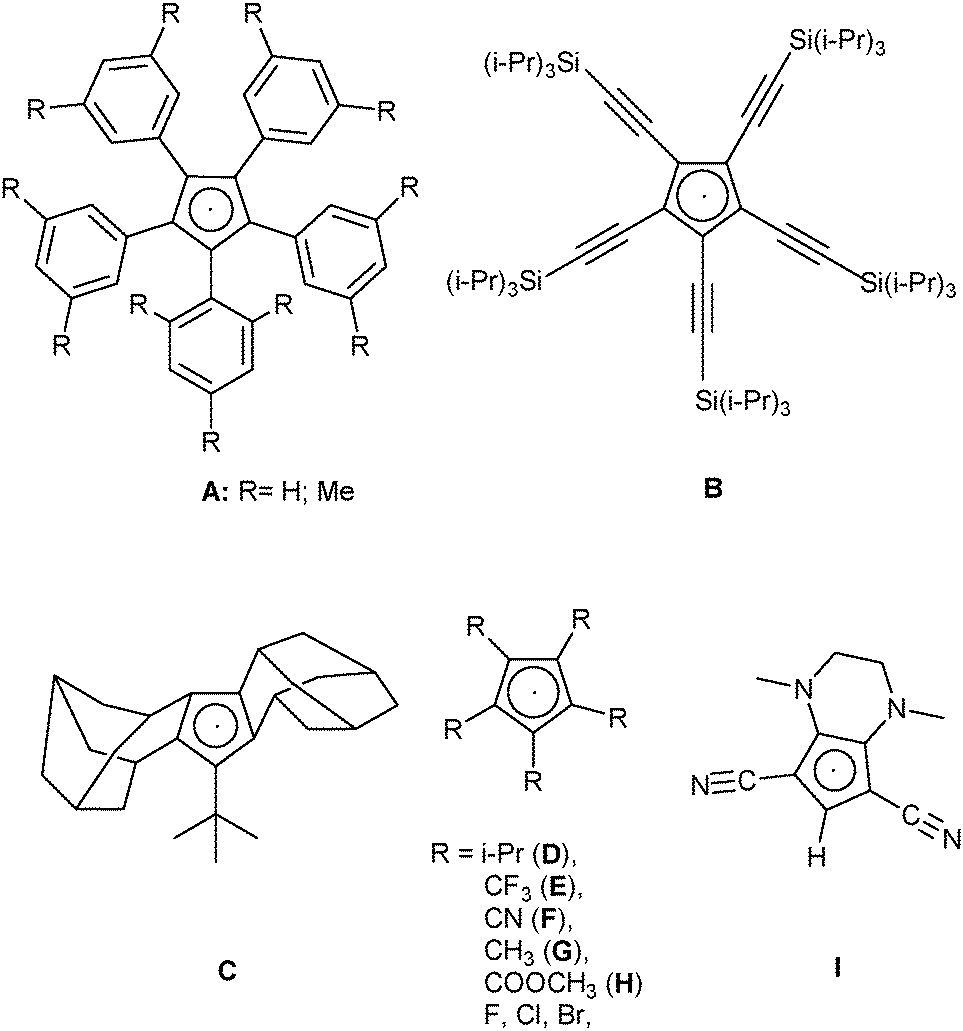 | ||
| Chart 1 Structures of known cyclopentadienyl radicals, including the presumed intermediate I. | ||
Radical chemistry in organic synthesis and physical applications has been a growing field of research over the past few years. This is particularly due to the rapidly growing field of photoredox catalysis.16,17 Within this field, two major approaches have developed: (1) use of metal-based catalysts, either with “noble”18 or “earth-abundant” metals19 and (2) use of purely organic compounds, mostly organic dyes.20 Especially in the latter field, the importance of “push–pull-chromophores” has been stressed.21 Conceptually related, although not yet tested in the context of photocatalysis, are coordination compounds with “non-innocent ligands”22 and charge-transfer salts of radical cations with polycyano anions.23
Considering the fact that (un-)substituted cyclopentadienyl systems are arguably the most important ligands in transition-metal organometallics, we reasoned that joining all the above-mentioned concepts by preparing coordination compounds with donor–acceptor-substituted cyclopentadienyl anions or radicals might lead to a new class of photoactive compounds. Possible candidates would be cyclopentadienides with donor groups like OMe or NR2 and acceptors like COOMe or CN, some of which are known for quite a while (e.g. the potassium salt precursor of radical I in Chart 1).24,25 In the course of our studies on the coordination chemistry of polynitriles we already had looked at the crystal structures of the Na and Mn(II) salts of the [C5(CN)4(NH2)] (“ATCC”) anion.26 In both structures, the ATCC anion coordinates only via one nitrile function and shows a slight distortion towards an “allylene” form. We decided to have a closer look at the ATCC system.
Results and discussion
Webster had shown in his 1971 patent (see Scheme 1) that the zwitterion [C5(CN)4(NH3)] (1a) is formed, when the diene [C5(CN)5NH2] was treated either by concentrated HI or HCl. Refluxing the diene in MeOH yielded the salt NH4+ ATCC− (1b) while refluxing in MeCN in the presence of KI gave the corresponding potassium salt 1c. According to this patent, treatment of 1a with NaHCO3 in water followed by addition of NEt4Br yielded the tetraethylammonium salt 1d.25b Our synthetic approach to the above-mentioned Na and Mn(II) salts involved the preparation of Ag(ATCC) (1e), followed by treatment with NaCl or MnCl2, respectively. When we tried to prepare analogously the corresponding Ca and Cu(II) salts, recrystallization of the obtained products X and Y yielded crystals of the neutral radical [C5(CN)4(NH2)] (2) or the ammonium salt 1b (see Scheme 2).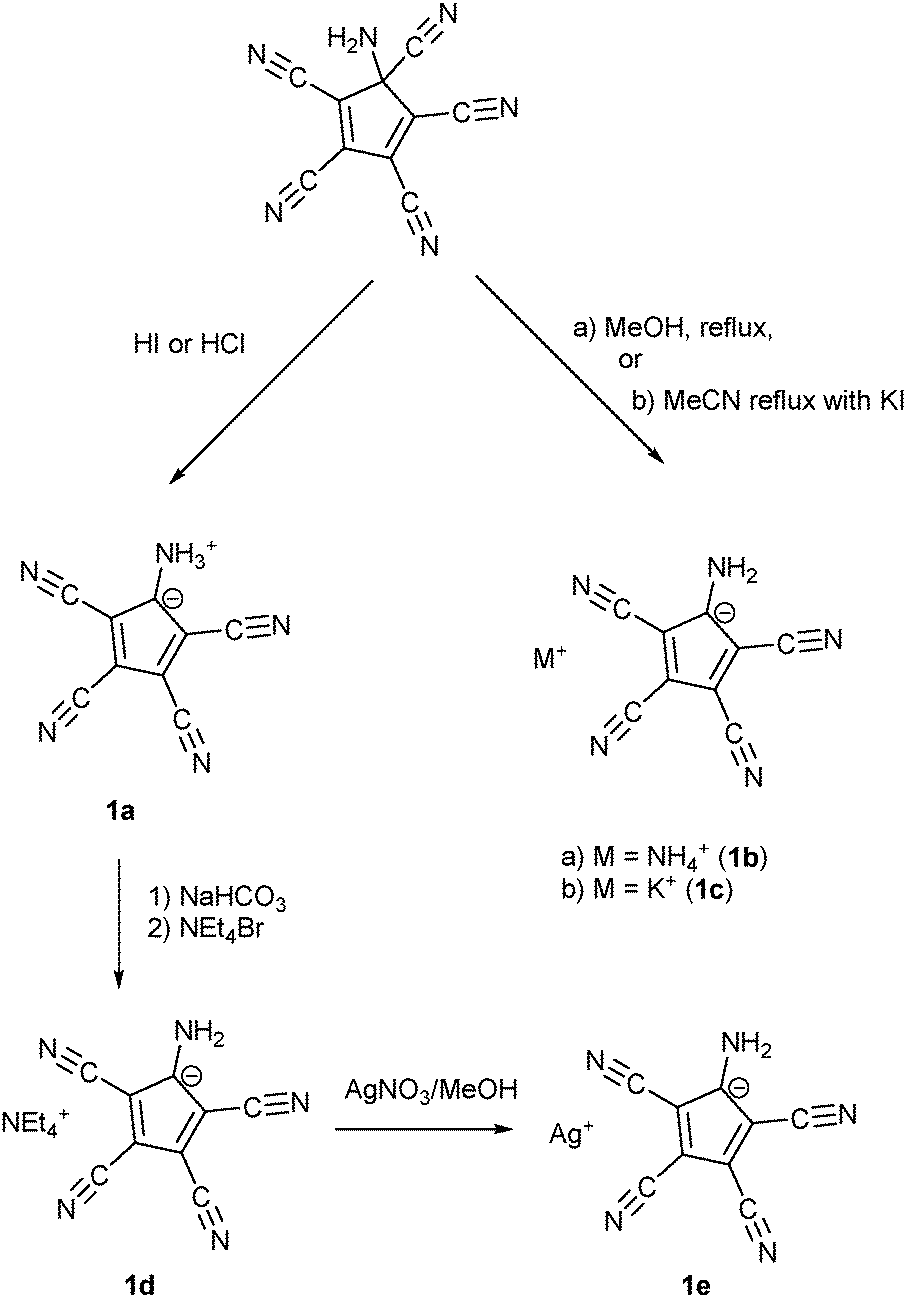 | ||
| Scheme 1 Synthesis of different salts of the aminotetracyanocyclopentadienide anion according to Webster. | ||
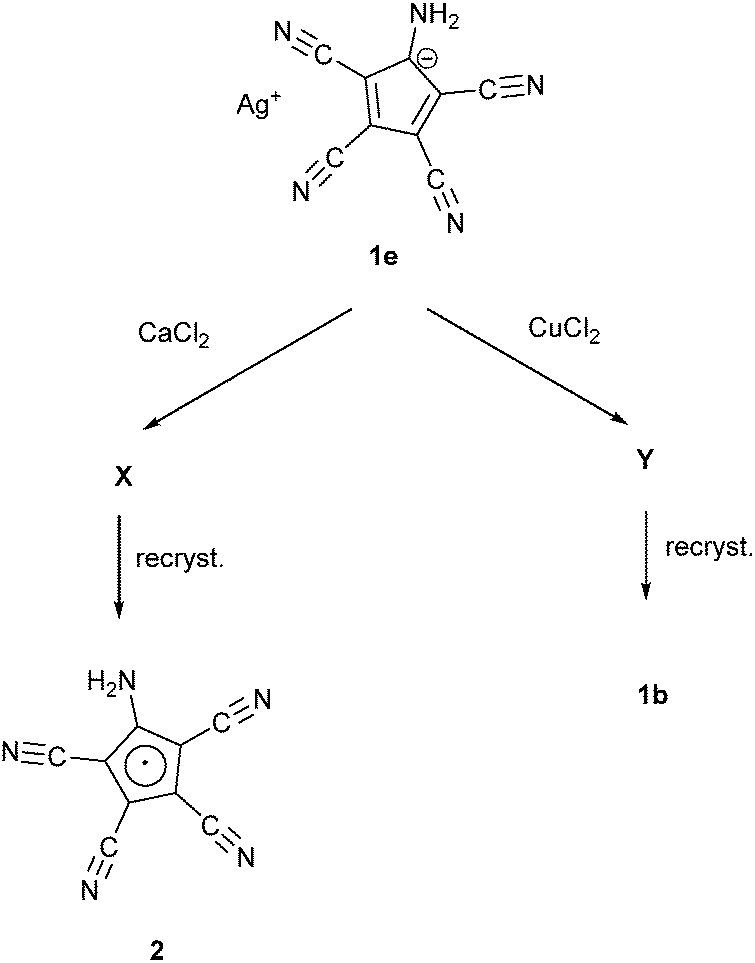 | ||
| Scheme 2 The reactions of 1e with CaCl2 and CuCl. | ||
Fig. 1 and 2 show the molecular structures of both compounds.
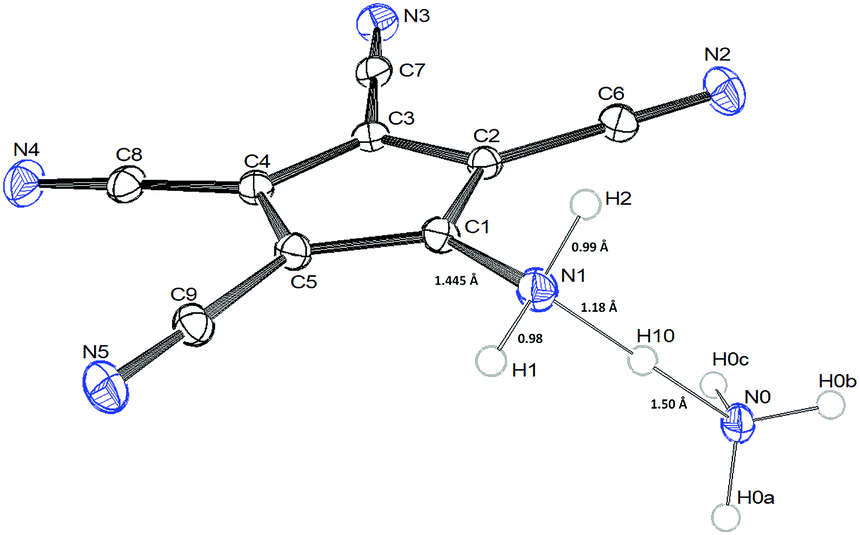 | ||
| Fig. 1 ORTEP-view of 1b showing important bond lengths [Å] of the N–H–N bridge. | ||
The crystals of 1b show large voids (ca. 80 Å3 per unit cell, corresponding to 15% of the volume) and the final structure refinements were performed using the “SQUEEZE” routine in “PLATON”. The NH4+ cation in 1b might have been formed by Cu(II) assisted hydrolysis of a CN bond during recrystallization in MeOH.25,27 The molecular structure of 1b shows an asymmetric hydrogen bridge between both ammonium nitrogens. The N1–H10 bond is significantly longer than N1–H1 and N1–H2, but also significantly shorter than the N0–H10 bond (Fig. 1). The three hydrogen atoms around N1 could be localized in difference Fourier synthesis and were freely refined, while the remaining hydrogen atoms at N0 were positioned with a riding model and then only a common thermal parameter at fixed positions was refined. It should, however, always be kept in mind that localization of hydrogen atoms from the X-ray data has large uncertainties, particularly when the data set is influenced by large voids as in this case. N1 shows typical tetrahedral angles (99–121°), while in the structure of 2 (Fig. 2) the angles around N1 correspond more to sp2 hybridization (angle sum: 355°). This is supported by a significantly shorter ring carbon bond to the amino nitrogen in 2 than in 1b.
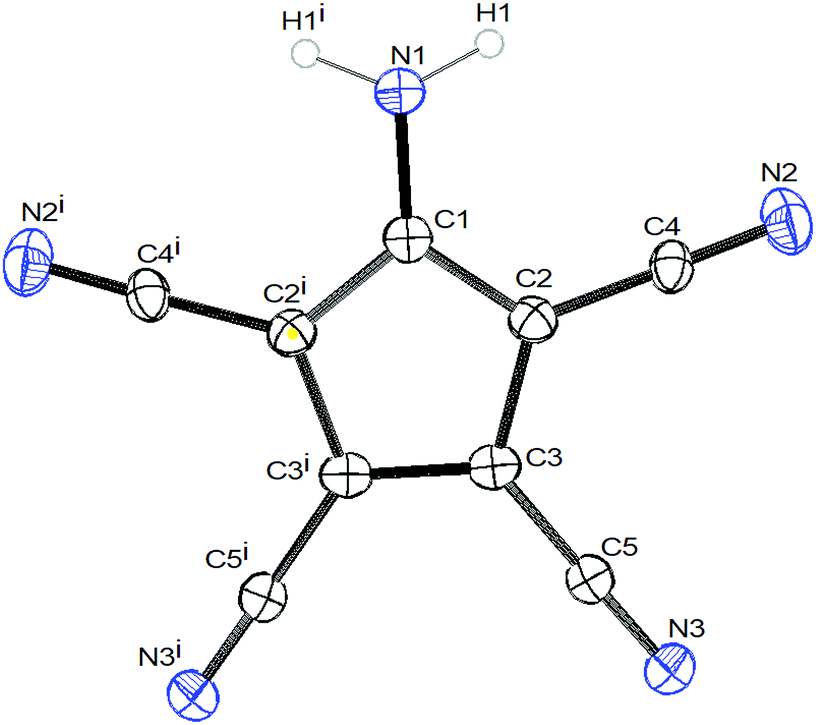 | ||
| Fig. 2 ORTEP view of 2. Symmetry operator: (i): x, 1/2 − y, z. | ||
Both compounds show the same distortion to an “allylene” system with the amino group bonded to the central allyl carbon (Table S3, ESI†), as it was observed in the Na and Mn(II) complexes studied by us earlier. When looking at the molecular packing, the presence of large solvent-accessible voids (15% in 1b, 40% in 2, see also Fig. S1 and S2, ESI†) becomes obvious. In 1b ATCC anions form one-dimensional chains in the b direction using H-bridges N1–H1–N3, and these chains are connected to inversion related chains via H-bridges N1–H2–N2, thus forming 12-membered rings. These double-chains are connected in the a direction via the NH4 cations using H-bridges N0–H0B–N5 (see Fig. S3 and S4, ESI†). In 2 the ATCC molecules also form one-dimensional chains parallel to c (base vector: 1 0 −1), using two H-bridges N1–H11–N3 (see Fig. 3).
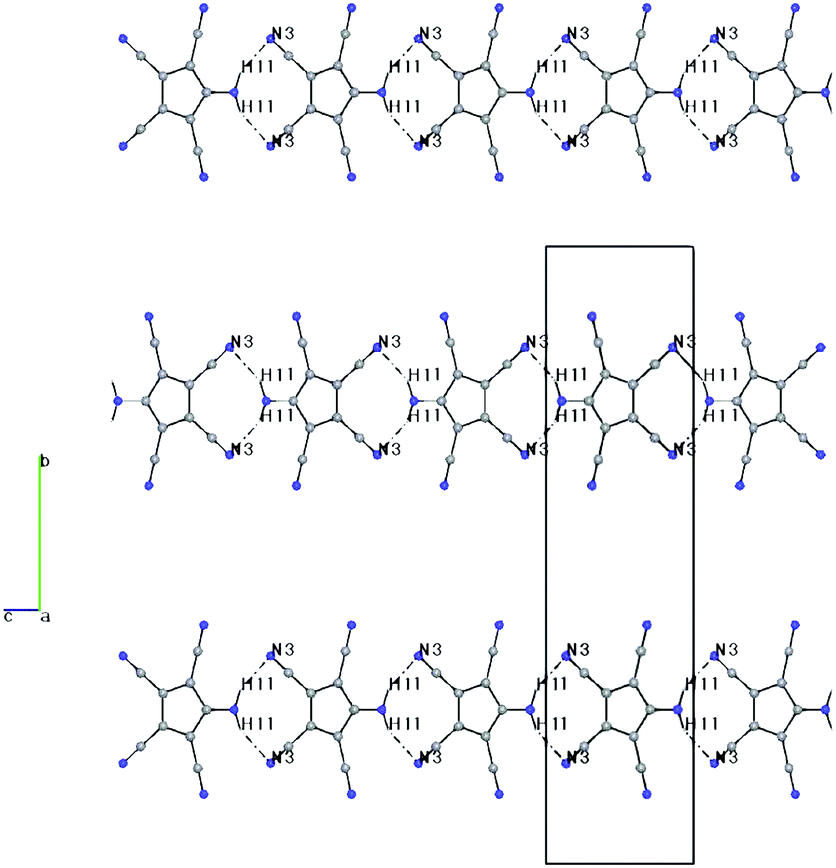 | ||
| Fig. 3 Packing diagram of 2, including H-bonds, viewed down a. H1–N3: 2.36(5) Å, N1–N3: 3.24(5) Å, N1–H1–N3: 153(4)°. | ||
However, parallel chains stack in such a manner that the amino nitrogens are 3.345 Å above the ring plane of the neighbouring chain (see Fig. 4 and Fig. S5, ESI†). A similar stacking was also observed in the structure of the NaATCC salt, published earlier26a by us (see Fig. S6 and S7, ESI†). The only difference in the stacking is that in 2 the projection of the NH2 nitrogen on the ring plane nearly coincides with the centroid (distance N–Ct 3.379 Å), while in NaATCC the corresponding projection is closer to the CNH2 ring carbon (distance N–Ct 3.431 Å vs. a perpendicular distance to the ring plane of 3.342 Å). This arrangement is reminiscent of the π-dimers/π-stacks observed in chloranil and TCNQ anion radicals.28
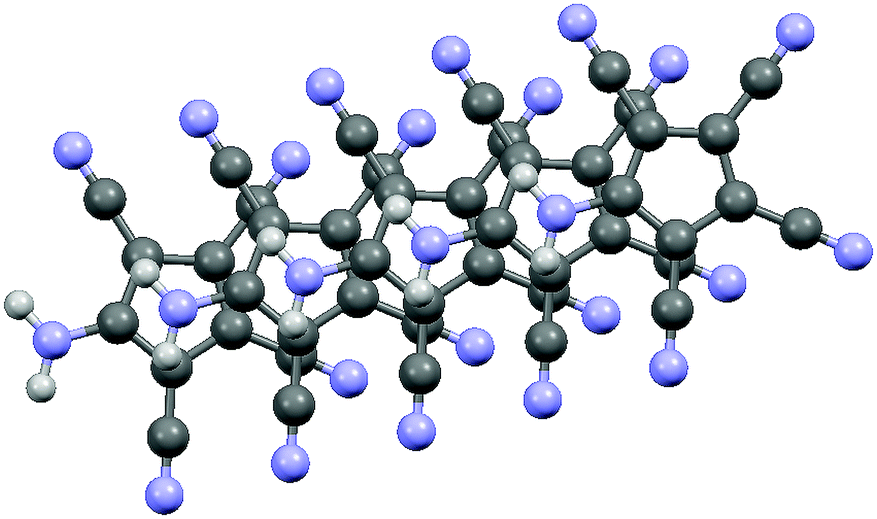 | ||
| Fig. 4 Stacking of the ATCC rings in 2, viewed perpendicular to the ring planes. | ||
ICP/AAS analysis of X shows only trace amounts of Ca2+ (0.37%) and no silver; the CHN elemental analysis corresponds to the composition [C5(CN)4(NH2)(O2)·3H2O]. This is similar to the findings in the sterically encumbered radicals A5 and D6 and shows that the compound is not air-stable. The oxygen does not show up in the crystal structure, however. There is a certain possibility that the oxidation product might be formulated as “[C5(CN)4(NH2)·2H2O2·H2O]”, which might contain H2O2 molecules together with water in the crystal voids, but this remains fully speculative.
We then looked at the mass spectra of 2. The EI spectrum shows besides the (M + H)+ peak also the (M + H + O)+, (M + H + CN)+, (M − HCN + O)+ and (M − HCN + O2)+ peaks, while the ESI(+) showed (M − H + O)+ signals as the base peak together with the (M − H + O2)+ peak and some unidentified higher-mass peaks. This is very similar to the mass spectra of A and D, showing again the oxidative instability of 2.5,6
Next we turned to the (solid-state) EPR spectra of 2 and – for comparison – also of the potassium and silver salts 1c and 1e (see Fig. 5). The crystals of 2 provide EPR signals at first, but they vanish over the course of hours. Very surprisingly, however, powdered samples of salts 1c and 1e, which had been exposed to sunlight, also showed EPR signals, with slightly lower g values. For comparison, the radicals A, C, D and G have g values in the range from 2.0013 to 2.0034.
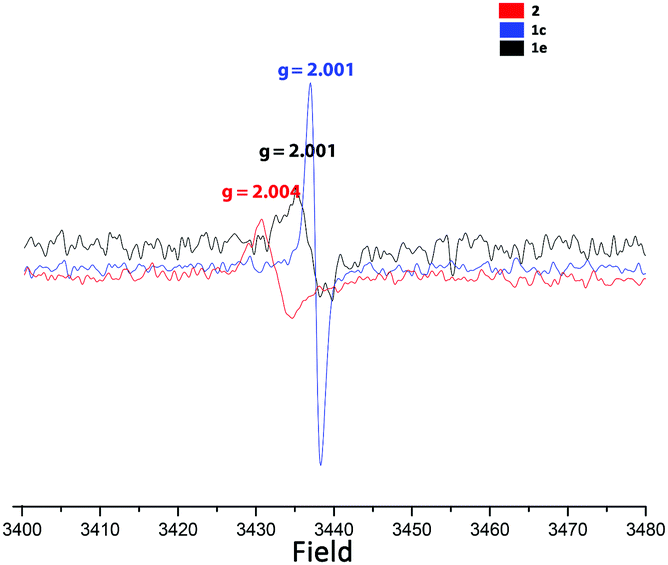 | ||
| Fig. 5 Solid-state EPR spectra of 1c, 1e and 2. | ||
As neither Ag+ nor [C5(CN)4(NH2)]− have unpaired electrons, the most probable explanation is that the anion acts as a “redox-non-innocent” ligand,22i.e. an equilibrium like:
| Ag+ + [C5(CN)4(NH2)]− ↔ Ag0 + [C5(CN)4(NH2)]0 |
For further clarification we looked at the electrochemical properties of 1c and X by cyclovoltammetry (Fig. 6 and Fig. S8, ESI†). Both compounds show two pairs of peaks at potentials around E1/2 of 0.45 V and 0.91 V vs. Ag/0.01 M AgNO3, respectively, which could be assigned to the Cp−/Cp0 redox pair and its subsequent oxidation to the Cp0/Cp+ at more positive potentials according to the literature convention. The detection of the corresponding reduction peaks in the reverse scan together with the similar height of the peak currents for reduction and oxidation indicates chemically reversible behavior, implying the high stability of the formed species under the conditions of the electrochemical experiment. A rather large peak-to-peak separation (100 mV and 120 mV for the first and the second pairs of peaks, respectively, without any IR-correction), however, points to a sluggish kinetics (quasi-reversible behavior) of the corresponding redox transformations. For comparison, the CV data of B–F and of I24a,29 are listed in Table 1. As can be seen from these data, the alkyl substituted radicals C and D are formed at very negative potentials and are easily oxidized to the corresponding cations. Both cyano-substituted radicals F and I apparently are easily oxidized to the cyclopentadienyl cations when formed in the electrochemical experiment from the corresponding anions and do not show up in the cyclovoltammograms. Only the CF3 substituted radical E shows similar electrochemical behaviour to our compounds, although it is more easily oxidized to the cyclopentadienyl cation.
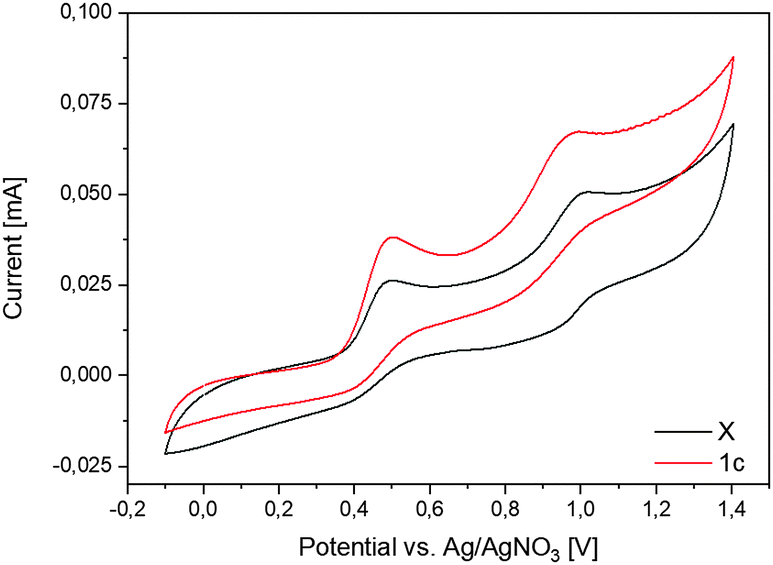 | ||
| Fig. 6 Cyclovoltammogram of 1c and X. The voltammograms were taken in MeCN on the Pt disk electrode. Ag/0.01 M AgNO3 in MeCN was used as a reference electrode. | ||
| Comp. | E (Cp−/Cp°)a | E (Cp°/Cp+)a | Ref. |
|---|---|---|---|
| a vs. FcH/FcH+; the data for D and I were reported related to SCE and were corrected by subtraction of 0.38 V. b In MeCN. c In CH2Cl2. d Irreversible. e Anodic peak potential (calc. vs. external Fc/Fc+). | |||
| B | +0.15c | 7 | |
| C | −1.78cd | +0.30c | 8 |
| D | −1.91b | +0.58b | 6b |
| E | +0.38 , | +0.52 , | 9 |
| F | 1.0b,d | 10 | |
| I | −0.92 , | 29 | |
| X | 0.36b,e | 0.88b,e | This work |
| 1c | 0.36b,e | 0.89b,e | |
Next, we had a look at the UV-Vis spectra of 1c. While in MeOH solution only a broad absorption ranging from 250 to 390 nm could be observed, the reflectance spectrum showed an even broader band ranging nearly over 300 nm, with weak maxima at 220, 277, 386 and 514 nm (Fig. 7, “1c_A”).
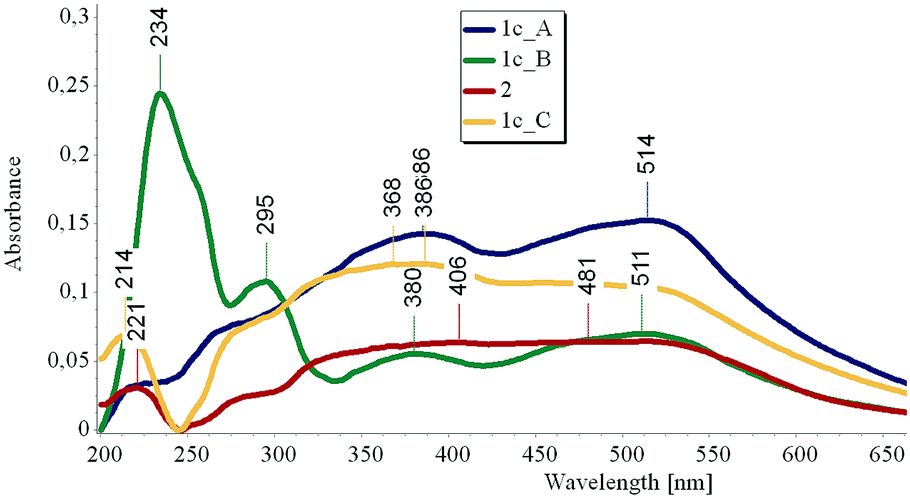 | ||
| Fig. 7 Reflectance UV-Vis spectra of 1c, taken in the dark (blue), after 3 h irradiation with a UV lamp operating at 254 nm (green) and after 8 h storage in the dark (yellow) and of 2, after storage in the dark (red). | ||
After 3 h irradiation at 254 nm, a strong band at 234 nm with a shoulder at 260 nm and a weaker band at 295 nm arise, together with two weak maxima at 380 and 511 nm (Fig. 7, “1c_B”). When the sample is stored in the dark for 8 hours, the absorption peaks at 234 and 295 nm disappear and a spectrum similar to the original one is obtained (Fig. 7, “1c_C”).
In an effort to find some explanations for the obtained results, we performed DFT calculations on the ATCC anion and the radical and zwitterion 1a. Based on recent DFT calculations on the [C5(CN)5] radical we decided to use the ωB97XD functional with a 6-311++G** basis set for calculations, both for the gas and solution phase (PCM model).30Fig. 8 shows a spin density plot calculated for the radical. Tables 2 and 3 show the most important results on the energetics of the anion and the radical.
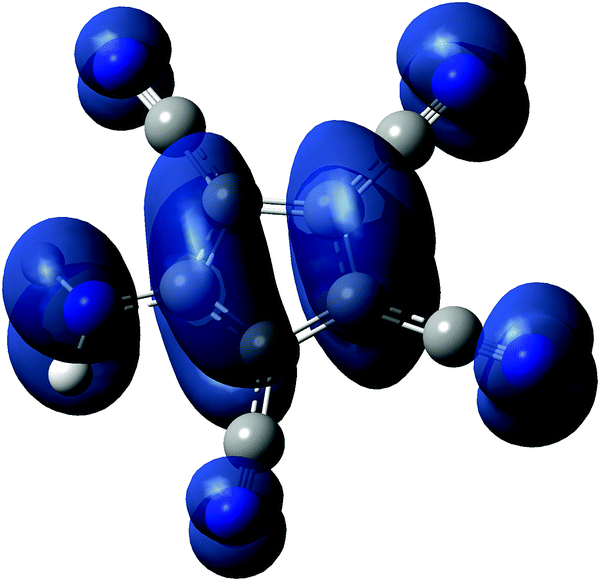 | ||
| Fig. 8 Spin density plot of radical 2. | ||
| Anion | Radical | |
|---|---|---|
| E tot (gas) [a.u.] | −617.891 | −617.738 |
| E HOMO (gas) [a.u.] | −0.168 | −0.342 |
| E LUMO (gas) [a.u.] | 0.120 | −0.047 |
| ΔE (gas) [a.u.] | 0.287 | 0.295 |
| ΔE (gas) [eV] | 7.82 | 8.02 |
| EAadi,g (radical) [a.u.] | 0.153 | |
| EAadi,g (radical) [eV] | 4.149 | |
| Anion | Radical | |
|---|---|---|
| E tot (solv) [a.u.] | −617.960 | −617.764 |
| E HOMO (solv) [a.u.] | −0.277 | −0.320 |
| E LUMO (solv) [a.u.] | 0.002 | −0.034 |
| ΔE (solv) [a.u.] | 0.279 | 0.285 |
| ΔE (solv) [eV] | 7.60 | 7.76 |
| EAadi,solv (radical) [a.u.] | 0.196 | |
| EAadi,solv (radical) [eV] | 5.333 | |
The adiabatic electron affinity can be calculated from the difference of the total energies of the radical and the anion.30c,31 The obtained gas-phase value of ca. 4.15 eV is significantly lower than the reported calculated values for [C5(CN)5] (5.55 ± 0.03 eV![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 30a,c,31) and [C5(CN)4H] (5.00 eV30c). Since 4.15 eV corresponds to a wavelength of ca. 299 nm, it appears plausible that radical 2 can be produced by irradiation of 1c with UV light at 256 nm. The electron affinity of the radical corresponds to the ionization potential of the anion, and is therefore correlated with the electrochemical oxidation potential of the anion. While exact calculations seem still impossible, most authors agree that a linear relationship of the type
30a,c,31) and [C5(CN)4H] (5.00 eV30c). Since 4.15 eV corresponds to a wavelength of ca. 299 nm, it appears plausible that radical 2 can be produced by irradiation of 1c with UV light at 256 nm. The electron affinity of the radical corresponds to the ionization potential of the anion, and is therefore correlated with the electrochemical oxidation potential of the anion. While exact calculations seem still impossible, most authors agree that a linear relationship of the type
| E1/2 = EA − ΔΔGsolv − Eref |
Comparison of the calculated and measured UV-Vis spectra (Fig. S12–S14, ESI†) shows a moderate agreement, which is not unexpected considering that the measured spectra were solid-state spectra and the calculations were performed on solution species. Similarly, comparison of the calculated and observed bond lengths shows poor agreement for 2, and moderate agreement for 1b with the calculated geometry for 1a (Table S3, ESI†). Again, this is not too surprising, since the solid state crystal structures showed both hydrogen bridging and π-stacking, which do not occur in the gas phase used for the calculations.
Conclusions
Taking all the results together, we believe that the “original” product X of the CaCl2/AgATCC reaction was zwitterion 1a. Upon exposure to light, this forms radical 2 presumably with concomitant formation of H2, similar to the formation of radicals F and G from irradiation of the corresponding dienes. A DFT calculation on the energy balance of this presumed formation reaction yields a slightly exergonic ΔGr value (Tables S4 and S5, ESI†). As a solid, the radical can survive for a certain time. When exposed to air, a dioxygen adduct 2*O2 of unknown structure is formed. The fact that the K+ salt 1c exhibits an EPR signal might arise from the partial hydrolysis of the ATCC anion (as all the ATCC salts appear to be quite hygroscopic) to afford 1a (which is reported to have a pKa of 2.025), which in turn forms radical 2 upon irradiation. Comparison of the crystal structures of 2 and the known NaATCC shows that the formation of the radical in the solid state needs only little structural rearrangements.Experimental
All solvents were of p.a. grade or higher and were used as provided. (Et4N)ATCC (1d) and Ag(ATCC) (1e) were prepared according to literature procedures.25b,26a5-Ammonium-1,2,3,4-tetracyanocyclopentadienide zwitterion (1a)
HCl (conc., 0.5 mL) was added dropwise to a solution of 1e (164.5 mg, ca. 0.56 mmol), in MeOH (10 mL) and the mixture was stirred for 1 h at room temperature. The solution was evaporated to a volume of 1 mL and filtered. Addition of CH2Cl2 to a yellow filtrate precipitated 1a as a red powder. Yield: 68 mg (48%).MS: m/z (DEI+): 180.9 [HATCC+]. 13C{1H} NMR (DMSO-d6): δ = 148.04 (C5-NH3), 116.10 (C1-CN), 115.68 (C2-CN), 94.82 (C2), 79.53 (C1). Elemental analysis (C9N5H3·H2O·1.37HCl): calc. C 43.40, N 28.10, H 2.58, found: C 43.32, N 27.81, H 2.59.
Ammonium 5-amino-1,2,3,4-tetracyanocyclopentadienide (1b)
A solution of 1e (158.7 mg, ca. 0.5 mmol) in MeOH (10 mL) was treated with CuCl2 (33.6 mg, 0.25 mmol). The mixture was stirred for 16 h at room temperature and the formed red solid was isolated by filtration. Extraction with MeCN, evaporation to ca. 1 mL and precipitation with CH2Cl2 yielded a red powder. Recrystallization from MeCN/toluene gave – besides a red powder – colourless crystals of NH4(ATCC) in small amounts.A solution of 1e (164.5 mg, ca. 0.56 mmol) in MeOH (10 mL) was treated with NH4Cl (26.7 mg, 0.50 mmol) in one portion. The mixture was stirred for 8 h at room temperature. The solution was evaporated to a volume of 1 mL and filtered. Addition of CH2Cl2 precipitated 1b as a red powder. Yield 15 mg (15%).
MS: m/z (FAB−): 180.4 [ATCC−]. Elemental analysis (C9N6H6·0.19CH4O·0.11H2O): calc. C 53.54, N 40.75, H 3.40; found C 53.79, N 40.78, H 3.41.
Potassium 5-amino-1,2,3,4-tetracyanocyclopentadienide (1c)
KCl (18.6 mg, 0.25 mmol) was added to a solution of 1e (92.2 mg, ca. 0.3 mmol) in MeOH (30 mL). The solution was stirred at RT and under exclusion of light for 16 h. After filtration (to remove AgCl) the solution was evaporated to a volume of 1 mL, filtered again and the product was isolated as a red powder by precipitation with CH2Cl2. Yield: 51 mg, 79%.MS: m/z (MALDI−) = 180.2 [ATCC−]. EA (K(C9N5H2)·0.75CH4O·H2O): calc. C 44.82, N 26.80, H 2.70, found C 44.99, N 26.90, H 2.45.
5-Amino-1,2,3,4-tetracyanocyclopentadienyl radical (2)
CaCl2 (8.6 mg, 0.08 mmol) was added to a solution of 1e (57 mg, ca. 0.19 mmol) in MeOH (10 mL) in one portion and the mixture was stirred for 8 h at room temperature. The solution was evaporated to a volume of 1 mL and the formed precipitate was removed by filtration. Addition of CH2Cl2 to a yellow filtrate yielded a red powder. Recrystallization from MeOH gave red needles of X-ray quality.MS: (DEI+): m/z = 168.9 [100%, ATCC + O-HCN], 180.9 [15%, HATCC+]; 185.0 [5%, ATCC + O2-HCN]; 196.1 [1%, HATCC + O]; 207.0 [5%, ATCC + HCN]; (ESI+): m/z 181.1 [10%, HATCC+]; 195.1 [100%, C9N5OH+]; 211.1 [20%, C9N5O2H+]; MS (ESI−): m/z 179.0 [100%, C9N5H−]; 180.0 [95%, ATCC−]; 181.0 [10%, HATCC−]. ICP-AAS Ag 0.0%, Ca 0.35%. Elemental analysis (C9N5H2·O2·3H2O): calc. C 40.61, N 26.302, H 3.03, found: C 40.76, N 26.31, H 2.99.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Ludwig-Maximilians-University of Munich for financial support.Notes and references
- (a) R. G. Hicks, Org. Biomol. Chem., 2007, 5, 1321 RSC; (b) A. D. Allen and T. T. Tidwell, Chem. Rev., 2001, 101, 1333 CrossRef CAS PubMed.
- K. Ziegler and B. Schnell, Justus Liebigs Ann. Chem., 1925, 445, 266 CrossRef CAS.
- C. Janiak, R. Weimann and F. Görlitz, Organometallics, 1997, 16, 4933 CrossRef CAS.
- S. Lamansky and M. E. Thompson, Chem. Mater., 2002, 14, 109 CrossRef CAS.
- J.-Y. Thepot and C. Lapinte, J. Organomet. Chem., 2002, 656, 146 CrossRef CAS.
- (a) H. Sitzmann and R. Boese, Angew. Chem., Int. Ed. Engl., 1991, 30, 971 CrossRef; (b) H. Sitzmann, H. Bock, R. Boese, T. Dezember, Z. Havlas, W. Kaim, M. Moscherosch and I. Zanathy, J. Am. Chem. Soc., 1993, 115, 12003 CrossRef CAS.
- N. Jux, K. Holczer and Y. Rubin, Angew. Chem., Int. Ed. Engl., 1996, 35, 1986 CrossRef CAS.
- T. Kitagawa, K. Ogawa and K. Komatsu, J. Am. Chem. Soc., 2004, 126, 9930 CrossRef CAS.
- R. D. Chambers, W. K. Gray, J. F. S. Vaughan, S. R. Korn, M. Medebiel-le, A. S. Batsanov, C. W. Lehmann and J. A. K. Howard, J. Chem. Soc., Perkin Trans. 1, 1997, 135 RSC.
- R. J. Less, B. Guan, N. M. Muresan, M. McPartlin, E. Reisner, T. C. Wil-son and D. S. Wright, Dalton Trans., 2012, 41, 5919 RSC.
- P. N. Culshaw, J. C. Walton, L. Hughes and K. U. Ingold, J. Chem. Soc., Perkin Trans. 2, 1993, 879 RSC.
- A. G. Davies, J. P. Goddard, M. B. Hursthouse and N. P. C. Walker, J. Chem. Soc., Dalton Trans., 1986, 1873 RSC.
- T. Chen, F. Graf and H. H. Günthard, Chem. Phys., 1983, 75, 165 CrossRef CAS.
- (a) P. Bachmann, F. Graf and H. H. Günthard, Chem. Phys., 1975, 9, 41 CrossRef CAS; (b) T. J. A. Wolf, O. Schalk, R. Radloff, G. Wu, P. Lang, A. Stolow and A.-N. Unterreiner, Phys. Chem. Chem. Phys., 2013, 15, 6673 RSC.
- F. Graf and H. H. Günthard, Chem. Phys. Lett., 1971, 8, 395 CrossRef CAS.
- (a) N. Corrigan, S. Shanmugam, J. Xu and C. Boyer, Chem. Soc. Rev., 2016, 45, 6165 RSC; (b) J. M. R. Naryaman and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102 RSC.
- A special issue of “Accounts of chemical research” has been devot-ed to this topic: (a) C. Stephenson and T. Yoon, Acc. Chem. Res., 2016, 49, 2059 CrossRef CAS; (b) D. Staveness, I. Bosque and C. R. J. Stephenson, Acc. Chem. Res., 2016, 49, 2295 CrossRef CAS; (c) J.-P. Goddard, C. Ollivier and L. Fensterbank, Acc. Chem. Res., 2016, 49, 1924 CrossRef CAS PubMed.
- T. Koike and M. Akita, Inorg. Chem. Front., 2014, 1, 562 RSC.
- C. B. Larsen and O. S. Wenger, Chem. – Eur. J., 2018, 24, 2039 CrossRef CAS PubMed.
- (a) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075 CrossRef CAS PubMed; (b) E. Speckmeier, T. Fischer and K. Zeitler, J. Am. Chem. Soc., 2018, 140, 15353 CrossRef CAS PubMed.
- (a) Z. Hlouskova and F. Bures, ARKIVOC, 2017,(pt iv), 330 Search PubMed; (b) Y. Zhao, C. Zhang, K. F. Chin, O. Pytela, G. Wei, H. Liu, F. Bures and Z. Jiang, RSC Adv., 2014, 4, 30062 RSC; (c) F. Bures, RSC Adv., 2014, 4, 58826 RSC.
- W. Kaim, Eur. J. Inorg. Chem., 2012, 343 CrossRef CAS.
- S. Benmansour, M. Marchivie, S. Triki and C. J. Gomez-Garcia, Crystals, 2012, 2, 306 CrossRef CAS.
- (a) R. Gompper and T. Geßner, Angew. Chem., Int. Ed. Engl., 1985, 24, 982 CrossRef; (b) E. Aquad, P. Leriche, G. Mabon, A. Gorgues, M. Allain, A. Ri-ou, A. Ellern and V. Khodorkovsky, Tetrahedron, 2003, 59, 5773 CrossRef.
- (a) O. W. Webster, J. Am. Chem. Soc., 1966, 88, 4055 CrossRef CAS; (b) O. W. Webster, US Pat., 3835943, 1974 Search PubMed.
- (a) P. R. Nimax and K. Sünkel, ChemistrySelect, 2018, 3, 3330 CrossRef CAS; (b) K. Sünkel, D. Reimann and P. Nimax, Z. Naturforsch., B: J. Chem. Sci., 2019, 74(1), 109 Search PubMed.
- (a) V. Y. Kukushkin and A. L. Pombeiro, Inorg. Chim. Acta, 2005, 358, 1 CrossRef CAS; (b) L.-L. Zheng, J.-D. Leng, W.-T. Liu, W.-X. Zhang, J.-X. Lu and M.-L. Tong, Eur. J. Inorg. Chem., 2008, 4616 CrossRef CAS; (c) B. Szafranowska and J. Beck, Z. Anorg. Allg. Chem., 2014, 640, 1960 CrossRef CAS.
- (a) J.-M. Lu, S. V. Rosokha and J. K. Kochi, J. Am. Chem. Soc., 2003, 125, 12161 CrossRef PubMed; (b) V. A. Starodub and T. N. Starodub, Russ. Chem. Rev., 2014, 83, 391 CrossRef.
- T. Geßner, PhD thesis, Ludwig-Maximilians University Munich, 1986.
- (a) B. Z. Child, S. Girib, S. Gronert and P. Jena, Chem. – Eur. J., 2014, 20, 4736 CrossRef CAS; (b) Y. Bando, Y. Haketa, T. Sakurai, W. Matsuda, S. Seki, H. Ta-kaya and H. Maeda, Chem. – Eur. J., 2016, 22, 7843 CrossRef CAS PubMed; (c) N. Ilawe, J. Fu, S. Ramanathan, B. M. Wong and J. Wu, J. Phys. Chem. C, 2016, 120, 27757 CrossRef CAS; (d) F.-Q. Zhou, R.-F. Zhao, J.-F. Li, W.-H. Xu, C.-C. Li, L. Luo, J.-L. Li and B. Yin, Phys. Chem. Chem. Phys., 2019, 21, 2804 RSC.
- R. L. Lord, S. E. Wheeler and H. F. Schaefer III, J. Phys. Chem., 2005, 109, 10084 CrossRef CAS PubMed.
- J. Calbo, R. Viruela, E. Orti and J. Arago, Chem. Phys. Chem., 2016, 17, 38 CrossRef PubMed.
- C. M. Cardona, W. Li, A. E. Kaifer, D. Stockdale and G. Bazan, Adv. Mater., 2011, 23, 2367 CrossRef CAS PubMed.
- A. V. Marenich, J. Ho, M. L. Coote, C. J. Cramer and D. G. Truhlar, Phys. Chem. Chem. Phys., 2014, 16, 15068 RSC.
- V. V. Pavlishchuk and A. W. Addison, Inorg. Chim. Acta, 2000, 298, 97 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1909579 and 1909580. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9nj04354c |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2020 |